
Natural organic matter facilitates formation and microbial methylation of mercury selenide nanoparticles†
Qing
Chang‡
,
Zhanhua
Zhang‡
,
Yunyun
Ji
,
Li
Tian
,
Wei
Chen
 and
Tong
Zhang
*
and
Tong
Zhang
*
College of Environmental Science and Engineering, Ministry of Education Key Laboratory of Pollution Processes and Environmental Criteria, Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Nankai University, Tianjin 300350, P.R. China. E-mail: zhangtong@nankai.edu.cn; Fax: +86 22 66229516; Tel: +86 22 66229516
First published on 9th December 2020
Abstract
Due to their extremely low solubility, mercury selenide (HgSe) minerals are long considered as environmental sinks of mercury that pose minimal risks to ecological systems. Thus, selenium amendments have been widely applied in agricultural soils to mitigate mercury contamination and produce Se-rich crops. Here, we demonstrate that co-precipitation of ubiquitous natural organic matter (NOM), divalent mercury and selenium leads to the formation of nanoscale tiemannite (HgSe) particles, which are rapidly converted into bioaccumulative neurotoxin, methylmercury, upon exposure to methylating bacteria. The methylation potential of these tiemannite nanoparticles decreases during aging and yet remains to be significantly greater than that of bulk-HgSe formed in the absence of NOM. Nano-tiemannite formed with fulvic acid appears to be smaller in size and more available for methylation, compared with nano-tiemannite formed with humic acid. This trend may be explained by the different extents of chemical binding, likely through inner-sphere coordination between surface mercury atoms of HgSe and NOM carboxyl moieties. Our findings point out the potential risks to food safety by the application of mercury–selenium antagonism in agricultural practices, and add new perspectives to understanding the occurrence and impact of an emerging group of mercury species, mercury-containing nanominerals, that may substantially contribute to mercury biogeochemistry.
Environmental significanceThis research informs the scientific community that tiemannite (HgSe) minerals, although previously recognized as inert sinks of mercury, are actually potential sources of methylmercury, when present as nanoparticles. An important pathway of the natural occurrence of nanoparticulate tiemannite is co-precipitation of divalent mercury and selenium with natural organic matter (NOM), and different types of NOM lead to different extents of methylmercury production from nanoparticulate tiemannite. This nanoscale process is likely dictated by the NOM–mercury binding that effectively mitigates tiemannite particle growth. Given that selenium amendment in soils is a recommended strategy for remediating mercury contamination and producing nutritious crops, immediate caution is needed for the potential risks of generating methylmercury that directly enters the food chain and endangers human health. |
Introduction
Selenium (Se) is an essential micronutrient for organisms1,2 and often co-exists with soft metals (e.g., mercury, Hg) as metal selenides in natural mineral phases.3,4 Many environmental settings with relatively large mercury abundances, such as mining-impacted areas and volcanic regions, are seleniferous geographic zones.4–6 Yet, Se-deficient conditions are commonly encountered in natural environments due to the uneven distribution of this element in the earth's crust. On the global scale, over 15% of the population from more than 40 countries are susceptible to nutritional Se-deficiency.2,7 As such, soil amendments with Se-enriched fertilizers are becoming routine agricultural practices in these areas to produce Se-rich crops. This is deemed particularly preferable in mercury-containing soils, in which dissolved mercury species in porewater are expected to be sequestered to form highly insoluble mercury selenide (HgSe).8,9 Therefore, the co-occurrence of Se with Hg, due to both natural processes and anthropogenic activities, was initially thought to diminish and prevent the bioavailable fractions of mercury from entering the food web.The concept of mercury–selenium antagonism for remediating mercury contamination is stemmed from the extraordinarily low solubility of the bulk-scale HgSe minerals (Ksp = 10−58)8,10,11 without considering the possibility of forming nanoparticulate HgSe. However, recent studies revealed that nanoscale particles containing Hg and Se were present in Se-amended agricultural soils,8,12,13 and the addition of laboratory-synthesized nano-HgSe to soil samples resulted in prominent accumulation of the bioaccumulative neurotoxin, methylmercury (MeHg).14 To date, it remains unknown how HgSe nanoparticles naturally occur and participate in microbial methylation, which is the predominant pathway of inorganic mercury converted into MeHg in the environment. As a matter of fact, precipitation of inorganic divalent mercury and sulfide in the presence of natural organic matter (NOM) led to the formation of nanoparticulate mercury sulfide (HgS, i.e., metacinnabar) that was available for microbial methylation.15–17 Considering that soil microorganisms can reduce selenate and selenite, i.e. the main components of Se-fertilizers, to selenide,18–21 co-precipitation of mercuric mercury, selenide and NOM may represent as a potential pathway for forming bioavailable mercury species, particularly nano-HgSe, for MeHg production that induces severe ecological and health risks.
This study aims to elucidate the mechanism of microbial MeHg formation from the co-occurrence and interactions of divalent mercury, selenium and the ubiquitous NOM, using fulvic acid and humic acid as model compounds. The presence of NOM facilitated the formation of tiemannite (HgSe) nanoparticle aggregates that were readily available for microbial methylation, whereas bulk-scale tiemannite with minimal methylation potential was formed in the absence of NOM. Particularly, NOM–Hg binding, likely through inner-sphere coordination with NOM carboxyl groups, played a critical role in modulating the size, aggregation state and methylation potential of tiemannite nanoparticles. These findings point out the unintended risks of Se-amendment in Hg-impacted areas and mercury contamination in seleniferous regions due to NOM-facilitated formation of nanoscale precursors for in situ MeHg production.
Materials and methods
Materials
A stock solution of inorganic divalent Hg was prepared by dissolving Hg(NO3)2 powder (Sinopharm, China) in 0.2 mol L−1 HNO3. A stock solution of inorganic divalent Se was prepared by dissolving Na2Se powder (Aladdin, China) in N2-purged nanopure water (>18.2 MΩ cm), which was kept refrigerated and utilized within 2 h of preparation. Suwannee River fulvic acid (SRFA, catalog number: 2S101F) and Suwannee River humic acid (SRHA, catalog number: 3S101H) were used as model compounds of NOM, and were purchased from the International Humic Substances Society (IHSS, Colorado, USA). A total organic carbon analyzer (multi N/C 3100, Analytik Jena AG, Jena, Germany) was used to determine the dissolved organic carbon (DOC) content of SRFA and SRHA.Reactions of Hg, Se and NOM
All experimental manipulations were performed in an anaerobic glove box (Coy Laboratory Products Inc., USA). Nanopure water (>18.2 MΩ cm) was boiled and then cooled down to room temperature in the glove box with an atmosphere of 95% (v/v) nitrogen and 5% (v/v) hydrogen. This deoxygenated water was used to prepare the buffer solution, wherein inorganic divalent mercury, selenium and NOM were reacted. Briefly, 50 μmol L−1 Hg(NO3)2 and 2 mg-C L−1 SRFA or SRHA were added into a solution of 0.1 mol L−1 NaNO3 and 5 mmol L−1 sodium 4-(2-hydroxyethyl)piperazine-1-ethanesulfonate (HEPES, double-filtered to <0.05 μm), prior to the addition of 50 μmol L−1 Na2Se. For assessing the pH effects on particle aggregation, the pH of this buffer solution was adjusted to 5.0, 7.0 and 9.0 using trace-metal grade HNO3 and NaOH, respectively. For the rest of the experiments, the pH of all reaction matrices was buffered at 7.0. The reaction products of NOM–Hg–Se were aged for 1 d, 8 d, and 15 d in the dark prior to use for the methylation bioassays. Bulk-scale HgSe was synthesized by mixing 50 μmol L−1 Hg(NO3)2 and 50 μmol L−1 Na2Se in the above-mentioned HEPES buffer in the absence of NOM. The precipitation product (i.e., bulk HgSe) was aged for 40 d in the dark at room temperature, and the aged suspension was sonicated for 30 min prior to use for the methylation bioassays.Characterization of the reaction products of Hg, Se and NOM
The size, morphology and chemical composition of the reaction products were characterized using transmission electron microscopy (TEM), high-resolution TEM (HR-TEM) and energy dispersive X-ray spectroscopy (EDX, JEM-2800, JEOL, Japan). The geometric diameters were estimated by measuring 100 particles from the TEM images with Nano Measurer software (version 1.2.0). X-ray diffraction (XRD, Ultima IV, Rigaku Inc, Japan) was utilized to assess the crystallographic structures, using a Rigaku diffractometer with Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS, Axis Ultra DLD, Kratos, Britain) was applied to analyze photoelectron binding energies. Zeta potential and hydrodynamic diameters of the reaction products were measured on a Zetasizer (Nano ZS90, Malvern, UK).Fluorescence quenching experiment
Three-dimensional excitation emission matrices (3D-EEM) of the 2 mg-C L−1 SRFA or SRHA samples before and after reacting with 50 μmol L−1 divalent mercury and selenium were analyzed with a fluorescence spectrophotometer (Hitachi F-7100, Tokyo, Japan) to quantitatively characterize the NOM–Hg binding. The fluorescence spectra were collected at 5 nm increments with a scanning speed of 1200 nm min−1. The range of the scanning wavelength was set at excitation wavelengths (Ex) of 200–450 nm and emission wavelengths (Em) of 250–550 nm. An absorbance-based method was used for correcting the inner-filter effects.22Fluorescence titration was performed to further assess the chemical binding of NOM–Hg. SRFA or SRHA (2 mg-C L−1) solutions were titrated with Hg(NO3)2 solution to reach mercury concentrations of 0–2000 μmol L−1. The fluorescence data were fitted to the Stern–Volmer equation:23,24
| F0/F = 1 + Kqτ0[Q] = 1 + KSV[Q] | (1) |
log[(F0 − F)/F] = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KA + nlog[Q] KA + nlog[Q] | (2) |
Culturing of mercury methylating bacteria
Desulfovibrio desulfuricans ND132 (provided by Cynthia C. Gilmour, Smithsonian Environmental Research Center) was utilized as a model mercury methylating bacterium. D. desulfuricans ND132 cells were first grown in a sulfate reduction medium, and then transferred to a sulfate-free fermentation medium before mercury methylation bioassays.27,28 Both media were buffered with 19.2 mmol L−1 3-(N-morpholino)propanesulfonic acid sodium salt (MOPS–Na) at pH 7.0–7.3. The cultures reached the exponential growth phase after 36 h of inoculation, and then the addition of the NOM–Hg–Se reaction products was carried out. All culturing practices were performed in the dark, at room temperature (23–26 °C) in an anaerobic chamber (Coy Laboratory Products Inc., USA).Mercury methylation bioassay
Different HgSe species, including nanoparticulate HgSe aged for 1 d, 8 d or 15 d and bulk-scale HgSe, were added to the exponential-phase cultures of D. desulfuricans ND132 at a total mercury concentration level of 7 nmol L−1. Two types of control groups were incubated under the same conditions as the above-mentioned test groups, which included “abiotic controls” (i.e., uninoculated media spiked with 7 nmol L−1 Hg(NO3)2) and “blank controls” (i.e., active cultures with no mercury addition). Triplicate cultures were periodically sacrificed and subsampled for measurements of cell growth, MeHg and total mercury concentrations. Cell growth was quantified by measuring the bacterial cell density using a flow cytometer (Accuri C6 Plus, BD, Singapore). MeHg and total Hg concentrations were analyzed in accordance with the USEPA method 163029 and method 1631,30 respectively.
Statistical analyses
One-way analysis of variance (ANOVA) was conducted to assess the differences in the results of material characterization and microbial methylation bioassays, using SPSS Statistics software (version 26.0). An independent two-sample t-test was conducted to assess the differences in the results of the fluorescence quenching experiments, using OriginPro software (version 2018). Statistically significant differences were recognized at p < 0.05.Results and discussion
Formation and characteristics of nano-tiemannite
Co-precipitation of mercuric ions and selenide ions occurred in the presence of SRFA or SRHA under anaerobic conditions, which led to the formation of nanoscale spherical particles (Fig. 1). According to the HR-TEM and XRD analysis (Fig. S1 and S2†), the crystalline phase of the NOM–Hg–Se co-precipitation products was tiemannite, and the dominant exposed facet was the tiemannite (111) facet with a crystal lattice spacing of 3.46–3.48 Å. The size and surface area of the tiemannite nanoparticles appeared to depend on the type of NOM present during co-precipitation (Fig. 1, Table S1†). For example, the geometric diameter and surface area of 1 d aged nano-HgSe formed with SRFA were 4.35 ± 0.64 nm and 167 ± 34.6 m2 g−1, respectively, while the particle size increased to 6.90 ± 0.81 nm and the corresponding surface area decreased to 106 ± 37.3 m2 g−1, in the presence of SRHA (Table S1†). Aging under environmentally relevant conditions (i.e., room temperature and neutral pH) from 1 d to 15 d did not significantly alter the morphology, size or surface area of these tiemannite nanoparticles (Fig. 1, Table S1†).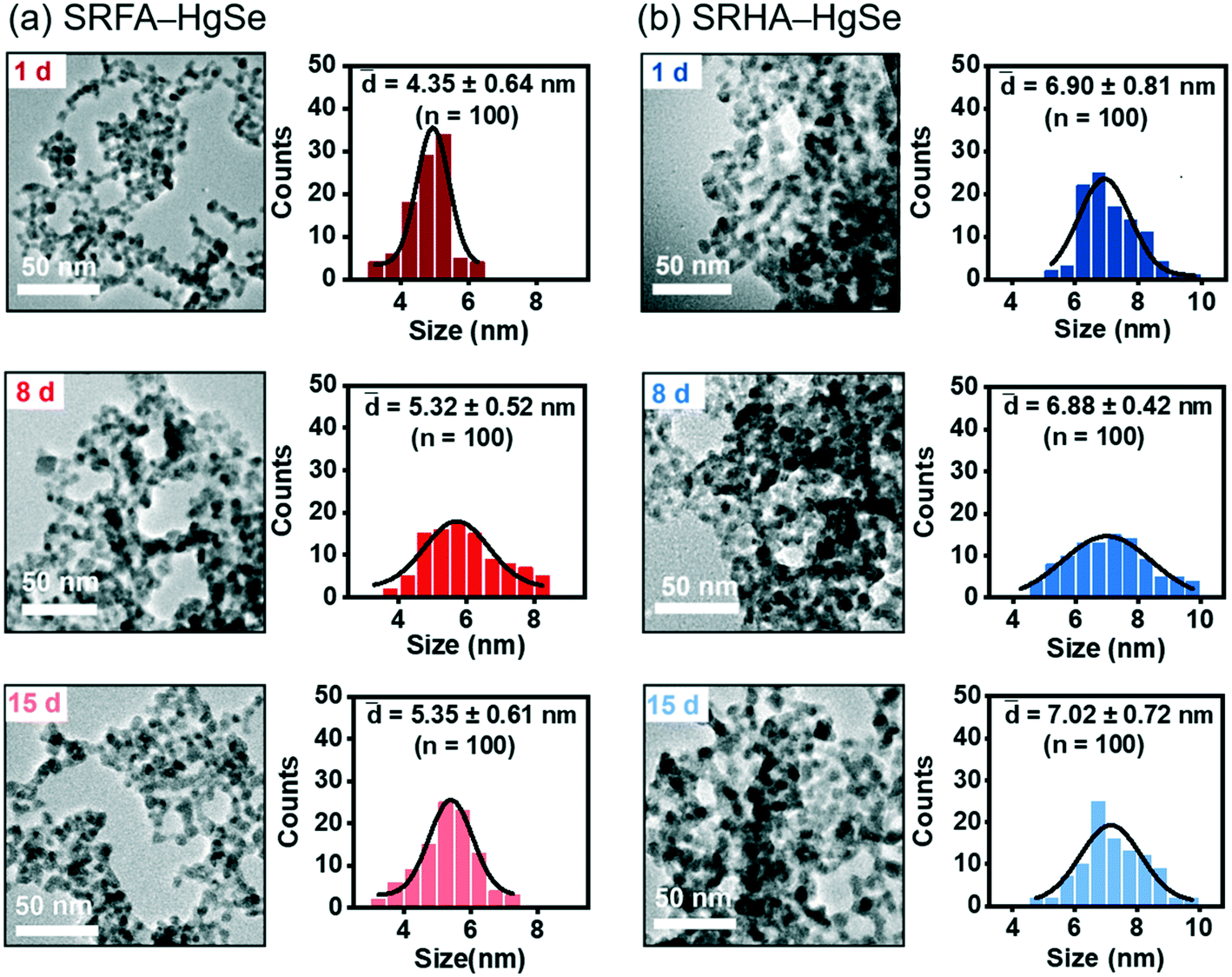 | ||
| Fig. 1 Transmission electron microscopy (TEM) images and particle size distributions of the reaction products of inorganic divalent Hg and Se in the presence of Suwannee River fulvic acid (SRFA) (a) or Suwannee River humic acid (SRHA) (b) aged for 1 d, 8 d and 15 d. | ||
Within the pH range that is commonly encountered in natural aquatic environments (pH 5.0–9.0), the co-precipitation products of NOM–Hg–Se rapidly agglomerated to form particle aggregates within the nanoscale (Fig. 2, Table S1†), whereas bulk-scale tiemannite was formed in the absence of NOM (Fig. S3†). The mitigating effect of NOM on nano-HgSe aggregation was, at least partially, caused by the enhanced particle–particle electrostatic repulsion, as the surface charge of the tiemannite particles became significantly more negative when NOM was present (Fig. S4†). In the absence of NOM, the hydrodynamic diameters of the precipitation products went beyond 1 μm shortly (~30 min) after initiating the reactions between mercuric ions and selenide ions (Fig. 2a–c). In contrast, NOM substantially stabilized the aggregation of nanoparticulate tiemannite (Fig. 2, Table S1†). The aggregates of nanoparticulate tiemannite were relatively stable during aging from 1 d to 15 d, with the hydrodynamic diameters changing from 179 ± 17 nm to 244 ± 20 nm for SRFA–HgSe, and from 346 ± 26 nm to 382 ± 50 nm for SRHA–HgSe (Table S1†). The hydrodynamic diameters measured during the initial 15 min of the NOM–Hg–Se reactions were fitted with respect to the reaction time (R2 > 0.95) in order to estimate the observed aggregation rates of the tiemannite nanoparticles, which appeared to be consistent over the pH range of 5–9 and significantly lower than the aggregation rates of the tiemannite particles formed without NOM (Fig. 2d).
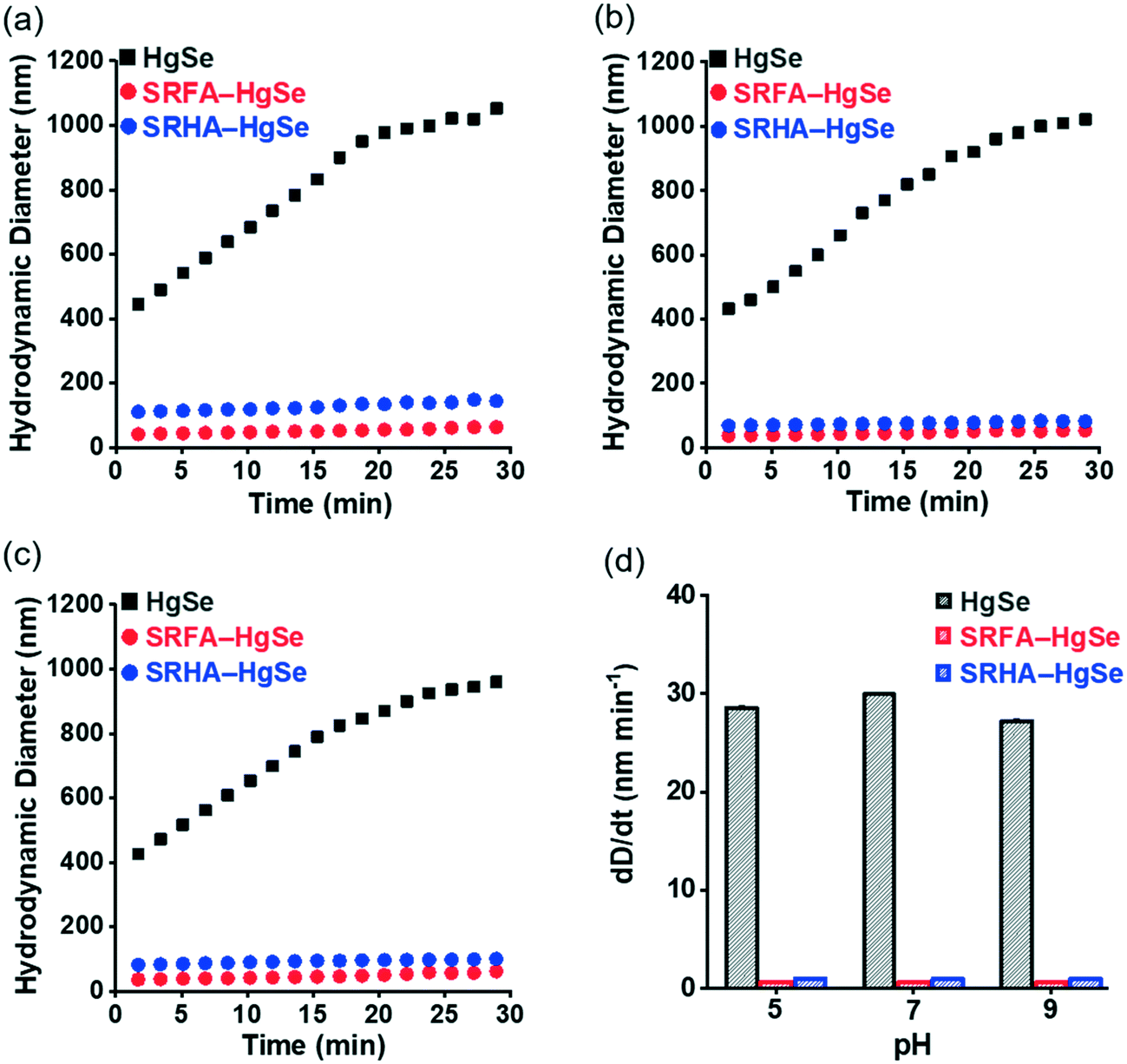 | ||
| Fig. 2 Time-dependent hydrodynamic diameter measurements assessed using dynamic light scattering (a–c) and observed aggregation rates (d) of the reaction products of inorganic divalent Hg and Se in the absence and presence of SRFA or SRHA at pH 5.0 (a), 7.0 (b) and 9.0 (c). Error bars in (d) represent ± 1 standard error of the linear regression of hydrodynamic diameter vs. precipitation time (initial 15 min, data points shown in a–c). | ||
Chemical binding between NOM and nano-tiemannite
SRFA is known to contain a larger density of functional groups, particularly oxygen-containing groups, compared with SRHA.31–33 The majority of these functional groups are prone to deprotonation under neutral pH conditions,32,34 and subsequently become available for metal complexation. Interactions between nanoparticulate tiemannite and NOM were assessed based on the peak shifting in the XPS spectra of the NOM–HgSe nanoparticles, relative to the free NOM molecules and bulk HgSe formed without NOM (Table S2, Fig. S5†). The three peaks in the C 1s spectra at 284.8, 286.5 and 288.5 eV are attributed to the aromatic rings (C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), hydroxyl/epoxy groups (C–O, C–O–C) and carboxyl groups (OC–O) of uncomplexed NOM, respectively.35–38 Upon NOM–HgSe interactions, the binding energy of the carboxyl groups shifted to larger values by 0.4 eV for SRFA (Table S2, Fig. S5a†) and by 0.1 eV for SRHA (Table S2, Fig. S5b†). Meanwhile, both mercury peaks, corresponding to Hg 4f5/2 and Hg 4f7/2, shifted to lower values, with SRFA inducing more prominent shifts than SRHA (Table S2, Fig. S5c†). These results suggest the formation of inner-sphere coordination bonds between the surface mercury atoms of nano-tiemannite and the carboxyl moieties of NOM,39–41 which occurred to a greater degree for SRFA than SRHA.
C), hydroxyl/epoxy groups (C–O, C–O–C) and carboxyl groups (OC–O) of uncomplexed NOM, respectively.35–38 Upon NOM–HgSe interactions, the binding energy of the carboxyl groups shifted to larger values by 0.4 eV for SRFA (Table S2, Fig. S5a†) and by 0.1 eV for SRHA (Table S2, Fig. S5b†). Meanwhile, both mercury peaks, corresponding to Hg 4f5/2 and Hg 4f7/2, shifted to lower values, with SRFA inducing more prominent shifts than SRHA (Table S2, Fig. S5c†). These results suggest the formation of inner-sphere coordination bonds between the surface mercury atoms of nano-tiemannite and the carboxyl moieties of NOM,39–41 which occurred to a greater degree for SRFA than SRHA.
The stronger chemical binding between nano-tiemannite and SRFA vs. SRHA was further demonstrated by the fluorescence quenching experiments (Fig. 3). Two peaks were readily identified from the 3D-EEM spectra of SRFA or SRHA at excitation/emission (Ex/Em) wavelengths of 265/450 nm (peak A) and 335/450 nm (peak B), corresponding to fulvic acid-like and humic acid-like substances, respectively.42,43 The fluorescence intensities of both peaks markedly declined upon reaction with divalent mercury and selenium. The SRFA–HgSe binding decreased the intensities of peak A and peak B by 56.8% and 58.2%, respectively, while the SRHA–HgSe binding decreased the intensities of peak A and peak B by 52.1% and 49.1%, respectively (Fig. 3a). According to the fluorescence titration data, all the fitted bimolecular quenching rate constants (Kq, Fig. 3b and S6†) were one order of magnitude higher than the maximum Kq value for diffusion-controlled quenching processes (2.0 × 1010 L mol−1 s−1),24,25 which substantiated the occurrence of static quenching led by inner-sphere complexation.44–46 Moreover, both Stern–Volmer quenching constants (KSV) and binding constants for static quenching (KA) were consistently larger for SRFA–Hg than for SRHA–Hg (Fig. 3b and S6†). These results corroborate the trends of the XPS data (Table S2, Fig. S5†), and are consistent with previous observations that the fluorescent components of NOM, corresponding to fulvic-like substances, exhibited strong complexation abilities toward soft and transition metals.47,48 More importantly, the different extents of NOM–Hg binding may explain the stronger inhibiting effect of SRFA vs. SRHA on the growth of tiemannite nanoparticles, as the chemical binding of surface modifiers is expected to mitigate the crystal growth of the initial precipitation products via reducing the surface energy.49–51
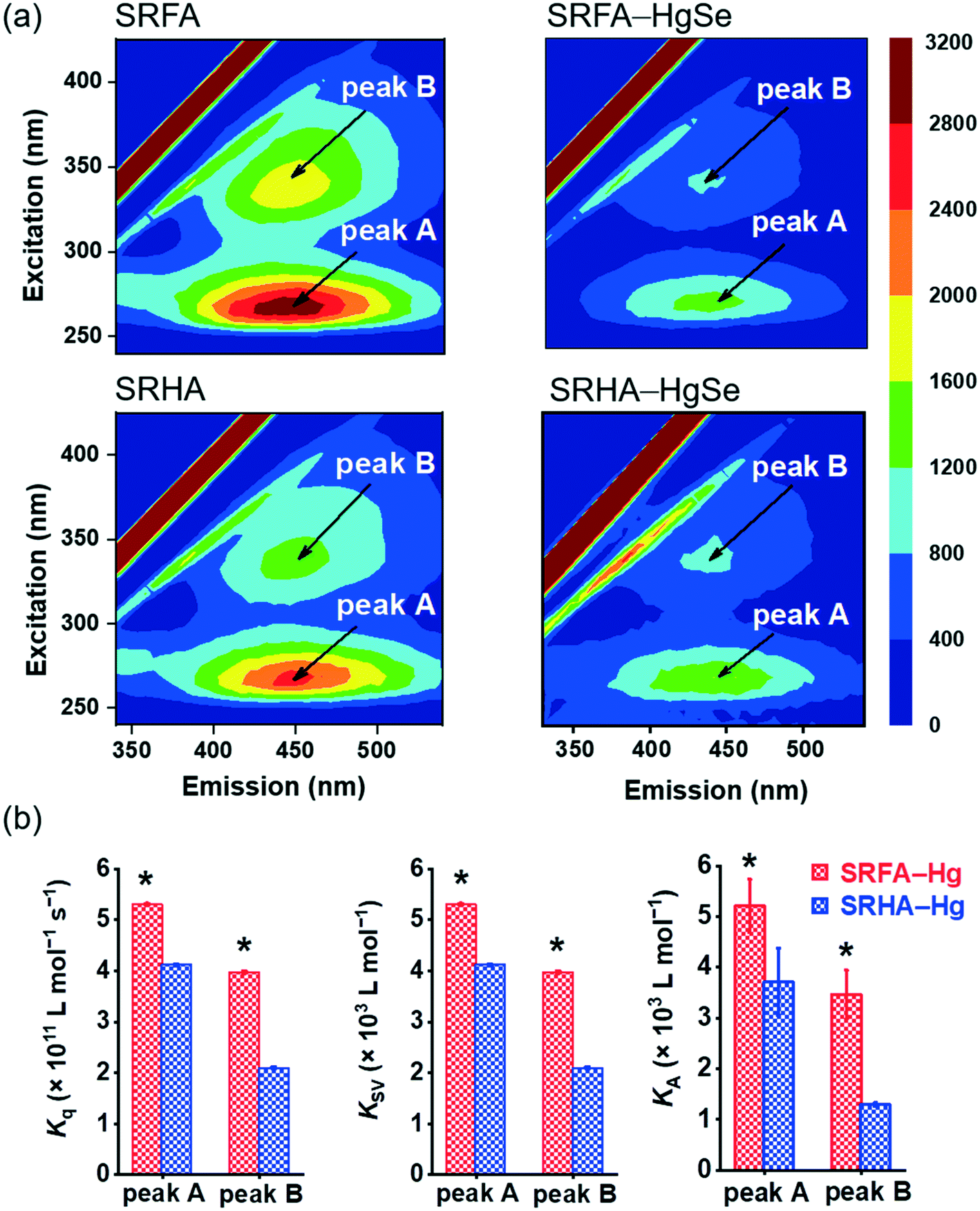 | ||
| Fig. 3 Three-dimensional excitation emission matrix (3D-EEM) fluorescence spectra of SRFA and SRHA before and after reacting with inorganic divalent Hg and Se (a). Bimolecular quenching rate constants (Kq), Stern–Volmer quenching constants (KSV) and binding constants for static quenching (KA) of peak A and peak B of SRFA and SRHA (b). Error bars in (b) represent ± 1 standard error of the linear regression for the extracting values of Kq, KSV and KA. | ||
Microbial methylation of nano-tiemannite
After the exposure of similar concentration levels of total inorganic mercury (Fig. S7†) to the methylating bacterium, D. desulfuricans ND132, nanoparticulate tiemannite exhibited significantly larger production of MeHg, relative to bulk-scale tiemannite, on a per cell basis (Fig. 4). MeHg production from SRFA–HgSe appeared to be consistently higher than that from SRHA–HgSe (Fig. 4). Given that growing evidence has pointed out the importance of cell interfacial reactions, such as ligand exchange, on dictating mercury bioavailability for methylation,52–54 the greater bioavailability of SRFA–HgSe was likely because of the sufficient particle–cell contact facilitated by the larger surface areas of the smaller nanoparticulate tiemannite formed in the presence of SRFA (Fig. 1 and 2, Table S1†).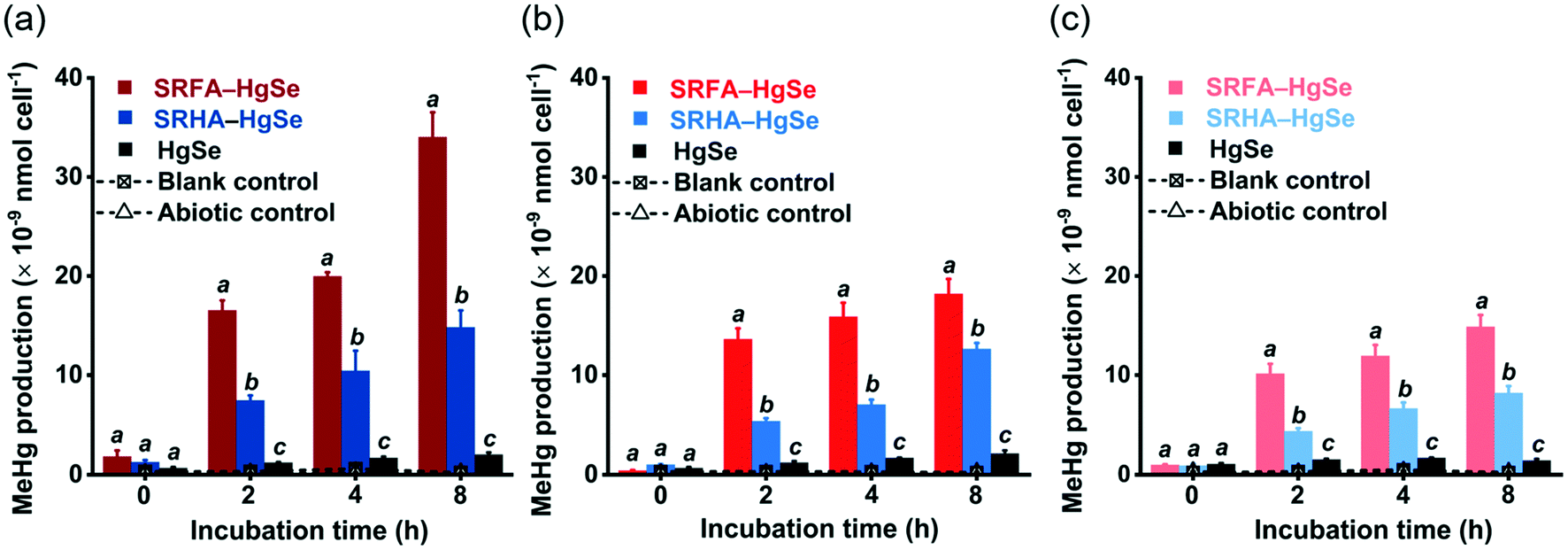 | ||
| Fig. 4 Methylmercury (MeHg) production from the cultures of Desulfovibrio desulfuricans ND132 after exposure to the reaction products of inorganic divalent Hg and Se in the absence and presence of SRFA or SRHA aged for 1 d (a), 8 d (b) and 15 d (c). Blank control represents the active cultures with no mercury addition. Abiotic control represents the uninoculated media spiked with the same amount of Hg(NO3)2. The MeHg production is normalized to the cell numbers of D. desulfuricans ND132. Error bars represent ± 1 standard deviation of the replicate samples (n = 3). | ||
It is also interesting to note that the methylation potential of nano-HgSe decreased with aging time (Fig. 4), yet in a slower pace than that of nanoparticulate metacinnabar (nano-HgS). In the bioassays using D. desulfuricans ND132, aging of nanoparticulate SRHA–HgS from 16 h to 7 d reduced its methylation potential by 78.5%;28 nevertheless, a longer aging period of nanoparticulate SRHA–HgSe (i.e., from 1 d to 15 d) reduced its methylation potential by 51.1% (Fig. 4). Hence, the environmental behavior and impact of nano-HgSe cannot be precisely predicted based on the information obtained from nano-HgS studies, and especially, nano-HgSe may cause more persistent contamination of MeHg that leads to greater ecological and health risks.
Conclusions
Overall, our results suggest that nanoparticulate tiemannite available for microbial methylation is expected to form when NOM, divalent mercury and selenium co-exist in aquatic environments. The methylation potential of these tiemannite nanoparticles decreases during aging and yet remains to be significantly greater than that of bulk-HgSe formed in the absence of NOM. Nano-tiemannite formed with fulvic acid appears to be smaller in size and more available for methylation, compared with nano-tiemannite formed with humic acid. This trend may be explained by the different extents of chemical binding, likely through inner-sphere coordination between surface mercury atoms of HgSe and NOM carboxyl moieties. These findings point to a new group of precursors for in situ MeHg production and will inform risk assessments of Se-amendment in mercury-impacted areas and mercury contamination in seleniferous regions. Agricultural fields, such as paddy soils and saturated soils for growing aquatic plants,4,55,56 may be particularly susceptible to the MeHg risks caused by the co-occurrence of divalent mercury, selenium and NOM. Given the important role of NOM in controlling the formation, characteristics and bioavailability of nano-tiemannite aggregates, caution is needed, primarily on the NOM properties related to mercury binding, in the scenarios of “new” NOM inputs induced by climate change, wildfire and altered landscape utilization (e.g., dissolved black carbon enriched with oxygen-containing functional groups and protein-like compounds with relatively low aromaticity).57–59Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2019YFC1804400 and 2018YFC1800705), the National Natural Science Foundation of China (21976095), and the Ministry of Education of China (T2017002). We thank Cynthia C. Gilmour for supplying D. desulfuricans ND132.References
- L. H. E. Winkel, C. A. Johnson, M. Lenz, T. Grundl, O. X. Leupin, M. Amini and L. Charlet, Environmental selenium research: from microscopic processes to global understanding, Environ. Sci. Technol., 2012, 46, 571–579 CrossRef CAS.
- L. C. Tan, Y. V. Nancharaiah, E. D. van Hullebusch and P. N. L. Lens, Selenium: environmental significance, pollution, and biological treatment technologies, Biotechnol. Adv., 2016, 34, 886–907 CrossRef CAS.
- D. Mercone, J. Thomson, I. W. Croudace and S. R. Troelstra, A coupled natural immobilisation mechanism for mercury and selenium in deep-sea sediments, Geochim. Cosmochim. Acta, 1999, 63, 1481–1488 CrossRef CAS.
- H. Zhang, X. Feng, J. Zhu, A. Sapkota, B. Meng, H. Yao, H. Qin and T. Larssen, Selenium in soil inhibits mercury uptake and translocation in rice (Oryza sativa L.), Environ. Sci. Technol., 2012, 46, 10040–10046 CAS.
- H. Zhang, X. Feng, C. Jiang, Q. Li, Y. Liu, C. Gu, L. Shang, P. Li, Y. Lin and T. Larssen, Understanding the paradox of selenium contamination in mercury mining areas: high soil content and low accumulation in rice, Environ. Pollut., 2014, 188, 27–36 CrossRef CAS.
- T. R. Kulp and L. M. Pratt, Speciation and weathering of selenium in upper cretaceous chalk and shale from South Dakota and Wyoming, USA, Geochim. Cosmochim. Acta, 2004, 68, 3687–3701 CrossRef CAS.
- G. F. Combs, Selenium in global food systems, Br. J. Nutr., 2001, 85, 517–547 CrossRef CAS.
- F. Dang, Z. Li and H. Zhong, Methylmercury and selenium interactions: mechanisms and implications for soil remediation, Crit. Rev. Environ. Sci. Technol., 2019, 49, 1737–1768 CrossRef CAS.
- J. Gerson, D. M. Walters, C. A. Eagles-Smith, E. S. Bernhardt and J. Brandt, Do two wrongs make a right? persistent uncertainties regarding environmental selenium-mercury interactions, Environ. Sci. Technol., 2020, 54, 9228–9234 CrossRef CAS.
- A. Bjösrnberg, L. Håkanson and K. A. Lundbergh, A theory on the mechanisms regulating the bioavailability of mercury in natural waters, Environ. Pollut., 1988, 49, 53–61 CrossRef.
- M. A. K. Khan and F. Wang, Mercury-selenium compounds and their toxicological significance: toward a molecular understanding of the mercury-selenium antagonism, Environ. Toxicol. Chem., 2009, 28, 1567–1577 CrossRef CAS.
- Y. Wang, F. Dang, J. Zhao and H. Zhong, Selenium inhibits sulfate-mediated methylmercury production in rice paddy soil, Environ. Pollut., 2016, 213, 232–239 CrossRef CAS.
- J. Zhao, X. Liang, N. Zhu, L. Wang, Y. Li, Y. Li, L. Zheng, Z. Zhang, Y. Gao and Z. Chai, Immobilization of mercury by nano-elemental selenium and the underlying mechanisms in hydroponic-cultured garlic plant, Environ. Sci.: Nano, 2020, 7, 1115–1125 RSC.
- W. Cai, J. Jin, F. Dang, W. Shi and D. Zhou, Mercury methylation from mercury selenide particles in soils, J. Hazard. Mater., 2020, 400, 123248 CrossRef CAS.
- T. Zhang, K. H. Kucharzyk, B. Kim, M. A. Deshusses and H. Hsu-Kim, Net methylation of mercury in estuarine sediment microcosms amended with dissolved, nanoparticulate, and microparticulate mercuric sulfides, Environ. Sci. Technol., 2014, 48, 9133–9141 CrossRef CAS.
- A. M. Graham, K. T. Cameron-Burr, H. A. Hajic, C. Lee, D. Msekela and C. C. Gilmour, Sulfurization of dissolved organic matter increases Hg–sulfide–dissolved organic matter bioavailability to a Hg-methylating bacterium, Environ. Sci. Technol., 2017, 51, 9080–9088 CrossRef CAS.
- Z. Zhang, R. Si, J. Lv, Y. Ji, W. Chen, W. Guan, Y. Cui and T. Zhang, Effects of extracellular polymeric substances on the formation and methylation of mercury sulfide nanoparticles, Environ. Sci. Technol., 2020, 54, 8061–8071 CrossRef CAS.
- J. P. Zehr and R. S. Oremland, Reduction of selenate to selenide by sulfate-respiring bacteria: experiments with cell suspensions and estuarine sediments, Appl. Environ. Microbiol., 1987, 53, 1365–1369 CrossRef CAS.
- D. C. Nelson, W. H. Casey, J. D. Sison, E. E. Mack, A. Ahmad and J. S. Pollack, Selenium uptake by sulfur-accumulating bacteria, Geochim. Cosmochim. Acta, 1996, 60, 3531–3539 CrossRef CAS.
- M. J. Herbel, J. S. Blum, R. S. Oremland and S. E. Borglin, Reduction of elemental selenium to selenide: experiments with anoxic sediments and bacteria that respire Se-oxyanions, Geomicrobiol. J., 2003, 20, 587–602 CrossRef CAS.
- C. I. Pearce, R. A. D. Pattrick, N. Law, J. M. Charnock, V. S. Coker, J. W. Fellowes, R. S. Oremland and J. R. Lloyd, Investigating different mechanisms for biogenic selenite transformations: Geobacter sulfurreducens, Shewanella oneidensis and Veillonella atypica, Environ. Technol., 2009, 30, 1313–1326 CrossRef CAS.
- D. N. Kothawala, K. R. Murphy, C. A. Stedmon, G. A. Weyhenmeyer and L. J. Tranvik, Inner filter correction of dissolved organic matter fluorescence, Limnol. Oceanogr.: Methods, 2013, 11, 613–630 Search PubMed.
- H. Boaz and G. Rollefson, The quenching of fluorescence. Deviations from the Stern-Volmer laws, J. Am. Chem. Soc., 1950, 72, 3435–3443 CrossRef CAS.
- W. Shou, F. Kang and J. Lu, Nature and value of freely dissolved EPS ecosystem services: insight into molecular coupling mechanisms for regulating metal toxicity, Environ. Sci. Technol., 2018, 52, 457–466 CrossRef CAS.
- X. Wang, W. Song, H. Qian, D. Zhang, X. Pan and G. M. Gadd, Stabilizing interaction of exopolymers with nano-Se and impact on mercury immobilization in soil and groundwater, Environ. Sci.: Nano, 2018, 5, 456–466 RSC.
- T. L. Hill, Cooperativity theory in biochemistry: steady-state and equilibrium systems, Springer-Verlag, New York, 1985 Search PubMed.
- C. C. Gilmour, D. A. Elias, A. M. Kucken, S. D. Brown, A. V. Palumbo, C. W. Schadt and J. D. Wall, Sulfate-reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation, Appl. Environ. Microbiol., 2011, 77, 3938–3951 CrossRef CAS.
- T. Zhang, B. Kim, C. Levard, B. C. Reinsch, G. V. Lowry, M. A. Deshusses and H. Hsu-Kim, Methylation of mercury by bacteria exposed to dissolved, nanoparticulate, and microparticulate mercuric sulfides, Environ. Sci. Technol., 2012, 46, 6950–6958 CrossRef CAS.
- Method 1630: Methyl Mercury in water by Distillation, Aqueous Ethylation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectroscopy, U.S, Environmental Protection Agency, Washington, DC, 2001.
- Method 1631, Revision D: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectroscopy, U.S, Environmental Protection Agency, Washington, DC, 2001.
- M. Ravichandran, G. R. Aiken, M. M. Reddy and J. N. Ryan, Enhanced dissolution of cinnabar (mercuric sulfide) by dissolved organic matter isolated from the Florida Everglades, Environ. Sci. Technol., 1998, 32, 3305–3311 CrossRef CAS.
- J. D. Ritchie and E. M. Perdue, Proton-binding study of standard and reference fulvic acids, humic acids, and natural organic matter, Geochim. Cosmochim. Acta, 2003, 67, 85–96 CrossRef CAS.
- J. S. Waples, K. L. Nagy, G. R. Aiken and J. N. Ryan, Dissolution of cinnabar (HgS) in the presence of natural organic matter, Geochim. Cosmochim. Acta, 2005, 69, 1575–1588 CrossRef CAS.
- D. S. Gamble, Titration curves of fulvic acid: the analytical chemistry of a weak acid polyelectrolyte, Can. J. Chem., 1970, 48, 2662–2669 CrossRef CAS.
- Y. Wang, K. Yang and D. Lin, Nanoparticulate zero valent iron interaction with dissolved organic matter impacts iron transformation and organic carbon stability, Environ. Sci.: Nano, 2020, 6, 1818–1830 RSC.
- P. L. Desbene, L. Silly, J. P. Morizur and M. Delamar, XPS analysis of humic and fuvic acid, Anal. Lett., 1986, 19, 2131–2140 CrossRef CAS.
- J. R. Araujo, B. S. Archanjo, K. R. de Souza, W. Kwapinski, N. P. S. Falcão, E. H. Novotny and C. A. Achete, Selective extraction of humic acids from an anthropogenic Amazonian dark earth and from a chemically oxidized charcoal, Biol. Fertil. Soils, 2014, 50, 1223–1232 CrossRef CAS.
- L. Wei, T. Hong, K. Cui, T. Chen, Y. Zhou, Y. Zhao, Y. Yin, J. Wang and Q. Zhang, Probing the effect of humic acid on the nucleation and growth kinetics of struvite by constant composition technique, Chem. Eng. J., 2019, 378, 122130 CrossRef CAS.
- M. Haitzer, G. R. Aiken and J. N. Ryan, Binding of mercury(II) to aquatic humic substances: influence of pH and source of humic substances, Environ. Sci. Technol., 2003, 37, 2436–2441 CrossRef CAS.
- T. Jiang, U. Skyllberg, S. Wei, D. Wang, S. Lu, Z. Jiang and D. C. Flanagan, Modeling of the structure-specific kinetics of abiotic, dark reduction of Hg(II) complexed by O/N and S functional groups in humic acids while accounting for time-dependent structural rearrangement, Geochim. Cosmochim. Acta, 2015, 154, 151–167 CrossRef CAS.
- X. Liang, X. Lu, J. Zhao, L. Liang, E. Y. Zeng and B. Gu, Stepwise reduction approach reveals mercury competitive binding and exchange reactions within natural organic matter and mixed organic ligands, Environ. Sci. Technol., 2019, 53, 10685–10694 CrossRef CAS.
- W. Chen, P. Westerhoff, J. A. Leenheer and K. Booksh, Fluorescence excitation−emission matrix regional integration to quantify spectra for dissolved organic matter, Environ. Sci. Technol., 2003, 37, 5701–5710 CrossRef CAS.
- W. Chen, C. Qian, K. Zhou and H. Yu, Molecular spectroscopic characterization of membrane fouling: a critical review, Chem, 2018, 4, 1492–1509 CAS.
- J. R. Lackowicz, Principles of fluorescence spectroscopy, Plenum Press, New York, 1983 Search PubMed.
- X. Lu and R. Jaffe, Interaction between Hg(II) and natural dissolved organic matter: a fluorescence spectroscopy based study, Water Res., 2001, 35, 1793–1803 CrossRef CAS.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, Investigation of the interaction between flavonoids and human serum albumin, J. Mol. Struct., 2004, 703, 37–45 CrossRef CAS.
- J. Wu, H. Zhang, P. He and L. Shao, Insight into the heavy metal binding potential of dissolved organic matter in MSW leachate using EEM quenching combined with PARAFAC analysis, Water Res., 2011, 45, 1711–1719 CrossRef CAS.
- Z. Shen, Z. Zhang, T. Li, Q. Yao, T. Zhang and W. Chen, Facet-dependent adsorption and fractionation of natural organic matter on crystalline metal oxide nanoparticles, Environ. Sci. Technol., 2020, 54, 8622–8631 CrossRef CAS.
- Y. Jun, M. F. Casula, J. H. Sim, S. Y. Kim, J. Cheon and A. P. Alivisatos, Surfactant-assisted elimination of a high energy facet as a means of controlling the shapes of TiO2 nanocrystals, J. Am. Chem. Soc., 2003, 125, 15981–15985 CrossRef CAS.
- J. H. Strehlau, M. S. Stemig, R. L. Penn and W. A. Arnold, Facet-dependent oxidative goethite growth as a function of aqueous solution conditions, Environ. Sci. Technol., 2016, 50, 10406–10412 CrossRef CAS.
- S. Guo, M. D. Ward and J. A. Wesson, Direct visualization of calcium oxalate monohydrate crystallization and dissolution with atomic force microscopy and the role of polymeric additives, Langmuir, 2002, 18, 4284–4291 CrossRef CAS.
- J. K. Schaefer, A. Szczuka and F. M. M. Morel, Effect of divalent metals on Hg(II) uptake and methylation by bacteria, Environ. Sci. Technol., 2014, 48, 3007–3013 CrossRef CAS.
- Y. Liu, X. Lu, L. Zhao, J. An, J. He, E. M. Pierce, A. Johs and B. Gu, Effects of cellular sorption on mercury bioavailability and methylmercury production by Desulfovibrio desulfuricans ND132, Environ. Sci. Technol., 2016, 50, 13335–13341 CrossRef CAS.
- X. Lu, A. Johs, L. Zhao, L. Wang, E. M. Pierce and B. Gu, Nanomolar copper enhances mercury methylation by Desulfovibrio desulfuricans ND132, Environ. Sci. Technol. Lett., 2018, 5, 372–376 CrossRef CAS.
- Y. Wang, F. Dang, H. Zhong, Z. Wei and P. Li, Effects of sulfate and selenite on mercury methylation in a mercury-contaminated rice paddy soil under anoxic conditions, Environ. Sci. Pollut. Res., 2016, 23, 4602–4608 CrossRef CAS.
- X. Guo, Q. Ji, M. Rizwan, H. Li, D. Li and G. Chen, Effects of biochar and foliar application of selenium on the uptake and subcellular distribution of chromium in Ipomoea aquatica in chromium-polluted soils, Ecotoxicol. Environ. Saf., 2020, 206, 111184 CrossRef CAS.
- E. Hood, J. Fellman, R. G. M. Spencer, P. J. Hernes, R. Edwards, D. D'Amore and D. Scott, Glaciers as a source of ancient and labile organic matter to the marine environment, Nature, 2009, 462, 1044–1048 CrossRef CAS.
- C. Santin, S. H. Doer, C. M. Preston and G. Gonzalez-Rodriguez, Pyrogenic organic matter production from wildfires: a missing sink in the global carbon cycle, Glob. Change Biol., 2015, 21, 1621–1633 CrossRef.
- C. J. Williams, P. C. Frost, A. M. Morales-Williams, J. H. Larson, W. B. Richardson, A. S. Chiandet and M. A. Xenopoulos, Human activities cause distinct dissolved organic matter composition across freshwater ecosystems, Glob. Change Biol., 2016, 22, 613–626 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0en00992j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |