
Tailoring the magnetic ordering of the Cr4O5/Fe(001) surface via a controlled adsorption of C60 organic molecules
Federico
Orlando
 a,
Guido
Fratesi
b,
Giovanni
Onida
b and
Simona
Achilli
*b
a,
Guido
Fratesi
b,
Giovanni
Onida
b and
Simona
Achilli
*b
aDipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, Via Celoria 16, I-20133 Milano, Italy
bETSF and Dipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, Via Celoria 16, I-20133 Milano, Italy. E-mail: simona.achilli@unimi.it
First published on 5th January 2021
Abstract
We analyse the spinterface formed by a C60 molecular layer on a Fe(001) surface covered by a two-dimensional Cr4O5 layer. We consider different geometries, by combining the high symmetry adsorption sites of the surface with three possible orientations of the molecules in a fully relaxed Density Functional Theory calculation. We show that the local hybridization between the electronic states of the Cr4O5 layer and those of the organic molecules is able to modify the magnetic coupling of the Cr atoms. Both the intra-layer and the inter-layer magnetic interaction is indeed driven by O atoms of the two-dimensional oxide. We demonstrate that the C60 adsorption on the energetically most stable site turns the ferromagnetic intra-layer coupling into an antiferromagnetic one, and that antiferromagnetic to ferromagnetic switching and spin patterning of the substrate could be possible by adsorption on other sites.
1 Introduction
The advancements in the field of spintronics are intimately related to the capability to control the magnetic properties of surfaces and interfaces. The magnetic switching of magnetic materials is usually controlled via externally applied magnetic field or by electric-field driven means,1 such as spin torque methods, that require large currents and complex setup and guarantee low efficiency in the process.2 On the other hand, magnetic behaviour at surfaces can be induced or modified through the adsorption of foreign species and molecules and by the growth of magnetic thin films.3–7 In past years it has been established that spin selective electron transfer through chiral molecules is able to magnetize the underlying substrate and induce a magnetic switching.8In recent times, organic semiconductors (OS) have proven intriguing possibilities in the field of spintronics. Devices realized on the basis of the so-called spinterfaces,9,10i.e., interfaces between a magnetic layer (ML) and an OS, show indeed enhanced spin lifetimes and conducting paths with respect to their inorganic counterparts.
It is known that the spin-transport process in such devices can be influenced by new electronic states arising at the interface, as a result of the hybridization between the d-band of the ML and the molecular orbitals of the OS.11 Magnetic character may be induced on non-magnetic molecules, or ferro-/antiferromagnetic molecule–substrate coupling could occur for magnetic ones.12–14 On the other hand, the OS may itself influence the magnetism of the surface to which it is coupled15,16 or induce a magnetic character into a surface that would be otherwise non-magnetic.17 Accordingly, achieving information and control over the electronic and magnetic properties of the interface, as well as over the occurring chemical interactions, may enable to better tailor such systems – via a suitable choice of the materials involved – in view of practical applications.
In recent works12,18,19 some of us focused on the C60/Fe(001) interface, both on the experimental and theoretical viewpoint. We demonstrated that the induced magnetic properties on the molecules depend on the spin polarization of the substrate states interacting with the molecule itself and by their decay toward vacuum. In particular we showed that the insertion of a thin two-dimensional Cr4O5 film at the spinterface is able to enhance the spin-injection process, inducing a larger spin polarization of the C60 molecules with respect to the clean substrate case.
Here we analyse the system in a different perspective, i.e., focusing on the capability of the organic layer to modify the magnetic properties of the Cr4O5/Fe(001) surface. The Cr atoms in an isolated Cr4O5 layer would naturally arrange with antiferromagnetic (AFM) ordering. The interaction with the Fe substrate turns the AFM intra-layer ordering into a ferromagnetic (FM) one18 leading to a magnetization direction of Cr atoms opposite to that of Fe ones (inter-layer AFM coupling). The magnetic ordering at the surface is intimately related to the local symmetry of the electronic states involved in the magnetic interaction. It is reasonable to expect that, by controlling the local symmetry of the spinterface states – for example by tuning the local substrate–molecule interaction – the magnetic ordering of the Cr atoms in the oxide layer could be modified. We verified that the induced magnetic properties on the molecule are mildly dependent on the adsorption geometry. On the other hand we cannot assume that the same holds for the magnetic properties of the oxide substrate layer.
Here we explore the possibility of inducing a rearrangement of the surface magnetic ordering by tailoring the C60 adsorption on Cr4O5/Fe(001). We base our study on ab initio calculations of a relaxed overlayer of C60 molecules in different high symmetry adsorption geometries.
We show that the adsorption of a C60 layer can indeed restore the AFM ordering in the underlying Cr4O5. Moreover, by moving the molecules onto different adsorption sites it is possible to switch the Cr4O5 ordering from AFM to FM, or to induce a nontrivial magnetic patterning involving the Cr atoms in the oxide layer. The possibility to control the adsorption geometry through experimental techniques is not a remote scenario: recent works demonstrate indeed that the local adsorption configuration of organic molecules can be controlled, e.g., by means of a STM tip.20–22
The total energy calculations performed in this work allow us to identify the most stable configurations that could lead to the desired magnetic pattern. The study of the possible adsorption configurations and the associated diffusion barriers can shed light also on the nucleation mechanism, as recently done for the clean Fe(100) surface, where the competitive adsorbate–substrate and adsorbate–adsorbate interactions give rise to a mixed layered growth.23
2 Results and discussion
The system under examination offers several possible adsorption configurations, depending on the adsorption site and the relative orientation between the C60 and the surface. We explore here nine high symmetry cases, by combining three possible adsorption sites – namely a surface vacancy, a Cr atom and an O atom – with three possible orientations of the fullerenes, facing towards the surface with a hexagonal ring, a pentagonal one, and a C–C covalent bond, respectively.Upon relaxation the system appears as reported in Fig. 1. Major details regarding the surface structure can be found in our previous studies.18,24 The configurations with adsorbed C60 reported in the two upper rows of Fig. 1 (adsorption via a hexagon or a pentagon) show a partial roto-translation of the molecules on the surface, implying that the hexagon/pentagon is not perfectly centred on the chosen adsorption site. The leading force influencing the fullerene roto-translation seems to be the interaction with O atoms: indeed, with the exception of the adsorption on O, the molecules shift in order to get two C atoms facing to the surface close to two O atoms of the substrate. Differently, when C60 faces the surface with a C–C bond the starting adsorption site remains almost unaltered upon relaxation.
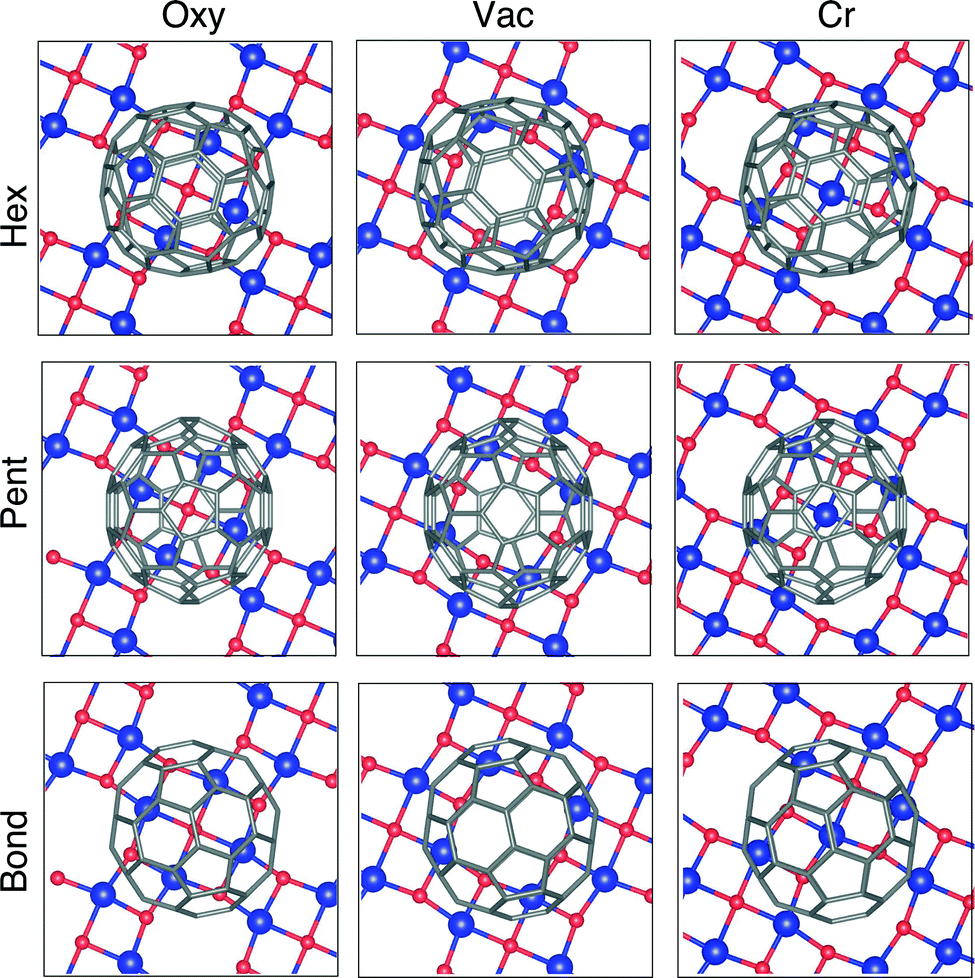 | ||
| Fig. 1 Top view of the relaxed geometries of the explored configurations, depending on adsorption site and relative orientation between C60 and Cr4O5. The colour code is as follows: Cr – blue, O – red and C – grey. The Fe substrate layers are not shown. | ||
The energetically most stable configuration turns out to be the central one in Fig. 1, where a vacancy acts as adsorption site and a pentagon faces to the surface (shortcut to Pent/Vac hereafter). The calculated adsorption energy of a C60 molecule for this configuration is −2.99 eV per unit cell. We take this one as a reference, and report in Table 1 the energy differences, ΔEads, of the other fully relaxed configurations.
| ΔEads [eV] | ΔErlx [eV] | d [Å] | Δq [10−2 e− per atom] | ||||
|---|---|---|---|---|---|---|---|
| Fe | Cr | O | C60 | ||||
| Pent/Vac | 0.00 | −3.40 | 2.89 | −1.24 | −1.83 | −9.01 | 2.92 |
| Pent/Oxy | 0.26 | −2.67 | 2.74 | −1.02 | −5.96 | −8.22 | 3.22 |
| Hex/Oxy | 0.41 | −2.97 | 2.67 | −1.17 | −6.38 | −9.02 | 3.47 |
| Bond/Cr | 0.61 | −0.97 | 2.64 | −1.28 | −2.56 | −8.99 | 3.02 |
| Hex/Cr | 0.72 | −2.94 | 3.15 | −1.09 | −3.31 | −7.73 | 2.64 |
| Hex/Vac | 1.12 | −1.60 | 2.97 | −1.25 | −1.96 | −9.17 | 2.94 |
| Pent/Cr | 1.59 | −3.22 | 3.06 | −0.79 | −5.18 | −6.99 | 2.75 |
| Bond/Vac | 1.95 | −0.32 | 2.78 | −1.12 | −1.90 | −8.83 | 2.83 |
| Bond/Oxy | 4.65 | −1.23 | 2.79 | −1.17 | −3.55 | −8.44 | 3.05 |
The energy gain related to the roto-translation of the molecules is reported in Table 1 (third column) as the energy difference ΔErlx between the fully relaxed systems and the same adsorption configurations in which the molecules were left free to relax only along the direction perpendicular to the surface.19 Notably, the relaxation of all the molecular degrees of freedom modifies the energetic order of the different configurations turning the Pent/Vac in the most stable equilibrium geometry, at odds with what reported in previous studies.12,19 Indeed, the Pent/Vac case displays the largest energy gain related to roto-translation, while the Bond/Vac and the Bond/Cr configurations are mildly modified by the full relaxation.
Among the relaxed systems we find no evident trend in the adsorption energy defining a preferential adsorption site or molecular orientation.
The equilibrium distance of the fullerenes from the surface, evaluated as the difference between the average ẑ coordinate of the C atoms in the pentagon/hexagon/bond and of the Cr layer, ranges between 2.67 and 3.15 Å. It is worth noting that the O atoms lie 0.25 Å further out from the Cr plane,24 resulting in a smaller distance between the molecule and the O atom acting as adsorption site. This confirms the tendency of the molecule to interact with oxygen. The most stable configuration is characterized by the intermediate distance of 2.89 Å; in this case, the average distance between the C atoms of the pentagon and the O atoms nearest to the vacancy amounts to 2.67 Å.
In Table 1 we also report the charge transfer induced by the C60 adsorption, averaged for each atomic species. In all the explored configurations there is a partial electron transfer from the surface to the molecule, whose entity is obviously related to the adsorption distance. The depletion of charge on the O atoms is larger than in the Cr atoms while the contribution from the underlying Fe substrate is smaller. The data reported in Table 1 are only indicative because the average procedure over the atoms in the unit cell is affected by a relatively large percent variation (up to 30%), due to the different behaviour of the atoms near the adsorption site with respect to those lying far away. For this reason we also report in Fig. 2 the charge transfer of the O and Cr atoms averaged over the nearest neighbours of the molecule (i.e., in the adsorption site, when occupied by an atom, and for the four atoms around it) together with the average charge transfer of the C atoms facing to the surface (five, six and two in the Hex, Pent and Bond configurations, respectively). In this case the percent variation of the reported values is lower than 3%. A detailed discussion of the results reported in Fig. 2 will be given below in the text.
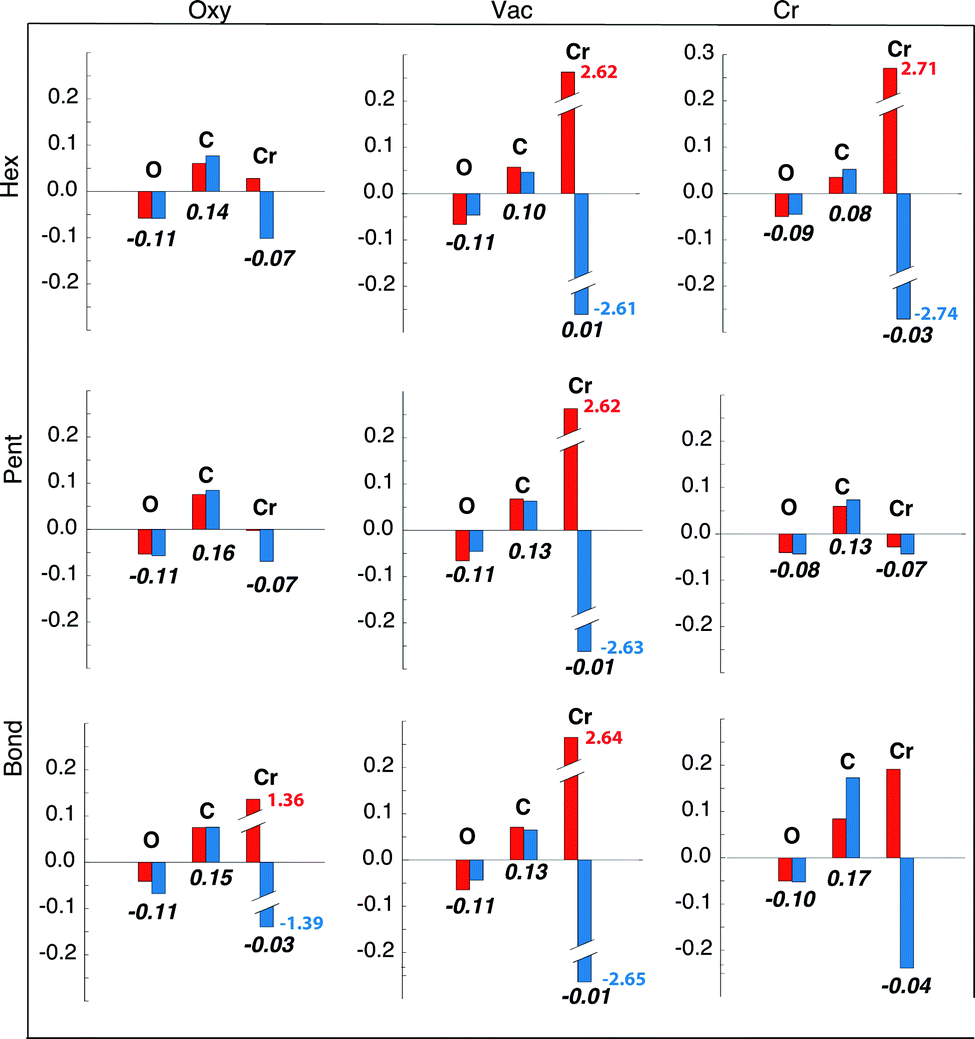 | ||
| Fig. 2 Spin resolved (red, majority; blue, minority) charge transfer averaged over the O and Cr atoms at and around the adsorption site (see the text for details) together with the average value on the C atoms facing to the surface. The total charge transfer (spin up + spin down) averaged over the same atoms is reported in bold below the block histograms. For the systems that display AFM patterns (except Bond/Cr) the spin dependent charge transfer of Cr is out of scale (see the text) and the actual value is reported in red and blue for spin up and spin down, respectively. | ||
The analysed configurations display different magnetic properties, in particular for what concerns the magnetic alignment of the surface Cr atoms, as can be observed in the spin densities reported in Fig. 3. Here we cut the three-dimensional spin distribution onto the plane passing through the Cr atoms in the Cr4O5; the position of Cr and O atoms, and the vacancies in the unit cell are indicated in the central panel of Fig. 3. They can be identified also on the basis of their different spin density: very large and flower-shaped on Cr atoms, small on O atoms and zero (white) in correspondence of the vacancies.
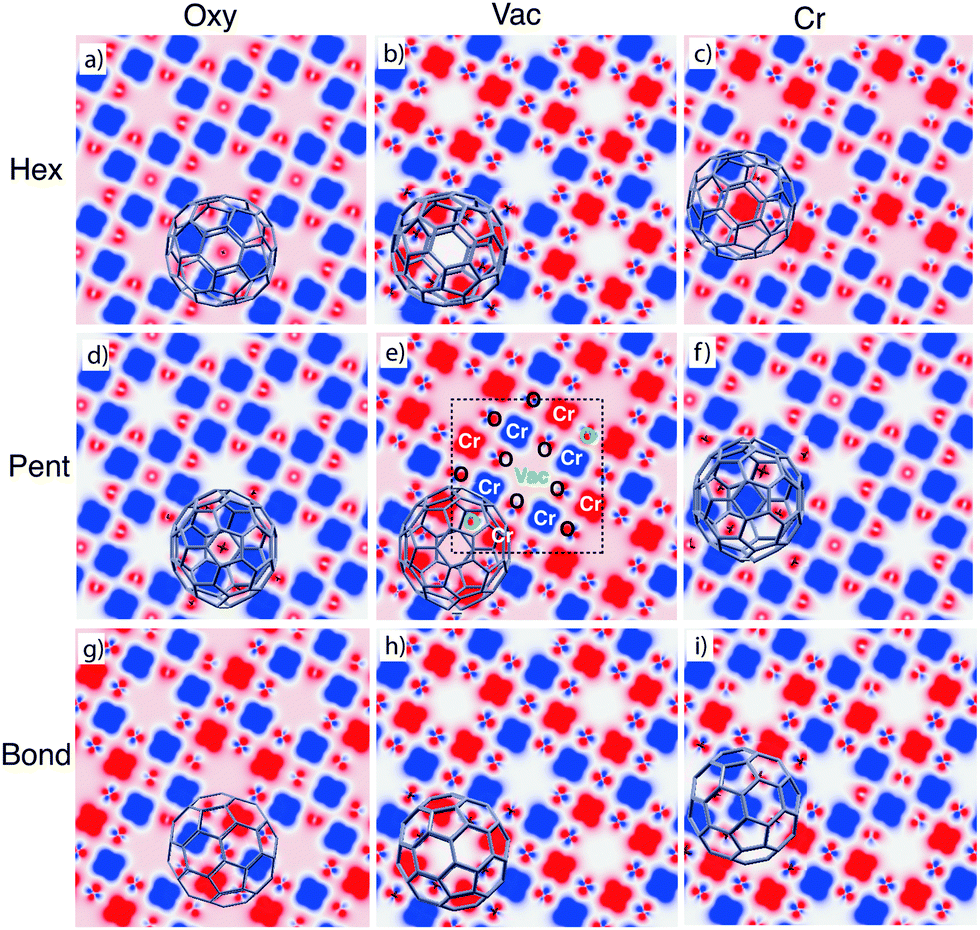 | ||
| Fig. 3 Spin polarization ρup–ρdown for each pattern (top view). A cut on the surface plane passing through the Cr atoms is reported with an isosurface value of 0.02 Å−3. Red and blue areas correspond to positive and negative values of spin polarization, respectively. | ||
We find that the adsorption of C60 in the most stable Pent/Vac configuration induces a recovery of the AFM ordering of the nearest neighbour Cr atoms (central panel in Fig. 3), which would be the preferred magnetic state in absence of the underlying Fe substrate. Indeed some of us have already shown12 that the interaction between the Cr4O5 overlayer and the Fe substrate destabilizes the AFM coupling in the oxide layer, leading to an intra-layer FM alignment of the Cr atoms, that are in turn AFM coupled to the Fe substrate.
The same AFM pattern, typical of the isolated oxide layer, occurs for the other two configurations featuring a vacancy as adsorption site (Hex/Vac and Bond/Vac, ordered by increasing energy). In three configurations (Pent/Oxy, Hex/Oxy and Pent/Cr) the final pattern is instead FM, i.e., with the magnetic moment of all the Cr atoms parallel to each other. For the remaining cases (Hex/Cr, Bond/Oxy and Bond/Cr), different kinds of magnetic patterning appear on the surface, characterized by the spinflip of a fraction of the Cr atoms in the cell (2/8 in Hex/Cr and 3/8 in Bond/Oxy and Bond/Cr).
We report in Table 2 the magnetic moments calculated for the different species. In particular we report the average values for the outermost Fe substrate layer, for the Cr and O atoms in the oxide layer and for the whole C60 molecule. In the non-FM cases, we also give the average values separately for the positively and negatively polarized Cr atoms in the cell (reported in parentheses). The percent variation of the magnetic moment of Cr atoms is lower than 1% in the perfectly AFM configurations, about 2% in FM configurations and smaller than 5% in the partial AFM patterning.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 only in the AFM configurations)
:1 only in the AFM configurations)
| μ [μB per atom] | μ [10−2μB per atom] | |||||
|---|---|---|---|---|---|---|
| Fe | Cr | O | C60 | ONN | CNN | |
| Pent/Vac | 2.25 | −0.31 (2.26/−2.88) | 1.5 | 0.1 | 0.9 | 0.5 |
| Pent/Oxy | 2.32 | −2.96 | 2.6 | −0.6 | 0.9 | −0.9 |
| Hex/Oxy | 2.32 | −2.93 | 2.3 | −0.4 | 0.6 | −1.6 |
| Bond/Cr | 2.29 | −0.87 (2.40/−2.84) | 1.8 | −0.1 | 1.8 | −0.2 |
| Hex/Cr | 2.32 | −1.58 (2.45/−2.92) | 2.4 | −0.1 | 1.8 | −6.7 |
| Hex/Vac | 2.25 | −0.31 (2.25/−2.88) | 1.5 | 0.2 | 0.9 | 1.1 |
| Pent/Cr | 2.32 | −3.00 | 2.7 | −0.4 | 2.6 | −1.4 |
| Bond/Vac | 2.24 | −0.29 (2.30/−2.88) | 1.4 | 0.1 | 0.8 | 0.6 |
| Bond/Oxy | 2.30 | −1.55 (2.46/−2.89) | 3.4 | −0.1 | 3.1 | −0.3 |
In the two columns on the right the average magnetic moment for the O atoms near the adsorption site (ONN) and for the C atoms facing to the surface (CNN) are also reported. As for the charge transfer these values are more representative of the variation induced by the molecule–surface interaction and are affected by a smaller percent variation (≈10% for O and ≈30% for C) with respect to the average on the whole cell.
The average magnetization of the Fe surface layer is about 2.3 μB showing a reduction with respect to the clean surface value (3.0 μB).
Notably, the net magnetization of the Cr atoms in the oxide layer is always opposite to the underlying Fe one, as in absence of molecules, confirming the antiferromagnetic coupling between the overlayer and the substrate evidenced by the experiments.12 Indeed, also in the configurations with intra-layer AFM ordering the magnetic moment of opposite Cr atoms is not identical, and the total magnetization of the metal atoms in the oxide does not cancel out.
The magnetic moment of the C atoms is three orders of magnitude smaller than that of the Cr atoms and its sign depends on the adsorption configuration. The largest magnetic moments on the molecule are found for the FM configurations and have the same sign of the magnetic moment of the Cr atoms. On the contrary, for the equilibrium configuration and the other cases with AFM ordering of the Cr atoms, the net magnetic moment on the molecule and on C atoms facing to the surface is opposite to that of the Cr4O5 layer, but has the same sign as that of the Cr atoms closest to the molecule (see Fig. 3).
In the FM systems the magnetic moment of single O atoms is opposite to that of the surrounding Cr atoms, resulting into a net positive magnetization. The magnetization of O atoms nearby the molecule is in general reduced and this effect is extended to all the O atoms in the cell when a AFM patterning is recovered, leading to a smaller positive net magnetic moment. It is worth noting that the configurations that display AFM ordered Cr atoms also display a peculiar symmetry of the spin density on the O atoms which resemble the in-plane p orbitals (see Fig. 3). Differently, in the FM cases such feature is absent, and the spin density on the O atoms shows an s-like symmetry. This peculiarity can be observed also in the freestanding Cr4O5 overlayer by switching the system from the AFM ground state to the high energy FM configuration (not shown).
This evidence suggests that the AFM coupling between the Cr atoms may be mediated by the O orbitals that tailor also the interaction with the molecule.
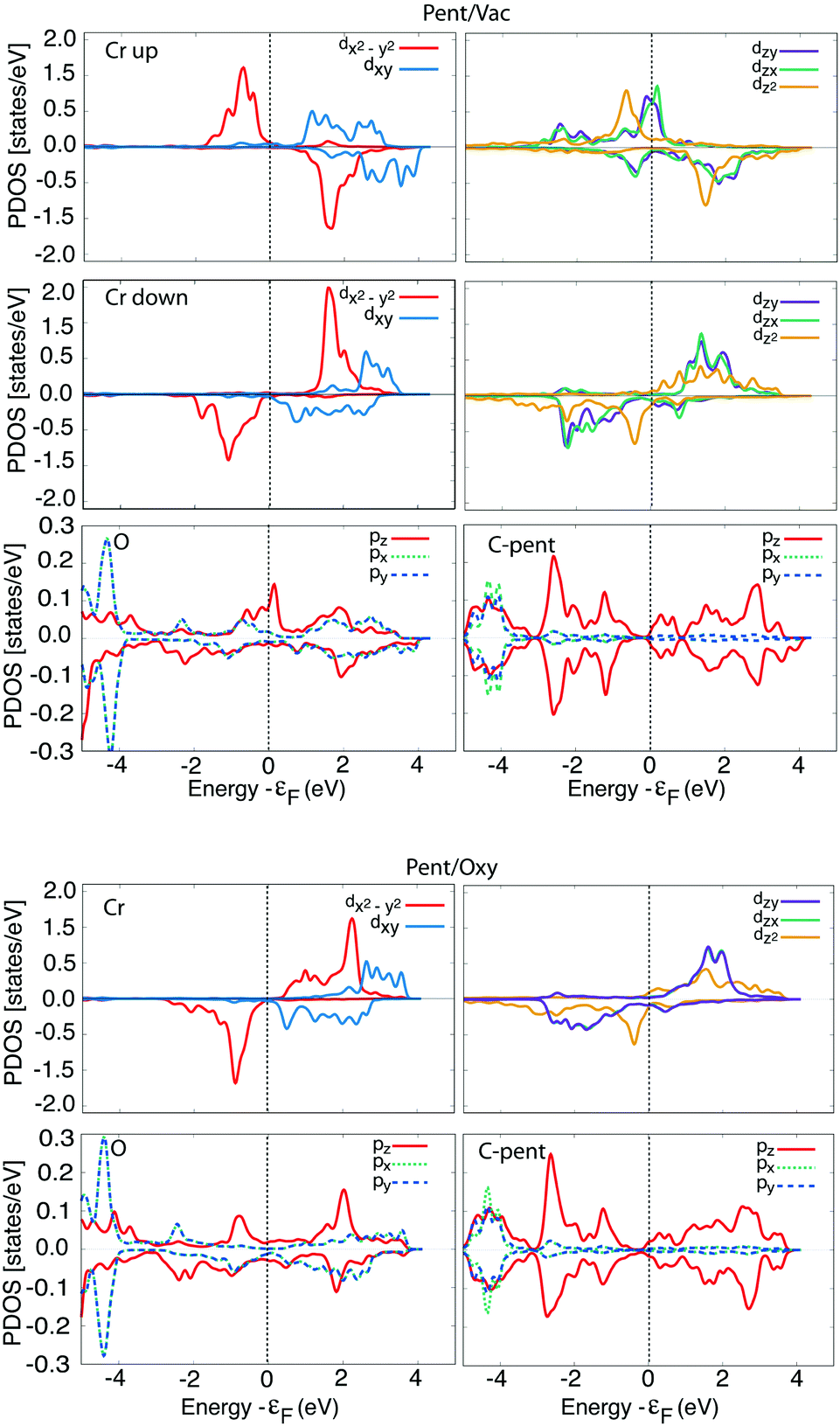
In order to give a deep insight into the induced magnetic properties at the interface, we consider the Projected Density of States (PDOS) of the different atomic species that, together with a detailed analysis of the spin and orbital dependent Löwdin populations, can help in understanding the driving mechanism for spin–flip of the Cr atoms.
In Fig. 4 we report the PDOS for the two lowest energy configurations, namely Pent/Vac and Pent/Oxy, that exhibit a different magnetic ordering of the Cr atoms in the oxide layer. We consider the average d component of the Fe surface layer and of the Cr atoms and the average p component of O atoms in the oxide layer, of the O atoms nearest neighbours of the molecule (black dashed line) and of C atoms facing to the surface.
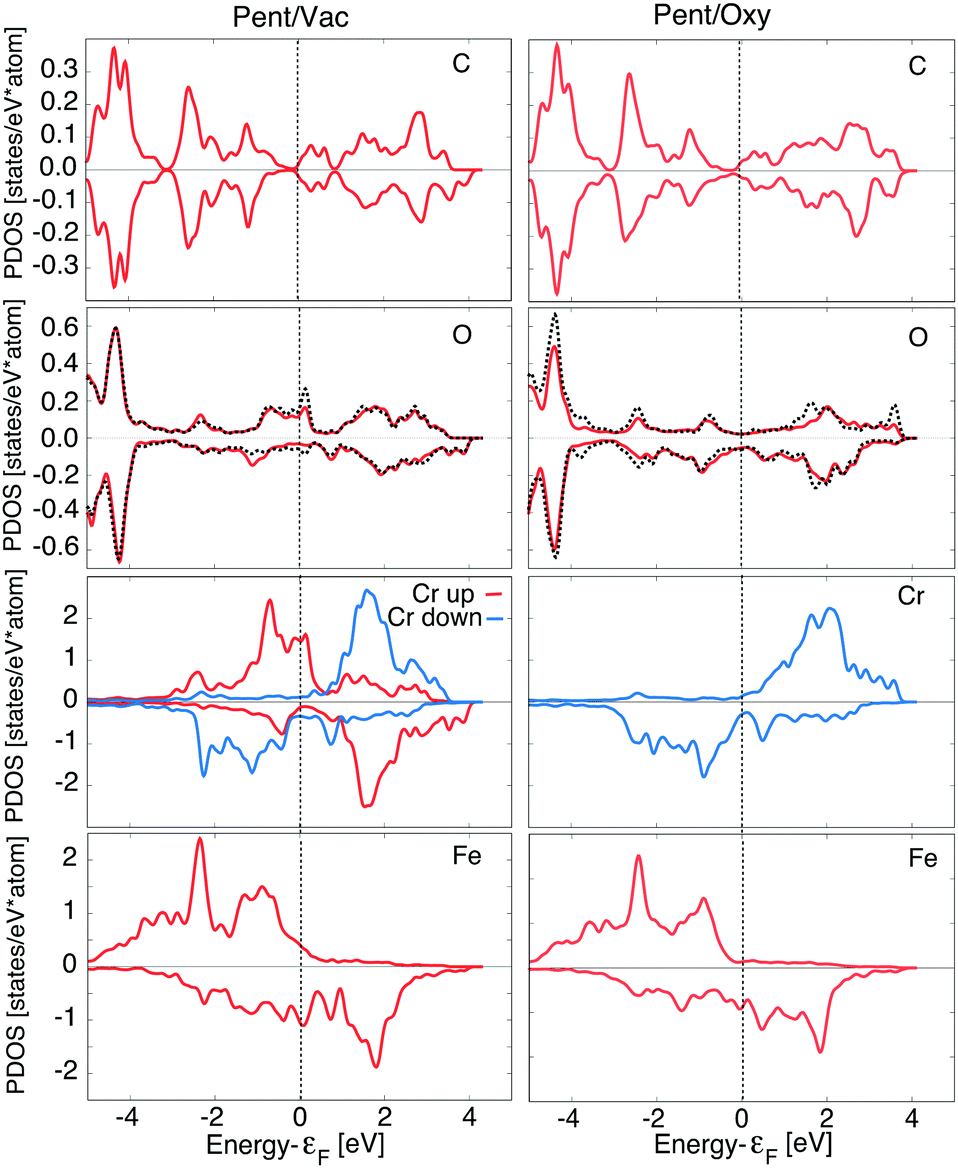 | ||
| Fig. 4 PDOS on Fe and Cr (d component), O and C atoms facing to the surface (p component) for the Pent/Vac (left) and Pent/Oxy (right) configurations. Two representative Cr atoms with opposite polarization are reported for the AFM configuration. For O we report as a red line the PDOS averaged over the cell, and as a black dashed line that averaged on the four nearest neighbours to C60 in the Pent/Vac configuration, and that of the O below C60 for the Pent/Oxy case. | ||
The Fe PDOS is similar in the two configurations, demonstrating that the underlying substrate is on average weakly affected by the different adsorption conditions. The spin polarization of the Cr atoms in the Pent/Oxy configuration is opposite to that of the Fe surface layer, as already evidenced in our past study.18 Differently, in the AFM configuration induced by C60 adsorption one half of the Cr atoms are spin-flipped, with magnetization oriented parallel to the substrate (Crup) while the others remain antiferromagnetically coupled to the substrate (Crdown). The asymmetry between filled states of Crup and Crdown atoms gives rise to the net negative magnetic moment reported in Table 2.
For what concerns the O atoms, they display a negligible spin polarization at the Fermi level in the Pent/Oxy configuration, which is enhanced in the Pent/Vac case due to the presence of majority spin states. This feature is mainly due to the changes of the spectral properties of the O atoms near the molecule (dashed line in Fig. 4). From the analysis of the m-resolved PDOS, reported in Fig. 5, we can assign these states mainly to pz orbitals, that are emptied due to the charge transfer toward the molecule. The majority spin O pz component (red line) in the Pent/Oxy configuration displays indeed an occupied feature at −0.8 eV which is shifted beyond the Fermi level in the Pent/Vac case.
 | ||
| Fig. 5 PDOS resolved in m components for Cr, O, and C atoms, for the Pent/Vac and Pent/Oxy configurations. For Cr and O we report unit-cell averages, while for C atoms we average values over the pentagon facing to the surface. | ||
This result is supported also by the analysis of the charge transfer evaluated only for the atoms participating in the molecule–substrate interaction which is reported in Fig. 2. The charge donated by O atoms to the C atoms in the pentagon is the major source of charge transfer to the molecule in the Pent/Vac configuration, bypassing the Cr contribution (see Table 1 and Fig. 2). Indeed in this configuration the spatial proximity between O atoms and the C atoms in the pentagon facilitates the interaction between the two species. Note that the very large charge transfer for the two spin populations in Cr reported in Fig. 2 for the AFM configurations is due to the spinflip of the Cr atoms surrounding the adsorption site. Indeed the reduction of the minority spin charge corresponds almost exactly to the increase of the majority spin one. This process is not indicative of a charge transfer to a different species, being an intra-atomic effect. For this reason only the total (spin up + spin down) value is relevant in the present analysis.
Also the in-plane majority spin p states of O atoms display a small increase of the spectral weight at the Fermi level with respect to the Pent/Oxy case (see Fig. 5), due to the charge transfer to the nearby Cr atoms. The majority spin character of the charge donated by O atoms explains the reduction of the magnetic moment of O in the AFM configurations with respect to the FM ones (see Fig. 2 and Table 2). Accordingly, the magnetic moment of the molecule is positive. Being the in-plane charge transfer strongly directional, the spin density displays the asymmetry observed in Fig. 3 with negative lobes pointing toward the Cr atoms. As a consequence the nearest neighbour Cr atoms undergo a spin–flip to stabilize the magnetic interaction, as in the freestanding overlayer.
Differently, in the Pent/Oxy configuration the charge transfer to the molecule comes from the Cr atoms due to their spatial proximity with the pentagon facing to the surface (see Fig. 2). The charge donated to the molecule has negative spin character, being transferred from the spin-down d states of Cr to the s states of the molecule. Some electronic charge is also donated by the O atoms, with the same amount for both spin up and down. The overall effect is a net negative magnetic moment of the molecule, while that of O atoms is unaltered with respect to the Cr4O5/Fe(001).
On the basis of these results we can infer that the AFM patterning of the Cr atoms in the oxide layer is stabilized by the interaction between O atoms and the molecule, that induces a spin asymmetry in the Cr4O5 plane able to decouple some Cr atoms from the substrate and to destroy the substrate-induced FM ordering between Cr atoms.
To conclude our analysis, we report some energetic considerations relative to the magnetic switching between the AFM and FM ordering in the Cr4O5 layer.
Once the C60 molecules are adsorbed in the most stable Pent/Vac configuration, the transition to Pent/Oxy and thus to FM ordering can be obtained with an energetic cost of 0.26 eV per cell containing eight Cr atoms. Compared to the energetic cost for the FM/AFM switching of the clean Cr4O5/Fe(001) surface (note that in this case the ground state is FM), amounting to 1.12 eV in the 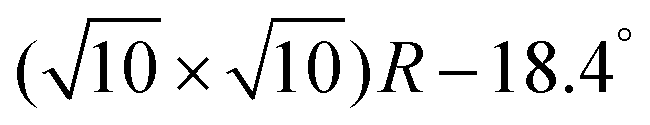 cell, this mechanism turns out to be energetically more convenient as well as easier to be realized.
cell, this mechanism turns out to be energetically more convenient as well as easier to be realized.
It is worth noting that the energetic cost reported above includes two contributions: the structural one, related to the shift onto a different adsorption site, and the magnetic cost associated to the spin–flip of certain Cr atoms. The difference in energy from the Pent/Oxy to the Pent/Vac adsorption site, evaluated keeping the FM ordering of the Cr atoms fixed, amounts to +0.04 eV. Such energy difference is anyway sufficiently high to prevent the displacement of C60 molecules to the high energy adsorption site simply by thermal annealing. Indeed also increasing the temperature to several hundreds of Kelvin would not allow a relevant probability of occupation of Pent/Oxy sites that could be achieved by using external means for a controlled adsorption.
On the other hand, the energy gain upon allowing the system in the Pent/Vac free to relax in the AFM ground state is −0.30 eV (the sum of these contributions gives indeed the value ΔEads = 0.26 eV, as reported in Table 1). The quite large absolute value of the magnetic contribution compared to the structural one demonstrates that the AFM ordering realized in the Pent/Vac configuration is stabilized by magnetic effects more than structural ones.
Furthermore, by comparing the absolute value of the FM/AFM energy difference with and without the molecule (0.30 eV versus 1.12 eV) it is evident that the presence of the molecule reduces the magnetic coupling between the Cr4O5 and the Fe substrate which is responsible of the forced magnetic ordering in the overlayer. Indeed, in the absence of the substrate the energy cost for the AFM/FM switching in the Cr4O5 layer would be extremely small (0.04 eV).
3 Conclusions
In conclusion, in the present work we demonstrated that the adsorption of a C60 layer on the Cr4O5/Fe(001) surface can tailor the intra-layer magnetic ordering between the Cr atoms restoring the AFM configuration proper of the freestanding oxide overlayer, destroyed by the interaction with the substrate. Moreover the AFM/FM switching is possible via a precise control on the adsorption site of the molecule that could be achieved by exploiting the ad hoc positioning via the STM tip or modified by thermal annealing. The effectiveness of an organic layer at the interface – be it C60 or something else – is a crucial feature which may be exploited to realize more efficient spintronic devices; furthermore, attaining a patterning on the surface by means of the adsorption of organic species is expected to be far easier than creating it ad hoc on the bare Cr4O5/Fe(001) by exploiting magnetic or electric means. Therefore, our results may be considered as a route towards the design of a desired magnetic patterning by means of adsorbed organic molecules.4 Computational methods
Ab initio calculations were performed in the framework of Density Functional Theory (DFT) following the scheme of our previous works.12,18,19We used a plane-wave ultrasoft pseudopotential method,25 as implemented in the PWSCF code of the Quantum ESPRESSO distribution.26,27 We treated the DFT exchange–correlation term by using the vdW-DF-c09x functional,28,29 also including van der Waals interaction between the C60s and the underlying surface. A high computational efficiency can be achieved with this choice, allowing us to perform accurate and extensive calculations, although we are aware that nonlocal functionals (such as hybrid30 or LDA+U31) would further improve the description of d-metal atoms.
The clean Cr4O5/Fe(001) substrate exhibits an experimentally-observed 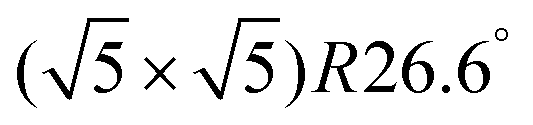 reconstruction,24 featuring a regular array of Cr vacancies. For the calculations, the introduction of a C60 overlayer calls for the employment of a larger periodically repeated cell: we employ a
reconstruction,24 featuring a regular array of Cr vacancies. For the calculations, the introduction of a C60 overlayer calls for the employment of a larger periodically repeated cell: we employ a 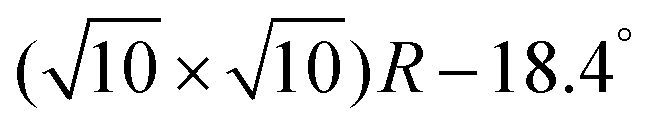 supercell, whose area is twice that of the clean surface. The Cr4O5 layer is supported by a four-layers Fe slab, separated from its replicas along the ẑ direction by a 25 Å-thick vacuum layer.
supercell, whose area is twice that of the clean surface. The Cr4O5 layer is supported by a four-layers Fe slab, separated from its replicas along the ẑ direction by a 25 Å-thick vacuum layer.
Equilibrium geometries were obtained by letting the C atoms of the fullerene molecules free to relax, up to the desired convergence threshold for the forces (0.001 Ry bohr−1). For the most stable configuration (Pent/Vac) we also performed a calculation in which the Cr-oxide layer and the two outermost Fe layers were also allowed to relax.
The magnetic properties and the charge transfer are very similar to those discussed in the present manuscript. In particular we found the same AFM patterning of the Cr atoms with magnetic moments differing by less than 3%. This justifies the use of a frozen substrate for the analysis reported in the text.
A Monkhorst-Pack grid32 was adopted for the surface Brillouin zone sampling, equivalent to a 14 × 14 mesh in the surface unit cell of Fe(001); the kinetic energy cutoffs were set to 55 Ry for the plane-wave expansion and 280 Ry for the effective potential and charge density.
To disentangle magnetic contributions to energy differences, we performed additional self-consistent calculations constraining the magnetic moment at specific atoms and keeping the geometry unchanged.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the CINECA award under the ISCRA initiative (grants IscrC-SPINOF2-HP10C3S9Z0 and IscrC-APOCAPOF-HP10CB0ZW2), for the availability of high performance computing resources and support.References
- O. O. Brovko, P. Ruiz-Díaz, T. R. Dasa and V. S. Stepanyuk, J. Phys.: Condens. Matter, 2014, 26, 093001 CrossRef.
- E. B. Myers, D. C. Ralph, J. A. Katine, R. N. Louie and R. A. Buhrman, Science, 1999, 285, 867 CrossRef CAS.
- I. Carmeli, J. Chem. Phys., 2003, 118, 10372 CrossRef CAS.
- M. Callsen, V. Caciuc, N. Kiselev, N. Atodiresei and S. Blügel, Phys. Rev. Lett., 2013, 111, 106805 CrossRef.
- A. Hernando, P. Crespo and M. A. García, Phys. Rev. Lett., 2006, 96, 57206 CrossRef CAS.
- S. Achilli, S. Caravati and M. I. Trioni, J. Phys.: Condens. Matter, 2007, 19, 305021 CrossRef.
- E. Del Castillo, F. Cargnoni, S. Achilli, G. Tantardini and M. Trioni, Surf. Sci., 2015, 634, 62 CrossRef CAS.
- O. Ben Dor, S. Yochelis, A. Radko, K. Vankayala, E. Capua, A. Capua, S. Yang, L. T. Baczewski, S. S. Papworth Parkin, R. Naaman and Y. Paltiel, Nat. Commun., 2017, 8, 14567 CrossRef CAS.
- S. Sanvito, Nat. Phys., 2010, 6, 562 Search PubMed.
- M. Cinchetti, V. A. Dediu and L. E. Hueso, Nat. Mater., 2017, 16, 507 CrossRef CAS.
- S. Delprat, M. Galbiati, S. Tatay, C. Quinard, B. Anf Barraud and F. Petroff, J. Phys. D: Appl. Phys., 2019, 51, 473001 CrossRef.
- A. Brambilla, A. Picone, D. Giannotti, A. Calloni, G. Berti, G. Bussetti, S. Achilli, G. Fratesi, M. I. Trioni, G. Vinai, P. Torelli, G. Panaccione, L. Duò, M. Finazzi and F. Ciccacci, Nano Lett., 2017, 17, 7440 CrossRef CAS.
- A. Calloni, M. Jagadeesh, G. Bussetti, G. Fratesi, S. Achilli, A. Picone, A. Lodesani, A. Brambilla, C. Goletti, F. Ciccacci, L. Duò, M. Finazzi, A. Goldoni, A. Verdini and L. Floreano, Appl. Surf. Sci., 2020, 505, 144213 CrossRef CAS.
- G. Fratesi, S. Achilli, A. Ugolotti, A. Lodesani, A. Picone, A. Brambilla, L. Floreano, A. Calloni and G. Bussetti, Appl. Surf. Sci., 2020, 530, 147085 CrossRef CAS.
- K. Bairagi, A. Bellec, V. Repain, C. Chacon, Y. Girard, Y. Garreau, J. Lagoute, S. Rousset, R. Breitwieser, Y.-C. Hu, Y. C. Chao, W. W. Pai, D. Li, A. Smogunov and C. Barreteau, Phys. Rev. Lett., 2015, 114, 247203 CrossRef CAS.
- M. Denk, D. Queteschiner, M. Hohage, A. Navarro-Quezada and P. Zeppenfeld, J. Appl. Phys., 2019, 125, 142902 CrossRef.
- F. A. MaMari, T. Moorsom, G. Teobaldi, W. Deacon, T. Prokscha, H. Luetkens, S. Lee, G. E. Sterbinsky, D. A. Arena, D. A. MacLaren, M. Flokstra, M. Ali, M. C. Wheeler, G. Burnell, B. J. Hickey and O. Cespedes, Nature, 2015, 524, 69–73 CrossRef CAS.
- A. Calloni, G. Fratesi, S. Achilli, G. Berti, G. Bussetti, A. Picone, A. Brambilla, P. Folegati, F. Ciccacci and L. Duò, Phys. Rev. B, 2017, 96, 085427 CrossRef.
- A. Brambilla, A. Picone, S. Achilli, G. Fratesi, A. Lodesani, A. Calloni, G. Bussetti, M. Zani, M. Finazzi, L. Duò and F. Ciccacci, J. Appl. Phys., 2019, 125, 142907 CrossRef.
- L. Liu, S. Liu, X. Chen, C. Li, J. Ling, X. Liu, Y. Cai and L. Wang, Sci. Rep., 2013, 3, 3062 CrossRef.
- J. Liu, C. Li, X. Liu, Y. Lu, F. Xiang, X. Qiao, Y. Cai, Z. Wang, S. Liu and L. Wang, ACS Nano, 2014, 8, 12734 CrossRef CAS.
- S. Wei, Z. Wang, J. Jin, H. Xu, Y. Lu and L. Wang, Nanotechnology, 2018, 29, 395301 CrossRef.
- L. Hu, R. Pang, P. Gong and X. Shi, J. Phys. Chem. C, 2019, 123, 15477 CrossRef CAS.
- A. Picone, G. Fratesi, M. Riva, G. Bussetti, A. Calloni, A. Brambilla, M. I. Trioni, L. Duò, F. Ciccacci and M. Finazzi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 085403 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. D. Jr, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. O. de-la Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- V. Cooper, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 161104 CrossRef.
- F. Tran, P. Blaha, K. Schwarz and P. Novák, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 155108 CrossRef.
- M. Cococcioni and S. De Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
| This journal is © the Owner Societies 2021 |