
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical oxidative Z-selective C(sp2)–H chlorination of acrylamides†‡
James
Harnedy§
a,
Mishra Deepak
Hareram§
a,
Graham J.
Tizzard
b,
Simon J.
Coles
 b and
Louis C.
Morrill
*a
b and
Louis C.
Morrill
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: MorrillLC@cardiff.ac.uk
bUK National Crystallographic Service, Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 11th November 2021
Abstract
An electrochemical method for the oxidative Z-selective C(sp2)–H chlorination of acrylamides has been developed. This catalyst and organic oxidant free method is applicable across various substituted tertiary acrylamides, and provides access to a broad range of synthetically useful Z-β-chloroacrylamides in good yields (22 examples, 73% average yield). The orthogonal derivatization of the products was demonstrated through chemoselective transformations and the electrochemical process was performed on gram scale in flow.
A diverse array of important compounds contain chlorine atoms, including pharmaceuticals, natural products, agrochemicals, and polymers.1 Furthermore, Z-chloroacrylic acid derivatives serve as useful building blocks to selectively access ubiquitous Z-configured olefins.2 Existing synthetic methods to access Z-β-chloroacrylic acid derivatives include Vilsmeier–Haack,3 Wittig,4 and chloropalladation/Heck approaches,5 in addition to hydrochlorination6 and carbochlorination7 of alkynes, which somewhat limits the accessible substitution patterns and some of these methods can exhibit poor E/Z selectivity. As such, the development of new synthetic methods that enable the direct and selective access to Z-chloroacrylic acid derivatives is an important and timely goal in organic synthesis. Recently, Besset and co-workers reported the Pd-catalyzed Z-selective chlorination of secondary acrylamides bearing an 8-aminoquinoline directing group (Scheme 1A), employing N-chlorosuccinimide as the chlorinating agent, representing a state-of-the-art approach.8
 | ||
| Scheme 1 State-of-the-art and outline of the electrochemical strategy. | ||
Organic electrochemistry presents vast opportunities for the development of efficient and selective synthetic methods.9 A variety of electrochemical methods have been developed for the chlorination of organic compounds, including alkene dichlorination,10 chlorofunctionalization of alkenes11 and alkynes,12 aromatic chlorination,13 α-chlorination of 1,3-dicarbonyl compounds,14 and deconstructive chlorination of cycloalkanols.15 As such, we envisaged the development of an electrochemical oxidative Z-selective C(sp2)–H chlorination of acrylamides to generate valuable Z-β-chloroacrylamides (Scheme 1B), and herein we report the successful realization of this approach.
To commence our studies, N-methyl-N-tosylacrylamide 1 was selected as the model substrate (Table 1).16 It was found that an electrochemical system composed of MgCl2 (5 equiv.) as both chloride source and electrolyte in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH (7:1, [1] = 0.05 M) using galvanostatic conditions (i = 10 mA, janode = 7.8 mA cm−2, Q = 2.5 F mol−1), a graphite anode and a platinum cathode at 25 °C for 2 h under N2, enabled the oxidative C(sp2)–H chlorination of 1, with Z-β-chloroacrylamide 2 formed in 97% NMR yield as a single stereoisomer (>95:<5 Z
:AcOH (7:1, [1] = 0.05 M) using galvanostatic conditions (i = 10 mA, janode = 7.8 mA cm−2, Q = 2.5 F mol−1), a graphite anode and a platinum cathode at 25 °C for 2 h under N2, enabled the oxidative C(sp2)–H chlorination of 1, with Z-β-chloroacrylamide 2 formed in 97% NMR yield as a single stereoisomer (>95:<5 Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :E) (entry 1).17 No conversion occurs in the absence of electricity (entry 2). Employing a constant cell potential (Ecell = 3.75 V) or variation of the current (i = 15 mA or 5 mA) lowered the NMR yield of 2 (entries 3–5), as did substituting the platinum foil cathode for graphite (entry 6). Employing LiCl or NaCl as the chloride source was detrimental to conversion, presumably due to the decreased solubility in MeCN:AcOH (entries 7 and 8). Using MeCN:MeOH (3.5:1) as solvent resulted in reduced conversion to 2. Lowering the quantity of MgCl2 to 3 equivalents reduced the NMR yield of 2 to 68%, which increased to 78% upon the addition of LiClO4 (2 equiv.) as a supporting electrolyte. A faradaic efficiency of 85% was obtained when 2 F mol−1 of charged was passed (entry 12), which indicated that most of the electricity passing through the cell is utilized productively. Replacing MgCl2 with MgBr2 resulted in recovery of 1 (>90%).
:E) (entry 1).17 No conversion occurs in the absence of electricity (entry 2). Employing a constant cell potential (Ecell = 3.75 V) or variation of the current (i = 15 mA or 5 mA) lowered the NMR yield of 2 (entries 3–5), as did substituting the platinum foil cathode for graphite (entry 6). Employing LiCl or NaCl as the chloride source was detrimental to conversion, presumably due to the decreased solubility in MeCN:AcOH (entries 7 and 8). Using MeCN:MeOH (3.5:1) as solvent resulted in reduced conversion to 2. Lowering the quantity of MgCl2 to 3 equivalents reduced the NMR yield of 2 to 68%, which increased to 78% upon the addition of LiClO4 (2 equiv.) as a supporting electrolyte. A faradaic efficiency of 85% was obtained when 2 F mol−1 of charged was passed (entry 12), which indicated that most of the electricity passing through the cell is utilized productively. Replacing MgCl2 with MgBr2 resulted in recovery of 1 (>90%).
|
|
||
|---|---|---|
| Entry | Variation from “standard” conditions | Yieldb (%) |
|
a Reactions performed using 0.3 mmol of acrylamide 1 using the ElectraSyn 2.0 batch electrochemical reactor. [1] = 0.05 M.
b Yield after 2 h as determined by 1H NMR analysis of the crude reaction mixture with 1,3,5-trimethylbenzene as the internal standard. Formed as a single stereoisomer (>95:<5 Z:E). Isolated yield given in parentheses.
c 96 minutes reaction time.
|
||
| 1 | None | 97 (92) |
| 2 | No electricity | <2 |
| 3 | E cell = 3.75 V | 91 |
| 4 | i = 15 mA, janode = 11.7 mA cm−2 | 91 |
| 5 | i = 5 mA, janode = 3.9 mA cm−2 | 85 |
| 6 | Graphite cathode instead of Pt foil | 83 |
| 7 | LiCl instead of MgCl2 | 83 |
| 8 | NaCl instead of MgCl2 | 25 |
| 9 | MeCN:MeOH (3.5:1) |
67 |
| 10 | MgCl2 (3 equiv.) | 68 |
| 11 | MgCl2 (3 equiv.) + LiClO4 (2 equiv.) | 78 |
| 12c | Q = 2 F mol−1 | 85 |
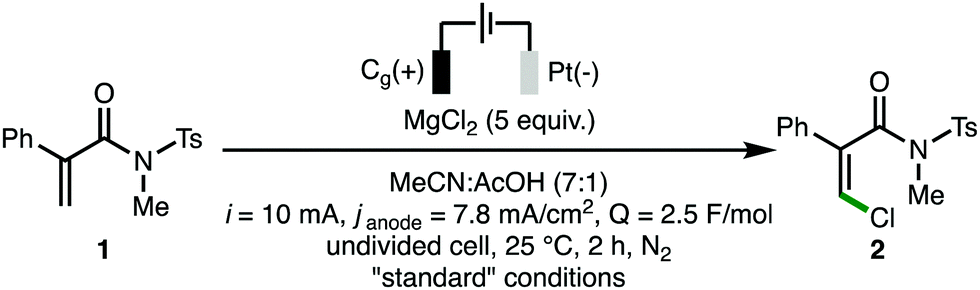
With optimized reaction conditions in hand (Table 1, entry 1), the full scope and limitations of the electrosynthetic method with respect to the acrylamide starting materials was evaluated (Scheme 2). Unless otherwise stated, only the Z-alkene isomer was observed for all substrates. Employing N-methyl-N-tosylacrylamide starting materials (Scheme 2A), it was found that various substituents could be incorporated into the C(2)-aromatic unit, including electron-releasing (e.g., 4-OMe), electron-withdrawing (e.g., 4-CF3) and halogen substituents, with the corresponding Z-β-chloroacrylamides products accessed in good yields (products 3–13). Introducing sterically demanding groups into the acrylamide starting materials did not negatively impact upon product formation, with products 8 and 9 formed in 74% and 83% isolated yields, respectively. The introduction of a C(3)-methyl substituent afforded tetrasubstituted Z-β-chloroacrylamide 14 in 75% isolated yield,18 whereas the C(2)-aryl group could be replaced with a C(2)-methyl substituent to access 15 as the major regioisomer (86:14 r.r.). Further variation of the acrylamide starting material revealed that a selection of N-alkyl, N-phenyl, and N-sulfonyl substituents could be incorporated to access products 16–20 in high isolated yields (Scheme 2B). Employing a Weinreb acrylamide substrate produced the corresponding vinyl chloride 21 as a 2:1 mixture of Z:E alkene isomers, which was isolated in a combined 61% yield. This lower observed selectivity may be attributed to increased C–N partial double-bond character within the Weinreb amide functionality, resulting in a greater steric effect, in comparison to the twisted amide (c.f., substrate 1).19N,N-Dialkylacrylamides could be employed as substrates, with Z-β-chloroacrylamides 22 and 23 isolated in 61% and 73% isolated yields, respectively. When N,N-diphenylacrylamide 24 was subjected to the optimized reaction conditions, oxindole 25 was accessed as the sole product (Scheme 2C), which can be formed via intramolecular trapping of a C-centered radical intermediate with a N-phenyl substituent.20 Substituted methyl acrylate 26 produced a mixture of Z-β-chloroacrylate 27 and dichloro ester 28 in 44% and 32% NMR yields, respectively. The formation of 28 may indicate that it is an intermediate on route to the formation of 27, or that the operative reaction mechanism for methyl acrylate substrate 26 is distinct to the tertiary acrylamide substrates predominantly employed. Secondary acrylamide 29 produced a complex reaction mixture whereas acrylamides 30 and 31 were found to be unreactive, presumably due to radical instability and steric encumbrance, respectively.
 | ||
| Scheme 2 Scope and limitations. Reactions performed using 0.3 mmol of starting material. All yields are isolated yields after chromatographic purification unless noted otherwise. a Isolated as a mixture of isomers. b 3.5 F mol−1. c As determined by 1H NMR analysis of the crude reaction mixture. | ||
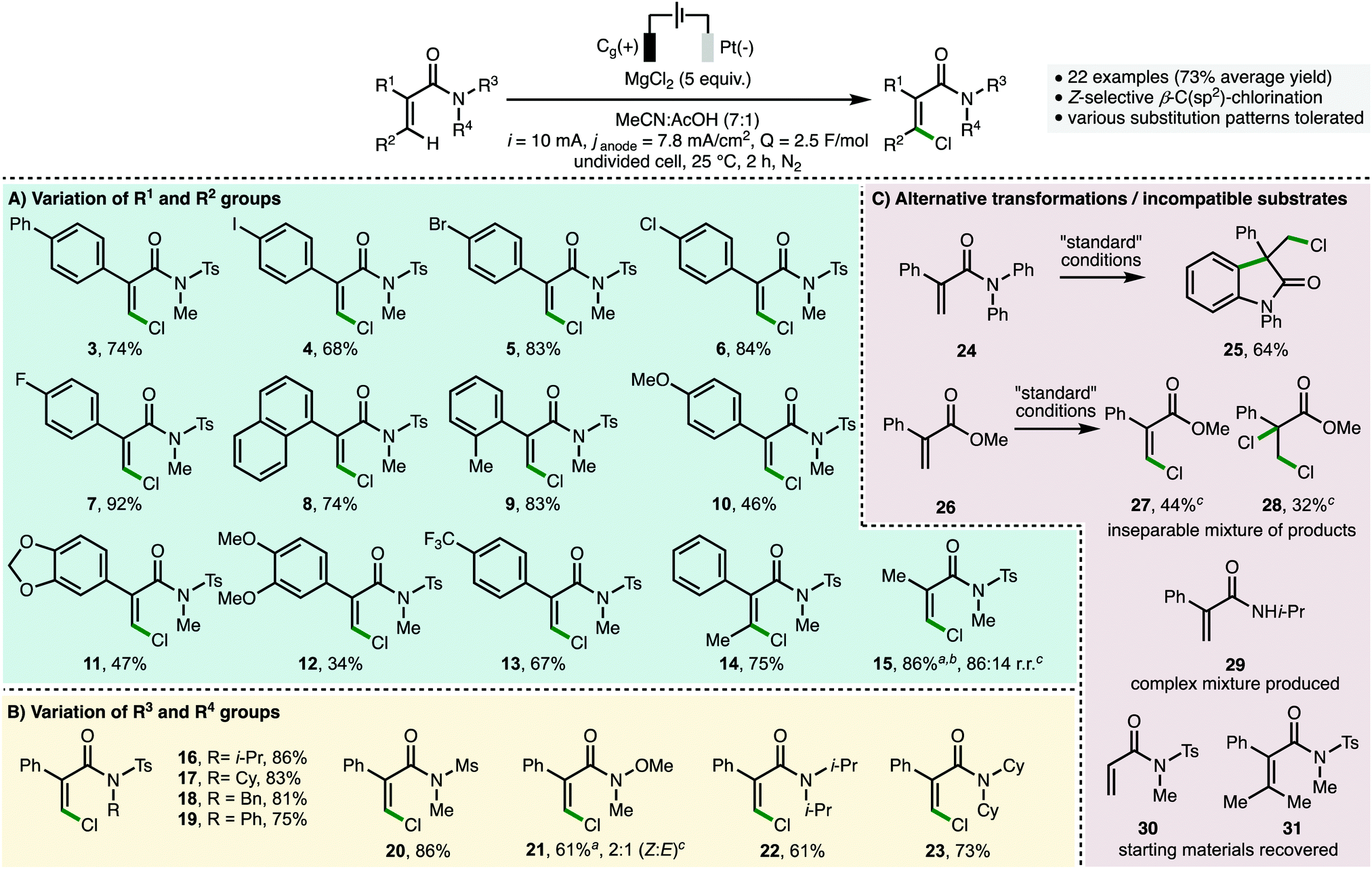
To demonstrate product utility, the orthogonal functionalization of Z-β-chloroacrylamide 2 was investigated (Scheme 3A). It was found that the C(sp2)–Cl functionality could serve as a coupling partner in a palladium-catalyzed Suzuki cross-coupling with 4-methoxyphenylboronic acid to access arylated alkene 32 in 49% isolated yield. Furthermore, the N-sulfonyl activated twisted amide functionality within 2 was hydrolyzed to give the corresponding carboxylic acid derivative 33 in 72% isolated yield, which can serve as a useful building block for further synthetic elaboration. To demonstrate scalability, the batch process was translated to a flow electrochemical setup (Scheme 3B).21 Employing a syringe pump in combination with commercially available Ammonite8 flow electroreactor (volume = 1 mL, i = 370 mA),22 4.4 mmol of acrylamide 1 was converted to Z-β-chloroacrylamide 2 in 91% isolated yield (1.40 g) in a single-pass.
 | ||
| Scheme 3 Product utility and reaction scale up in flow. | ||
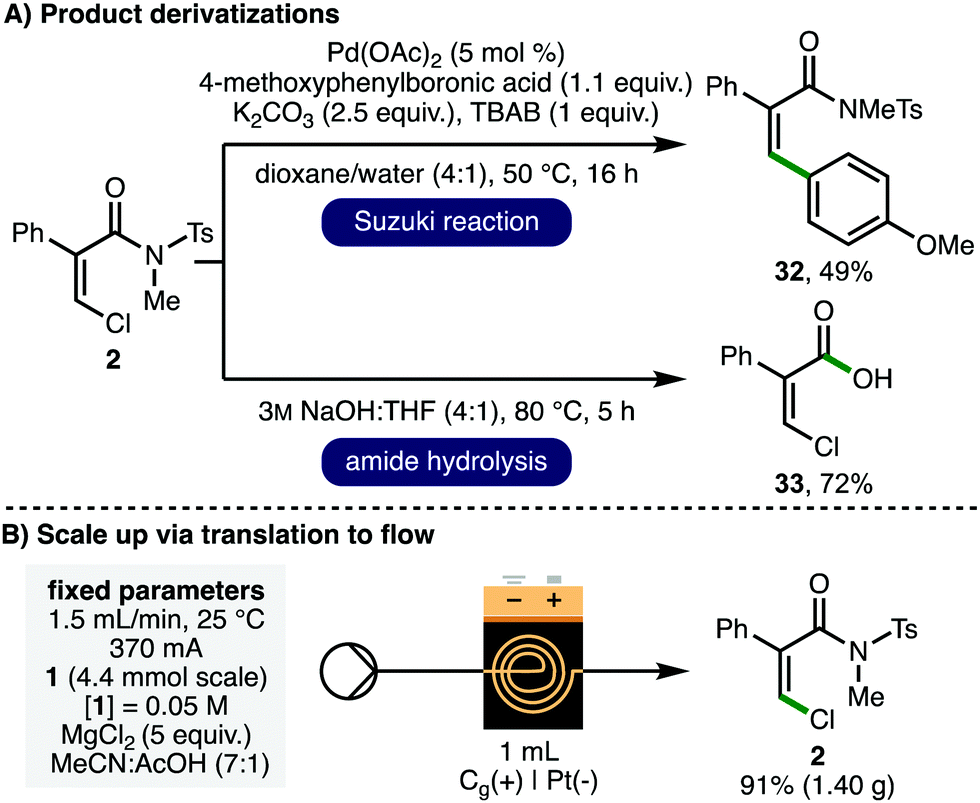
To obtain insight into the reaction mechanism, acrylamide 34, containing a C(2)-cyclopropyl substituent, was synthesized and subjected to the optimized electrochemical reaction conditions as a radical clock mechanistic probe (Scheme 4A). It was found that ring-opened dichlorinated acrylamide 35 was obtained as a 1:1 mixture or Z:E alkene isomers in 44% NMR yield, signifying the likelihood of radical intermediates within the operative reaction mechanism. Cyclic voltammetry studies indicated that chloride ions (Eox = 1.10 V vs. Fc/Fc+) should undergo preferential oxidation in the presence of electron-deficient acrylamide 1 (Eox = 1.31 V vs. Fc/Fc+). It was also found that replacing MgCl2 with N-chlorosuccinimide in the absence of electricity resulted in quantitative recovery of 1. In line with these observations, and previous related investigations,23 a plausible reaction mechanism would initiate with anodic chloride oxidation to form a chlorine radical,11d,12b,13c,13d which undergoes regioselective addition to the electron-deficient acrylamide 1 to furnish tertiary C-centered radical 36. Further anodic oxidation would form carbocation 37, with subsequent deprotonation providing access to the observed Z-β-chloroacrylamide 2 (path a). Substitution at C(2) is crucial for reactivity (c.f.Scheme 2C, starting material 30), presumably due to the requirement to stabilize intermediates 36 and 37. Deprotonation of carbocation 37 proceeds via the conformation that minimizes steric repulsion between the chlorine atom and the phenyl ring to afford the Z-isomer as the major product stereoisomer (Scheme 4c). Alternatively, carbocation 37 or radical 36 could be intercepted by a chloride anion (path b) or a chlorine radical (path c), respectively, to form dichlorinated compound 38, which could subsequently generate 2via loss of HCl. Dichlorinated species such as 38 have not been directly observed using acrylamide substrates, however, we cannot unambiguously rule out their intermediacy. Hydrogen gas is generated via proton reduction at the cathode.
 | ||
| Scheme 4 Reaction mechanism. a As determined by 1H NMR analysis of the crude reaction mixture. | ||
In conclusion, we have developed a new electrochemical method for the oxidative Z-selective C(sp2)–H chlorination of acrylamides. The method is applicable across various substituted tertiary acrylamides, accessing a broad range of synthetically useful Z-β-chloroacrylamides (22 examples). The orthogonal derivatization of the products was demonstrated through chemoselective transformations, and the electrochemical process was performed on gram scale in flow.
We would like to thank Cardiff University for studentship funding for James Harnedy and the EPSRC for a standard research grant (EP/R006504/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed; (b) B. R. Smith, C. M. Eastman and J. T. Njardarson, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed; (c) H. Swada in Encylopedia of Polymeric Nanomaterials, S. Kobayashi, K. Müllen, ed., Springer, Berlin, Heidelberg, 2015, pp. 1–10 Search PubMed.
- (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed; (b) S. Tang and K. L. Erickson, J. Nat. Prod., 2008, 71, 898–901 CrossRef CAS PubMed; (c) A. Français, A. Leyva-Pérez, G. Etxebarria-Jardi, J. Peña and S. V. Ley, Chem. – Eur. J., 2011, 17, 329–343 CrossRef PubMed.
- K. U. Krishnaraj and K. S. Devaky, Tetrahedron, 2014, 70, 6450–6456 CrossRef CAS.
- C. J. Moody, M. Pass, C. W. Rees and G. Tojo, J. Chem. Soc., Chem. Commun., 1986, 1062–1063 RSC.
- J.-M. Huang, Y. Dong, X.-X. Wang and H.-C. Luo, Chem. Commun., 2010, 46, 1035–1037 RSC.
- S. Ma, X. Lu and Z. Li, J. Org. Chem., 1992, 57, 709–713 CrossRef CAS.
- (a) C. M. Le, T. Sperger, R. Fu, X. Hou, Y. H. Lim, F. Schoenebeck and M. Lautens, J. Am. Chem. Soc., 2016, 138, 14441–14448 CrossRef CAS PubMed; (b) H. Koo, H. Y. Kim and K. Oh, Org. Chem. Front., 2019, 6, 1868–1872 RSC.
- M.-Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Org. Lett., 2020, 22, 7556–7561 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (c) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (d) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (e) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (f) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (g) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- (a) N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed; (b) X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507–514 CrossRef CAS PubMed.
- (a) H.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef PubMed; (b) X. Sun, H.-X. Ma, T.-S. Mei, P. Fang and Y. Hu, Org. Lett., 2019, 21, 3167–3171 CrossRef CAS PubMed; (c) C. Chen, J.-C. Kang, C. Mao, J.-W. Dong, Y.-Y. Xie, T.-M. Ding, Y.-Q. Tu, Z.-M. Chen and S.-Y. Zhang, Green Chem., 2019, 21, 4014–4019 RSC; (d) X. Sun, H.-X. Ma, T.-S. Mei, P. Fang and Y. Hu, Org. Lett., 2019, 21, 3167–3171 CrossRef CAS PubMed; (e) K. Liu, Y. Deng, W. Song, C. Song and A. Lei, Chin. J. Chem., 2020, 38, 1070–1074 CrossRef CAS.
- (a) K.-Y. Ye, Z. Song, G. S. Sauer, J. H. Harenberg, N. Fu and S. Lin, Chem. – Eur. J., 2018, 24, 12274–12279 CrossRef CAS PubMed; (b) X. Meng, Y. Zhang, J. Luo, F. Wang, X. Cao and S. Huang, Org. Lett., 2020, 22, 1169–1174 CrossRef CAS PubMed.
- (a) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nichiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed; (b) M. Konishi, K. Tsuchida, K. Sano, T. Kochi and F. Kakiuchi, J. Org. Chem., 2017, 82, 8716–8724 CrossRef CAS PubMed; (c) Q. Liu, B. Sun, Z. Liu, Y. Kao, B.-W. Dong, S.-D. Jiang, F. Li, G. Liu, Y. Yang and F. Mo, Chem. Sci., 2018, 9, 8731–8737 RSC; (d) Y. Yuan, A. Yao, Y. Zheng, M. Gao, Z. Zhou, J. Qiao, J. Hu, B. Ye, J. Zhao, H. Wen and A. Lei, iScience, 2019, 12, 293–303 CrossRef CAS PubMed; (e) Y. Liang, F. Lin, Y. Adeli, R. Jin and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 4566–4570 CrossRef CAS PubMed; (f) F. Liu, N. Wu and X. Cheng, Org. Lett., 2021, 23, 3015–3020 CrossRef CAS PubMed.
- K. Tsuchida, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2013, 2, 935–937 CrossRef CAS.
- (a) N. I. Kapustina, L. L. Sokova and H. I. Nikishin, Russ. Chem. Bull., 1996, 45, 124601248 Search PubMed; (b) B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS PubMed.
- See the ESI‡ for full experimental details.
- The Z-alkene configuration within 2 was unambiguously confirmed by X-ray crystal structure analysis. The stereoisomer of all other Z-β-chloroacrylamide products was assigned by analogy.
- Only the Z isomer of product 14 was observed upon varying the acrylamide substrate E:Z ratio from 10:1 to 1:1.
- C. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121(20), 12746–12783 CrossRef PubMed.
- Y. Su, L. Cao, Y. Shi, Y. Feng, W. Xue, G. Cao, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Synthesis, 2019, 2331–2338 CAS.
- (a) G. Laudadio, W. de Smet, L. Struik, Y. Cao and T. Noël, J. Flow Chem., 2018, 8, 157–165 CrossRef PubMed; (b) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed; (c) M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef CAS PubMed.
- R. A. Green, R. C. D. Brown, D. Pletcher and B. A. Harji, Org. Process Res. Dev., 2015, 19, 1424–1427 CrossRef CAS.
- For the electrochemical difluoromethylation of electron-deficient alkenes, see: H.-H. Xu, J. Song and H.-C. Xu, ChemSusChem, 2019, 12, 3060–3063 CrossRef CAS PubMed.
Footnotes |
| † Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2021.0143191217. |
| ‡ Electronic supplementary information (ESI) available: Optimization data, experimental procedures, characterization of new compounds and spectral data. CCDC 2104458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05824j |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |