
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis base-free thiophosphonium ion: a cationic sulfur atom transfer reagent†
Pawel
Löwe
 a,
Tim
Witteler
a and
Fabian
Dielmann
*ab
a,
Tim
Witteler
a and
Fabian
Dielmann
*ab
aInstitute of Inorganic and Analytical Chemistry, Westfälische Wilhelms-Universität Münster, Corrensstrasse 28-30, 48149 Münster (Germany), Germany. E-mail: Fabian.Dielmann@uibk.ac.at
bInstitute of General, Inorganic and Theoretical Chemistry, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, 6020 Innsbruck (Austria), Austria
First published on 16th April 2021
Abstract
Phosphorus(V) sulfide and Lawesson's reagent are commonly used thionating reagents which are considered to operate after dissociation into highly reactive dithiophosphorane fragments. We report the synthesis and properties of a monomeric thiophosphonium ion [R2P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S]+. The highly electrophilic species reacts with carbonyls in oxo-for-sulfido exchange reactions at room temperature and undergoes phosphorus–chalcogen bond metathesis reactions with phosphine chalcogenides.
S]+. The highly electrophilic species reacts with carbonyls in oxo-for-sulfido exchange reactions at room temperature and undergoes phosphorus–chalcogen bond metathesis reactions with phosphine chalcogenides.
The chemical conversion of carbonyls to thiocarbonyls using sulfur atom transfer reagents is an important method in synthetic organic chemistry.1 Phosphorus(V) sulfide (P4S10) and Lawesson's reagent (I, Scheme 1) are most commonly used in this context.2 Their cyclic structures of alternating sulfur and phosphorus atoms require thermal cleavage into reactive dithiophosphoranes (R-PS2), which can then react with carbonyls via four-membered ring intermediates to give the corresponding thiocarbonyls and metathiophosphonates.3,4
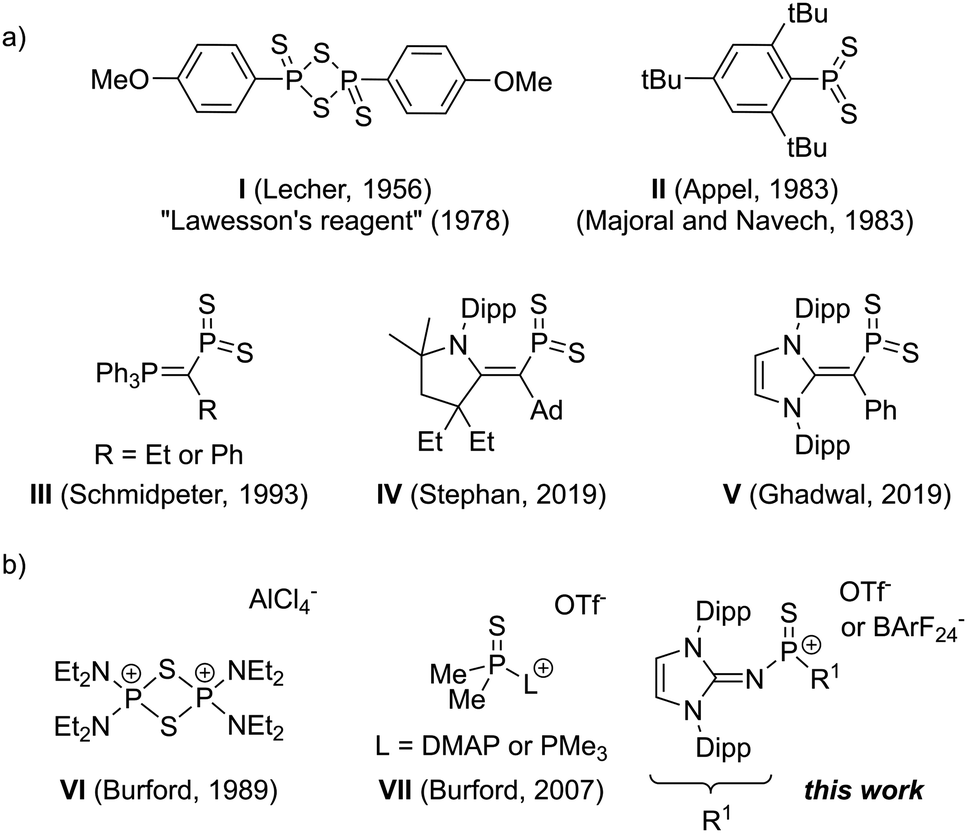 | ||
| Scheme 1 (a) Dimeric and monomeric dithiophosphoranes. (b) Thiophosphonium dimer VI, Lewis base-stabilised thiophosphonium ion VII and the Lewis base-free thiophosphonium presented in this work. | ||
Trigonal planar dithiophosphoranes have a natural tendency to dimerise or trimerise forming dithiadiphosphetanes or trithiatriphosphinanes, in which phosphorus adopts a tetrahedral binding mode. Since the discovery of the potential of dithiophosphoranes as thionating reagents,5 scientists were eager to isolate the monomeric and thus more reactive species. In 1983, the groups of Appel,6 as well as Majoral and Navech,7 synthesised the first monomeric dithiophosphorane II (Scheme 1) by using a very bulky 2,4,6-tri-tert-butylphenyl group, which prevents dimerisation upon blocking both sides of the PS2 plane but decomposes at 110 °C via isomerisation.8,9 The group of Yoshifuji extended the scope of monomeric phenyl-substituted dithiophosphoranes by using amino groups to prevent dimerisation.10 Furthermore, the group replaced one tert-butyl group adjacent to phosphorus in II with a methoxy group, resulting in a dithiophosphorane that is monomeric in solution but dimeric in the solid state.4 Ylide-substituted monomeric dithiophosphoranes III were reported by Schmidpeter and coworkers.11 Very recently, the group of Mardyukov trapped the monomeric form of the Lawesson's reagent in argon matrices at 10 K12 and the groups of Stephan and Ghadwal reported two examples for vinyl-substituted dithiophosphoranes IV and V, respectively.13
The reactivity of main group carbonyl analogues (R2EO) has recently gained considerable interest,14 fueled by the successful isolation of monomeric, Lewis base-free representatives, such as neutral germanones15 and silanones,16 cationic oxophosphonium17 and anionic boryloxy18 compounds. However, heavier monomeric phosphathiocarbonyls, namely thiophosphonium cations [R2PS]+, have not yet been isolated. These cationic species should exhibit a highly electrophilic phosphorus center and a more polar PS double bond compared with the neutral dithiophosphoranes. In fact, the group of Burford showed that these properties translate into a pronounced tendency to form dimers VI19,20 and Lewis-base adducts VII.21 Herein, we report the synthesis and characterisation of a Lewis base-free thiophosphonium ion, which can be regarded as a monomeric, more reactive form of the Lawesson's reagent, as indicated by the efficient transfer of the sulfur atom to both carbonyls and phosphorus compounds.
The synthesis of thiophosphonium salts [4][X] ([X]− = trifluoromethanesulfonate, [OTf]− or tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, [BArF24]−) is based on our synthetic protocol for oxophosphonium salts (Scheme 2).17 Bulky π-donating 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-imine substituents (R1) were employed to provide both kinetic and thermodynamic stabilisation of the electrophilic PS double bond. The reaction of PSCl3 with (R1)SiMe3 (1) upon elimination of Me3SiCl gave the monosubstituted thiophosphoryl chloride (R1)PSCl2 (2). The subsequent reaction of 2 with a second equivalent of 1 gave the disubstituted thiophosphoryl chloride (R1)2PSCl (3) with an overall isolated yield of 71% over both steps. A single-crystal X-ray diffraction (XRD) study of 3 (Fig. 1, left) confirmed the formation of the desired species. Treatment of 3 with AgOTf or NaBArF24 resulted in precipitation of AgCl or NaCl, respectively, and the formation of the corresponding thiophosphonium salts [4][OTf] or [4][BArF24]. The Lewis base-free, trigonal-planar nature of thiophosphonium ion [4]+ is confirmed by the shift of the 31P NMR signal (116.6 ppm) to higher frequency with respect to the thiophosphoryl chloride precursor 3 (25.9 ppm). The choice of the anion (OTf− or BArF24−) does not affect the 31P NMR shift, which suggests that the anion is separated from the phosphorus center in solution. The phosphorus atom of [4]+ is deshielded compared with other imidazoline-2-imine substituted trigonal planar phosphorus(V) cations (iminophosphonium: 79.9 ppm,22 oxophosphonium: 59.1 ppm,17 phosphorandiylium: 50.7 ppm23) but appears at lower frequency compared to the dithiophosphorane II (295.3 ppm).6
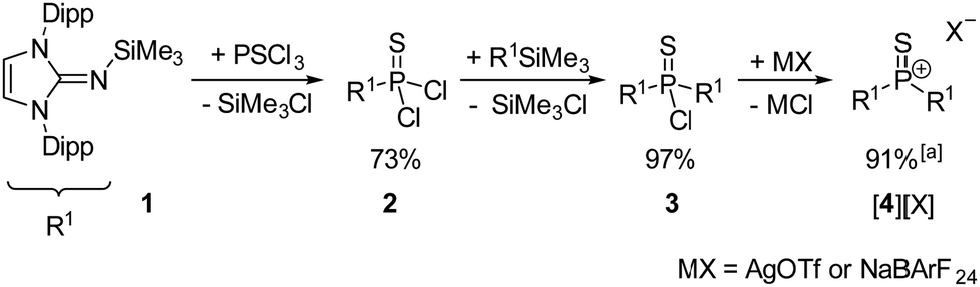 | ||
| Scheme 2 Synthesis of [4][OTf] and [4][BArF24]. Dipp = 2,6-diisopropylphenyl, OTf− = trifluoromethanesulfonate, BArF24− = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate. ayield for MX = NaBArF24. | ||
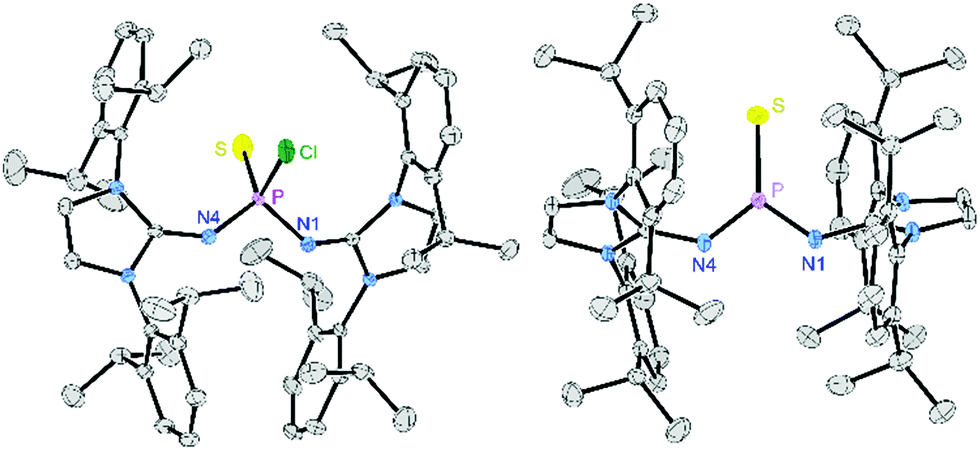 | ||
| Fig. 1 Solid-state structure of 3 (left) and [4][BArF24] (right). Hydrogen atoms, solvent molecules and the anion of [4][BArF24] are omitted for clarity. Ellipsoids are drawn at 50% probability. Only one of the two crystallographically independent molecules of 3 and only one part of the disordered isopropyl group and N2PSCl moiety is depicted. Selected bond lengths [Å] and angles [°] of [4][BArF24]: P–N1 1.5578(12), P–N4 1.5606(12), P–S 1.8967(5), N1–P–N4 108.02(6), N1–P–S 124.79(5), N4–P–S 127.17(5). | ||
An XRD study of single crystals obtained from a concentrated CH2Cl2 solution confirmed that [4][BArF24] is a monomeric, Lewis base-free thiophosphonium salt (Fig. 1, right). In the solid-state structure, the electrophilic N2PS unit is surrounded by the bulky 2,6-diisopropylphenyl (Dipp) substituents and thus well separated from the BArF24− anions. The phosphorus atom is in a perfectly planar environment (sum of angles: 360°) comprising a short P–S bond (1.897 Å), which is in the range of the P–S bonds in dithiophosphorane II (1.90 Å)6 and III (RPh, 1.92 Å and 1.93 Å),11 and shorter than that of the thiophosphonium dimer VI (2.11 Å).19
Density functional theory calculations at the B3LYP-D3BJ/def2-TZVP level of theory were performed on [(R1)2PS]+ ([4]+) and [(R1)2PSe]+ ([5b]+). The results from our previous computations on [(R1)2PO]+ ([5a]+) are included in the following discussion for comparison.17 The frontier orbitals (see Fig. 2 for [5a]+ and [4]+; see S42 in the ESI† for all three analogues) of the three chalcogenophosphonium ions are similar with an increasing contribution of the chalcogen lone pairs to the HOMO for the heavier congeners. The LUMOs of [4]+ and [5b]+ have π* character with equal contributions from the P and S/Se atoms. According to the Wiberg bond indices (WBI) (Table 1), the phosphorus–chalcogen bond in [4]+ has the highest double bond character in this series, which can be rationalised by the similar atom size and thus better orbital overlap of sulfur and phosphorus. The natural bond orbital analysis reveals a concomitant decreasing positive charge for the phosphorus and negative charge for the chalcogen atom in the order O > S > Se, which is associated with a lower polarity of the phosphorus–chalcogen bond. As a common metric to determine the Lewis acidity, the fluoride ion affinities (FIA) were calculated using Christe's method.24 The FIA correlate with the electronegativity of the chalcogen atom and give smaller values for [4]+ (616 kJ mol−1) and [5b]+ (614 kJ mol−1) compared to [5a]+ (634 kJ mol−1),17 ranking the electrophilicity of chalcogenophosphonium ions in the range of phosphenium cations.25 Our attempt to experimentally determine the Lewis acidity of [4]+ using the Gutmann-Beckett parameter26 was unsuccessful since no adduct between POEt3 and [4]+ was detected.
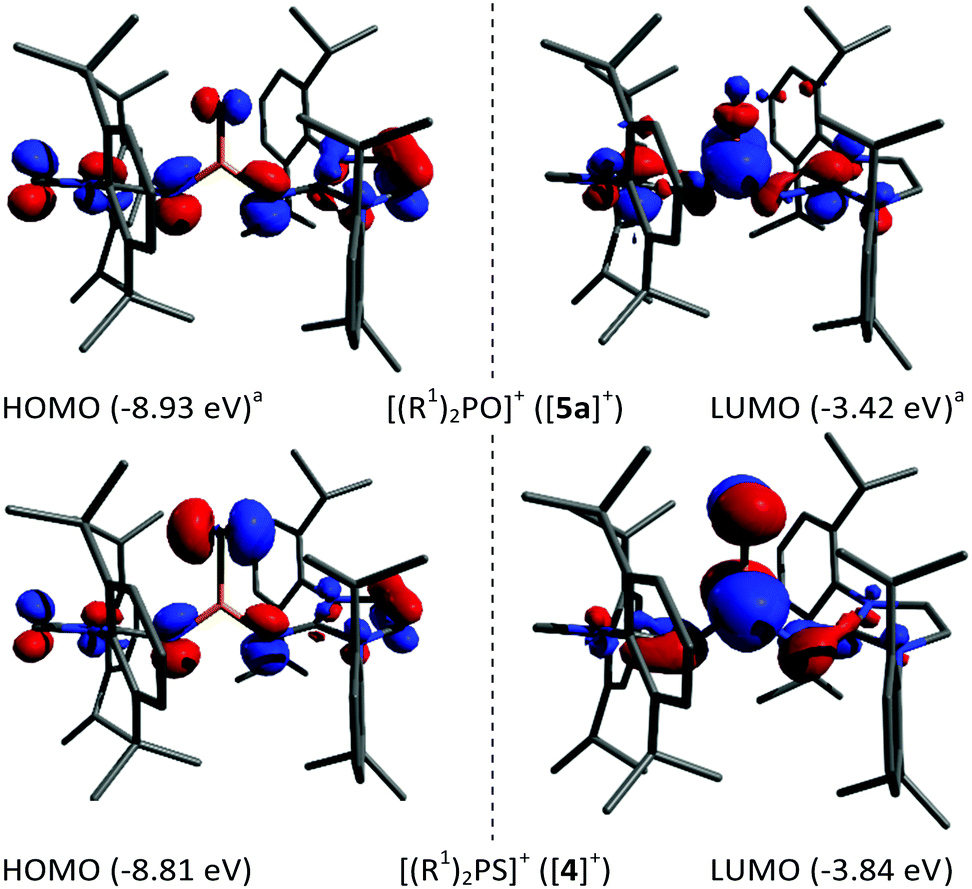 | ||
| Fig. 2 Selected molecular orbitals of [5a]+ (top)17 and [4]+ (bottom) at the B3LYP-D3BJ/def2-TZVP level of theory (±0.05 isosurface). aliterature data.17 | ||
Inspired by the structural similarity of the trigonal planar thiophosphonium ion [4]+ with the monomeric form of the Lawesson's reagent, we wanted to explore the ability of [4]+ to act as thionating reagent.
Preliminary studies were performed by treating [4][BArF24] with different amounts of benzaldehyde which gave mixtures of phosphorus containing products as indicated by 1H and 31P NMR spectroscopy. The stoichiometric reaction resulted in incomplete conversion of [4][BArF24] suggesting that the oxophosphonium ion is formed as an intermediate, which then undergoes subsequent reactions with benzaldehyde or thiobenzaldehyde. The reaction between the literature-known oxophosphonium salt [5a][BArF24]17 and benzaldehyde was confirmed to give a complex mixture of products in a separate experiment (Fig. S29 in the ESI†). To avoid this subsequent reaction, triethylphosphine oxide was used as a trapping reagent, because it readily forms the Lewis base adduct [6a]+ with [5a]+,17 but does not react with [4]+ at room temperature. Indeed, adding one equivalent of benzaldehyde to a solution containing [4][BArF24] and an excess of POEt3 resulted in sulfur atom transfer and formation of [6a][BArF24] in more than 70% yield determined by 31P{1H} NMR (Scheme 3). Thiobenzaldeyde could not be detected by NMR spectroscopy and GC/MS analysis owing to its tendency to oligomerise.27 The thionation of benzaldehyde with [4]+ proceeds within minutes at room temperature, while the same reaction requires refluxing in toluene for 15 hours with the Lawesson's reagent.27 Although this indicates that the thiophosphonium ion [4]+ is very reactive, the carbonyl substrate scope turned out to be rather narrow. N-Methyl-2-pyrrolidinone undergoes thionation to 1-methylpyrrolidine-2-thione at room temperature in 59% yield determined by 31P{1H} NMR. However, other carbonyl compounds such as benzophenone, methyl benzoate, γ-butyrolactone and acetone did not react with [4]+ at room temperature. The limited substrate scope can be rationalised by the steric shielding of the reactive PS double bond. The thionation reacions with the thiophosphonium cation [4]+ are complementary to those with monomeric aryl-substituted dithiophosphoranes reported by the groups of Navech9 and Yoshifuji.4 The reaction is assumed to proceed via a four-membered thiadiphosphetane rings, which is equally conceivable for the cationic system.
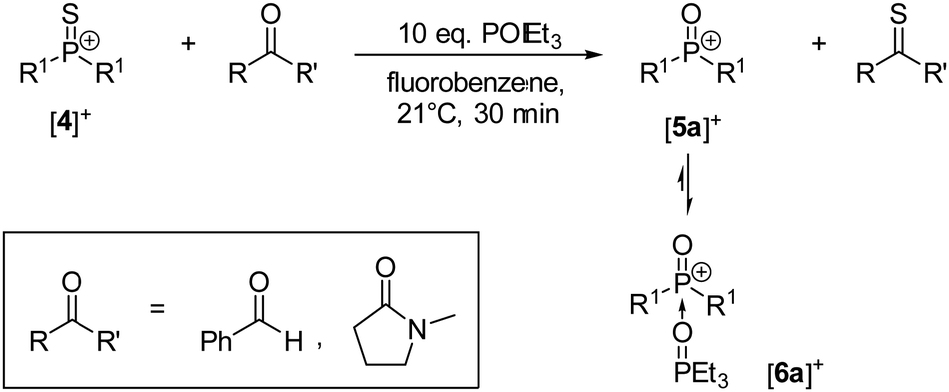 | ||
| Scheme 3 Conversion of carbonyls to thiocarbonyls using [4][BArF24] and POEt3 as trapping reagent. [BArF24]− anions are omitted for clarity. | ||
Given the polar nature of the P–S bond in [4]+, we were wondering if it would undergo bond metathesis reactions with other polar P–X (XO, Se, Te) bonds such as those of Et3PX (Scheme 4).
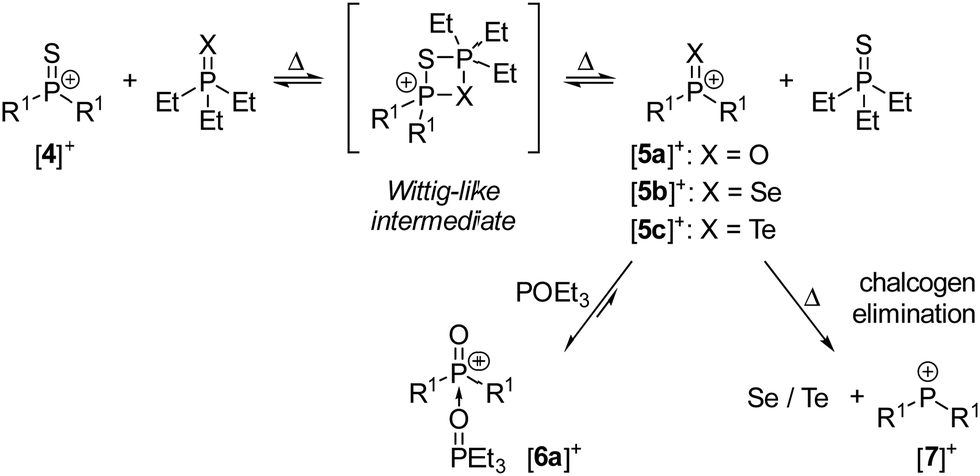 | ||
| Scheme 4 Phosphorus–chalcogen bond metathesis reactions. [BArF24]− anions are omitted for clarity. | ||
The reaction between [4]+ and Et3PX required prolonged heating, consistent with the limited steric access of the P–S bond and the fact that four-membered P2SX ring intermediates are assumed to be formed in the chalcogen exchange process. Heating a fluorobenzene solution of [4][BArF24] and two equivalents of Et3PO at 180 °C for 29 hours resulted in 96% conversion to [6a][BArF24] and Et3PS. The conversion did not change significantly upon further heating of the mixture, suggesting an equilibrium state. Indeed, the same set of compounds was obtained by reacting [5a][BArF24] with Et3PS under equal conditions, which confirms the reversibility of this phosphorus–chalcogen bond metathesis.
The metathesis reaction between [4][BArF24] and Et3PSe gave the selenophosphonium salt [5b][BArF24] and Et3PS. [5b][BArF24] is the second example of a Lewis base-free selenophosphonium salt28 and was unambiguously identified by its 31P NMR resonance at 104.2 ppm, which is in the same range like that of [4]+ (116.6 ppm) and shows characteristic 77Se satellites with an exceptionally large 1JPSe coupling constant of 999 Hz. The corresponding doublet in the 77Se NMR spectrum appears at 119.8 ppm. Schmidpeter and co-workers reported the first selenophosphonium salt, which is stabilised by phosphonium ylidyl groups and has a significantly smaller 1JPSe coupling constant of 847 Hz.28 [5b][BArF24] could not be isolated because it decomposes thermally into the phosphenium ion [7]+ and elemental selenium, as indicated by the characteristic 31P NMR resonance of [7]+ at 307.5 ppm29 and the precipitation of gray selenium. This observation confirms the reported trend for phosphine chalcogenides of decreasing phosphorus–chalcogen bond energies O > S > Se.30 In line with this trend, [4][BArF24] is reduced by Et3PTe to give the phosphenium salt [7][BArF24] and Et3PS with concomitant precipitation of elemental tellurium in quantitative yield. The intermediate formation of tellurophosphonium [5b]+ was not observed since it appears to decompose rapidly at 120 °C.
In conclusion, we have synthesised and isolated the first Lewis base-free thiophosphonium ion [4]+ by using bulky, electron-donating N-heterocyclic imine substituents attached to the phosphorus atom. [4]+ contains a highly electrophilic, trigonal-planar phosphorus center and a polar PS double bond that undergoes thionation reactions with benzaldehyde and N-methyl-2-pyrollidinone at room temperature. The mild reaction conditions of this process also support the notion that Lawesson's reagent requires the dissociation into reactive dithiophosphorane fragments prior to its reaction with carbonyls. In addition, the chalcogen exchange reactions between [4]+ and triethylphosphine chalcogenides demonstrate the potential of trigonal planar phosphorus cations in bond metathesis reactions, which is under current investigation in our laboratory.
This work was funded by the DFG (Emmy Noether program: DI 2054/1-1, IRTG 2027).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. P. Cava and M. I. Levinson, Tetrahedron, 1985, 41, 5061–5087 CrossRef CAS.
- (a) T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2010, 110, 3419–3478 CrossRef CAS PubMed; (b) V. Polshettiwar and M. P. Kaushik, J. Sulfur Chem., 2006, 27, 353–386 CrossRef CAS.
- (a) T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS PubMed; (b) M. Jesberger, T. P. Davis and L. Barner, Synthesis, 2003, 1929–1958 CrossRef CAS.
- M. Yoshifuji, D.-L. An, K. Toyota and M. Yasunami, Tetrahedron Lett., 1994, 35, 4379–4382 CrossRef CAS.
- B. S. Pedersen, S. Scheibye, K. Clausen and S.-O. Lawesson, Bull. Soc. Chim. Belg., 1978, 87, 293–297 CrossRef CAS.
- R. Appel, F. Knoch and H. Kunze, Angew. Chem., Int. Ed. Engl., 1983, 22, 1004–1005 CrossRef.
- J. Navech, J. Majoral and R. Kraemer, Tetrahedron Lett., 1983, 24, 5885–5886 CrossRef CAS.
- J. Navech, M. Revel and R. Kraemer, Tetrahedron Lett., 1985, 26, 207–210 CrossRef CAS.
- J. Navech, M. Revel, R. Kraemer and S. Mathieu, Phosphorus Sulfur Relat. Elem., 1986, 26, 83–89 CrossRef CAS.
- K. Kamijo, A. Otoguro, K. Toyota and M. Yoshifuji, Bull. Chem. Soc. Jpn., 1999, 72, 1335–1342 CrossRef CAS.
- G. Jochem, H. Nöth and A. Schmidpeter, Angew. Chem., Int. Ed. Engl., 1993, 32, 1089–1091 CrossRef.
- A. Mardyukov, D. Niedek and P. R. Schreiner, Chem. Commun., 2018, 54, 2715–2718 RSC.
- (a) L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed; (b) D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. – Eur. J., 2019, 25, 8127–8134 CrossRef PubMed.
- Y. K. Loh and S. Aldridge, Angew. Chem., Int. Ed., 2020, 8626–8648 Search PubMed.
- L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Nat. Chem., 2012, 4, 361–365 CrossRef CAS PubMed.
- (a) I. Alvarado-Beltran, A. Rosas-Sánchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CrossRef CAS PubMed; (b) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS PubMed; (c) R. Kobayashi, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2019, 58, 9425–9428 CrossRef CAS PubMed; (d) A. Rosas-Sánchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 15916–15920 CrossRef PubMed; (e) D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS PubMed.
- M. A. Wünsche, T. Witteler and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef PubMed.
- Y. K. Loh, K. Porteous, M. Á. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CAS.
- N. Burford, R. E. v. H. Spence and R. D. Rogers, J. Am. Chem. Soc., 1989, 111, 5006–5008 CrossRef CAS.
- N. Burford, R. E. v. H. Spence and R. D. Rogers, Dalton Trans., 1990, 3611–3619 RSC.
- J. J. Weigand, N. Burford, D. Mahnke and A. Decken, Inorg. Chem., 2007, 46, 7689–7691 CrossRef CAS PubMed.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808–1815 RSC.
- (a) M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS; (b) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS; (c) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS; (d) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC.
- A. A. El-Kateb and N. M. Abd El-Rahman, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 249–254 CrossRef CAS.
- A. Schmidpeter, G. Jochem, K. Karaghiosoff and C. Robl, Angew. Chem., Int. Ed. Engl., 1992, 31, 1350–1352 CrossRef.
- O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858 RSC.
- M. Bollmark and J. Stawinski, Chem. Commun., 2001, 771–772 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, mass spectrometry data, crystallographic data, and computational details. CCDC 2060578, 2057624 and 2057623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc01273h |
| This journal is © The Royal Society of Chemistry 2021 |