
Atomic defects in monolayer ordered double transition metal carbide (Mo2TiC2Tx) MXene and CO2 adsorption†
Rasoul
Khaledialidusti
 *a,
Abhishek Kumar
Mishra
b and
Afrooz
Barnoush
ac
*a,
Abhishek Kumar
Mishra
b and
Afrooz
Barnoush
ac
aDepartment of Mechanical and Industrial Engineering, Norwegian University of Science and Technology (NTNU), 7491 Trondheim, Norway. E-mail: rasoul.khaledialidusti@ntnu.no
bDepartment of Physics, School of Engineering, University of Petroleum and Energy Studies, Bidholi via Premnagar, Dehradun 248007, India
cCurtin Corrosion Centre, WASM-MECE, Curtin University, Australia
First published on 18th February 2020
Abstract
Transition metal carbides (MXenes) with formula Mn+1CnTx (n = 2 and 3) have been emerging as a new family of two-dimensional (2D) materials that have great potential in electronic applications and CO2 conversion catalysts. It has been already found that the electronic and electrochemical properties of Ti3C2Tx MXenes can be tuned by replacing the two outer titanium layers with molybdenum layers. Similar to other 2D materials, intrinsic defects can be formed in the synthesized MXene flakes and the formation of defects can influence the performance of these materials. Herein, we systematically study the effect of the different types of structural defects on the structural stability, electronic behavior, and electrochemical properties of ordered Mo2TiC2Tx terminated with the specific surface functional groups of fluorine, oxygen, and hydroxide. The calculated defect formation energies imply that the formation of defects is dependent on the surface terminations, where the O-terminated MXenes demand more energy than the F- and OH-terminated MXenes. We found that defect formation is more feasible in the outer molybdenum layers than in the inner titanium layer. Our results predicted that the CO2 molecule adsorbs on the defective surfaces through a spontaneous and exothermic process that is critical to its capture, while the perfect surface weakly attracts the molecule through a nonspontaneous and endothermic process. Thus, our study predicts that the electronic and electrochemical properties of Mo2TiC2Tx can be tuned by forming specific defects and these MXenes could be promising materials for CO2 adsorption and conversion.
1 Introduction
MXenes are 2D materials that have shown great promise in energy storage applications,1 including lithium-ion batteries2,3 and supercapacitors,4 and catalysis.5 Quasi-two-dimensional (2D) Mo2TiC2Tx is an ordered double transition metal MXene, synthesized in 2015,6 in which the outer titanium layers of Ti3C2Tx (the first MXene discovered in 20117) are replaced with molybdenum layers. In this MXene, the titanium layer is sandwiched by the molybdenum layers and carbon atoms occupy the octahedral sites between these two transition metals, as shown in Fig. 1a. It was shown that the electrochemical and electronic properties of the ordered Mo2TiC2Tx MXene are different from those of Ti3C2Tx MXene.6,8,9 While Ti3C2Tx is a metallic-like conductor,8–10 it was confirmed that Mo2TiC2Tx exhibits semiconductor-like transport behavior.6,10 It was predicted that the ordered Mo2TiC2Tx MXene terminated by the F group would be more semiconducting than that terminated by the OH group, while –O termination would provide a metallic behavior.10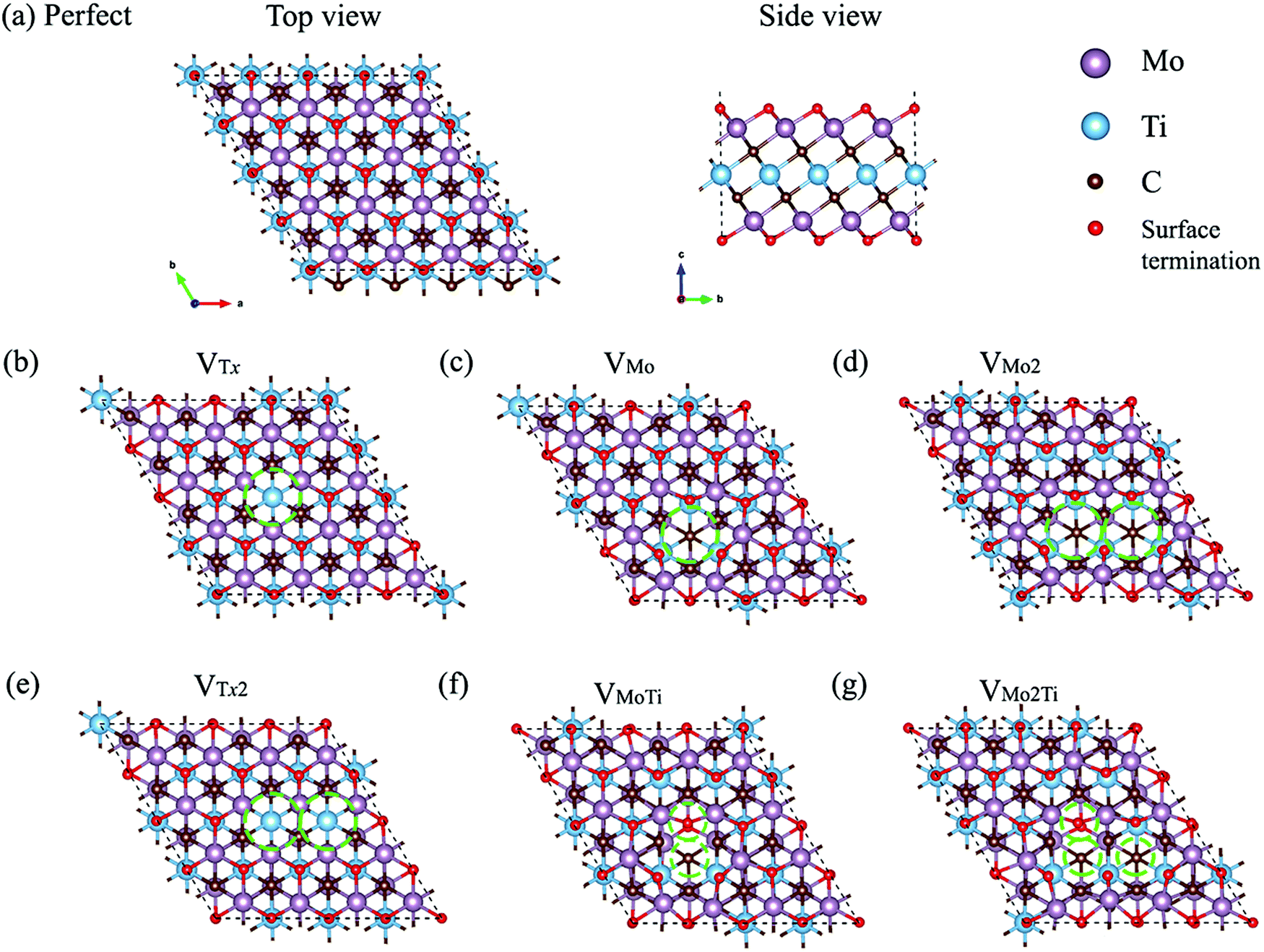 | ||
| Fig. 1 Fully relaxed structural models of different types of defects considered in monolayer ordered Mo2TiC2Tx. (a) Top and side view of the perfect MXene. Top views of (b) VTx, (c) VMo, (d) VMo2, (e) VTx2, (f) VMoTi, and (g) VMo2Ti. | ||
Defect formation was observed in 2D materials, such as graphene,14,20,21 BN,22–24 and MoS2.13,25 It has been well documented that the presence of defects affects the electrical, electrochemical, electronic, optoelectronic, and mechanical properties of these 2D materials.13,14,20,21,23 Intrinsic defects such as atomic vacancies and vacancy clusters were also observed using scanning tunneling microscopy (STM) images in synthesized MXene flakes,16,17 which the synthesis conditions might affect the defect concentrations of MXene flakes.16,18,19 Therefore, the formation of different kinds of defects is unavoidable in MXenes. Similar to other 2D materials, the presence of intrinsic defects might alter the electronic and electrochemical properties of monolayer Mo2TiC2Tx MXenes.
Structural defects in 2D materials could be unfavorable. For example, the presence of defects leads to weakening of the mechanical strength21,26 and the electronic performance of graphene.27 However, certain types of defects could be favorable and provide applicable functionalities by adjusting the local properties. For example, the mechanical properties of graphene could be also increased by controlled defect creation21 or the properties of MoS2 could be tailored by controlled defect formation.13 Compared to these 2D materials, while intrinsic defects were previously observed in MXene flakes,16,17 the influence of defects on the electronic and electrochemical properties of MXenes has not extensively been investigated. From the rare investigations,11,12,16 it was confirmed that defects have unfamiliar effects on the MXene properties compared to other 2D materials depending on the type of defects, reconstruction of the structure, and local chemical bonding.11,12,16 It was predicted that titanium vacancies in Ti3C2Tx MXenes could not alter the metallic conductivity considerably; however, they affect the surface morphology and terminations.16 The effects of carbon vacancies in Ti2CTx indicated the improvement of electronic conductivity and the enhancement of mechanical flexibility.11 It was also investigated that the intrinsic defects in Ti2XTx (X = C, N) MXenes could change the electronic properties from metallic to semiconducting or from semiconducting to metallic depending on the type of defect and the system.12
Previous studies indicate that various MXenes with different transition metals and functional groups might exhibit quite different behavior toward defects because of the comparatively complex structures and variety of chemical bonding between atoms. Despite tuning the electronic and electrochemical properties of Ti3C2Tx MXenes by manipulating the outer transition metal layers,6 which are important for electronics, energy storage, sensors, and other applications, the presence of defects might significantly change the properties of ordered Mo2TiC2Tx MXenes. Therefore, a detailed atomistic understanding of the influence of different types of defects on the electronic and electrochemical properties of ordered Mo2TiC2Tx MXenes is important to explore their real-device applications, in which understanding these properties is crucial.
In this study, we performed first-principles calculations based on density functional theory (DFT) to explore the effects of different types of defects on the structural stability, electronic behavior, and electrochemical properties of ordered Mo2TiC2Tx (T = O, F, and OH). The formation of defects on the top and bottom molybdenum sublayers is more feasible because of the direct contact with the etchant solution; however, the presence of defects in the titanium layer is also possible, which are inherited from the MAX phase. Moreover, cluster-defects were observed within the same sublayer in Ti3C2Tx MXenes.16Fig. 1 shows the optimized defect structures of the seven different types of defects considered in this study, including a single Tx vacancy (VTx) and two adjacent Tx vacancies (VTx2) on both sides of the flake, a single molybdenum vacancy (VMo), two adjacent molybdenum vacancies (VMo2) within the same sublayer, a vacancy complex of a single molybdenum vacancy and one nearby titanium vacancy (VMoTi), a vacancy complex of two adjacent molybdenum vacancies forming within the same sublayer and one nearby titanium vacancy (VMo2Ti), and two molybdenum vacancies forming within two different sublayers (VMoMo). The top view of VMoMo is the same as VMo and is not shown in Fig. 1. The structural stability of all types of defects is investigated through their formation energies. The electronic properties of these defected Mo2TiC2Tx MXenes are also analyzed to highlight the influence of these effects on the conductance of MXenes terminated with different functional groups. The effects of these defects on the electrochemical properties of MXenes are also studied by evaluating the CO2 adsorption performance on Mo2TiC2O2.
2 Computational methods
First-principles calculations based on density functional theory (DFT) are performed using the Vienna ab initio Simulation Package (VASP).28 We employed the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE)29 functional to describe the exchange and correlation of electrons. The projected augmented-wave (PAW)30 potential is applied to take the interaction between the ion-core and valence electrons into consideration and a plane wave basis set with a sufficient energy cutoff of 520 eV is employed to consider valence electrons. A 4 × 4 × 1 supercell is used to model 2D Mo2TiC2Tx (T = F, O, and OH) MXene monolayers to provide at least a 10 Å distance between the point defects. A large vacuum of at least 20 Å is used to avoid any undesirable interaction between an MXene sheet and its periodically repeated images along the c-axis. The Brillouin zone is sampled with 5 × 5 × 1 Monkhorst–Pack31k-points for structural optimizations and with a 10 × 10 × 1 mesh for electronic structure analysis. The atomic positions and lattice constants are fully optimized using the conjugate gradient method. The geometries of the MXenes are relaxed until the forces acting on each atom become less than 0.001 eV Å−1. The criterion for energy convergence is 10−6 eV per cell. The partial occupancies are determined using the Methfessel–Paxton smearing scheme with a smearing width of 0.1 eV.32 We employed the spin-dependent GGA plus Hubbard U (GGA + U)33 methodology to apply the correlation effects in transition metals of Mo and Ti with a value of U = 4 eV, which has been applied for Mo2TiC2Tx MXenes10 and extensively applied for their oxides.34,35 We performed spin-polarized DFT calculations with the antiferromagnetic (AFM) configurations predicted by Anasori et al.10 for the perfect Mo2TiC2Tx MXenes chemically terminated by different terminations. We also studied the probable effect of the defects on the AFM configurations of the MXenes. However, our results show that the considered defects cannot change the AFM configurations predicted for the perfect MXenes.We calculated the formation energies of defects as 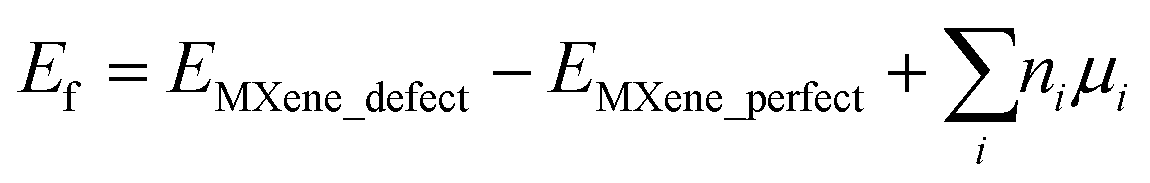 , where EMXene_defect and EMXene_perfect are the total energies of the MXene supercell with specific defects and related perfect structure without defects, respectively, ni is the number of removed atoms, and μi is the chemical potential of the removed atom(s). Since it is not straightforward to obtain the exact value of μi, which is dependent on pressure and temperature,36 we calculated Ef with the chemical potential between −2.0 and 0.0 eV.
, where EMXene_defect and EMXene_perfect are the total energies of the MXene supercell with specific defects and related perfect structure without defects, respectively, ni is the number of removed atoms, and μi is the chemical potential of the removed atom(s). Since it is not straightforward to obtain the exact value of μi, which is dependent on pressure and temperature,36 we calculated Ef with the chemical potential between −2.0 and 0.0 eV.
The adsorption energy of the CO2 molecule is calculated as Eads = EMxene+mol − (EMXene + Emol) by taking van der Waals (vdW) interactions into account by employing the dispersion-corrected DFT-D2 scheme,37 which is essential for the accurate description of the interaction between CO2 and a surface.38–40EMxene+mol is the total energy of the adsorbate–substrate system; EMXene is the energy of the MXene monolayer; and Emol is the energy of the isolated CO2 molecule. We investigated the CO2 adsorption on the MXene by placing the molecule close to different sites of the monolayer parallel and perpendicular in different possible directions to find the largest adsorption energy. Negative (positive) values of the adsorption energy indicate an exothermic (endothermic) process.
3 Results and discussion
3.1 Formation energy of defects
We calculated the formation energy of different types of defects considered (see Fig. 1) in both non-terminated and terminated ordered MXenes to better understanding the energetic costs of the formation of defects. We first evaluated these energies for non-terminated Mo2TiC2 to gain more information about the feasibility of forming defects during the first step of exfoliation when the flakes are not terminated. Then, those energies are presented for terminated MXenes with specific functional groups. Since a defect formation energy is expected to be positive, it costs (consumes) energy to make a defect.Our results for the non-terminated MXene (Fig. 2a) indicate that the formation energies of VMo are lower than the other types of defects, varying from 6.65 to 8.65 eV with the chemical potential between −2.0 and 0.0 eV. The formation of VMo2 consumes approximately the same amount of energy as VMoMo and almost twice as much energy as VMo, which indicates a linear relationship between the formation energy of defects and the number of VMo. This linear relationship was also found for titanium vacancy clusters with up to six titanium vacancies from the outer sublayer of the Ti3C2 MXene.16 Moreover, VMoTi and VMo2Ti defect formation is more energy consuming than that of other types of defects. The results show that defect formation in the titanium sublayer is energetically more difficult than within the outer molybdenum sublayers. Our obtained results can be confirmed by the study of Sang et al.,16 where they calculated the formation energy of a single titanium vacancy from the outer (2.85 eV) and inner (6.49 eV) sublayer of a 4 × 5 × 1 supercell of the Ti3C2 MXene.
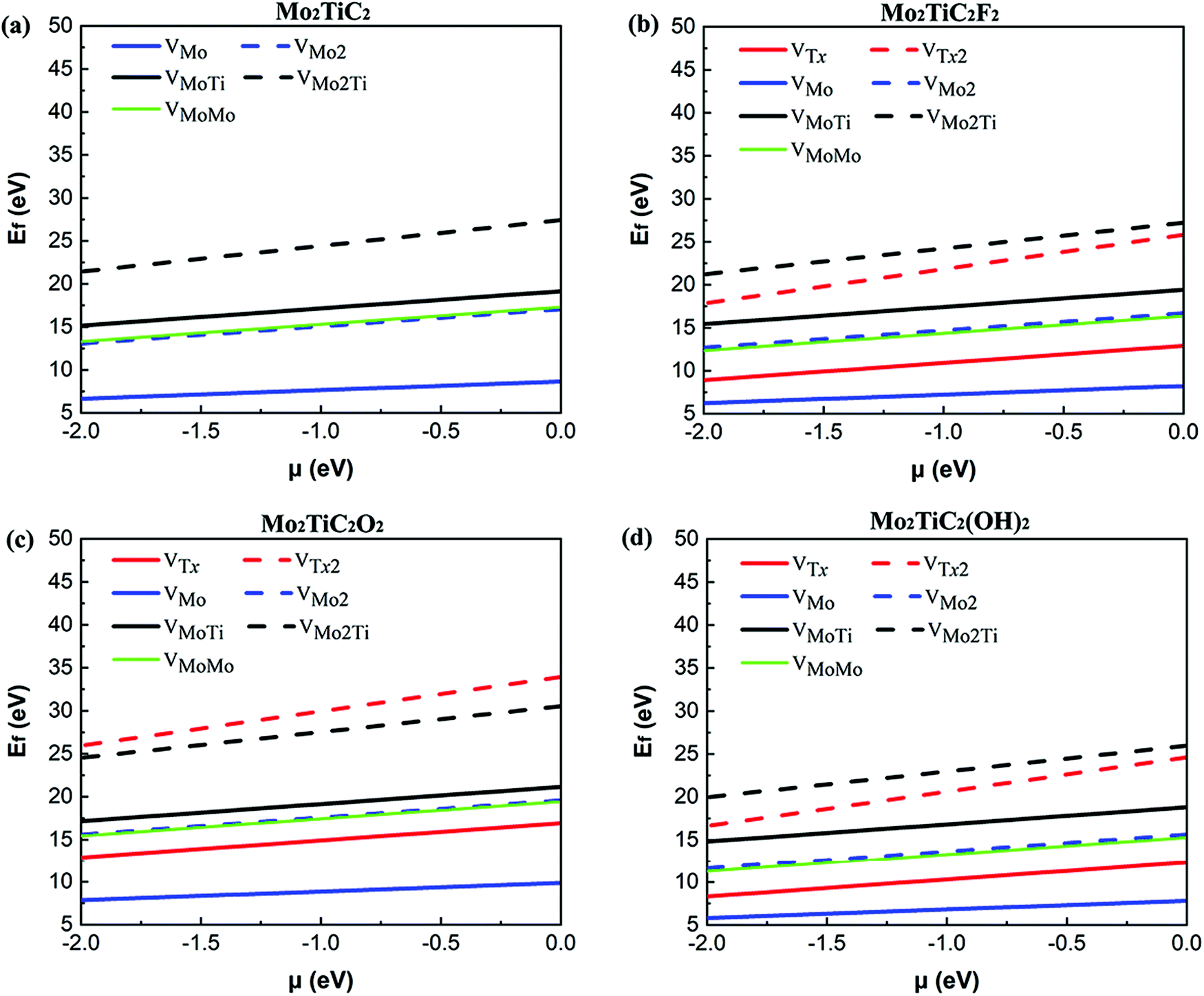 | ||
| Fig. 2 Formation energies of all types of defects as a function of the chemical potential of the removed atom(s) in the range of −2 eV < μ < 0 eV in (a) Mo2TiC2, (b) Mo2TiC2F2, (c) Mo2TiC2O2, (d) and Mo2TiC2(OH)2. | ||
The effect of the surface functions on the calculated defect formation energies (Fig. 2b–d) indicates relatively the same behavior as determined for the pristine Mo2TiC2 MXene. The formation of VMo, VMo2, VMoTi, VMo2Ti, and VMoMo on the MXene terminated by oxygen (Fig. 2c) is more energy consuming than that for the pristine non-terminated MXene (Fig. 2a), while the energetic costs of these defects on the MXenes terminated by fluorine and hydroxide (Fig. 2b and d) are comparable with those on the pristine MXene (Fig. 2a). This mainly originates from the effect of the surface functions on the bonding strength of the MXenes. It was comprehensively predicted that the bond strength between the atoms in the MXenes terminated by F and OH is relatively similar since the degree of covalency in both surface functions is the same (one electron), but the binding between atoms is stiffer in the MXenes functionalized by O atoms than those terminated by F or OH.8 This is mainly because O atoms share two electrons to form a covalent bond with the transition metals. Therefore, the F and O atoms make effectively single and double bonds with the surface, respectively, and that is why O atoms make stronger bonds with the surface. F and OH groups receive similarly one electron from the surface and that is why the bond strength of M–F and M–OH bonds is almost of the same order. The higher formation energy of a titanium vacancy on Tin+1CnTx (n = 1, 2) MXenes terminated by oxygen than that for pristine and –F and –OH terminated MXenes was also predicted.12,16 Moreover, the calculated formation energies demonstrate that fluorine and hydroxide defect formation are comparable and are much more feasible than oxygen defect formation. It is worth mentioning that the formation of functional group atoms is considered on both sides of the flake because of the direct contact of the flakes with the etchant solution.
To further explore the effect of the replacement of the outer titanium layers in Ti3C2Tx MXenes by molybdenum layers on the defect formation energies, we compared our calculated formation energies of VMo on a 4 × 4 × 1 supercell of Mo2TiC2Tx MXenes with the calculated formation energies of VTi from the outer titanium sublayers of a 4 × 5 × 1 supercell of Ti3C2Tx MXenes.16 We scaled linearly the calculated formation energies of VTi to provide the same vacancy concentration considered in our study since a linear relationship between the formation energies of VTi clusters and the number of VTi was predicted. The scaled formation energies of VTi in the pristine Ti3C2 MXene (Ti3C2–VTi: 3.56 eV) and terminated Ti3C2Tx MXenes (Ti3C2F2–VTi: 4.53, Ti3C2O2–VTi: 9.68, and Ti3C2(OH)2–VTi: 4.34) indicate that the replacement of the outer titanium layers in Ti3C2Tx MXenes by molybdenum layers makes the formation of defects more difficult in the non-terminated Mo2TiC2 MXene and MXenes terminated by fluorine and hydroxide; however, the formation of defects in the MXene terminated by oxygen functional groups is comparable. This is because of the fact that Mo–Mo bonds are stronger than Ti–Ti bonds.41 Thus, based on our study, the energetic costs of the formation of defects become higher by manipulating the outer titanium layers in Ti3C2Tx MXenes by molybdenum layers because of the intrinsic bond strengths in these two MXene systems and VMo is the most probable defect to form during the synthesis of MXene monolayers.
3.2 Electronic properties
Before evaluating the electronic properties of defected Mo2TiC2Tx MXenes, we present the electronic properties of perfect Mo2TiC2Tx MXenes with different terminations. The total densities of states (DOS) of the perfect MXenes are displayed in Fig. 3. Similar to all predicted pristine mono-M element MXenes (M is a transition metal), which are metallic,8 the results indicate that the pristine ordered Mo2TiC2 MXene is also metallic (Fig. 3a). It was predicted that some of the mono-M element MXenes with surface terminations could exhibit semiconducting behavior depending upon the participating components and surface terminations.8 Here, we consider a specific surface termination (e.g., pure –F, –O or –OH) and our results show that ordered Mo2TiC2Tx MXenes terminated by F and OH become semiconducting (Fig. 3b and d) and the band-gap of the OH-terminated MXene is smaller than that of the F-terminated MXene, while the O-terminated MXene exhibits metallic behavior (Fig. 3c). Our results are in reasonable agreement with an earlier study by Anasori et al.,10 where they also investigated the electronic structure of MXenes with mixed terminations since it was observed from EDX analysis that the synthesized surface has a mixture of –F, –O, and –OH terminations. Their results indicated that the synthesized Mo2TiC2Tx MXenes have a semiconducting behavior, except for the MXene terminated by high concentrations of oxygen.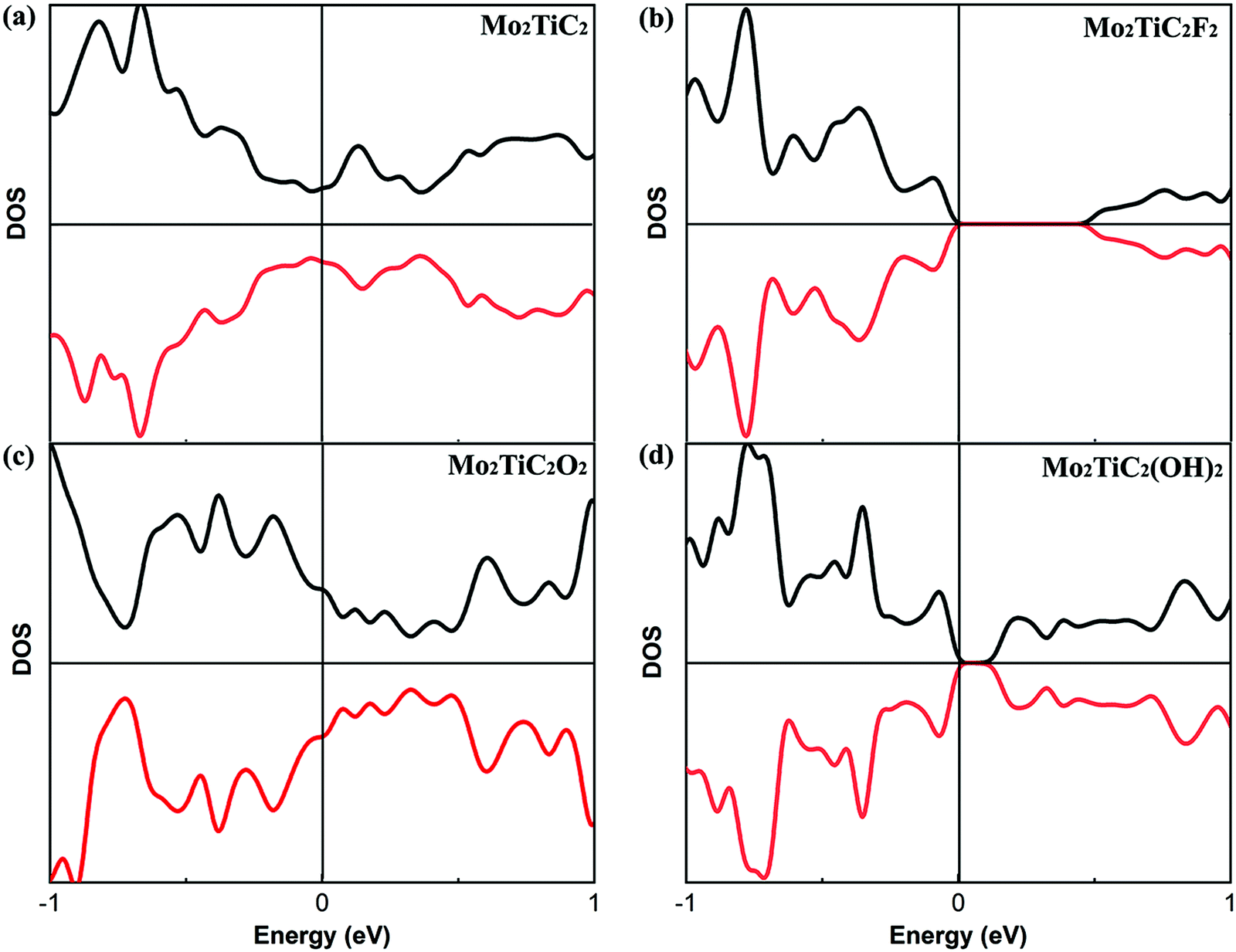 | ||
| Fig. 3 Calculated total density of states (DOS) of perfect MXenes. (a) Pristine MXene, (b) Mo2TiC2F2, (c) MoTiC2O2, and (d) Mo2TiC2(OH)2. Black and red lines show spin-up and -down states, respectively. | ||
The effect of different types of defects on the electronic properties of non-terminated and terminated ordered MXenes is evaluated. Our calculated results of the total DOS of the non-terminated MXene (Fig. S1, ESI†) indicate that the defective Mo2TiC2 MXene is still metallic. Fig. 4 presents the DOS of the different types of defected MXenes under investigation, while for all different surface terminated MXenes (F, O and OH), it is very clear that the VTx vacancy makes these MXenes again metallic as we can observe states due to vacancies crossing the Fermi energy levels in these structures (Fig. 4a, e and i). For VMO and VMoMo vacancies, we do not observe any significant changes in the electronic DOS of the structures; for F and OH terminations, the MXenes are semiconducting, while for O terminations the structure retains its metallic nature. From the other defected MXenes, we can conclude that there are very minor changes in the electronic states, making the structures more semiconducting due to shifting of the bands away from the Fermi energy level.
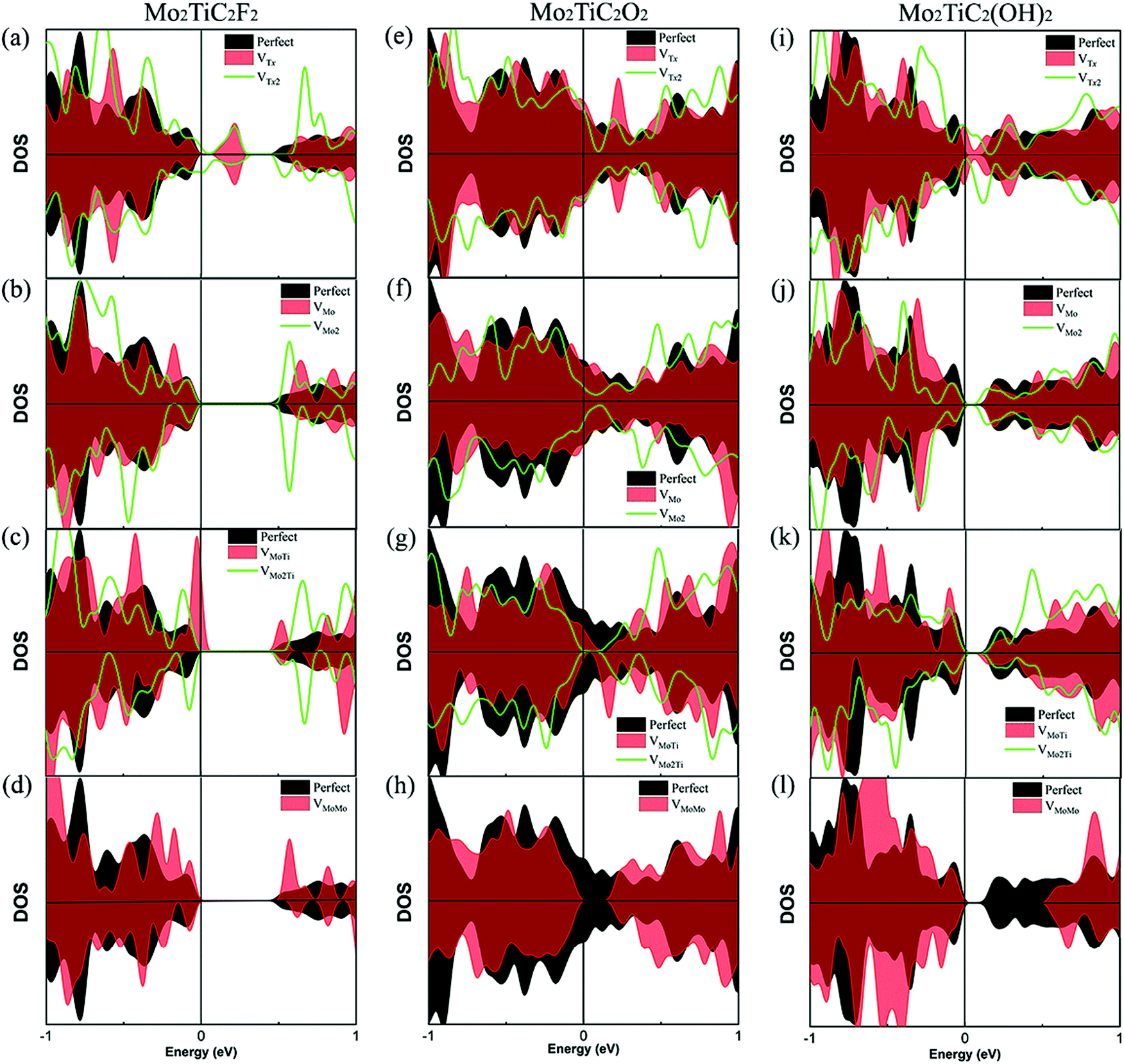 | ||
| Fig. 4 Effect of different types of defects on the DOS of MXenes with different terminations. (a–d) Mo2TiC2F2, (e–h) Mo2TiC2O2, and (i–l) Mo2TiC2(OH)2. The type of defect is shown in the different panels. | ||
3.3 CO2 adsorption on Mo2TiC2O2 defects
The interactions of different molecules with defective structures such as TiC2,15 TiO2,42 graphene and silicene,43 BCN,44 and MoS245–49 demonstrated that the molecular adsorption on the surfaces could be enhanced in the presence of defects. Defects act as electron trap sites and materials with defects have been found to have stronger adsorption energies for CO2 molecules. Very recently, we theoretically studied the adsorption of CO2 on BCN and graphene and found that a CO2 molecule binds with an adsorption energy of −18.91 kJ mol−1 and −13.19 kJ mol−1 for these two 2D materials, respectively.50 We also found that B-rich BCN samples exhibit significantly enhanced adsorption energies of CO2. Earlier, we investigated the effect of Stone–Wales (SW) defects on the adsorption energy of CO2 on BCN and found that SW defects act as strong adsorption sites for CO2.44 In this section, the influence of the considered defects on the reactivity and electrochemical transport properties of Mo2TiC2Tx MXenes is investigated by modeling the interaction of a CO2 molecule on the monolayer. The synthesized MXenes have a mixture of –O, –F, and –OH terminations and the type of surface terminations and the percentage of each type depend on the etching and delamination conditions. Here, we only consider the MXene terminated by oxygen surface functions since the MXenes are more likely functionalized with a higher percentage of oxygen than fluorine and hydroxide.6,10 We first evaluate the capability of a 3 × 3 × 1 supercell of the clean Mo2TiC2O2 MXene for CO2 adsorption. Then, we discuss the reactivity of defected Mo2TiC2O2 MXenes with a CO2 molecule.Our calculated results indicate that the CO2 molecule is physisorbed on the perfect Mo2TiC2O2 MXene, with a nonspontaneous reaction energy of 0.21 eV (see Fig. 5a). The carbon atom of CO2 interacts with the oxygen surface functions and the molecule is placed at a distance of 2.89 Å while it is not significantly tilted. Moreover, a comparative nonspontaneous Gibbs free reaction energy (at 298.15 K) of 0.23 eV for the physisorption of a CO2 molecule on the Mo3C2O2 MXene was calculated.51 Thus, we can realize that Mo-containing MXenes are weakly reactive for CO2 capture.
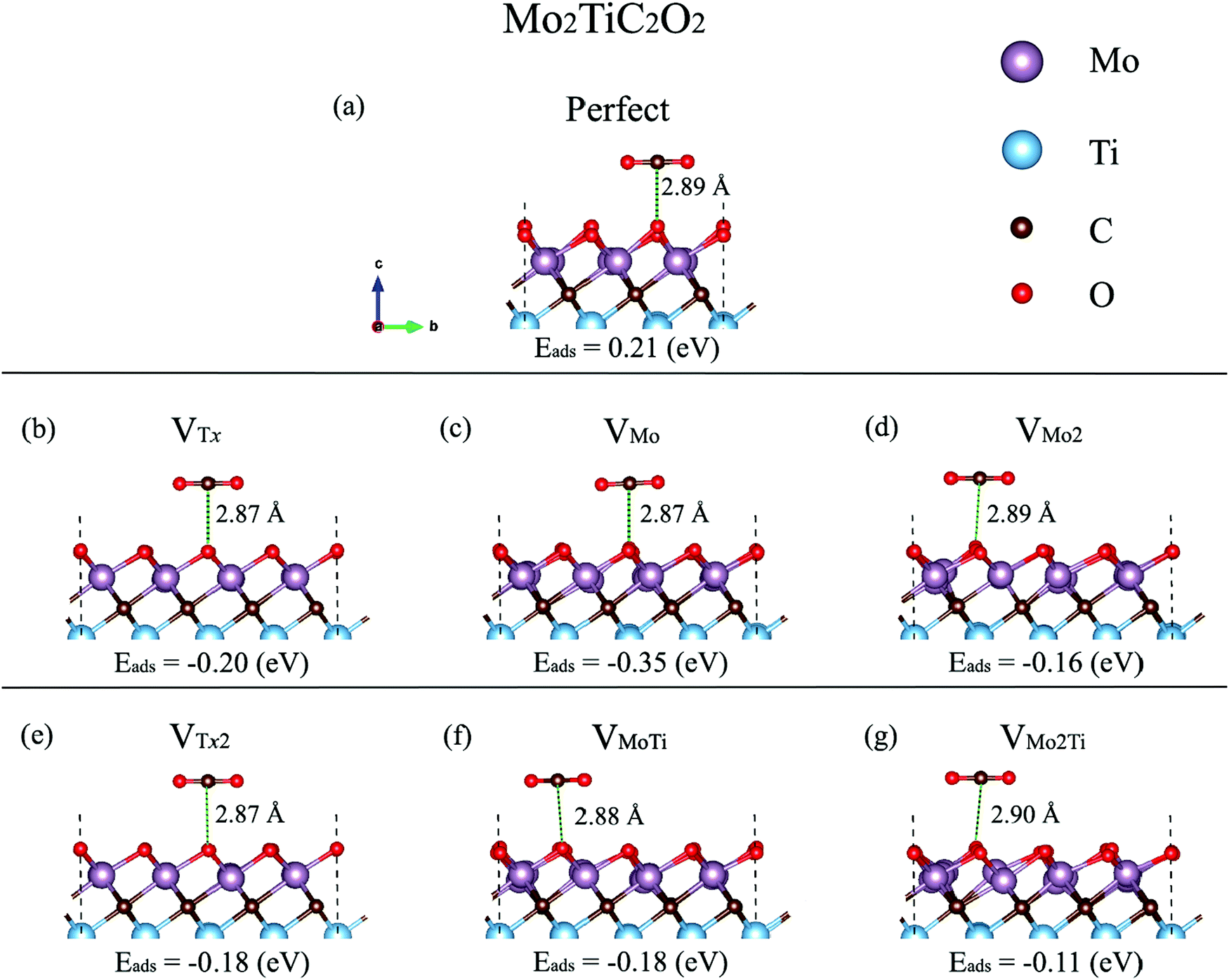 | ||
| Fig. 5 Relaxed adsorption configurations of a CO2 molecule on the perfect and defected Mo2TiC2O2 MXene. (a) Perfect MXene, (b) MXene–VTx, (c) MXene–VMo, (d) MXene–VMO2, (e) MXene–VTx2, (f) MXene–VMoTi, and (g) MXene–VMo2Ti. | ||
We further evaluated the proficiency of defected Mo2TiC2O2 MXenes for CO2 adsorption. Before that, we analyzed the electron localization function (ELF) over the surfaces, which provides a qualitative understanding of the empirical concept of electron localization and the behavior of the electrons over the defective surfaces, as shown in Fig. 6. The calculated ELFs for the non-terminated MXene and MXenes terminated by fluorine and hydroxide are shown in Fig. S2–S4 (ESI†), respectively. The ELF is defined in the range between 0 and 1, where the upper limit of ELF = 1 exhibits perfect localization and ELF = 0.5 corresponds to a uniform electron gas. Here, we showed ELFs in the range between 0 and 0.5 since perfect localization was not detected over the surfaces. As it is shown in Fig. 6, electrons are generally localized on the surface functional groups over the MXene. In the case of VTx and VTx2, electrons are completely delocalized on the place of the removed oxygen atom(s) and freely move to other oxygen atoms on the surface. However, in other types of defects, electrons are partially delocalized on the oxygen atoms close to the removed atoms and move to other places. Based on the ELF analysis, we placed the CO2 molecule close to different sites of the monolayer, in which electrons are more localized, parallel and perpendicular in different possible directions to find the largest adsorption energy in defective MXenes.
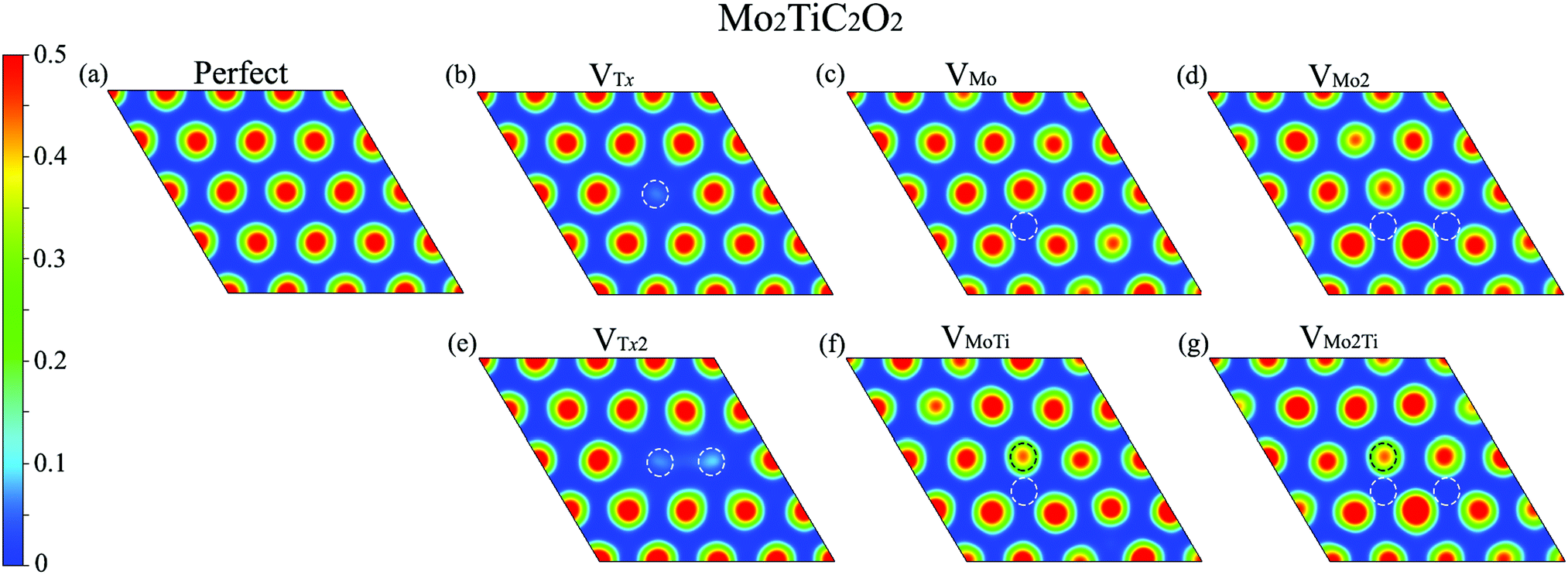 | ||
| Fig. 6 Electron localization function (ELF) on the plane perpendicular to the c-axis at a close distance on top of surface terminations of the defected Mo2TiC2O2 MXenes. Dashed circle lines highlight the place of the removed atoms. | ||
Our calculated adsorption energies (Fig. 5) imply that the molecule spontaneously adsorbs on the defective surfaces with adsorption energies between −0.11 and −0.35 eV, hence indicating that the molecule is trapped in the defect. The molecule is placed at distances between 2.87 and 2.90 Å and is not considerably tilted, relatively similar to the case of the perfect MXene. The adsorption energies on the defective surfaces including VTx and VTx2 are comparative (−0.20 and −0.18 eV, respectively). The molecule is highly attracted by the defect formed by a single Mo atom with a spontaneous reaction energy of −0.35 eV, while our results show a spontaneous reaction energy of −0.16 eV for MXene–VMo2. Moreover, the results indicate that a Ti vacancy decreases the proficiency of MXene–VMo and MXene–VMo2 in CO2 adsorption from −0.35 to −0.18 eV in MXene–VMo and from −0.16 to −0.11 eV in MXene–VMo2. We also performed Bader charge analyses of the perfect and defected MXene–CO2 systems to quantify the electron transfer from the MXenes to the CO2 molecule, but we observed that the charge transfer to the CO2 molecule is quite negligible since the molecule is not chemisorbed on the surfaces. The electron density difference isosurface plot for the case of the MXene–VTx surface, as an example, is shown in Fig. S5 (ESI†).
4 Conclusion
In the present study, we have investigated atomic defects in ordered Mo2TiC2Tx terminated with specific surface functional groups of fluorine, oxygen or hydroxide. Different atomic vacancy defects in the top and sublayer of the MXenes have been studied. Through defect formation energy calculations, we conclude that the defects are dependent on the surface terminations, where the O-terminated MXenes demand more energy than the F- and OH-terminated MXenes; also the defect formation is more feasible in the outer molybdenum layers than in the inner titanium layer. Meanwhile, DOS calculations indicate that there are minor changes in the electronic states with a shift in states away from the Fermi energy level, making the system more semiconducting.Our results of the CO2 molecule adsorption indicate the potential catalytic properties of the Mo2TiC2Tx MXenes and the catalytic properties of the MXenes can be enhanced by forming defects in the monolayer. While a CO2 molecule adsorbs on the perfect O-terminated MXene through a nonspontaneous and endothermic process, defect formation on the monolayer leads to direct CO2 interactions with the surfaces through a spontaneous and exothermic process that is crucial to its capture. Our study shows that defect formation on the monolayer cannot activate the CO2 molecule since the bond angle ∠O–C–O is not considerably changed. Our study unveils a route to the possibility of enhancing the catalytic properties of Mo2TiC2Tx MXenes by the formation of specific defects in the monolayer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge that they greatly appreciate the financial support from VISTA which is a basic research program in collaboration between the Norwegian Academy of Science and Letters, and Equinor. The authors would also like to thank the Department of Mechanical and Industrial Engineering at the Norwegian University of Science and Technology (NTNU). The authors also acknowledge generous grants of high-performance computer time from both Vilje and UNINETT Sigma. One of the authors (AKM) acknowledges the SEED grant from the University of Petroleum and Energy Studies (UPES).References
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- M. Zhao, M. Torelli, C. E. Ren, M. Ghidiu, Z. Ling, B. Anasori, M. W. Barsoum and Y. Gogotsi, Nano Energy, 2016, 30, 603–613 CrossRef CAS.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- Y. Peng, B. Akuzum, N. Kurra, M. Zhao, M. Alhabeb, B. Anasori, E. C. Kumbur, H. N. Alshareef, M. Ger and Y. Gogotsi, Energy Environ. Sci., 2016, 9, 2847–2854 RSC.
- G. Fan, X. Li, Y. Ma, Y. Zhang, J. Wu, B. Xu, T. Sun, D. Gao and J. Bi, New J. Chem., 2017, 41, 2793–2799 RSC.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Khazaei, A. Ranjbar, M. Arai and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- R. Khaledialidusti, B. Anasori and A. Barnoush, Phys. Chem. Chem. Phys., 2020, 22(6), 3414–3424 RSC.
- B. Anasori, C. Shi, E. J. Moon, Y. Xie, C. A. Voigt, P. R. C. Kent, S. J. May, S. J. L. Billinge, M. W. Barsouma and Y. Gogotsi, Nanoscale Horiz., 2016, 1, 227–234 RSC.
- T. Hu, X. Wang and X. Wang, Phys. Chem. Chem. Phys., 2017, 19, 31773–31780 RSC.
- A. Bandyopadhyay, D. Ghosh and S. K. Pati, Phys. Chem. Chem. Phys., 2018, 20, 4012–4019 RSC.
- W. Zhou, X. Zou, S. Najmaei, Z. Liu, Y. Shi, J. Kong and J. Lou, Nano Lett., 2013, 13, 2615–2622 CrossRef CAS PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- W. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef PubMed.
- X. Sang, Y. Xie, M. Lin, M. Alhabeb, K. L. Van Aken, Y. Gogotsi, P. R. C. Kent, K. Xiao and R. R. Unocic, ACS Nano, 2016, 10, 9193–9200 CrossRef CAS PubMed.
- L. H. Karlsson, J. Birch, J. Halim, M. W. Barsoum and P. O. Å. Persson, Nano Lett., 2015, 15, 4955–4960 CrossRef CAS PubMed.
- J. Halim, S. Kota, M. R. Lukatskaya, M. Naguib, M. Zhao, E. J. Moon, J. Pitock, J. Nanda, S. J. May, Y. Gogotsi and M. W. Barsoum, Adv. Funct. Mater., 2016, 2, 3118–3127 CrossRef.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- L. Vicarelli, S. J. Heerema, C. Dekker and H. W. Zandbergen, ACS Nano, 2015, 9, 3428–3435 CrossRef CAS PubMed.
- G. López-polín, C. Gómez-navarro, V. Parente, M. I. Katsnelson, F. Pérez-murano and J. Gómez-herrero, Nat. Phys., 2015, 11, 26 Search PubMed.
- D. Wong, J. V. Jr, L. Ju, J. Lee, S. Kahn, H. Tsai, C. Germany, T. Taniguchi, K. Watanabe, A. Zettl, F. Wang and M. F. Crommie, Nat. Nanotechnol., 2015, 10, 949–953 CrossRef CAS PubMed.
- Y. Liu, X. Zou and B. I. Yakobson, ACS Nano, 2012, 6, 7053–7058 CrossRef CAS PubMed.
- R. Bourrellier, S. Meuret, A. Tararan, O. Ste, M. Kociak, L. H. G. Tizei and A. Zobelli, Nano Lett., 2016, 16, 4317–4321 CrossRef CAS PubMed.
- M. Amani, D. Lien, D. Kiriya, J. Xiao, A. Azcatl, J. Noh, S. R. Madhvapathy, R. Addou, M. Dubey, K. Cho, R. M. Wallace, S. Lee, J. He, X. Zhang, E. Yablonovitch and A. Javey, Science, 2015, 350, 1065–1068 CrossRef CAS PubMed.
- Y. Wei, J. Wu, H. Yin, X. Shi, R. Yang and M. Dresselhaus, Nat. Mater., 2012, 11, 759–763 CrossRef CAS PubMed.
- O. V. Yazyev and S. G. Louie, Nat. Mater., 2010, 9, 806–809 CrossRef CAS PubMed.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 11–19 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 5188–5192 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. Nolan, S. D. Elliott, J. S. Mulley, R. A. Bennett and M. Basham, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235424 CrossRef.
- G. Hautier, S. P. Ong, A. Jain, C. J. Moore and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155208 CrossRef.
- W.-X. Li, C. Stampfl and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 165412 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Khaledialidusti, A. K. Mishra and A. Barnoush, ACS Omega, 2019, 4(14), 15935–15946 CrossRef CAS PubMed.
- R. Khaledialidusti, A. K. Mishra and A. Barnoush, J. Chem. Phys., 2018, 149, 224702 CrossRef PubMed.
- A. K. Mishra, A. Roldan and N. H. De Leeuw, J. Phys. Chem. C, 2016, 120, 2198–2214 CrossRef CAS.
- M. Khazaei, A. Ranjbar, D. Bogdanovski and R. Dronskowski, Phys. Chem. Chem. Phys., 2018, 20, 8579–8592 RSC.
- O. Bikondoa, C. L. Pang, R. Ithnin, C. A. Muryn, H. Onishi and G. Thornton, Nat. Mater., 2006, 5, 189–192 CrossRef CAS.
- H. H. Gürel, V. O. O. Zçelik and S. Ciraci, J. Phys. Chem. C, 2014, 118, 27574–27582 CrossRef.
- N. Kumar, K. Moses, K. Pramoda, S. N. Shirodkar, A. K. Mishra, U. V. Waghmare, A. Sundaresana and C. N. R. Rao, J. Mater. Chem. A, 2013, 1, 5806–5821 RSC.
- S. Cho, S. J. Kim, Y. Lee, J. Kim, W. Jung, H. Yoo, J. Kim, H. Jung, B. Engineering, A. Technology, A. Development and D. Development, ACS Nano, 2015, 9, 9314–9321 CrossRef PubMed.
- L. Qi, Y. Wang, L. Shen and Y. Wu, Appl. Phys. Lett., 2016, 1089, 063103 CrossRef.
- B. Akdim, R. Pachter and S. Mou, Nanotechnology, 2016, 27, 185701 CrossRef PubMed.
- H. Li, M. Huang and G. Cao, Phys. Chem. Chem. Phys., 2016, 18, 15110–15117 RSC.
- C. Gonzalez, B. Biel and Y. Dappe, Phys. Chem. Chem. Phys., 2017, 19, 9485 RSC.
- A. Kumar and S. Mishra, J. Mol. Graphics Modell., 2019, 93, 107446 CrossRef PubMed.
- N. Li, X. Chen, W. Ong, D. R. Macfarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc06046d |
| This journal is © The Royal Society of Chemistry 2020 |