
Stretchable and tough conductive hydrogels for flexible pressure and strain sensors
Zhenwu
Wang
 ab,
Yang
Cong
c and
Jun
Fu
*a
ab,
Yang
Cong
c and
Jun
Fu
*a
aSchool of Materials Science and Engineering, Sun Yat-sen University, 135 Xingang Road West, Guangzhou 510275, China. E-mail: fujun8@mail.sysu.edu.cn
bInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology, Hermann-von-Helmholtz Platz 1, 76344, Eggenstein-Leopoldshafen, Germany. E-mail: zhenwu.wang@kit.edu
cCollege of Materials Science and Chemical Engineering, Ningbo University of Technology, 201 Fenghua Road, Ningbo 315211, China
First published on 28th January 2020
Abstract
Flexible pressure and strain sensors have great potential for applications in wearable and implantable devices, soft robotics and artificial skin. Compared to flexible sensors based on filler/elastomer composites, conductive hydrogels are advantageous due to their biomimetic structures and properties, as well as biocompatibility. Numerous chemical and structural designs provide unlimited opportunities to tune the properties and performance of conductive hydrogels to match various demands for practical applications. Many electronically and ionically conductive hydrogels have been developed to fabricate pressure and strain sensors with different configurations, including resistance type and capacitance type. The sensitivity, reliability and stability of hydrogel sensors are dependent on their network structures and mechanical properties. This review focuses on tough conductive hydrogels for flexible sensors. Representative strategies to prepare stretchable, strong, tough and self-healing hydrogels are briefly reviewed since these strategies are illuminating for the development of tough conductive hydrogels. Then, a general account on various conductive hydrogels is presented and discussed. Recent advances in tough conductive hydrogels with well designed network structures and their sensory performance are discussed in detail. A series of conductive hydrogel sensors and their application in wearable devices are reviewed. Some perspectives on flexible conductive hydrogel sensors and their applications are presented at the end.
 Zhenwu Wang | Zhenwu Wang obtained his BSc in materials science and engineering from the Zhejiang University of Technology in 2016, and his master's degree in polymer chemistry and physics from Shanghai University in 2019. He was a joint master student at the Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, from 2016 to 2019 supervised by Prof. Jun Fu. He is currently pursuing his PhD degree in chemistry at the Karlsruhe Institute of Technology. His research interests include functional hydrogels for flexible electronics. |
 Yang Cong | Yang Cong is an Associate Professor of the College of Materials Science and Chemical Engineering at the Ningbo University of Technology. She obtained her BSc degree at the Jilin Institute of Technology in 1999, and her PhD degree from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, in 2006. She was a postdoc in Professor Manfred Schmidt's group in Mainz University from 2006 to 2007. Her current research covers functional polymer hydrogels and block copolymer self-assemblies. |
 Jun Fu | Dr Jun Fu is a Professor in the School of Materials Science and Engineering at Sun Yat-sen University (SYSU). He obtained his BSc degree in applied chemistry at Wuhan University in 1999, and his PhD degree in polymer chemistry and physics from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (CAS), in 2005. He was a visiting scholar at the Max Planck Institute for Polymer Research from 2005 to 2007, and a research fellow at the Massachusetts General Hospital/Harvard Medical School from 2007 to 2010. He was appointed a professor at the Ningbo Institute of Materials Technology and Engineering, CAS, in 2010, and then moved to SYSU in 2019. His research focuses on high performance and functional hydrogels for biomimetic, wearable and implantable devices. |
1. Introduction
Flexible sensors have recently attracted great research attention due to their applications in wearable devices,1–3 soft robotics,4–7 and artificial skin.8–11 Inspired by biological sensors that convert external stimuli into bio-electronic signals to neurons, the brain, and muscles, numerous flexible sensors based on soft conductive materials have been recently developed. Elastomers (e.g., polydimethylsiloxane, silk fibroin, polyurethane, and polyethylene terephthalate)12–16 composited with conductive fillers (e.g., carbon materials,17–22 metal particles,23–25 liquid metals,26 and conductive polymers27,28) are sensitive to strain, pressure, or temperature changes.29,30 The elastomers serve as flexible matrices31 to impart deformations to the network of conductive fillers, leading to changes in their conductivity or resistance. Thus, external pressure and strain are converted into electronic outputs. The performance of such sensors relies on the properties of the matrix and the network, as well as the interactions between them. In comparison to conventional sensors based on rigid semiconductors, metals and ceramics, elastomers are advantageous in their stretchability, which is of utmost importance for wearable applications. Moreover, elastomers have low moduli, close to those of biotissues. However, the compatibility between fillers and elastomers has remained a challenge. Frequently, very high filler contents have to be used to establish a conductive network in the elastomer matrix to achieve acceptable conductivity. Consequently, such conductive composite materials show low stretchability (fracture strain < 100%). Moreover, most elastomers are bioinert and incompatible with biotissues. It is desired to develop flexible sensors with biomimetic mechanical properties, biocompatibility, high sensitivity, and reliability for wearable and implantable applications.29,32–34Conductive hydrogels35–38 are promising candidate materials for biosensors, flexible sensors, wearable devices, implantable sensors and electronic skin,39 because of their excellent biocompatibility40 resulting from their high water content and molecular similarity to natural soft tissues.41,42 Conductive hydrogels can be prepared by either compositing with conductive fillers, or incorporating ionic pendant groups or salts.43 Polymer hydrogels are a family of bioinspired, biomimetic and biofunctional soft materials comprised of crosslinked hydrophilic polymer networks containing a large amount of water. It is convenient to tune their chemistry, network structure, mechanical properties, and biofunctionalities. The hydrogel modulus can be tuned over a very wide range to match those of biotissues from the brain to bone. Polymer hydrogels can show very large stretchability and diverse functionalities. Some hydrogels are able to self-heal after damage.
Within a hydrogel network, conductive fillers establish a second network that conducts electrons, yielding an electron conductive material. In many cases, however, a high filling content is needed. Phase separation between fillers and matrixes usually deteriorates the stretchability, toughness, and fatigue resistance. Modification of fillers is usually needed to improve the affinity to the hydrogel matrix and depress phase separation. Many hydrogel matrices reported to date are intrinsically weak and could not sustain cyclic loadings. Surface modified fillers may improve their strength and toughness, but it has been difficult to compensate for their intrinsic weakness. Self-healing conductive hydrogels are able to autonomously repair after damage in terms of conductivity and mechanical properties,44 but this is usually time-consuming and relies on external stimuli. Therefore, in order to prepare conductive composite hydrogels with desired mechanical properties, substantial strategies are needed to consider both matrix and conductive networks.
Transparency is important for biomedical applications, particularly for devices in direct contact with tissues.45–47 Most electron conductive hydrogels reported so far are not transparent, primarily due to the intrinsic colour of fillers.
Ion conduction is ubiquitous in biosystems. The biomimetic high-water content in hydrogel networks provides an ideal environment for ion conduction. Polyelectrolyte hydrogels with immobilized charges on the chains and mobile ions in water have been widely studied as ion conductive materials for stress and strain sensors. Under an electric field, the free ion distribution and conductivity are sensitive to the pressure and strain.
For non-ionic polymer hydrogels, ion conduction can be achieved and manipulated by incorporating salts (e.g., LiCl, NaCl, or KCl) into their highly hydrated network. The migration of ions could be reversible under a cyclically reversed electric field. Ion conductive hydrogels with salts are believed to be ideal candidate materials for the fabrication of implantable sensors.
However, for practical applications, sophisticated challenges remain for hydrogel sensors. Electronic skin and wearable devices should work under very dynamic conditions (Fig. 1). The sensors and materials should survive cyclic loadings and maintain reliable and outstanding sensory performances. For in vivo applications or implantable devices (Fig. 1), flexible sensors should be made of biocompatible materials and conformably attach to and remain stable on highly dynamic organs like the heart, stomach, and lung. Moderate moduli close to target organs and excellent adhesion to biotissues are needed. Therefore, the development of conductive hydrogels with ideal moduli, strengths, toughnesses, and tissue adhesion is of special significance.
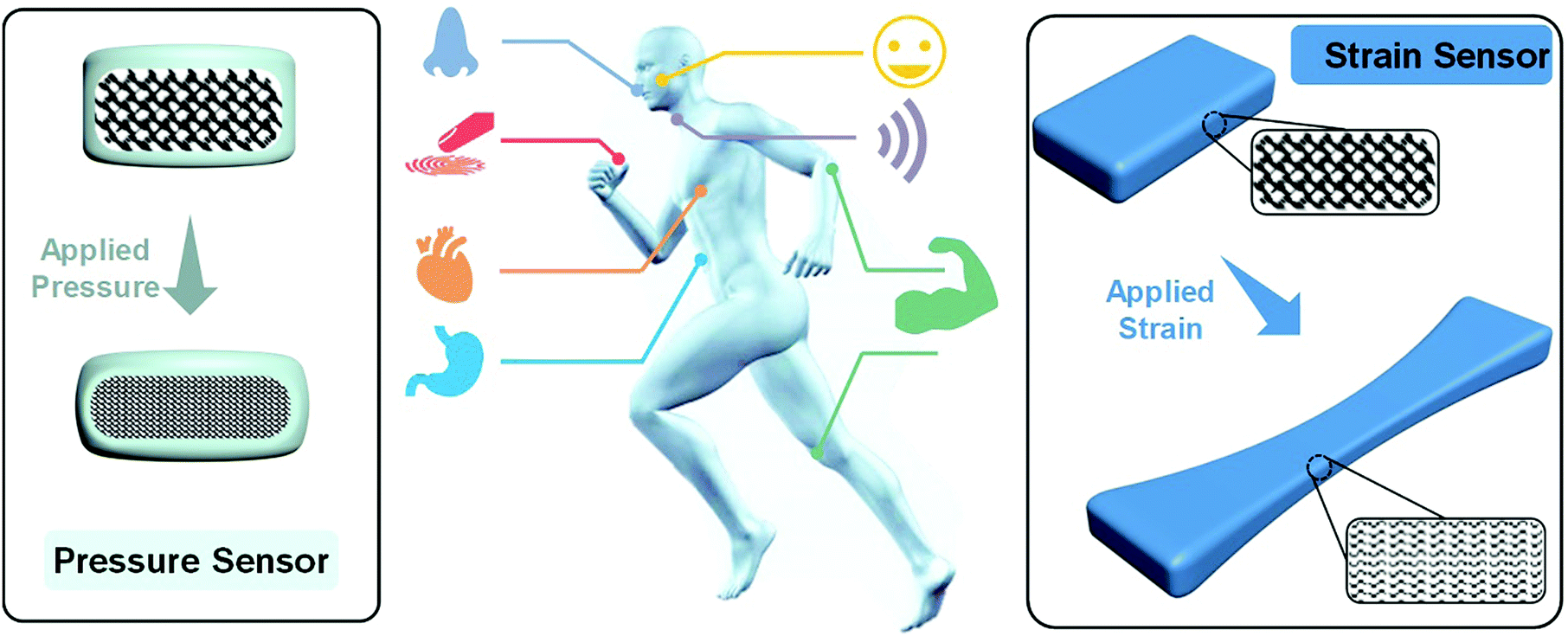 | ||
| Fig. 1 Schematic illustration of potential applications of flexible pressure and strain sensors for implantable and wearable devices. | ||
Advances in strong and tough hydrogels in the past few decades48–50 include double network hydrogels,51,52 nanocomposite hydrogels,53 micelle-crosslinked hydrogels,54 and polyampholyte hydrogels.55 Sacrificial mechanisms based on rigid networks52 or non-covalent interactions56 have been utilized to dissipate energy and thus improve the strength and toughness of hydrogels. Ionic or zwitterionic monomers have been used to synthesize stretchable, tough, and self-healing hydrogels with well-designed network structures. Moreover, interpenetrating conductive polymer chains throughout the hydrogel matrix, or establishing specific affinities or interactions between conductive networks and polymer matrixes, appears to be promising in controlling both the mechanical properties and conductivity. Interestingly, the network design is found to be beneficial for the sensitivity and working range of conductive hydrogel sensors.37
Many recent Review articles mainly focus on the fascinating fabrication and application of hydrogel sensors and flexible devices57–60 in flexible electronics, electronic skin, and flexible energy systems. Less attention is focused on how the network design influences the mechanical properties, conductivity and sensory performances of conductive hydrogels and sensors. We hope this review article can provide researchers a systematic understanding of tough conductive hydrogels, especially the network design, for flexible sensors, which we believe is inspiring for novel tough conductive hydrogels and flexible sensors toward practical applications.
This review aims to provide a comprehensive account on the latest advances in tough conductive hydrogels and their applications in flexible sensors and devices. It starts with a brief introduction to representative strategies to prepare tough hydrogels. Second, recent advances in electronically and ionically conductive hydrogels are reviewed and discussed. Third, conductive hydrogels with well-designed network structures, ultrastretchability, and toughness are presented in Section 4, focusing on how their network structures determine their mechanical properties, conductivity and sensory performance. Section 5 gives an account of representative updates on pressure and strain sensors based on tough conductive hydrogels. It ends with a brief conclusion and perspectives on this rapidly developing and promising field of flexible sensors and devices.
2. Representative tough hydrogels
Conventional polymer hydrogels are mostly fragile and weak with chemically crosslinked inhomogeneous networks.61 The heterogeneous chemical networks have poor capability to dissipate energy during loadings. As a result, the hydrogels usually fracture at low strain and show very low fracture strength or toughness.Strategies based on homogeneous networks,62,63 double networks,52,55,64–66 nanocomposites,67–72 and micelle cross-links56,73 have been widely developed in the past few decades to produce hydrogels with outstanding strength, toughness, and self-healing and recovery properties. Double network hydrogels comprised of two interpenetrating and contrasting networks are the first with extremely high strength and toughness.66 Hydrogels crosslinked by non-covalent interactions, including physical adsorption, hydrophobic association, ion crosslinking, and supramolecular recognition, have emerged as another important family. The reversible non-covalent interactions serve as an energy dissipation mechanism and allows for recovery or self-healing.
The strategies to create tough hydrogels are instructive for the preparation of tough conductive hydrogels, either ionic or electronic, to meet a broad spectrum of demands for wearable and implantable flexible devices. Fundamentals to double network and nanocomposite hydrogels are briefly introduced here although many comprehensive reviews74 can be found in the literature.
2.1. Double network hydrogels
Double network (DN) hydrogels are composed of two interpenetrating polymer networks with contrasting mechanical properties (Fig. 2a).75 The first network is highly stretched and densely cross-linked. In contrast, the second network is sparsely cross-linked and flexible. Upon loadings, the internal fracture of the brittle network dissipates substantial amounts of energy at large deformations, while the second network provides hidden elasticity to maintain the integrity of hydrogels.75 Based on this concept, double network hydrogels comprised of poly(2-acrylamido-2-methylpropane-sulfonic acid) (PAMPS) and poly(acrylamide) (PAAm) networks were first reported by Gong et al.66 The rigid and densely crosslinked first PAMPS network was used to host free radical polymerization of AAm to form a loosely crosslinked second network interpenetrating through the first one. The double network hydrogels show a very high compressive strength (17.2 MPa or even higher52), or more than 20 times that for each single network. The inter/intramolecular interactions and structures within and between two networks are important for optimizing their mechanical properties.76 This successful DN concept has been generalized to many other polymers, including neutral polymers, thermo-responsive polymers, and natural polymers. Functionalization of one or both of the networks enables diverse manipulation of the properties and functionalities of tough polymer hydrogels.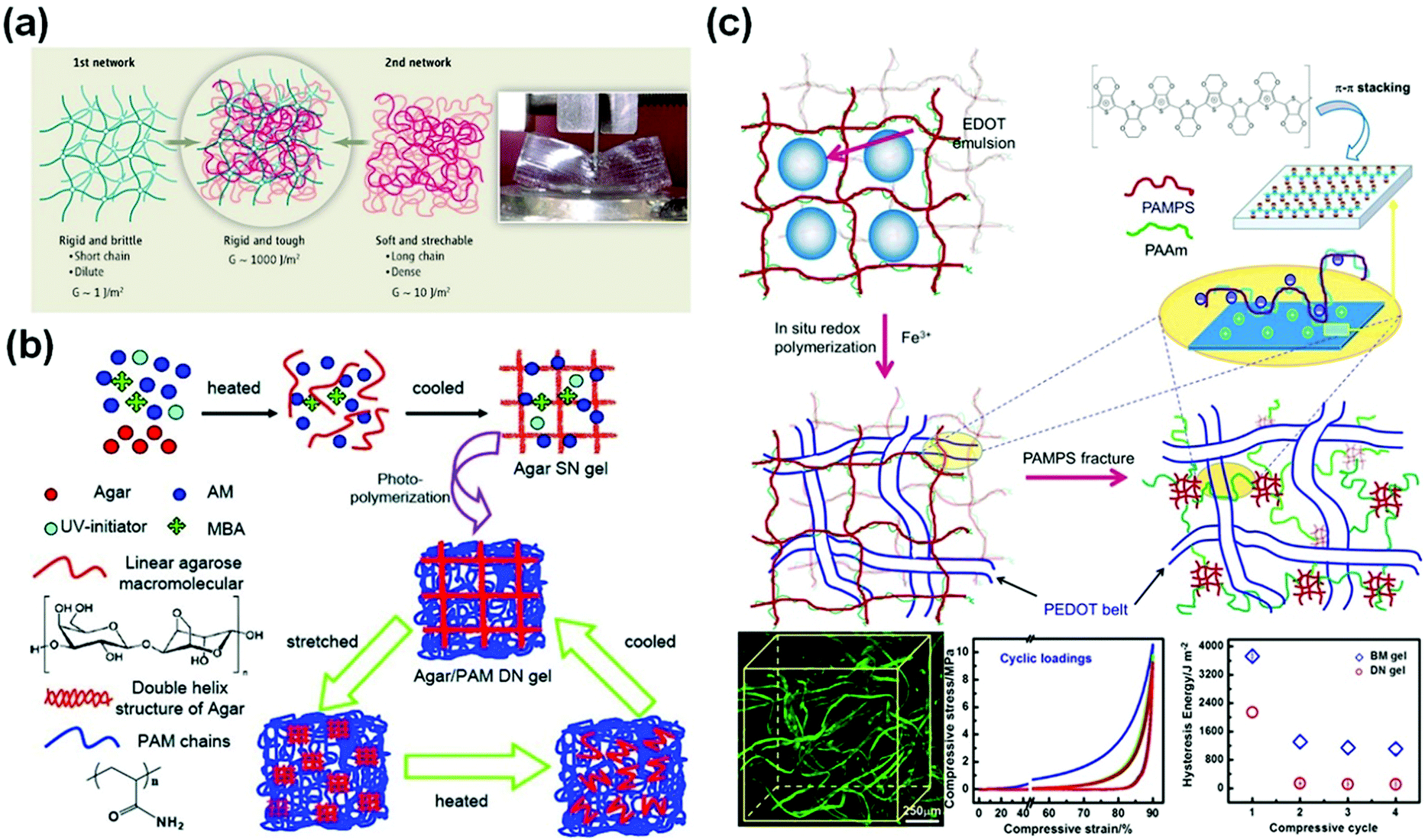 | ||
| Fig. 2 Double network hydrogels. (a) Schematic illustration of a classic double network hydrogel. Reprinted from ref. 75 with permission. Copyright 2014 AAAS. (b) Double network hydrogels based on agar and polyacrylamide. Reprinted from ref. 77 with permission. Copyright 2013 Wiley. (c) Schematic illustration of the preparation of cartilage extracellular matrix-like double network hydrogels, with interpenetrated structures observed by using confocal laser scanning microscopy and outstanding fatigue resistance and biomimetic toughness upon cyclic loadings. Reprinted from ref. 79 with permission. Copyright 2014 American Chemical Society. | ||
However, the destruction of the first network is irreversible, and thus the DN gels have poor resistance to cyclic loadings. Chen and co-workers used agar as the first network and PAAm as the second network.77 The agar chains form double stranded helices, which decompose at high temperatures and re-form after cooling, experiencing reversible thermo-responsive sol–gel transitions. The agar/polyacrylamide DN hydrogels show a tensile strength of 1.0 MPa, a fracture strain of 2000%, and a toughness of 9 MJ m−3. After thermal treatments at 100 °C for 10 min, the tested hydrogels could completely recover their original toughness (Fig. 2b).77
The intrinsic interactions between both networks are important for their strength and toughness. Inter-network interactions are ubiquitous in the extracellular matrix (ECM) of biotissues. For example, cartilage ECM is comprised of a collagen fibril network and an interlacing proteoglycan mesh with charges. The electrostatic interaction and hydrogen bonding between both networks play critical roles in the outstanding toughness and fatigue resistance of cartilage.78 By using a PAMPS/PAAm DN hydrogel as a template, in situ oxidation polymerization of EDOT:NaPSS (3,4-ethylenedioxythiophene/poly(sodium 4-styrenesulfonate) yields PEDOT within the hydrogels (Fig. 2c)). In the confined geometry, the conjugate PEDOT chains self-assemble into long belts interpenetrating through the hydrogel matrix.79 Besides, the positively charged PEDOT chains have strong electrostatic attractions with the PAMPS chains carrying negative charges. The interpenetrating structure and electrostatic interactions provide extraordinary compressive strength (>70 MPa), toughness, and fatigue resistance against cyclic loadings.79 After cyclic loadings, the hydrogels show a compressive toughness of 1000 J m−2, close to that of articular cartilage.78
Replacing chemical crosslinking with ion crosslinking makes DN hydrogels more dynamic and stretchable. Suo et al.80 used calcium ion crosslinked alginate as the first network and a sparsely crosslinked PAAm network as the second network (Fig. 3a). The ion-crosslinked alginate network dissipates energy through an unzipping mechanism. The unzipping of Ca2+-alginate coordinates at the crack front blunts the crack tips and prevents crack propagation. The obtained hydrogels show a fracture elongation of 2000% and a fracture energy of 9000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Jm−2. The notches in the hydrogels are rapidly blunted during stretching, showing very high stretchability (beyond 17 times). This concept has been easily extended to biocompatible polymer hydrogels for cell encapsulation.81
Jm−2. The notches in the hydrogels are rapidly blunted during stretching, showing very high stretchability (beyond 17 times). This concept has been easily extended to biocompatible polymer hydrogels for cell encapsulation.81
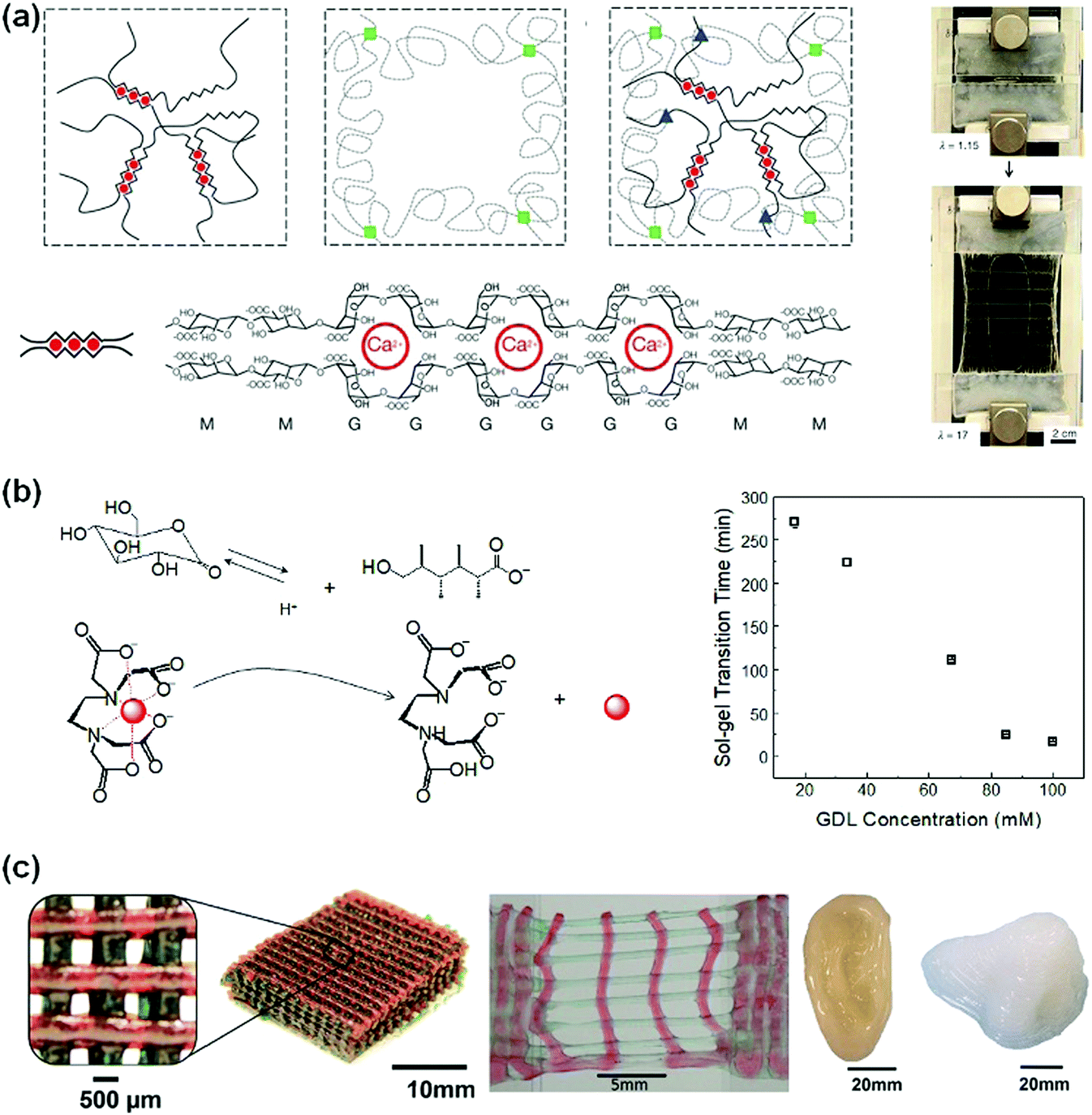 | ||
| Fig. 3 (a) Alginate/PAAm hydrogels crosslinked by Ca2+ show notch-insensitive stretchability. Reprinted from ref. 80 with permission. Copyright 2012 Springer Nature. (b) Controlled release of multi-valent metal ions to mediate the gelation kinetics of the alginate/PAAm DN gels. Reprinted from ref. 83 with permission. Copyright 2015 Royal Society of Chemistry. (c) 3D printed scaffolds, ear and nose from alginate/PEG hydrogels with high stretchability. Reprinted from ref. 81 with permission. Copyright 2015 Wiley. | ||
Biomedical applications of alginate-based hydrogels52,53 require control over their gelation kinetics and network structures. However, the very rapid chelation of multi-valent ions with alginate in very high viscosity solutions usually results in heterogeneous crosslinking.82 To tackle this problem, a protect-and-release strategy has been devised to achieve homogeneous distribution and controlled release and chelation of multivalent ions. Ca2+, for example, is chelated and protected with EDTA. The ion/EDTA coordinates serve as soluble ion reservoirs to form a homogeneous mixture with viscous sodium alginate solutions. Glucono delta-lactone (GDL) has a high dissociation rate to slowly release H+, which triggers the decomposition of the ion/EDTA coordinates to release Ca2+ (Fig. 3b). The released Ca2+ ions crosslink alginate homogeneously. This strategy is feasible for many multivalent ions (i.e., Ca2+, Co2+, Cu2+, La3+ and Ce4+).83 The gelation kinetics can be controlled by using different GDL concentrations. The obtained hydrogels show very high stretchability (up to 3500%) and notch-insensitivity. Such hydrogels can be used as inks for three-dimensional printing of biomedical scaffolds. With optimal viscosity and shear-thinning behaviour, such hydrogel inks have been used to print 3D structures that sustain stretching and compression.81 The outstanding mechanical properties are advantageous for the fabrication of supportive or load-bearing tissue scaffolds (e.g., nose, ear, and cartilage, Fig. 3c).
2.2. Nanocomposite hydrogels
Nanoparticles have been used to crosslink hydrogels,71 to adsorb polymer chains,84 or to impart new properties to hydrogel networks.85 Chemical bonds, hydrogen bonding, hydrophilic adsorption, hydrophobic association, and ionic interactions between polymer chains and nanoparticles have been used to prepare nanocomposite hydrogels.Haraguchi et al. pioneered nanocomposite hydrogels by using exfoliated clay nanosheets (LAPONITE® XLG, [Mg5.34Li0.66Si8O20(OH)4]Na0.66) to adsorb and crosslink polymer chains, including poly(N-isopropyl acrylamide), PNIPAAm,86 poly(N,N′-dimethyl acrylamide),87 and polyacrylamide.88 Upon stretching, the chain detachment and re-orientation of clay nanosheets work synergistically to dissipate energy.89 The nanocomposite hydrogels showed a tensile elongation of up to 3000% and a maximum fracture strength of 109 kPa. The detached chains are able to re-adsorb to clay surfaces, leading to recovery and self-healing.90 The nanocomposite hydrogels are able to self-heal at their fresh cut surfaces. The polymer chains are believed to migrate across the interface and adsorb on clay nanosheets to achieve strong adhesion at the interface.
The clay dimensions have a critical influence on the stretchability, toughness, and self-healing of the nanocomposite hydrogels. Sodium montmorillonite, NaMMT, is a layered silicate clay with a larger aspect ratio91 than LAPONITE® XLG and able to adsorb more organic molecules and polymer chains on their surfaces (Fig. 4a).92,93 During loadings, the polymer chains desorb from the clay surfaces to dissipate energy and toughen the hydrogels (Fig. 4b). The hydrogels show typical yielding, necking, and strain hardening, as well as fracture elongations up to 11800% and fracture toughnesses up to 10.1 MJ m−3 (Fig. 4c and d).94 This desorption is reversible. The stretched hydrogels after storage at room temperature for several hours can fully recover their strength, toughness, and stretchability (Fig. 4e). Moreover, the cut hydrogels could heal under mild conditions with the assistance of water.
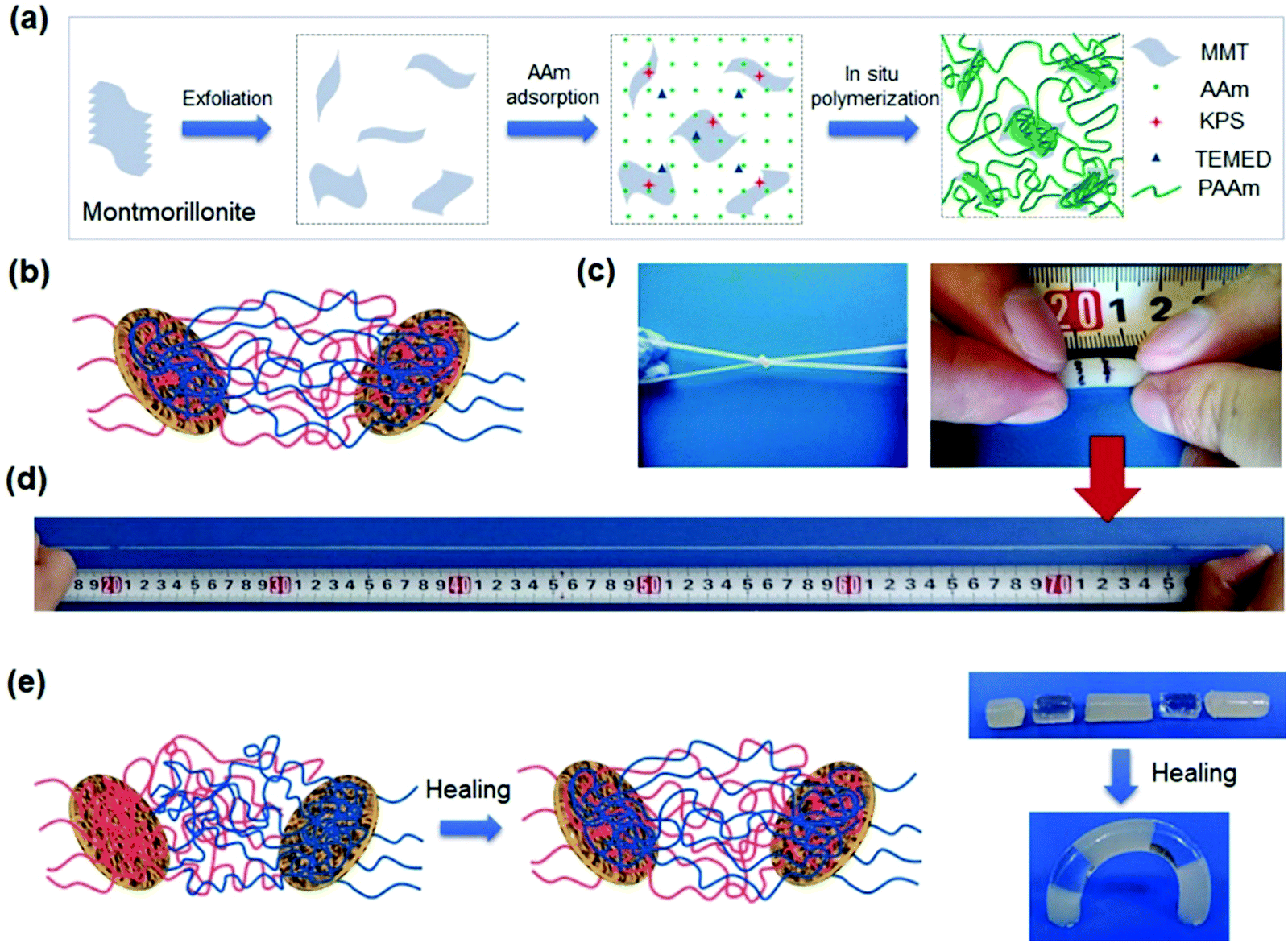 | ||
| Fig. 4 Montmorillonite nanosheet crosslinked hydrogels. (a) In situ exfoliation and free radical polymerization to synthesize nanocomposite hydrogels. (b) Polymer chains are adsorbed on clay nanosheets. (c) The obtained hydrogels are very flexible and (d) stretchable to very large strain (11800%). (e) The polymer chains are able to migrate across the interface of two hydrogels in contact, leading to complete healing. Adapted from ref. 94 with permission. Copyright 2015 American Chemical Society. | ||
On the other hand, spherical nanoparticles95–99 are used to crosslink polymer chains through covalent bonding or physical adsorption to generate highly stretchable and tough hydrogels. Surface functionalized silica nanoparticles can adsorb polymer chains through hydrogen bonding and covalent bonding with polymer chains to form dually crosslinked hydrogels.96 The obtained nanocomposite hydrogels show fracture strains larger than 3700% and a fracture strength of 120 kPa. The cut hydrogel can self-heal under ambient conditions for at least 20 cycles.99 Such nanocomposite hydrogels show an ionic conductivity of 0.15 mS cm−1,100 which is superior to conventional PVA-based acidic polyelectrolyte hydrogels. Compositing functionalized silica nanospheres with DN gels can further improve their strength and toughness by more than three times. The adsorption and chemical bonding of polymer chains to silica nanospheres allow for partial recovery of the hydrogels after cyclic tests.71
3. Conductive hydrogels
Conductive hydrogels can be roughly divided into electronically conductive and ionically conductive hydrogels. This section discusses and summarizes recent research studies on conductive hydrogels (Fig. 5), mainly focusing on the design and preparation of conductive hydrogels and their electronic properties. It begins with electronically conductive hydrogels, including conductor hydrogels, conductive composite hydrogels and interpenetrating network conductive hydrogels.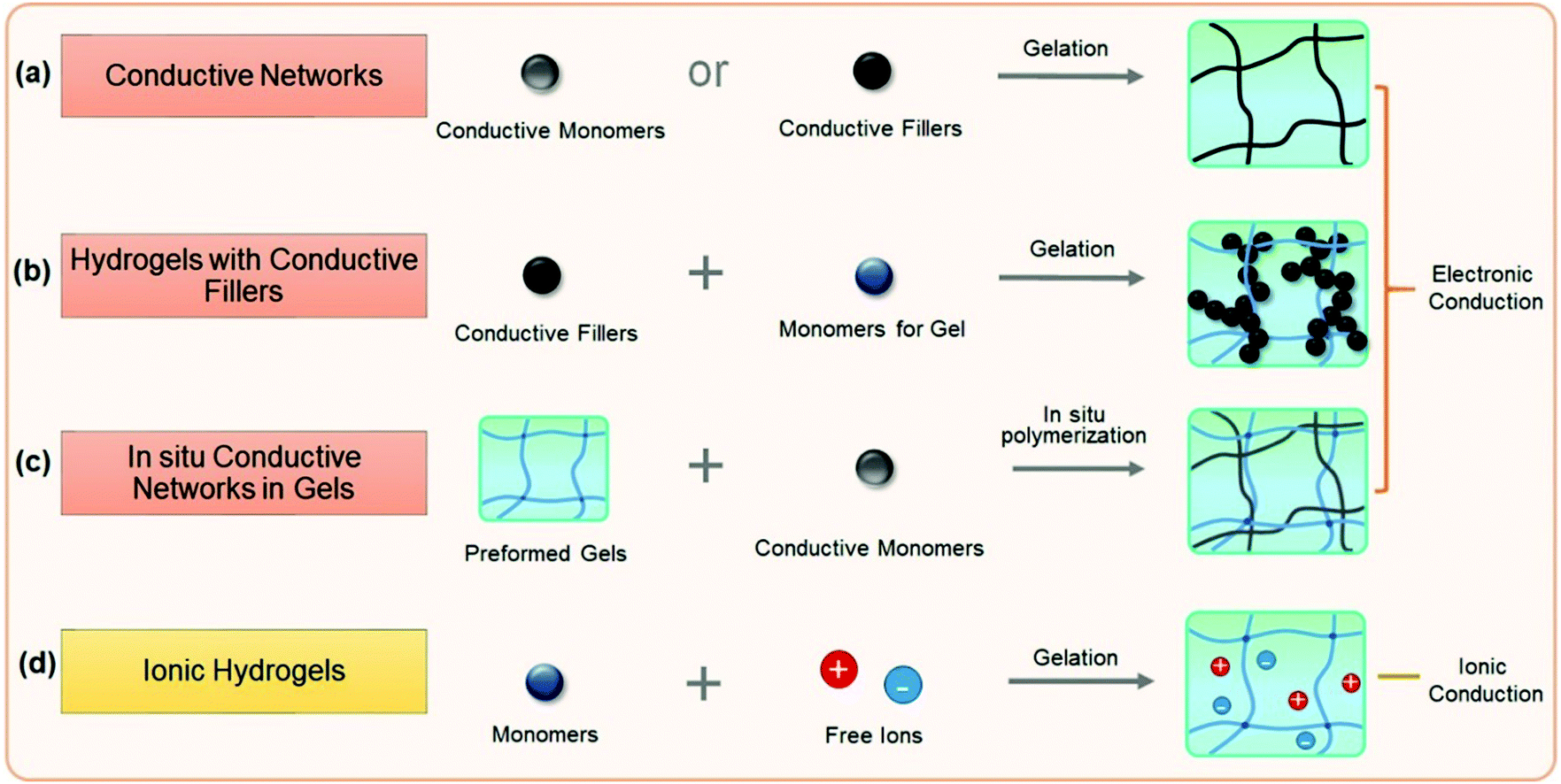 | ||
| Fig. 5 Major strategies to prepare conductive hydrogels. | ||
3.1. Electronically conductive hydrogels (ECHs)
Electronically conductive hydrogels (ECHs) are prepared by compositing or hybridizing with conductive fillers or conductive polymers. Some conductive polymers have been used to synthesize hydrogels with very high conductivity, and conductive particles can be glued together to achieve high conductivity (Fig. 5a). In most cases, conductive hydrogels reported are comprised of a conductive network and a hydrogel network, with the former providing conductivity and the latter serving as a stretchable and deformable matrix (Fig. 5b). The conductive network can be conductive fillers or in situ formed conductive polymers. Polymer hydrogel networks are utilized as matrices or templates to host in situ polymerization of monomers to generate a conductive network throughout the hydrogel (Fig. 5c). These strategies share some common aspects with nanocomposite hydrogels and double network hydrogels. For example, carbon nanomaterials and metal nanoparticles101 are composited with hydrogels to both improve the stretchability of and impart conductivity to hydrogels. In situ synthesis of conductive polymers within tough hydrogels has produced strong, tough, and fatigue resistant conductive hydrogels. The hydrogels show very good conductivity and high sensitivity, and sustain thousands of cyclic loadings. This section reviews some important recent advances in this topic and discusses how the network structures influence the performance of sensors.Graphene, with a flexible two-dimension structure and exceptional electronic properties, has been used as versatile building blocks for self-assembling into hydrogels by a facile one-step hydrothermal method.102–104 Graphene nanosheets overlap or coalesce into physically crosslinked 3D porous networks (Fig. 6a). The obtained graphene hydrogels contain about 2.6 wt% graphene and 97.4 wt% water, and show electronic conductivities as high as 0.5 S m−1.102
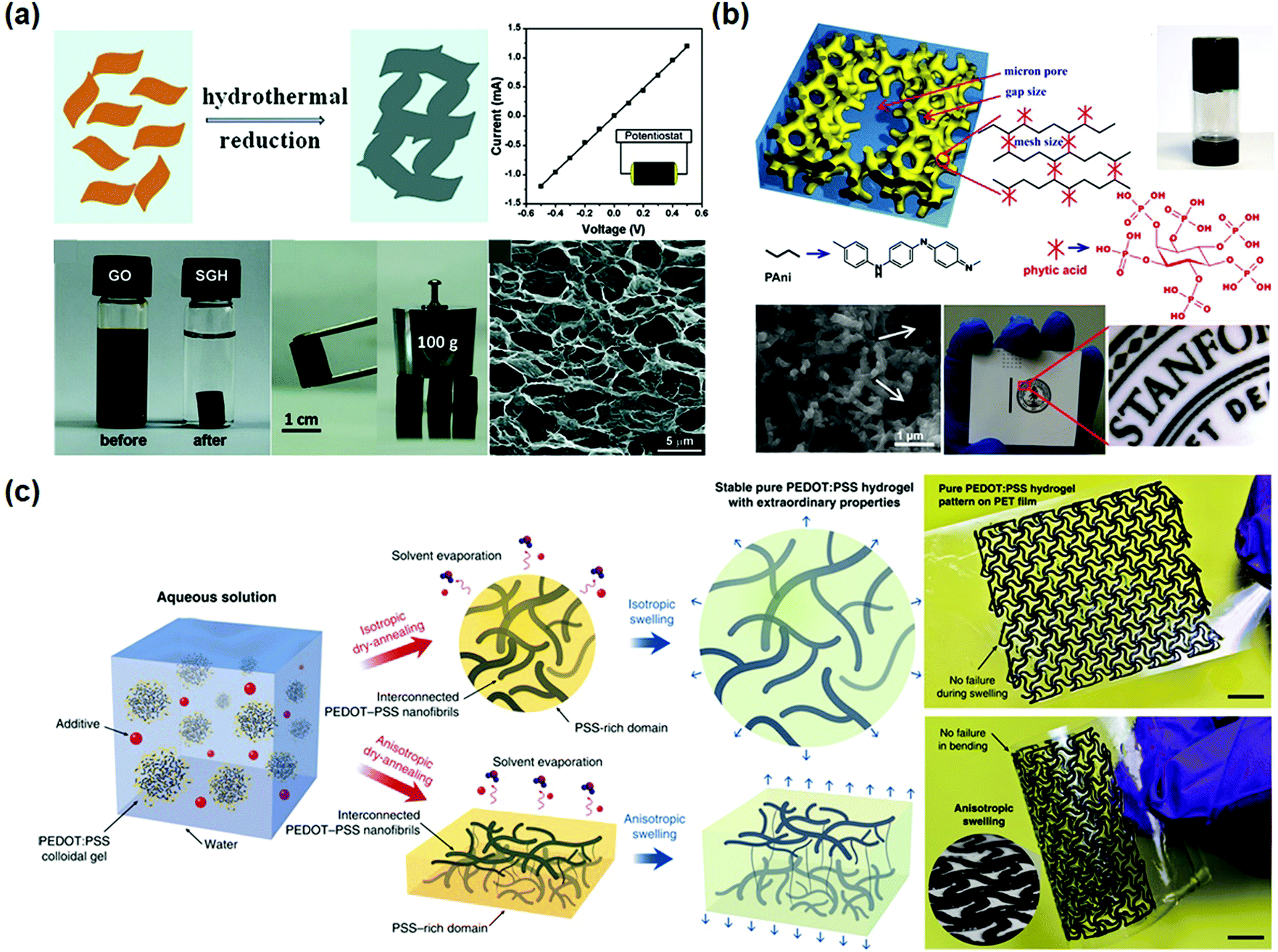 | ||
| Fig. 6 Conductor hydrogels. (a) Graphene oxide hydrogels prepared by hydrothermal reduction show excellent mechanical and electrical properties. Reprinted with permission from ref. 102. Copyright 2010 American Chemical Society. (b) Polyaniline hydrogels crosslinked by phytic acid are comprised of a three-dimensional network of nanoparticles. Reprinted with permission from ref. 110. Copyright 2012 National Academy of Sciences. (c) Preparation of pure PEDOT:PSS hydrogels and patterns, showing superb processability and flexibility. Reprinted with permission from ref. 117. Copyright 2019 Springer Nature. | ||
Polydopamine is used to reduce and functionalize graphene oxides to produce graphene hydrogels.105 Graphene nanosheets are assembled into a highly interconnected 3D network and serve as the supporting framework for hydrogels. Graphene hydrogels can be fabricated by vacuum filtration of dispersed chemically converted graphene colloidal solution through a filter membrane.106–109 A uniform black film is immediately formed as driven by vacuum. The obtained film is subsequently hydrated into conductive hydrogels containing about 92 wt% water.109
On the other hand, conductive polymers can form hydrogels. Polyaniline (PANI), a well-known conductive polymer, could become water soluble after protonation, and has been used to build conductive hydrogels. For example, polyaniline was cross-linked by phytic acid to form a conductive hydrogel with a high conductivity of 11 S m−1 (Fig. 6b).110 The PANI chains are connected with phytic acid through hydrogen bonds. Herein, each phytic acid molecule can interact with multiple PANI chains, forming three-dimensional hydrogel networks. The PANI hydrogels show foam-like nanostructures consisting of coral-like dendritic nanofibers with diameters of 60–100 nm. The large open channels of the micro/nano-scale pores within the foam-like structures provide relatively short diffusion paths for electrolyte ions to access the electroactive surface of PANI.111 The hydrogels can be used to fabricate micropatterns by ink-jet printing and spray coating. Inspired by this study, a series of conductive hydrogels based on conductive polymers have been developed.112–116
PEDOT has been directly crosslinked into hydrogels with very high conductivity.117,118 Pure PEDOT hydrogels can be obtained by treating commercially available poly(3,4-ethylenedioxythiophene):poly(4-styrene sulfonate) (PEDOT:PSS) suspension in 0.1 mol L−1 H2SO4 at 90 °C. The obtained hydrogels have an extremely high water content of 99.22 wt% and conductivity of 46 S m−1. After further treatment with concentrated H2SO4, the conductivity can be extremely increased to 880 S m−1.118 However, the involvement of sulfuric acid makes the procedure unsuitable for biomedical applications. Alternatively, pure PEDOT:PSS hydrogels are prepared by adding a non-volatile solvent, dimethyl sulfoxide (DMSO), into aqueous PEDOT:PSS solutions (Fig. 6c),117 followed by dry-annealing in a controlled manner, and subsequent reswelling in water. The obtained pure PEDOT:PSS hydrogels show electronic conductivities of 2000 S cm−1 in phosphate buffered saline and 4000 S cm−1 in water. The conductive hydrogels show a Young's modulus of 2 MPa and a stretchability over 35%. The hydrogels can be used as inks to fabricate patterned electrodes with complex geometries for bioelectronic devices.45,119,120
Recently, a combination of conductive nanosheets with a conductive polymer produced a conductive hydrogel with very high conductivity.121 Lu and co-workers developed a green and simple strategy to create a self-assembly of PEDOT on a polydopamine-reduced and sulfonated graphene oxide (PSGO) template, generating a PSGO-PEDOT nanosheet hydrogel with a conductivity of 829.6 S m−1, or 440 times that of the PEDOT hydrogel (1.8 S m−1). Therein, the functional groups on the PSGO template may act as dopants to PEDOT. Besides, the PEDOT nanosheets significantly improve the mechanical properties of the hydrogel, which is attributed to the intrinsic GO nanosheet stiffness and noncovalent interaction between the nanosheets and polymer chains.
Graphene oxide (GO) nanosheets contain abundant hydroxyl, epoxy and carboxyl groups on their surfaces,126 and have been used as cross-linkers to prepare conductive composite hydrogels.121,123,127,128 GO/PAAm nanocomposite hydrogels, for example, show a very high fracture strain of over 3000% (Fig. 7a). The GO nanosheets form an interconnected network for electronic conductivity. The GO/PAAm hydrogels are highly resilient.129 No obvious damage occurs during cyclic stretching and compression tests. Conductive reduced graphene oxide (rGO)/PAAc hydrogels can form additional PAAc-Fe3+ chelation,130–133 which further imparted reversible self-healing capability, high stretchability (larger than 600%), and high strength (400 kPa) to the conductive hydrogels.130
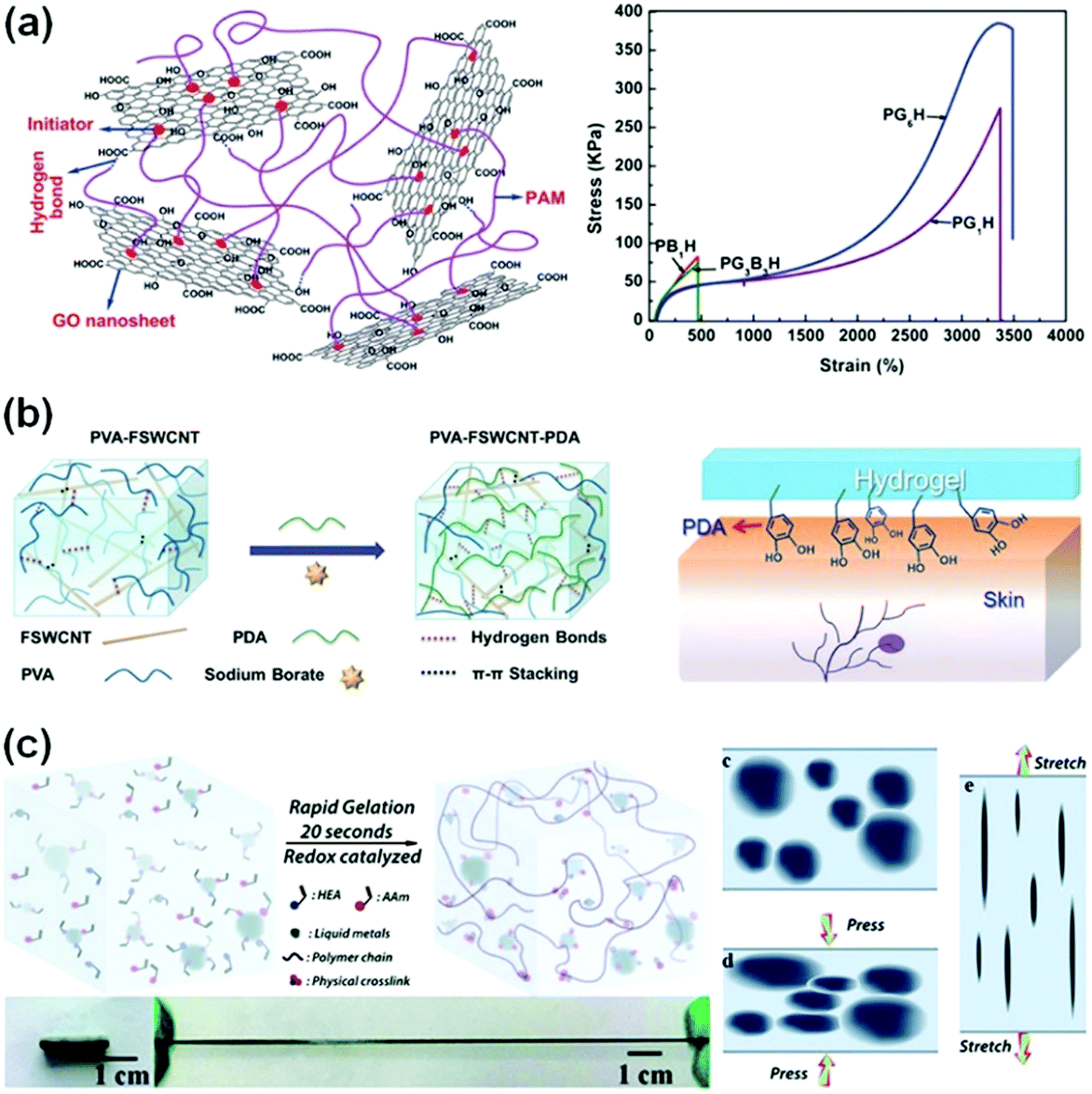 | ||
| Fig. 7 Conductive composite hydrogels. (a) Chemically crosslinked graphene oxide hydrogels show ultra-stretchability. Reprinted with permission from ref. 123. Copyright 2012 Royal Society of Chemistry. (b) Supramolecular crosslinked PVA/FSWCNT nanocomposite hydrogels. Reprinted with permission from ref. 36. Copyright 2017 Wiley. (c) Liquid metal/polymer composite hydrogels, where the deformation of liquid metal in the network provides outstanding toughness. Reprinted with permission from ref. 124. Copyright 2019 Royal Society of Chemistry. | ||
Moreover, pre-formed conductive graphene networks have been used to composite with hydrogels.125 Graphene aerogels were used as conductive networks and templates for in situ polymerization of hydrophilic polymer chains, leading to the formation of binary networks. The hydrogels show intrinsic high conductivity, while the compressive strength and toughness of the hydrogels with graphene aerogels are 6–8 times and even 40–300 times higher than those of the matrix hydrogel and graphene aerogels.
Carbon nanotubes (CNTs) are very attractive as conductive fillers for hydrogels due to their high aspect ratio, conductivity and strength.134,135 CNTs in hydrogel networks provide highly efficient electronic transport channels to achieve high conductivity of hydrogels.35,36,136 Low CNT contents (1–5 wt%) in polymer matrices have significantly improved their electronic conductivity.137,138 However, the random distribution and easy aggregation of CNTs limit the performance of the incorporated hydrogels. Modification of CNTs is key to overcoming the inherent poor compatibility of CNTs with polymer matrices.139 Polydopamine (PDA) was introduced into a functionalized single-walled carbon nanotube (FSWCNT) and PVA mixture solution (Fig. 7b). Hydrogen bonding and π–π stacking among PDA, FSWCNT and PVA36 impart good conductivity (about 0.04 S m−1) to the composite hydrogels. In the presence of PDA, the composite hydrogels exhibit excellent and repeatable adhesiveness to various surfaces, including glass, polypropylene and porcine skin. Moreover, carboxyl-functionalized multi-walled carbon nanotubes (c-MWCNTs) are composited with a chitosan network, generating nanocomposite hydrogels. The carboxylic groups chelate with multivalent ions, showing excellent conductivity, puncture resistance and self-healing capability.140
The combination of liquid metals with flexible polymer hydrogels opens a new pathway to develop highly conductive and stretchable hydrogels for sensors with very high sensitivity.26,124,141,142 Liquid metals can catalyse free radical polymerization and highly accelerate the gelation process to complete in about 20 seconds (Fig. 7c).124 Moreover, liquid metals significantly enhance the stretchability and toughness of hydrogels. Liquid metal/PVA hydrogels show an elongation at break of 1500%, much higher than those of liquid metal/elastomer composites. During tensile or compressive loadings, the liquid metal phase deforms accordingly, and thus converts the deformation into electronic signals (Fig. 7c). The conductive networks are able to sense extremely weak pressure changes, achieving a high sensitivity of 0.25 kPa−1, which is more than 20 times that of the hydrogel matrix. Furthermore, the hydrogels with liquid metals sustain more than 1000 cycle loadings and show little changes in the sensory performance with a rapid resistive response (180 ms). These performances are critical for sensory applications. To enhance the electronic stability under cyclic loadings, the liquid metal is designed into a three-dimensional helix in hydrogels. The structured hydrogel electronics present excellent mechanical and electrical robustness against cyclic loadings (only 10% resistance change with 300 cycles).143
Infiltration of conductive polymer precursors into a hydrogel network for subsequent in situ polymerization is efficient in creating a conductive network interpenetrating through the hydrogel matrix. EDOT was dissolved in agarose solution, and then electro-polymerized into PEDOT in the agarose gel (Fig. 8a).144 The distribution/patterning of PEDOT in the agarose hydrogel could be precisely defined by using electrode patterns or arrays. In situ oxidation polymerization of pyrrole in an agarose hydrogel produces interpenetrating network conductive hydrogels with stretchabilities up to 35% and self-healing capability (Fig. 8b).145 The obtained hydrogels show conductivities from 20 to 70 S m−1. The polypyrrole/agarose hydrogels can be painted and patterned directly on biological surfaces for bioelectronics. Recently, many conductive polymers have been introduced into different hydrogel networks in this way, providing a continuous electronic transmission network.128,146–149 However, these hydrogels exhibit poor mechanical properties or low sensitivity. This may result from the weak connection between the hydrogel network and conductive polymer.
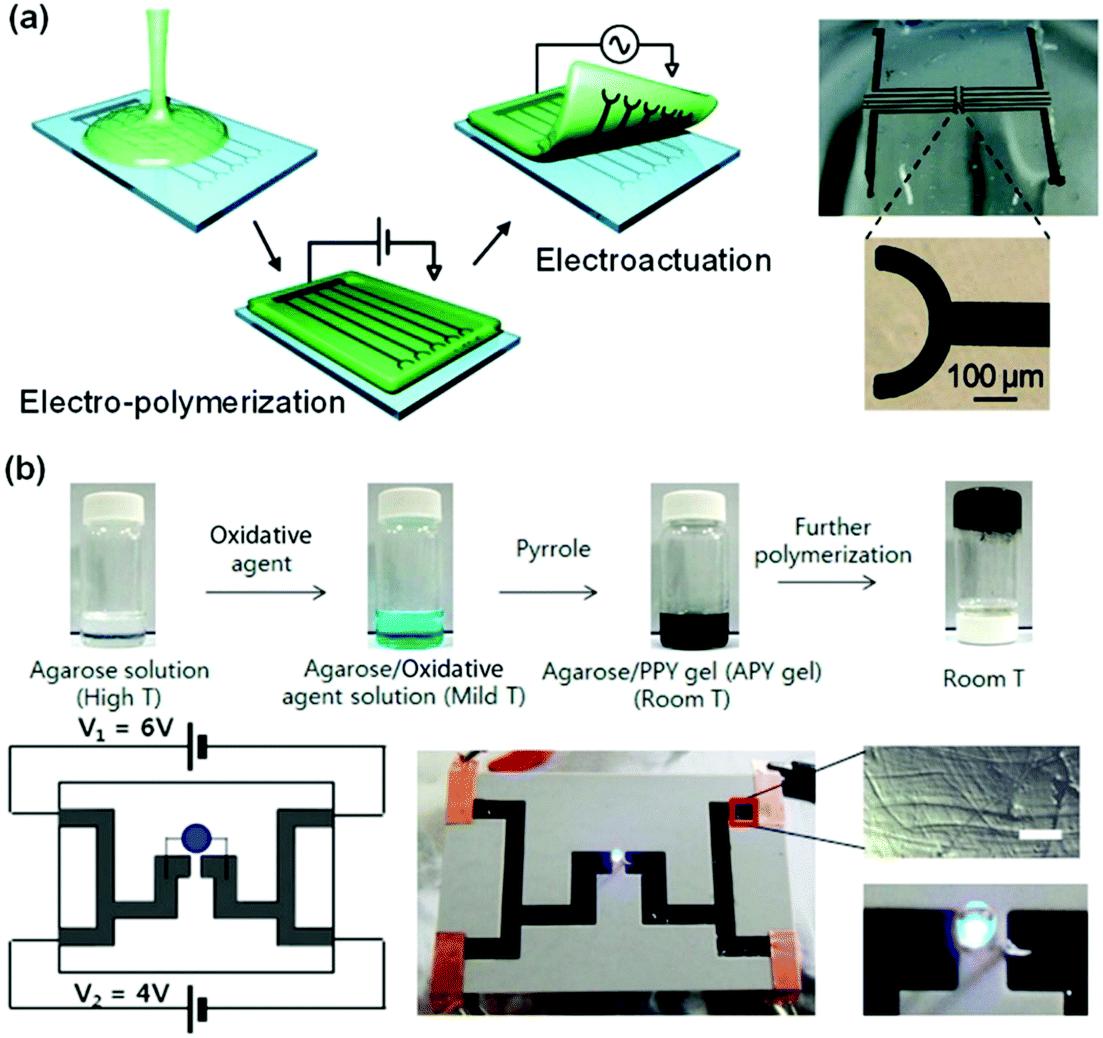 | ||
| Fig. 8 Interpenetrating network conductive hydrogels. (a) Electro-polymerization of an interpenetrating PEDOT network in an agarose matrix. Conductive hydrogel patterns are defined by the electrodes. Reprinted from ref. 144 with permission. Copyright 2010 American Chemical Society. (b) In situ oxidation polymerization of pyrrole in an agarose hydrogel matrix to form conductive hydrogels. Reprinted from ref. 145 with permission. Copyright 2014 American Chemical Society. | ||
3.2. Ionically conductive hydrogels (ICHs)
In biosystems, ionic conduction through free ion transportation is ubiquitous and extremely important.150 Ionically conductive hydrogels contain a large number of free ions in their three-dimensional polymer networks, which have flexibility similar to biosystems, making them ideal candidates for soft matrices of wearable or implantable devices.32,151–154 For ionically conductive hydrogels, the conductivity comes from directional transportation of free ions.155–157 There are two common ways to enhance their ionic conductivity: (1) fabricating polyelectrolyte networks to enhance transportation of free ions158 and (2) constructing ion channels in hydrogel networks for ion transportation.159In comparison to most electronic conductive hydrogels, ionic conductive hydrogels are usually transparent, stretchable, tissue-adhesive, and self-healable.54,160–163 Polyelectrolyte hydrogels are favourable for ion transportation.164 Sun and co-workers fabricated polyampholyte (PA) hydrogels with high ionic conductivity by one-step random polymerization of positively charged imidazolium-based ionic liquid monomers with urea groups and negatively charged 3-sulfopropyl methacrylate potassium salt (Fig. 9a).160 The electrostatic interactions between imidazolium and sulfonate groups dramatically improve the mechanical strength of the hydrogels. The PA hydrogels exhibit a tensile strength of 1.3 MPa and a fracture toughness of 6.7 MJ m−3, and a conductivity of 3 S m−1. The PA gels are able to self-heal at room temperature to restore their mechanical properties and conductivity after damage.
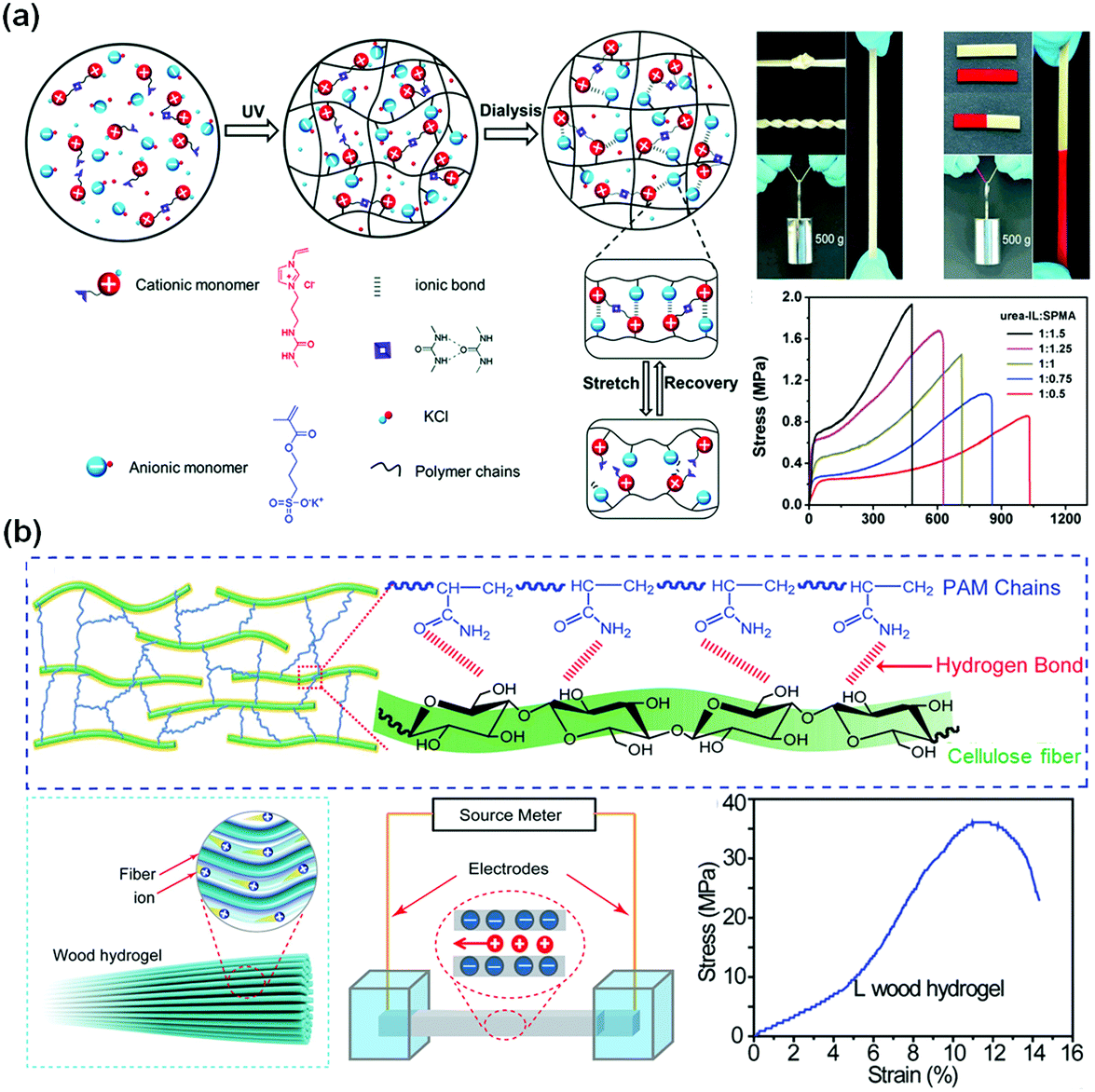 | ||
| Fig. 9 Ionic conductive hydrogels. (a) Poly(ionic liquid) hydrogel with high stretchability and self-healing capability with ionic and hydrogen bonds. Reprinted from ref. 160 with permission. Copyright 2018 Wiley. (b) Aligned cellulose fiber/polyacrylamide composite hydrogels with a low concentration of negative charges. Reprinted from ref. 162 with permission. Copyright 2018 Wiley. | ||
Biotissues consist of biological gels with well-ordered structures with channels for ion transportation. Introducing highly ordered nanostructures as ion transportation channels into hydrogel networks would be an effective method to obtain superb conductive hydrogels for biomedical engineering applications. Hu and co-workers developed a strong, anisotropic wood hydrogel by combining strong and stiff aligned wood nanofibers with a weak but flexible polyacrylamide hydrogel (Fig. 9b).162 The wood hydrogel exhibits a high tensile strength of 36 MPa along the longitudinal direction due to the strong bonding and cross-linking between the aligned cellulose nanofibers and the polyacrylamide chains. The wood hydrogel is 5 times and 500 times stronger than bacterial cellulose hydrogels (7.2 MPa) and a polyacrylamide hydrogel (0.072 MPa). The negatively charged aligned cellulose nanofibers at low concentrations serve as excellent nanofluidic conduits with an ionic conductivity of up to 0.05 S m−1 for highly selective ion transport, akin to that of biological muscle tissue. Similarly, the ion transportation channel is also useful to enhance the conductivity of polyelectrolyte hydrogels. It is proposed that the interconnected polymer-rich domains result in a slush-like ice formation that helps improve the ionic conductivity of the hydrogel at freezing temperatures.159
Metal ions can provide both conductivity and network crosslinking.165–167 Fe3+-cross-linked cellulose nanocrystal (CNC-Fe3+) networks are combined with polymer networks to generate robust, stretchable, and strain-sensitive hydrogels.165 With applied loadings, the dynamic CNC-Fe3+ coordinates serve as sacrificial bonds to dissipate energy. The hydrogels show very high tensile strength, stretchability, and self-healing capability. The rapid ionic coordination between CNCs and Fe3+ makes autonomous self-healing possible in only 5 min. Such ion conductive hydrogels showed stable sensing behaviour to applied strain, including finger joint motions, breathing, and even wrist pulses.
4. Tough conductive hydrogels
Practical applications of hydrogel sensors frequently require adequate mechanical properties and fatigue resistance against cyclic loadings.46,47 Despite the plenty of conductive hydrogels being used for flexible sensors, delicate design of network structures is still needed to produce hydrogels with outstanding conductivity and mechanical performance. The distribution of conductive networks throughout the matrixes is extremely important for their conductivity, mechanical strength and toughness, and sensory performance. To gain high strength and toughness, tough hydrogels could be used as matrixes to host conductive fillers or polymer networks. Another strategy utilises the conductive network as an energy dissipation mechanism to improve strength and toughness. This section discusses some advances in tough conductive hydrogels.4.1 Conductive double network hydrogels
Inspired by the double network hydrogel concept, conductive hydrogels are synthesized by replacing one network with conductive materials. The first network could be a tough or a flexible matrix, which hosts the polymerization of a conductive second network. It is advantageous to use a very small number of conductive monomers/polymers to construct the second conductive network. Usually, the second network has a good distribution throughout the first one. Specific interactions between both networks may help in improving their mechanical properties. Upon loadings, the immediate response of the conductive network may result in high sensitivity.Tough polymer hydrogels can serve as ideal matrixes for in situ synthesis of a second conductive network. Zhang et al. used chitosan microspheres (CSMs) to synthesize microparticle-crosslinked PAAm hydrogels (Fig. 10a).113 The CSMs serve as crosslinking and energy dissipation centres. The obtained hydrogels are immersed into aniline/phytic acid aqueous solution for in situ synthesis of polyaniline, generating conductive hydrogels with extremely high strength, toughness, and fatigue resistance. The phytic acid not only serves as a doping agent and crosslinker for PANI, but also bonds to chitosan through electrostatic interaction. Thus, the phytic acid strongly bridges chitosan with polyaniline, leading to a compact structure. The connection between the conductive network and hydrophilic polymer chain enables immediate responses to external stimuli, especially at low strains. The conductive DN gels exhibit an unexpectedly high tensile fracture strength of 879 kPa and fracture strain of 626%. Moreover, the conductivity of the hydrogels reaches 5 S m−1 with only 0.1 mol L−1 aniline.
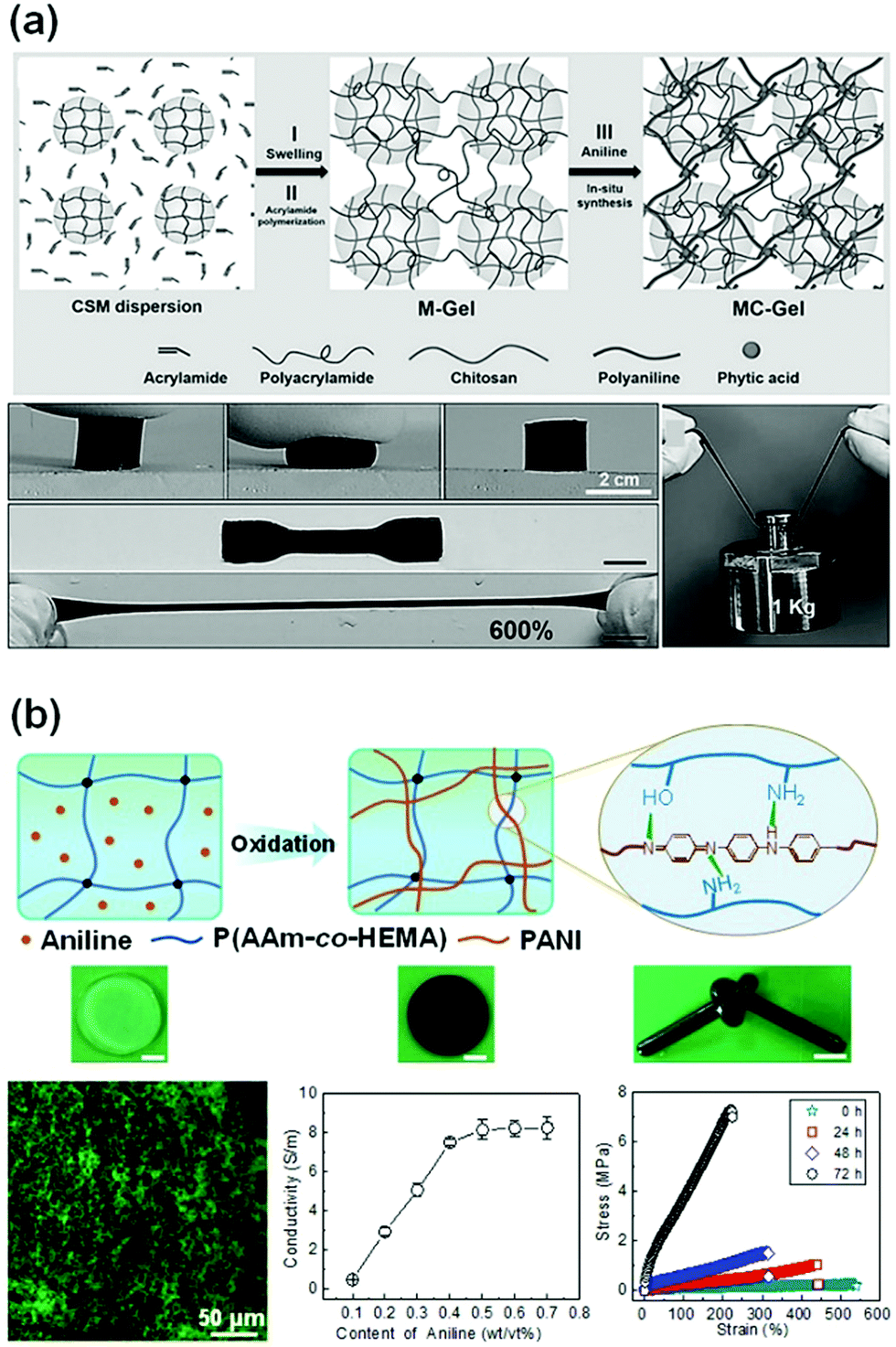 | ||
| Fig. 10 Conductive double network hydrogels. (a) In situ polymerization of aniline in chitosan microsphere-crosslinked hydrogels, producing stretchable and tough conductive hydrogels. Reprinted from ref. 113 with permission. Copyright 2017 Wiley. (b) In situ polymerization of aniline in a poly(acrylamide-co-2-hydroxyethyl methacrylate) network, generating a double network hydrogel with very high strength, toughness, and conductivity. Reprinted from ref. 37 with permission. Copyright 2018 American Chemical Society. | ||
Another class of conductive DN gels are prepared by one-pot synthesis of conductive polymers in a hydrogel matrix.37 Flexible poly(acrylamide-co-hydroxyethyl methyl acrylate) (P(AAm-co-HEMA)) hydrogels are used as the matrixes for in situ oxidation polymerization of aniline, yielding PANI/P(AAm-co-HEMA) DN gels (Fig. 10b). Scanning electron microscopy and confocal laser scanning microscopy confirmed that the PANI chains form a continuous network interpenetrating throughout the P(AAm-co-HEMA) matrix. Therein, PANI and P(AAm-co-HEMA) chains may form hydrogen bonds. The formation of a second PANI network significantly enhances the strength and toughness of the hydrogels, showing a tensile fracture toughness of 9.19 MJ m−3, in contrast to that (0.77 MJ m−3) of pristine P(AAm-co-HEMA) hydrogels. The hydrogels sustain a compressive strength of 17.00 MPa. Within the DN gels, PANI chains form continuous networks around a threshold value of only 0.5 wt/vol%, which leads to a conductivity of 8.14 S m−1. This conductivity is quite close to that of pure PANI hydrogels.110 The tight connection between PANI and P(AAm-co-HEMA) networks enables the PANI network to deform immediately under strain, resulting in very high sensitivity to strain (1.48), and linearity over large strains.
4.2 Conductive nanocomposite hydrogels
Conductive nanoparticles, nanorods, or nanosheets are composited with hydrogel matrices to produce conductive nanocomposite hydrogels. The conductive nanoparticles not only impart conductivity to the hydrogels, but also promote their strength and toughness. The toughening and energy dissipation mechanisms, and conducting performance are explored in this section by introducing several typical examples.165,166,168First, PANI nanoparticles are used to form tough hydrogels with polyvinyl alcohol (PVA).114 Ma and co-workers copolymerized 3-aminophenylboronic acid (ABA) and aniline to incorporate boronic acid groups onto PANI particles (Fig. 11a). Then, the boronic groups crosslink PVA and PANI through supramolecular interaction between boronic acid groups on PANI and hydroxy groups on PVA to form conductive hydrogels.114 The hydrogels are robust and have a tensile Young's modulus of 27.9 MPa, a tensile strength of 5.3 MPa, and an elongation at break of 250%. The hydrogels are recoverable immediately after repeated stretching or compression. In the presence of pre-formed PANI particles, the hydrogels show a conductivity of 10 S m−1.
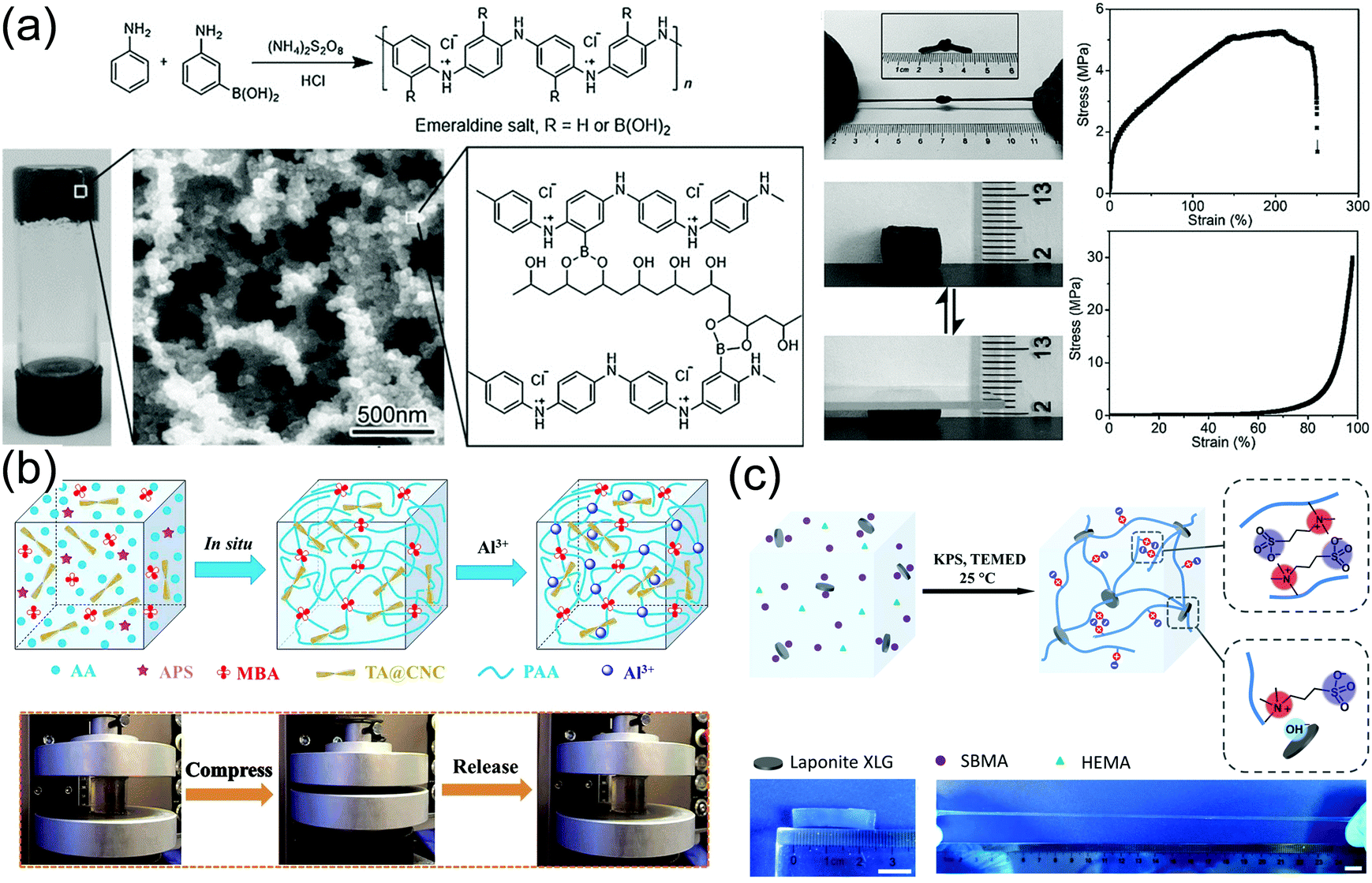 | ||
| Fig. 11 Conductive nanocomposite hydrogels. (a) Borate functionalized polyaniline/PVA nanocomposite hydrogels with supramolecular interactions between PANI nanoparticles and PVA chains. Reprinted from ref. 114 with permission. Copyright 2016 Wiley. (b) Poly(acryl acid) hydrogel crosslinked and reinforced by tannin acid-modified cellulose nanocrystals. Coordination among TA@CNC, PAA, and Al3+ provides additional ionic crosslinking and ionic conductivity. Reprinted from ref. 166 with permission. Copyright 2018 American Chemical Society. (c) Clay nanosheet crosslinked polyzwitterionic hydrogels with high stretchability. Reprinted from ref. 161 with permission. Copyright 2019 American Chemical Society. | ||
Cellulose nanocrystals (CNCs)169,170 with unique hierarchical structures can form hydrogen bonds with polymer chains to toughen hydrogels. Yang and co-workers constructed synergistic interfacial dynamic coordination bonds among poly(acrylic acid) chains, tannic acid-coated CNCs and metal ions in a covalent polymer network to prepare tough ionically conductive hydrogels (Fig. 11b).166 The tannic acid-coated CNCs serve as dynamic cross-linkers to generate a hierarchical porous network and metal ion coordinates to achieve an outstanding toughness of 5.60 MJ m−3. The reversible coordination and hydrogen bonds enable fast and reliable mechanical and electronic self-healing (healing efficiency 88% in 20 min). The catechol groups from tannic acid make the hydrogels adhesive to various substrates including human skin (adhesion strength ∼ 5 kPa).
Strong adhesion of zwitterionic monomers and polymers to clay nanosheets results in tough ionically conductive hydrogels. LAPONITE® XLG nanosheets are able to adsorb zwitterionic monomers for in situ free radical polymerization to prepare conductive nanocomposite hydrogels (Fig. 11c).161 The association between zwitterionic polymers and LAPONITE® XLG nanosheets provides reversible physical cross-linking to improve the mechanical properties of the hydrogels. The hydrogels can be stretched to 2000% at a fracture strength of up to 270 kPa. The high dipole moment in zwitterionic molecules171 imparts excellent adhesion of the hydrogels to many surfaces including skin, glass, silicone rubbers, and nitrile rubbers through ion–dipole or dipole–dipole interactions.172,173 Importantly, the zwitterionic moieties can assist the ion transportation along the highly polarized skeleton to promote ion conduction, showing a conductivity of 0.24 S m−1. The reversible non-covalent interactions enable quick self-healing of the gels in electronic conductivity and excellent self-healing in mechanical properties after cutting them apart at room temperature.
5. Conductive hydrogel sensors
Conductive hydrogels have shown great promise for applications as strain and pressure sensors. The changes in the electrical performances of hydrogels upon external stimulation are related to changes in the conductive network structures. When a pressure is applied to the hydrogel, the conductive network becomes denser, and the resistance decreases. The resistance change ratio is used as an indicator of the pressure. On the other hand, when the conductive hydrogel experiences stretching or compressive deformation, the internal conductive network deforms to change the resistance or conductivity. For both pressure and strain sensors, a synergistic effect of conductive networks and hydrogel networks is very important for their sensing performance.The sensitivity of hydrogel sensors is critical for the performance of devices and strongly dependent on the network structures. Table 1 summarizes recent reports on strain and pressure sensors based on tough conductive hydrogels (including hydrogel network, conductive type, sensing type, gauge factor, conductivity and working range). Interestingly, there is not much correlation between the conductivity and gauge factor of hydrogels. Actually, the former property comes from the density or integrity of conductive networks in hydrogels. However, the gauge factor benefits from the change in the conductive network upon exposure to external stimuli, which is closely related to the breaking or deformation of the conductive network. Thus, the network design for flexible sensors should put them together into consideration for a wonderful balance. Besides, hydrogel capacitors have been widely fabricated as pressure or strain sensors. Changes in capacitance upon loadings resulted in excellent sensitivity. Various conductive hydrogels have been used to develop sensors for applications in human activity monitoring and health monitoring. Many subtle physiological signals, such as breath, finger touch, heartbeat, peristole, expression changes, vocal-cord vibration, and joint bending can be detected in a timely manner and accurately.174–177
| Application | Hydrogel network | Conductive type | Sensing type | Gauge factor | Conductivity (S m−1) | Working range | Ref. |
|---|---|---|---|---|---|---|---|
| Strain sensor | PANI/P(AAm-co-HEMA) | Electron | Resistance | 1.48 | 8.14 | 300% | 37 |
| PVA/PVP/CNC-Fe3+ | Electron | Resistance | 0.478 (0–200%) | — | 200% | 165 | |
| SWCNT/PVA | Electron | Resistance | 0.24 (100%), 1.51 (1000%) | — | 1000% | 35 | |
| Ti3C2Tx/PVA | Electron | Resistance | 25 | — | 40% | 184 | |
| Graphene/PAAm | Electron | Resistance | 12 | 0.0075 | 30% | 185 | |
| PAA/PANI | Electron | Resistance | 0.6 (0–800%), 1.053 (800–1130%) | 5.12 | 1130% | 115 | |
| SA/PAAm | Ion | Resistance | 0–2.0 (0–200%), 2.0–2.7 (200–1800%) | — | 1800% | 186 | |
| PSBMA/PVA | Ion | Resistance | 1.5 | 0.051 | 300% | 164 | |
| ACC/PAA/alginate | Ion | Capacitance | 1 | — | 100% | 187 | |
| PAAm/NaCl | Ion | Capacitance | 0.65 | — | 500% | 188 | |
| PAAM/LiCl | Ion | Resistance | 0.84 | 10.4 | 50% | 189 | |
| Allyl cellulose | Ion | Resistance | 0.297 | 0.0018 | 230% | 190 | |
| k-Carrageenan/PAAm | Ion | Resistance | 0.63 | — | 1000% | 191 | |
| P(AM-co-AA)-Fe3+/CS-SO42− | Ion | Resistance | 6 | 3.2 | 700% | 192 | |
| PDA-talc-PAAm | Ion | Resistance | 0.125 (100%), 0.693 (1000%) | — | 1000% | 193 | |
| P(AAc-co-SBMA) | Ion | Capacitance | 1 | — | 100% | 152 | |
| PVA/FSWCNT/PDA | Electron | Resistance | About 2.0 | 0.05 | — | 36 | |
| HPC/PVA | Ion | Capacitance | 1.87 | 3.4 S m−1 | 400% | 163 | |
| PAAm/CS-c-MWCNT | Electron | Resistance | 1.65 (0–100%), 3.2 (100–500%) | 0.95 | 500% | 140 | |
| Allyl cellulose | Ion | Resistance | 0.30 | 0.016 | 100% | 194 | |
| Cellulose/PAAm/DES | Ion | Resistance | 4.1 | 0.0924 | 60% | 195 | |
| Poly α-lipoic acid | Ion | Resistance | 1.46 (0–50%), 2.30 (50–150%), 3.69 (150–300%) | 0.0028 | 300% | 196 | |
| PAA/TA@CNC | Ion | Resistance | 0.23 (0–40%), 0.76 (40–65%), 4.90 (65–75%) | — | 1000% | 166 | |
| PVA/PAANa-Tb3+ | Ion | Resistance | 0.8 | 3.04 | 150% | 197 | |
| Pressure sensor | ACC/PAA/alginate | Ion | Capacitance | 0.17 kPa−1 | — | 1 kPa | 187 |
| P(AAc-co-SBMA) | Ion | Capacitance | 0.09 kPa−1 | 5 kPa | 152 | ||
| HPC/PVA | Ion | Capacitance | 0.003 kPa−1 | 3.4 | — | 163 | |
| MWCNT/PEDOT:PSSS/PAA-Fe3+ | Electron | Resistance | 0.243 kPa−1 (4.67 kPa), 0.041 kPa−1 (4.67–12.67 kPa) and 0.011 kPa−1 (12.67–26 kPa) | — | 26 kPa | 198 | |
| PAAm/PVA | Ion | Resistance | 0.05 kPa−1 (0–3.27 kPa), 0.02 kPa−1 (3.27–6.83 kPa) | — | 6.83 kPa | 177 | |
| PEG/Na2SO4 | Ion | Resistance | 0.006 kPa−1 | 0.238 | — | 199 | |
| PAAm/LiCl | Ion | Capacitance | 0.006 kPa−1 | 30 kPa | 200 | ||
| SWCNT/alginate | Electron | Resistance | 0.176 kPa−1 (10 Pa) | 0.004 | — | 201 | |
| SA/PAAm | Ion | Resistance | 1.45 kPa−1 (0–1.5 kPa), 0.02 kPa−1 (1.5–6.0 kPa) | — | 6.0 kPa | 186 | |
| PAAm/LiCl | Ion | Triboelectricity | 0.013 kPa−1 | — | 1.3 kPa | 202 | |
| CSM/PAAm/PANI | Electron | Resistance | 0.35 kPa−1 (0–1 kPa), 0.05 kPa−1 (1–10 kPa), 10−5 kPa−1 (>500 kPa) | 5 | 1 MPa | 113 |
5.1 Strain sensors
Strain sensors are basically used to transform strain or deformation into electronic signals, which can precisely capture real-time signals from joints, the epidermis, and cardiac tissue for further analysis and diagnosis.33,177–182 The performance parameters of strain sensors, including their sensitivity and working range, are largely dependent on their network design and sensor configurations.Highly stretchable hydrogels composited with carbon nanotubes, graphene, and silver nanowires can serve as piezoresistive strain sensors.35 The extremely high stretchability allows for a very large working range to more than 1000% strain. For such strain sensors, the resistance change ratio (ΔR = |R − R0|/R0), where R is the resistance with applied strain and R0 is the original resistance, usually increases with increasing strain before the hydrogels are fractured. The sensitivity, or the gauge factor, is defined by (|R − R0|/R0)/ε (where ε is strain). Although hydrogels free of conductive fillers may show some resistance changes during stretching (probably due to the presence of a trace number of ions in water), the presence of conductive fillers significantly enhances the sensitivity of strain sensors. Hydrogels composited with carbon nanotubes, for example, may show a relatively lower sensitivity at low strain (e.g., 0.24 at 100% strain) and a much higher one at high strain (e.g., 1.51 at 1000%).35 The nonlinear dependence of ΔR on strain and low sensitivity are largely related to the poor formation of a conductive network and its poor affinity with the hydrogel network. In fact, most stretchable sensors based on conductive fillers/hydrogel composites show low sensitivity at small strains (0–100%). In general, the fillers have poor affinity with the hydrogel matrix. Upon stretching, particularly at low strains, the deformation of the hydrogel networks is much larger than rigid conductive fillers.
Linearity and high sensitivity have been pursued for hydrogel sensors but have rarely been reported.14,183 Recently, a conductive polymer network interpenetrating a flexible hydrogel matrix was constructed.37 Therein, polyaniline (PANI) networks are in situ prepared within the matrix of P(AAm-co-AMPS) hydrogels. The polyaniline (PANI) chains serve as both conductive and toughening networks that provide conductivity and improve the strength and toughness of the hydrogels. The PANI network percolates at a very low concentration (0.5 wt/vol%) to form a continuous conductive network and provide a high conductivity. Moreover, the positively charged PANI chains have electrostatic interactions and/or hydrogen bonding with the P(AAm-co-AMPS) network. Hydrogel deformations induce a rapid response of the PANI network even at very low strains (Fig. 12a). The hydrogel sensor shows a very high sensitivity of 5.7 at very low strain (0.3%), and a plateau value (1.48) from 40% to 300% strain (Fig. 12b). The hydrogel sensor attached to the wrist can accurately capture the bending of the wrist, providing electric signals directly related to the bending angle (Fig. 12c). Moreover, the hydrogel sensor sustains cyclic stretching/unloading tests, showing repeatable and reliable sensing outputs for hundreds of cycles (Fig. 12d). The high sensitivity enables the capture of very weak wrist pulse signals. As the hydrogel sensor is mounted on the wrist, the weak vibrations drive significant changes in resistance. The resistance change ratio even accurately records two diacritical peaks and a late systolic augmentation shoulder (Fig. 12e), which is a diagnostic indicator of arterial stiffness. Moreover, the sensor is able to obtain very clear signals with high signal-to-noise ratios during phonating, showing high-resolution detections on the vocal cord vibration (Fig. 12f).
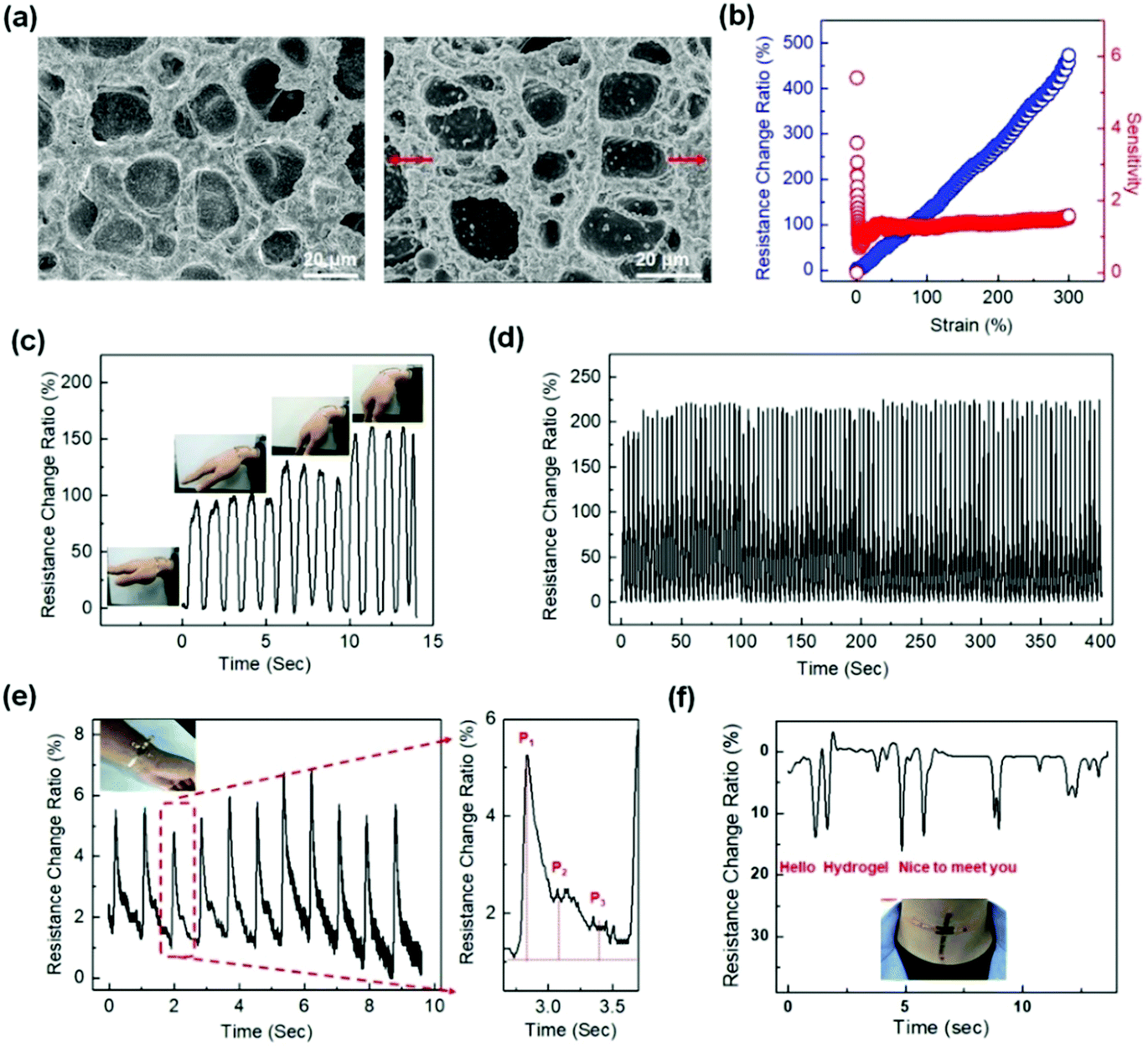 | ||
| Fig. 12 Representative strain sensors based on conductive interpenetrating PANI/P(AAm-co-AMPS) hydrogels. (a) SEM images of the IPN structures before and after strain. (b) Linear strain sensing. (c) Sensing the wrist bending. (d) 100 cycle sensing tests with 100% strain. (e) Arterial pulse signals collected at a volunteer's wrist with a hydrogel sensor. (f) Vocal signals collected at the throat of a volunteer speaking “hello”, “hydrogel”, and “nice to meet you”. Reproduced from ref. 37 with permission. Copyright 2018 American Chemical Society. | ||
For applications in health monitors, human–machine interfaces, and implantable sensors, a robust adhesion to biotissues are vitally important.164 The conformal adhesion to the substrate ensures accurate acquisition of deformation signals of the substrate. Wan and co-workers introduced mussel-inspired polydopamine materials into conductive SWCNT/PVA composite hydrogels, generating strain sensors that are adhesive to porcine skin.36 The sensor attaches onto human skin without the help of any other additional adhesive tapes for recording human respiration and pulses. However, most of these methods usually involve the use of polyphenols to achieve robust adhesion, which may result in unwanted colour and complicated preparation procedures. Novel methods are desired to achieve a robust self-adhesion of transparent flexible sensors to substrates including biotissues.
5.2. Pressure sensors
Flexible pressure sensors are able to convert the applied pressure into electronic signals.186,201,203 Pressure sensing has great potential for applications ranging from artificial intelligence to physiological signal monitoring.30,175,176,204 In this section, we introduce representative tough conductive hydrogels as pressure sensors, and emphatically focus on their measurement range, sensitivity and applications.29,177,186Wu and co-workers used amorphous calcium carbonate nanoparticles to physically crosslink alginate and polyacrylic acid chains to produce self-healing ionically conductive nanocomposite hydrogels (Fig. 13a).187 The amorphous calcium carbonate nanoparticles serve as Ca2+ reservoirs. The hydrogel was assembled into a capacitor with a dielectric spacing layer (Fig. 13a). The capacitance C = εS/4πkd, where ε is the dielectric constant of the dielectric layer, S is the effective area of the conducting layer, k is the electrostatic constant, and d is the thickness of the dielectric layer. When the capacitor is pressed, d becomes smaller, and S increases. Consequently, C is significantly increased.205 The hydrogel based capacitive sensor shows an almost linear response to pressure (0–1 kPa) with a sensitivity of 0.17 kPa−1. Moreover, the hydrogel sensor can fully recover its capacitive touch response, even after 10 drying–gel–swelling cycles. Although its sensitivity is very high compared with those of other reported pressure sensors,188,206 the working range is limited, primarily due to its poor mechanical properties.
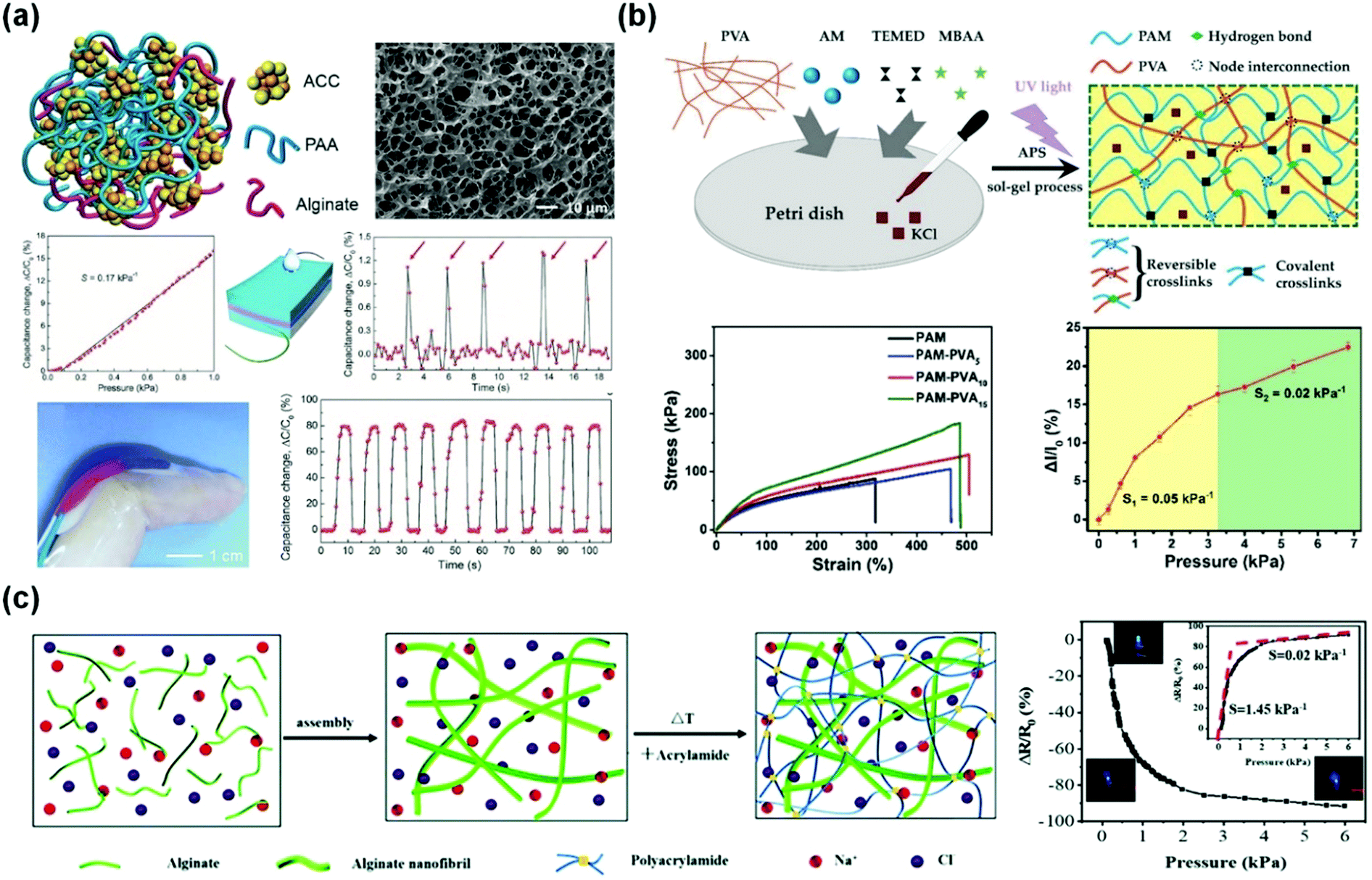 | ||
| Fig. 13 Pressure sensors. (a) A capacitive pressure sensor based on alginate/polyacrylic acid hydrogel crosslinked by amorphous calcium carbonate particles. Reprinted from ref. 187 with permission. Copyright 2017 Wiley. (b) Interpenetrating polyacrylamide/polyvinyl alcohol hydrogels with high stretchability and two-regime pressure sensitivity. Reprinted from ref. 177 with permission. Copyright 2018 Wiley. (c) Ion conductive alginate/polyacrylamide nanocomposite hydrogels with high pressure sensitivity. Reprinted from ref. 186 with permission. Copyright 2019 Royal Society of Chemistry. | ||
Microstructures on the surfaces of hydrogel films are beneficial for high sensitivity and stimuli-responsive behaviors.207–210 The micro-architectures may help in improving the signal-to-noise ratio of hydrogel-based sensors.211 Dong and co-workers obtained hierarchically wrinkled micro-architectures and interconnected ridges on self-patterned hydrogel surfaces (Fig. 13b).177 The patterns significantly increase the contact area and thus improve the sensitivity (0.05 kPa−1 for 0–3.27 kPa) and precise sensing of dynamic pressures. The hydrogels also present fast response (150 ms) and a reliable durability against 500 loading/unloading cycles.
Outstanding mechanical properties and fatigue resistance are needed for hydrogel-based pressure sensors with high sensitivity.212 Sui and co-workers reported a nature-inspired ionic double network hydrogel with superior mechanical and sensing properties (Fig. 13c).186 This hydrogel is comprised of sodium alginate nanofibrils and chemically crosslinked polyacrylamide, and mimics the structure of the dermis.213 The hydrogel shows excellent mechanical properties and reasonable conductivity (0.01 S m−1) and a sensitivity of 1.45 kPa−1 at low pressure (below 1.5 kPa). Conductive double network PANI/chitosan/polyacrylamide hydrogels show outstanding strength and toughness, and work as pressure sensors with a large working range of over 500 kPa and sensitivities of 0.35 kPa−1 at pressures <1 kPa, 0.05 kPa−1 at medium pressures (<10 kPa), and 10−5 kPa−1 at high pressures (>500 kPa).113
Besides resistance and capacitance, triboelectricity has recently been introduced into flexible sensors,202,214–216 which enables both biomechanical energy harvesting and touch sensing. This novel design is achieved by using an elastomer (PDMS, or 3M VHB) film as the electrification layer and an ionic hydrogel (PAAm/LiCl hydrogel) as the electrode. Once an object touches the elastomer film of the sensor, electrification occurs at the interface and generates charges with opposite polarities at the surface of the dielectric film and the elastomer. When the two separated surfaces move away, the static charges on the surface of the insulating elastomer will induce the movement of ions in the hydrogel to balance the static charges, forming a layer of excessive ions at the interface. Meanwhile, the electrical double layer formed at the ionic hydrogel interface will be polarized, forming the same amount of excessive negative ions at the interface and positive charges in the metal wire.202 Based on this novel device design, the sensitivity of hydrogel based sensors is up to 0.013 kPa−1. This approach avoids the dependence of conventional sensors on external energy, which allows an conversion from human motions to electricity and thus provides a new method for flexible sensors in the future.
5.3 Electronic devices based on hydrogel sensors
Tough conductive hydrogels are ideal candidate materials for flexible sensors, which are intrinsically flexible materials with biocompatibility, high conductivity, and so on.60,217,218 Electronic devices based on conductive hydrogels are on the rise as indispensable means to develop wearable devices for bioapplications.54,57 Here, the current status of electronic devices based on hydrogel sensors and potential applications are introduced briefly.Due to the increasingly important human–machine interactions, touch panels may require stretchability and biocompatibility to integrate with the human body. However, most touch panels have been developed based on stiff and brittle electrodes.59,219 As shown in Fig. 14a, an ionic touch panel based on a polyacrylamide hydrogel with lithium chloride salts was reported recently.220 The panel is flexible, and able to sustain a large deformation. The surface-capacitive sensor system is adopted to feel finger touch. The panel can freely transmit light information, resulting from the transparency of the hydrogel (98% transmittance for visible light). Moreover, this panel can be operated under more than 1000% areal strain without sacrificing performances.
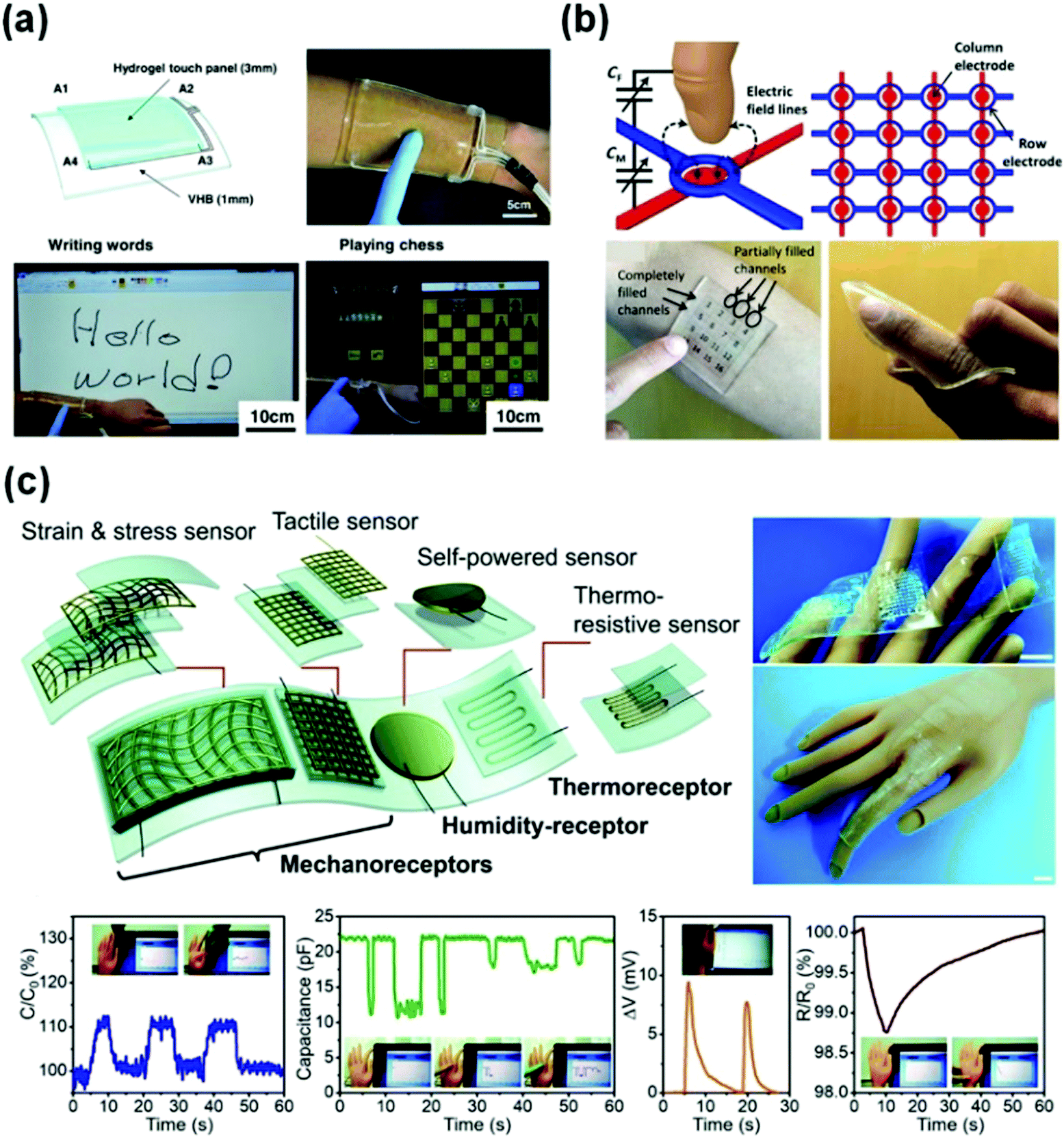 | ||
| Fig. 14 Representative flexible devices based on conductive hydrogel sensors. (a) Wearable ionic hydrogel touch panels. Reprinted from ref. 220 with permission. Copyright 2016 AAAS. (b) A 4 × 4 cross-grid sensor array. Reprinted from ref. 137 with permission. Copyright 2017 AAAS. (c) Integration of a strain and stress sensor, a tactile sensor, a self-powered sensor and a thermo-resistive sensor and receptors into single devices. Reprinted from ref. 153 with permission. Copyright 2019 Royal Society of Chemistry. | ||
More complex flexible tough panels are developed by using a 4 × 4 cross-grid sensor array with a 5 mm pitch (Fig. 14b).137 The finger hovering path on the touch pad is easily detected and accurately recorded by the array. The touch panel is able to recognize the locations of multiple fingers. A light touch generates a localized drop in capacitance by 15%. The applied pressure sensing performance is not affected by the bending or stretch of the panel.
Usually, electronic devices integrate multiple function sensors into one single device.9,13,39,221 Major challenges include the contrasting properties of soft and rigid materials and the limited processing techniques.8,12 A flexible device with various functional units can be integrated by 3D printing (Fig. 14c).153 Therein, different types of sensors use capacitance, voltage and resistance signals to mimic different receptors of human skin. Multiple information perception networks are built by using a hybrid circuit design, where the self-powered voltage, capacitance, and resistance signals imitate the humidity-receptor, mechanoreceptor, and thermo-receptor of human skin, respectively. The obtained device enables a plastic hand to feel a wide scope of stimuli. It reports decreasing capacitance with bare finger or gloved finger touches, shows increasing capacitance when the hand bends, senses breaths of a human via voltage output triggered by moisture, and detects a slight temperature increase caused by human finger contact based on decreasing resistive signals. This study not only contributes new concepts to the development of deformable conductors, but also enables the fabrication of human–machine interfaces with much more sophisticated bionic intelligence.
6. Conclusions and outlook
In this review, we summarized recent progress in tough conductive hydrogels for flexible sensors, including design of tough hydrogels, fabrication of conductive hydrogels, and applications of flexible hydrogel sensors. Tough conductive hydrogels are ideal candidate materials for flexible sensors, due to the perfect combination of various properties, such as conductivity, biocompatibility and flexibility. Significant advances in hydrogel-based flexible sensors for wearable devices or electronic skin have been recently achieved, which can be attached to human skin for recording physical health information including pulses, joint motion, and voice.Despite the excellent advances in tough hydrogels for flexible sensors in recent years, there are still urgent and unmet demands that stimulate further studies in this field.
First of all, the water loss of hydrogels has to be solved. The high-water content provides hydrogels with various unparalleled properties, including natural flexibility, tissue similarity, and even superb mechanical properties. Without water, the hydrogels would be hard and brittle, and show non-sensing properties. Plenty of studies have been devoted to avoiding water loss of hydrogels, such as modification of hydrogel surfaces and solvent replacement. However, the former would influence the mechanical properties of conductive hydrogels, and the latter may even change their electronic and sensing properties. For practical applications, the stability of flexible sensors under long-term work is necessary and crucial.
The integration of conductive hydrogel sensors and device units is another open issue. Different from traditional electronic materials, for example metals, conductive hydrogels show low moduli and contain lots of water, which would cause poor connection of the interface between sensors and circuits. There are few integrated multifunctional sensor platforms based on hydrogels that effectively combine various units with different sensing types.
Moreover, the micromachining of conductive hydrogels for the miniaturization and lightweight of devices is also a challenge. Although 3D microprinting is an effective technology to realize microscopic three-dimensional structure materials, a suitable hydrogel system is very rare. More efforts have to be directed towards promoting novel tough, conductive and 3D printable hydrogels to fabricate biomimetic devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51873224 and 21574145), the Ningbo Natural Science Foundation (2017A610232), and the MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Zhejiang University (2018MSF04).Notes and references
- D. J. Lipomi, Adv. Mater., 2016, 28, 4180–4183 CrossRef CAS PubMed
.
- J. A. Fan, W.-H. Yeo, Y. Su, Y. Hattori, W. Lee, S.-Y. Jung, Y. Zhang, Z. Liu, H. Cheng, L. Falgout, M. Bajema, T. Coleman, D. Gregoire, R. J. Larsen, Y. Huang and J. A. Rogers, Nat. Commun., 2014, 5, 3266 CrossRef PubMed
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603 CrossRef CAS PubMed
- S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger and R. Schwödiauer, Adv. Mater., 2014, 26, 149–162 CrossRef CAS PubMed
- Y. Kim, H. Yuk, R. Zhao, S. A. Chester and X. Zhao, Nature, 2018, 558, 274–279 CrossRef CAS PubMed
- G. Gu, J. Zou, R. Zhao, X. Zhao and X. Zhu, Sci. Rob., 2018, 3, eaat2874 CrossRef
- Y. Kim, G. A. Parada, S. Liu and X. Zhao, Sci. Rob., 2019, 4, eaax7329 CrossRef
- Y. Liu, K. He, G. Chen, W. R. Leow and X. Chen, Chem. Rev., 2017, 117, 12893–12941 CrossRef CAS PubMed
- Z. Ma, S. Li, H. Wang, W. Cheng, Y. Li, L. Pan and Y. Shi, J. Mater. Chem. B, 2019, 7, 173–197 RSC
- X. Wang, Z. Liu and T. Zhang, Small, 2017, 13, 1602790 CrossRef PubMed
- S. Wang, J. Y. Oh, J. Xu, H. Tran and Z. Bao, Acc. Chem. Res., 2018, 51, 1033–1045 CrossRef CAS PubMed
- B. Zhu, H. Wang, W. R. Leow, Y. Cai, X. J. Loh, M.-Y. Han and X. Chen, Adv. Mater., 2016, 28, 4250–4265 CrossRef CAS PubMed
- S.-K. Kang, J. Koo, Y. K. Lee and J. A. Rogers, Acc. Chem. Res., 2018, 51, 988–998 CrossRef CAS PubMed
- S. Zhao, J. Li, D. Cao, G. Zhang, J. Li, K. Li, Y. Yang, W. Wang, Y. Jin, R. Sun and C.-P. Wong, ACS Appl. Mater. Interfaces, 2017, 9, 12147–12164 CrossRef CAS PubMed
- Y. Zang, F. Zhang, C.-a. Di and D. Zhu, Mater. Horiz., 2015, 2, 140–156 RSC
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig, J. Lopez, T. Lei, S.-K. Kwon, Y. Kim, A. M. Foudeh, A. Ehrlich, A. Gasperini, Y. Yun, B. Murmann, J. B. H. Tok and Z. Bao, Nature, 2018, 555, 83 CrossRef CAS PubMed
- M. Chen, L. Zhang, S. Duan, S. Jing, H. Jiang and C. Li, Adv. Funct. Mater., 2014, 24, 7548–7556 CrossRef CAS
- Y. R. Jeong, H. Park, S. W. Jin, S. Y. Hong, S.-S. Lee and J. S. Ha, Adv. Funct. Mater., 2015, 25, 4228–4236 CrossRef CAS
- Z. Liu, D. Qi, P. Guo, Y. Liu, B. Zhu, H. Yang, Y. Liu, B. Li, C. Zhang, J. Yu, B. Liedberg and X. Chen, Adv. Mater., 2015, 27, 6230–6237 CrossRef CAS PubMed
- C. Yan, J. Wang, W. Kang, M. Cui, X. Wang, C. Y. Foo, K. J. Chee and P. S. Lee, Adv. Mater., 2014, 26, 2022–2027 CrossRef CAS PubMed
- D. J. Lipomi, M. Vosgueritchian, B. C. K. Tee, S. L. Hellstrom, J. A. Lee, C. H. Fox and Z. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS PubMed
- T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296–301 CrossRef CAS PubMed
- M. Amjadi, A. Pichitpajongkit, S. Lee, S. Ryu and I. Park, ACS Nano, 2014, 8, 5154–5163 CrossRef CAS PubMed
- S. Gong, D. T. H. Lai, B. Su, K. J. Si, Z. Ma, L. W. Yap, P. Guo and W. Cheng, Adv. Electron. Mater., 2015, 1, 1400063 CrossRef
- X. Xiao, L. Yuan, J. Zhong, T. Ding, Y. Liu, Z. Cai, Y. Rong, H. Han, J. Zhou and Z. L. Wang, Adv. Mater., 2011, 23, 5440–5444 CrossRef CAS PubMed
- Y. Yu, F. Liu, R. Zhang and J. Liu, Adv. Mater. Technol., 2017, 2, 1700173 CrossRef
- M. Li, H. Li, W. Zhong, Q. Zhao and D. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 1313–1319 CrossRef CAS PubMed
- B. Sun, Y.-Z. Long, S.-L. Liu, Y.-Y. Huang, J. Ma, H.-D. Zhang, G. Shen and S. Xu, Nanoscale, 2013, 5, 7041–7045 RSC
- Z. Lei, Q. Wang, S. Sun, W. Zhu and P. Wu, Adv. Mater., 2017, 29, 1700321 CrossRef PubMed
- C.-L. Choong, M.-B. Shim, B.-S. Lee, S. Jeon, D.-S. Ko, T.-H. Kang, J. Bae, S. H. Lee, K.-E. Byun, J. Im, Y. J. Jeong, C. E. Park, J.-J. Park and U. I. Chung, Adv. Mater., 2014, 26, 3451–3458 CrossRef CAS PubMed
- Y. Yang and W. Gao, Chem. Soc. Rev., 2019, 48, 1465–1491 RSC
- Z. Lei, Q. Wang and P. Wu, Mater. Horiz., 2017, 4, 694–700 RSC
- C. Yeom, K. Chen, D. Kiriya, Z. Yu, G. Cho and A. Javey, Adv. Mater., 2015, 27, 1561–1566 CrossRef CAS PubMed
- C. Yan, J. Wang, W. Kang, M. Cui, X. Wang, Y. Foo Ce, J. Chee Kenji and S. Lee Pooi, Adv. Mater., 2013, 26, 2022–2027 CrossRef PubMed
- G. Cai, J. Wang, K. Qian, J. Chen, S. Li and S. Lee Pooi, Adv. Sci., 2016, 4, 1600190 CrossRef PubMed
- M. Liao, P. Wan, J. Wen, M. Gong, X. Wu, Y. Wang, R. Shi and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703852 CrossRef
- Z. Wang, J. Chen, Y. Cong, H. Zhang, T. Xu, L. Nie and J. Fu, Chem. Mater., 2018, 30, 8062–8069 CrossRef CAS
- C. Yan and P. S. Lee, Small, 2014, 10, 3443–3460 CrossRef CAS PubMed
- H.-R. Lee, C.-C. Kim and J.-Y. Sun, Adv. Mater., 2018, 30, 1704403 CrossRef PubMed
- L. Han, X. Lu, M. Wang, D. Gan, W. Deng, K. Wang, L. Fang, K. Liu, W. Chan Chun, Y. Tang, L. T. Weng and H. Yuan, Small, 2016, 13, 1601916 CrossRef PubMed
- N. Annabi, S. R. Shin, A. Tamayol, M. Miscuglio, M. A. Bakooshli, A. Assmann, P. Mostafalu, J.-Y. Sun, S. Mithieux, L. Cheung, X. Tang, A. S. Weiss and A. Khademhosseini, Adv. Mater., 2016, 28, 40–49 CrossRef CAS PubMed
- D. Seliktar, Science, 2012, 336, 1124 CrossRef CAS PubMed
- W. Zhang, P. Feng, J. Chen, Z. Sun and B. Zhao, Prog. Polym. Sci., 2019, 88, 220–240 CrossRef CAS
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L.-T. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372 CrossRef CAS
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed
- Z. Jia, Y. Zeng, P. Tang, D. Gan, W. Xing, Y. Hou, K. Wang, C. Xie and X. Lu, Chem. Mater., 2019, 31, 5625–5632 CrossRef CAS
- A. Alam, Y. Zhang, H.-C. Kuan, S.-H. Lee and J. Ma, Prog. Polym. Sci., 2018, 77, 1–18 CrossRef CAS
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC
- J. Fu, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1336–1350 CrossRef CAS
- J. Fu and M. in het Panhuis, J. Mater. Chem. B, 2019, 7, 1523–1525 RSC
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS
- J. Y. Sun, C. Keplinger, M. Whitesides George and Z. Suo, Adv. Mater., 2014, 26, 7608–7614 CrossRef CAS PubMed
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed
- Y.-n. Sun, G.-r. Gao, G.-l. Du, Y.-j. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS
- H. Yuk, B. Lu and X. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC
- M. I. Svechtarova, I. Buzzacchera, B. J. Toebes, J. Lauko, N. Anton and C. J. Wilson, Electroanalysis, 2016, 28, 1201–1241 CrossRef CAS
- W. A. D. M. Jayathilaka, K. Qi, Y. Qin, A. Chinnappan, W. Serrano-García, C. Baskar, H. Wang, J. He, S. Cui, S. W. Thomas and S. Ramakrishna, Adv. Mater., 2019, 31, 1805921 CrossRef PubMed
- C. G. Wiener, C. Wang, Y. Liu, R. A. Weiss and B. D. Vogt, Macromolecules, 2017, 50, 1672–1680 CrossRef CAS
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS
- K. Oshima, T. Fujimoto, E. Minami and Y. Mitsukami, Macromolecules, 2014, 47, 7573–7580 CrossRef CAS
- A. Nakayama, A. Kakugo, J. P. Gong, Y. Osada, M. Takai, T. Erata and S. Kawano, Adv. Funct. Mater., 2004, 14, 1124–1128 CrossRef CAS
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS
- K. Haraguchi, Polym. J., 2011, 43, 223 CrossRef CAS
- D. W. Thompson and J. T. Butterworth, J. Colloid Interface Sci., 1992, 151, 236–243 CrossRef CAS
- M.-H. Ling and Y.-J. Lin, J. Biomater. Sci., Polym. Ed., 2009, 20, 637–652 CrossRef PubMed
- Q. Wang, R. Hou, Y. Cheng and J. Fu, Soft Matter, 2012, 8, 6048–6056 RSC
- A. K. Gaharwar, S. A. Dammu, J. M. Canter, C.-J. Wu and G. Schmidt, Biomacromolecules, 2011, 12, 1641–1650 CrossRef CAS PubMed
- Y. Li, Y. Sun, Y. Xiao, G. Gao, S. Liu, J. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2016, 8, 26326–26331 CrossRef CAS PubMed
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC
- J. P. Gong, Science, 2014, 344, 161 CrossRef CAS PubMed
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv.
Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed
-
Articular Cartilage Tissue Engineering, ed. K. A. Athanasiou, E. M. Darling and J. C. Hu, Morgan & Claypool Publishers, 2010 Search PubMed
- G. Du, G. Gao, R. Hou, Y. Cheng, T. Chen, J. Fu and B. Fei, Chem. Mater., 2014, 26, 3522–3529 CrossRef CAS
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed
- S. Hong, D. Sycks, H. F. Chan, S. Lin, G. P. Lopez, F. Guilak, K. W. Leong and X. Zhao, Adv. Mater., 2015, 27, 4035–4040 CrossRef CAS PubMed
- J. Cui, M. Wang, Y. Zheng, G. M. Rodríguez Muñiz and A. del Campo, Biomacromolecules, 2013, 14, 1251–1256 CrossRef CAS PubMed
- G. Du, F. Wu, Y. Cong, L. Nie, S. Liu, G. Gao and J. Fu, Chem. Commun., 2015, 51, 15534–15537 RSC
- Z. Zhao, R. Fang, Q. Rong and M. Liu, Adv. Mater., 2017, 29, 1703045 CrossRef PubMed
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339 CrossRef CAS PubMed
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS
- K. Haraguchi, R. Farnworth, A. Ohbayashi and T. Takehisa, Macromolecules, 2003, 36, 5732–5741 CrossRef CAS
- K. Haraguchi, H.-j. Li, H.-y. Ren and M. Zhu, Macromolecules, 2010, 43, 9848–9853 CrossRef CAS
- K. Haraguchi and H.-J. Li, Macromolecules, 2006, 39, 1898–1905 CrossRef CAS
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253–1258 CrossRef CAS PubMed
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80 CrossRef CAS PubMed
- P. Chen and L. Zhang, Biomacromolecules, 2006, 7, 1700–1706 CrossRef CAS PubMed
- H. Heinz, H. Koerner, K. L. Anderson, R. A. Vaia and B. L. Farmer, Chem. Mater., 2005, 17, 5658–5669 CrossRef CAS
- G. Gao, G. Du, Y. Sun and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 5029–5037 CrossRef CAS PubMed
- C. He, K. Jiao, X. Zhang, M. Xiang, Z. Li and H. Wang, Soft Matter, 2011, 7, 2943–2952 RSC
- F.-K. Shi, X.-P. Wang, R.-H. Guo, M. Zhong and X.-M. Xie, J. Mater. Chem. B, 2015, 3, 1187–1192 RSC
- M. Takafuji, S.-y. Yamada and H. Ihara, Chem. Commun., 2011, 47, 1024–1026 RSC
- J. Yang, X.-P. Wang and X.-M. Xie, Soft Matter, 2012, 8, 1058–1063 RSC
- M. Zhong, X.-Y. Liu, F.-K. Shi, L.-Q. Zhang, X.-P. Wang, A. G. Cheetham, H. Cui and X.-M. Xie, Soft Matter, 2015, 11, 4235–4241 RSC
- Y. Huang, M. Zhong, Y. Huang, M. Zhu, Z. Pei, Z. Wang, Q. Xue, X. Xie and C. Zhi, Nat. Commun., 2015, 6, 10310 CrossRef CAS PubMed
- B. C. K. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825 CrossRef CAS PubMed
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed
- K.-x. Sheng, Y.-x. Xu, C. Li and G.-q. Shi, New Carbon Mater., 2011, 26, 9–15 CrossRef CAS
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CrossRef CAS PubMed
- W. Guo, C. Cheng, Y. Wu, Y. Jiang, J. Gao, D. Li and L. Jiang, Adv. Mater., 2013, 25, 6064–6068 CrossRef CAS PubMed
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z.-F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef CAS PubMed
- Q. Cheng, M. Wu, M. Li, L. Jiang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 3750–3755 CrossRef CAS PubMed
- L. Pan, G. Yu, D. Zhai, H. R. Lee, W. Zhao, N. Liu, H. Wang, B. C. K. Tee, Y. Shi, Y. Cui and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287 CrossRef CAS PubMed
- Z. R. Tian, J. Liu, J. A. Voigt, H. Xu and M. J. McDermott, Nano Lett., 2003, 3, 89–92 CrossRef CAS
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed
- J. Duan, X. Liang, J. Guo, K. Zhu and L. Zhang, Adv. Mater., 2016, 28, 8037–8044 CrossRef CAS PubMed
- W. Li, F. Gao, X. Wang, N. Zhang and M. Ma, Angew. Chem., Int. Ed., 2016, 55, 9196–9201 CrossRef CAS PubMed
- Z. Wang, H. Zhou, J. Lai, B. Yan, H. Liu, X. Jin, A. Ma, G. Zhang, W. Zhao and W. Chen, J. Mater. Chem. C, 2018, 6, 9200–9207 CAS
- S. Bai, C. Sun, P. Wan, C. Wang, R. Luo, Y. Li, J. Liu and X. Sun, Small, 2014, 11, 306–310 CrossRef
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Nat. Commun., 2019, 10, 1043 CrossRef PubMed
- B. Yao, H. Wang, Q. Zhou, M. Wu, M. Zhang, C. Li and G. Shi, Adv. Mater., 2017, 29, 1700974 CrossRef PubMed
- C. Kleber, M. Bruns, K. Lienkamp, J. Rühe and M. Asplund, Acta Biomater., 2017, 58, 365–375 CrossRef CAS PubMed
- D.-H. Kim, N. Lu, R. Ma, Y.-S. Kim, R.-H. Kim, S. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T.-i. Kim, R. Chowdhury, M. Ying, L. Xu, M. Li, H.-J. Chung, H. Keum, M. McCormick, P. Liu, Y.-W. Zhang, F. G. Omenetto, Y. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838 CrossRef CAS PubMed
- D. Gan, Z. Huang, X. Wang, L. Jiang, C. Wang, M. Zhu, F. Ren, L. Fang, K. Wang, C. Xie and X. Lu, Adv. Funct. Mater., 2019, 1907678 Search PubMed
- H. Zhou, W. Yao, G. Li, J. Wang and Y. Lu, Carbon, 2013, 59, 495–502 CrossRef CAS
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC
- H. Peng, Y. Xin, J. Xu, H. Liu and J. Zhang, Mater. Horiz., 2019, 6, 618–625 RSC
- L. Qiu, D. Liu, Y. Wang, C. Cheng, K. Zhou, J. Ding, V.-T. Truong and D. Li, Adv. Mater., 2014, 26, 3333–3337 CrossRef CAS PubMed
- A. Lerf, H. He, T. Riedl, M. Forster and J. Klinowski, Solid State Ionics, 1997, 101-103, 857–862 CrossRef CAS
- C. Pan, L. Liu, Q. Chen, Q. Zhang and G. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 38052–38061 CrossRef CAS PubMed
- K. Ren, Y. Cheng, C. Huang, R. Chen, Z. Wang and J. Wei, J. Mater. Chem. B, 2019, 7, 5704–5712 RSC
- J. Liu, C. Chen, C. He, J. Zhao, X. Yang and H. Wang, ACS Nano, 2012, 6, 8194–8202 CrossRef CAS PubMed
- X. Jing, H.-Y. Mi, X.-F. Peng and L.-S. Turng, Carbon, 2018, 136, 63–72 CrossRef CAS
- Z. Tang, L. Gao, Y. Wu, T. Su, Q. Wu, X. Liu, W. Li and Q. Wang, J. Mater. Chem. B, 2013, 1, 5393–5397 RSC
- H.-P. Cong, J.-H. Qiu and S.-H. Yu, Small, 2015, 11, 1165–1170 CrossRef CAS PubMed
- Y. Wang, Y. Xiao, G. Gao, J. Chen, R. Hou, Q. Wang, L. Liu and J. Fu, J. Mater. Chem. B, 2017, 5, 511–516 RSC
- P. M. Ajayan, Chem. Rev., 1999, 99, 1787–1800 CrossRef CAS PubMed
- R. A. MacDonald, C. M. Voge, M. Kariolis and J. P. Stegemann, Acta Biomater., 2008, 4, 1583–1592 CrossRef CAS PubMed
- X. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS PubMed
- M. S. Sarwar, Y. Dobashi, C. Preston, J. K. M. Wyss, S. Mirabbasi and J. D. W. Madden, Sci. Adv., 2017, 3, e1602200 CrossRef PubMed
- A. Kyritsis, P. Pissis and J. Grammatikakis, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1737–1750 CrossRef CAS
- K. Guo, D.-L. Zhang, X.-M. Zhang, J. Zhang, L.-S. Ding, B.-J. Li and S. Zhang, Angew. Chem., Int. Ed., 2015, 54, 12127–12133 CrossRef CAS PubMed
- S. Xia, S. Song, F. Jia and G. Gao, J. Mater. Chem. B, 2019, 7, 4638–4648 RSC
- T. Shay, O. D. Velev and M. D. Dickey, Soft Matter, 2018, 14, 3296–3303 RSC
- K. Liu, L. Han, P. Tang, K. Yang, D. Gan, X. Wang, K. Wang, F. Ren, L. Fang, Y. Xu, Z. Lu and X. Lu, Nano Lett., 2019, 19, 8343–8356 CrossRef CAS PubMed
- H. Liu, M. Li, C. Ouyang, T. J. Lu, F. Li and F. Xu, Small, 2018, 14, 1801711 CrossRef PubMed
- S. Sekine, Y. Ido, T. Miyake, K. Nagamine and M. Nishizawa, J. Am. Chem. Soc., 2010, 132, 13174–13175 CrossRef CAS PubMed
- J. Hur, K. Im, S. W. Kim, J. Kim, D.-Y. Chung, T.-H. Kim, K. H. Jo, J. H. Hahn, Z. Bao, S. Hwang and N. Park, ACS Nano, 2014, 8, 10066–10076 CrossRef CAS PubMed
- V. Guarino, M. A. Alvarez-Perez, A. Borriello, T. Napolitano and L. Ambrosio, Adv. Healthcare Mater., 2013, 2, 218–227 CrossRef CAS PubMed
- Y. Wu, Y. X. Chen, J. Yan, S. Yang, P. Dong and P. Soman, J. Mater. Chem. B, 2015, 3, 5352–5360 RSC
- S. Hu, L. Zhou, L. Tu, C. Dai, L. Fan, K. Zhang, T. Yao, J. Chen, Z. Wang, J. Xing, R. Fu, P. Yu, G. Tan, J. Du and C. Ning, J. Mater. Chem. B, 2019, 7, 2389–2397 RSC
- D. Gan, L. Han, M. Wang, W. Xing, T. Xu, H. Zhang, K. Wang, L. Fang and X. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 36218–36228 CrossRef CAS PubMed
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984 CrossRef CAS PubMed
- Z. Lei and P. Wu, Nat. Commun., 2019, 10, 3429 CrossRef PubMed
- Z. Lei and P. Wu, ACS Nano, 2018, 12, 12860–12868 CrossRef CAS PubMed
- Z. Lei and P. Wu, Mater. Horiz., 2019, 6, 538–545 RSC
- Z. Lei and P. Wu, Nat. Commun., 2018, 9, 1134 CrossRef PubMed
- A. Keller, J. Pham, H. Warren and M. in het Panhuis, J. Mater. Chem. B, 2017, 5, 5318–5328 RSC
- L. Guan, S. Yan, X. Liu, X. Li and G. Gao, J. Mater. Chem. B, 2019, 7, 5230–5236 RSC
- X.-Y. Yin, Y. Zhang, X. Cai, Q. Guo, J. Yang and Z. L. Wang, Mater. Horiz., 2019, 6, 767–780 RSC
- C.-J. Lee, H. Wu, Y. Hu, M. Young, H. Wang, D. Lynch, F. Xu, H. Cong and G. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 5845–5852 CrossRef CAS PubMed
- X. Li, H. Charaya, G. M. Bernard, J. A. W. Elliott, V. K. Michaelis, B. Lee and H.-J. Chung, Macromolecules, 2018, 51, 2723–2731 CrossRef CAS
- T. Long, Y. Li, X. Fang and J. Sun, Adv. Funct. Mater., 2018, 28, 1804416 CrossRef
- L. Wang, G. Gao, Y. Zhou, T. Xu, J. Chen, R. Wang, R. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 3506–3515 CrossRef CAS PubMed
- W. Kong, C. Wang, C. Jia, Y. Kuang, G. Pastel, C. Chen, G. Chen, S. He, H. Huang, J. Zhang, S. Wang and L. Hu, Adv. Mater., 2018, 30, 1801934 CrossRef PubMed
- Y. Zhou, C. Wan, Y. Yang, H. Yang, S. Wang, Z. Dai, K. Ji, H. Jiang, X. Chen and Y. Long, Adv. Funct. Mater., 2019, 29, 1806220 CrossRef
- Z. Wang, J. Chen, L. Wang, G. Gao, Y. Zhou, R. Wang, T. Xu, J. Yin and J. Fu, J. Mater. Chem. B, 2019, 7, 24–29 RSC
- Y.-J. Liu, W.-T. Cao, M.-G. Ma and P. Wan, ACS Appl. Mater. Interfaces, 2017, 9, 25559–25570 CrossRef CAS PubMed
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS
- M. X. Wang, Y. M. Chen, Y. Gao, C. Hu, J. Hu, L. Tan and Z. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 26610–26617 CrossRef CAS PubMed
- L. Han, X. Lu, M. Wang, D. Gan, W. Deng, K. Wang, L. Fang, K. Liu, C. W. Chan, Y. Tang, L.-T. Weng and H. Yuan, Small, 2017, 13, 1601916 CrossRef PubMed
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC
- M. Chau, K. J. De France, B. Kopera, V. R. Machado, S. Rosenfeldt, L. Reyes, K. J. W. Chan, S. Förster, E. D. Cranston, T. Hoare and E. Kumacheva, Chem. Mater., 2016, 28, 3406–3415 CrossRef CAS
- Y. Chevalier, Y. Storet, S. Pourchet and P. Le Perchec, Langmuir, 1991, 7, 848–853 CrossRef CAS
- C. K. Roy, H. L. Guo, T. L. Sun, A. B. Ihsan, T. Kurokawa, M. Takahata, T. Nonoyama, T. Nakajima and J. P. Gong, Adv. Mater., 2015, 27, 7344–7348 CrossRef CAS PubMed
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7, 11782 CrossRef CAS PubMed
- Q. Trung Tran, S. Ramasundaram, B. U. Hwang and N. E. Lee, Adv. Mater., 2015, 28, 502–509 CrossRef PubMed
- Z. Wang, X. Guan, H. Huang, H. Wang, W. Lin and Z. Peng, Adv. Funct. Mater., 2019, 29, 1807569 CrossRef
- B. C. K. Tee, A. Chortos, R. R. Dunn, G. Schwartz, E. Eason and Z. Bao, Adv. Funct. Mater., 2014, 24, 5427–5434 CrossRef CAS
- G. Ge, Y. Zhang, J. Shao, W. Wang, W. Si, W. Huang and X. Dong, Adv. Funct. Mater., 2018, 28, 1802576 CrossRef
- J. Chossat, Y. Park, R. J. Wood and V. Duchaine, IEEE Sens. J., 2013, 13, 3405–3414 CAS
- S. Gong, W. Schwalb, Y. Wang, Y. Chen, Y. Tang, J. Si, B. Shirinzadeh and W. Cheng, Nat. Commun., 2014, 5, 3132 CrossRef PubMed
- D. J. Lipomi, M. Vosgueritchian, B. C. K. Tee, S. L. Hellstrom, J. A. Lee, C. H. Fox and Z. Bao, Nat. Nanotechnol., 2011, 6, 788 CrossRef CAS PubMed
- T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296 CrossRef CAS PubMed
- Y. Liu, Y. Hu, J. Zhao, G. Wu, X. Tao and W. Chen, Small, 2016, 12, 5074–5080 CrossRef CAS PubMed
- M. Amjadi, K.-U. Kyung, I. Park and M. Sitti, Adv. Funct. Mater., 2016, 26, 1678–1698 CrossRef CAS
- Y.-Z. Zhang, K. H. Lee, D. H. Anjum, R. Sougrat, Q. Jiang, H. Kim and H. N. Alshareef, Sci. Adv., 2018, 4, eaat0098 CrossRef PubMed
- J. M. González-Domínguez, C. Martín, Ó. J. Durá, S. Merino and E. Vázquez, ACS Appl. Mater. Interfaces, 2018, 10, 1987–1995 CrossRef PubMed
- X. Zhang, N. Sheng, L. Wang, Y. Tan, C. Liu, Y. Xia, Z. Nie and K. Sui, Mater. Horiz., 2019, 6, 326–333 RSC
- Z. Lei, Q. Wang, S. Sun, W. Zhu and P. Wu, Adv. Mater., 2017, 29, 1700321 CrossRef PubMed
- J.-Y. Sun, C. Keplinger, G. M. Whitesides and Z. Suo, Adv. Mater., 2014, 26, 7608–7614 CrossRef CAS PubMed
- K. Tian, J. Bae, S. E. Bakarich, C. Yang, R. D. Gately, G. M. Spinks, M. in het Panhuis, Z. Suo and J. J. Vlassak, Adv. Mater., 2017, 29, 1604827 CrossRef PubMed
- R. Tong, G. Chen, D. Pan, J. Tian, H. Qi, R. a. Li, F. Lu and M. He, ACS Sustainable Chem. Eng., 2019, 7, 14256–14265 CrossRef CAS
- S. Liu and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 26429–26437 CrossRef CAS PubMed
- Y. Liang, L. Ye, X. Sun, Q. Lv and H. Liang, ACS Appl. Mater. Interfaces, 2020, 12, 1577–1587 CrossRef CAS PubMed
- X. Jing, H.-Y. Mi, Y.-J. Lin, E. Enriquez, X.-F. Peng and L.-S. Turng, ACS Appl. Mater. Interfaces, 2018, 10, 20897–20909 CrossRef CAS PubMed
- R. Tong, G. Chen, D. Pan, H. Qi, R. a. Li, J. Tian, F. Lu and M. He, Biomacromolecules, 2019, 20, 2096–2104 CrossRef CAS PubMed
- S. Hong, Y. Yuan, C. Liu, W. Chen, L. Chen, H. Lian and H. Liimatainen, J. Mater. Chem. C, 2020, 8, 550–560 RSC
- C. Dang, M. Wang, J. Yu, Y. Chen, S. Zhou, X. Feng, D. Liu and H. Qi, Adv. Funct. Mater., 2019, 29, 1902467 CrossRef
- J. Lai, H. Zhou, M. Wang, Y. Chen, Z. Jin, S. Li, J. Yang, X. Jin, H. Liu and W. Zhao, J. Mater. Chem. C, 2018, 6, 13316–13324 RSC
- G. Ge, W. Yuan, W. Zhao, Y. Lu, Y. Zhang, W. Wang, P. Chen, W. Huang, W. Si and X. Dong, J. Mater. Chem. A, 2019, 7, 5949–5956 RSC
- S. Zhao, P. Tseng, J. Grasman, Y. Wang, W. Li, B. Napier, B. Yavuz, Y. Chen, L. Howell, J. Rincon, F. G. Omenetto and D. L. Kaplan, Adv. Mater., 2018, 30, 1800598 CrossRef PubMed
- C. Larson, B. Peele, S. Li, S. Robinson, M. Totaro, L. Beccai, B. Mazzolai and R. Shepherd, Science, 2016, 351, 1071 CrossRef CAS PubMed
- Y. Tai, M. Mulle, I. Aguilar Ventura and G. Lubineau, Nanoscale, 2015, 7, 14766–14773 RSC
- X. Pu, M. Liu, X. Chen, J. Sun, C. Du, Y. Zhang, J. Zhai, W. Hu and Z. L. Wang, Sci. Adv., 2017, 3, e1700015 CrossRef PubMed
- Z. Lou, S. Chen, L. Wang, K. Jiang and G. Shen, Nano Energy, 2016, 23, 7–14 CrossRef CAS
- G. Y. Bae, J. T. Han, G. Lee, S. Lee, S. W. Kim, S. Park, J. Kwon, S. Jung and K. Cho, Adv. Mater., 2018, 30, 1803388 CrossRef PubMed
- A. Chortos and Z. Bao, Mater. Today, 2014, 17, 321–331 CrossRef CAS
- S. C. B. Mannsfeld, B. C. K. Tee, R. M. Stoltenberg, C. V. H. H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed
- Y. Pang, K. Zhang, Z. Yang, S. Jiang, Z. Ju, Y. Li, X. Wang, D. Wang, M. Jian, Y. Zhang, R. Liang, H. Tian, Y. Yang and T.-L. Ren, ACS Nano, 2018, 12, 2346–2354 CrossRef CAS PubMed
- Q. Wang, M. Jian, C. Wang and Y. Zhang, Adv. Funct. Mater., 2017, 27, 1605657 CrossRef
- S. Jung, J. H. Kim, J. Kim, S. Choi, J. Lee, I. Park, T. Hyeon and D.-H. Kim, Adv. Mater., 2014, 26, 4825–4830 CrossRef CAS PubMed
- S.-J. Park, J. Kim, M. Chu and M. Khine, Adv. Mater. Technol., 2016, 1, 1600053 CrossRef
- C. Pang, J. H. Koo, A. Nguyen, J. M. Caves, M.-G. Kim, A. Chortos, K. Kim, P. J. Wang, J. B. H. Tok and Z. Bao, Adv. Mater., 2015, 27, 634–640 CrossRef CAS PubMed
- M. L. Hammock, A. Chortos, B. C. K. Tee, J. B. H. Tok and Z. Bao, Adv. Mater., 2013, 25, 5997–6038 CrossRef CAS PubMed
- M. Nishita, S.-Y. Park, T. Nishio, K. Kamizaki, Z. Wang, K. Tamada, T. Takumi, R. Hashimoto, H. Otani, G. J. Pazour, V. W. Hsu and Y. Minami, Sci. Rep., 2017, 7, 1 CrossRef CAS PubMed
- S. W. Chen, X. Cao, N. Wang, L. Ma, H. R. Zhu, M. Willander, Y. Jie and Z. L. Wang, Adv. Energy Mater., 2017, 7, 1601255 CrossRef
- X. Wang, H. Zhang, L. Dong, X. Han, W. Du, J. Zhai, C. Pan and Z. L. Wang, Adv. Mater., 2016, 28, 2896–2903 CrossRef CAS PubMed
- R. Fu, L. Tu, Y. Zhou, L. Fan, F. Zhang, Z. Wang, J. Xing, D. Chen, C. Deng, G. Tan, P. Yu, L. Zhou and C. Ning, Chem. Mater., 2019, 31, 9850–9860 CrossRef CAS
- M. Amjadi, K. U. Kyung, I. Park and M. Sitti, Adv. Funct. Mater., 2016, 26, 1678–1698 CrossRef CAS
- T. R. Ray, J. Choi, A. J. Bandodkar, S. Krishnan, P. Gutruf, L. Tian, R. Ghaffari and J. A. Rogers, Chem. Rev., 2019, 119, 5461–5533 CrossRef CAS PubMed
- D. Chen and Q. Pei, Chem. Rev., 2017, 117, 11239–11268 CrossRef CAS PubMed
- C.-C. Kim, H.-H. Lee, K. H. Oh and J.-Y. Sun, Science, 2016, 353, 682–687 CrossRef CAS PubMed
- C. S. Luo, P. Wan, H. Yang, S. A. A. Shah and X. Chen, Adv. Funct. Mater., 2017, 27, 1606339 CrossRef
| This journal is © The Royal Society of Chemistry 2020 |