
Assessing the range of enzymatic and oxidative tunability for biosensor design

Abstract
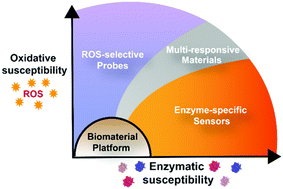
Development of multi-functional materials and biosensors that can achieve an in situ response designed by the user is a current need in the biomaterials field, especially in complex biological environments, such as inflammation, where multiple enzymatic and oxidative signals are present. In the past decade, there has been extensive research and development of materials chemistries for detecting and monitoring enzymatic activity, as well as for releasing therapeutic and diagnostic agents in regions undergoing oxidative stress. However, there has been limited development of materials in the context of enzymatic and oxidative triggers together, despite their closely tied and overlapping mechanisms. With research focusing on enzymatically and oxidatively triggered materials separately, these systems may be inadequate in monitoring the complexity of inflammatory environments, thus limiting in vivo translatability and diagnostic accuracy. The intention of this review is to highlight a variety of enzymatically and oxidatively triggered materials chemistries to draw attention to the range of synthetic tunability available for the construction of novel biosensors with a spectrum of programmed responses. We focus our discussion on several types of macromolecular sensors, generally classified by the causative material response driving ultimate signal detection. This includes sensing based on degradative processes, conformational changes, supramolecular assembly/disassembly, and nanomaterial interactions, among others. We see each of these classes providing valuable tools toward coalescing current gaps in the biosensing field regarding specificity, selectivity, sensitivity, and flexibility in application. Additionally, by considering the materials chemistry of enzymatically and oxidatively triggered biomaterials in tandem, we hope to encourage synthesis of new biosensors that capitalize on their synergistic roles and overlapping mechanisms in inflammatory environments for applications in disease diagnosis and monitoring.
- This article is part of the themed collection: Biosensors
 Please wait while we load your content...
Please wait while we load your content...

