
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the stability of organic radicals: from covalent approaches to non-covalent approaches
Bohan
Tang
,
Jiantao
Zhao
,
Jiang-Fei
Xu
 and
Xi
Zhang
*
and
Xi
Zhang
*
Key Laboratory of Organic Optoelectronics & Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: xi@mail.tsinghua.edu.cn
First published on 4th January 2020
Abstract
Organic radicals are important species with single electrons. Because of their open-shell structure, they are widely used in functional materials, such as spin probes, magnetic materials and optoelectronic materials. Owing to the high reactivity of single electrons, they often serve as a key intermediate in organic synthesis. Therefore, tuning the stability of radicals is crucial for their functions. Herein, we summarize covalent and non-covalent approaches to tune the stability of organic radicals through steric effects and tuning the delocalization of spin density. Covalent approaches can tune the stability of radicals effectively and non-covalent approaches benefit from dynamicity and reversibility. It is anticipated that the further development of covalent and non-covalent approaches, as well as the interplay between them, may push the fields forward by enriching new radical materials and radical mediated reactions.
1. Introduction
Organic radicals are molecular entities possessing an unpaired electron. In 1900, Gomberg discovered the first stable organic radical, the triphenylmethyl radical.1 From then on, the radical chemistry was built up step by step. Because of the open-shell structure of organic radicals, they possess special magnetic, optical and redox properties, which can be applied in functional materials, such as spin probes, magnetic materials and optoelectronic materials.2–7 In addition, organic radicals are often highly reactive species undergoing single-electron redox processes. Therefore, organic radicals serve as key intermediates in a number of organic reactions, including radical polymerization and organic photocatalysis.8–13Tuning the stability of radicals is crucial for their functions. For radical based functional materials, stability is indispensable.3 For radical mediated reactions, both persistent radicals and transient radicals play important roles, respectively.14,15 Many approaches depending on covalent modification are developed to modulate the stability of radicals.16,17 Because most organic radicals are transient, previous studies were mostly focused on the stabilization of radicals. Through different strategies, various organic radicals, including neutral radicals, radical ions, diradicals and so on, can be air-stable, water-stable, isolatable and even thermodynamically stable.1,18,19
In addition to covalent approaches, non-covalent approaches to tune the stability of organic radicals are emerging.20,21 Benefiting from the dynamicity and reversibility of non-covalent interactions, non-covalent approaches can be utilized to fabricate radicals with tunable stability. On the one hand, non-covalent approaches may avoid complicated synthesis to some extent.22 On the other hand, the stability of radicals can be reversibly controlled and radicals can also respond to external stimuli, thus providing novel possibilities, such as switchable properties, smart functional materials and adaptive radical systems.23–25
In general, the principles of tuning the stability of organic radicals can be summarized as steric protection and tuning the delocalization of spin density.15,16,26 In this perspective article, we will introduce covalent approaches and non-covalent approaches to modulate the stability of radicals from the viewpoint of these two principles (Scheme 1).
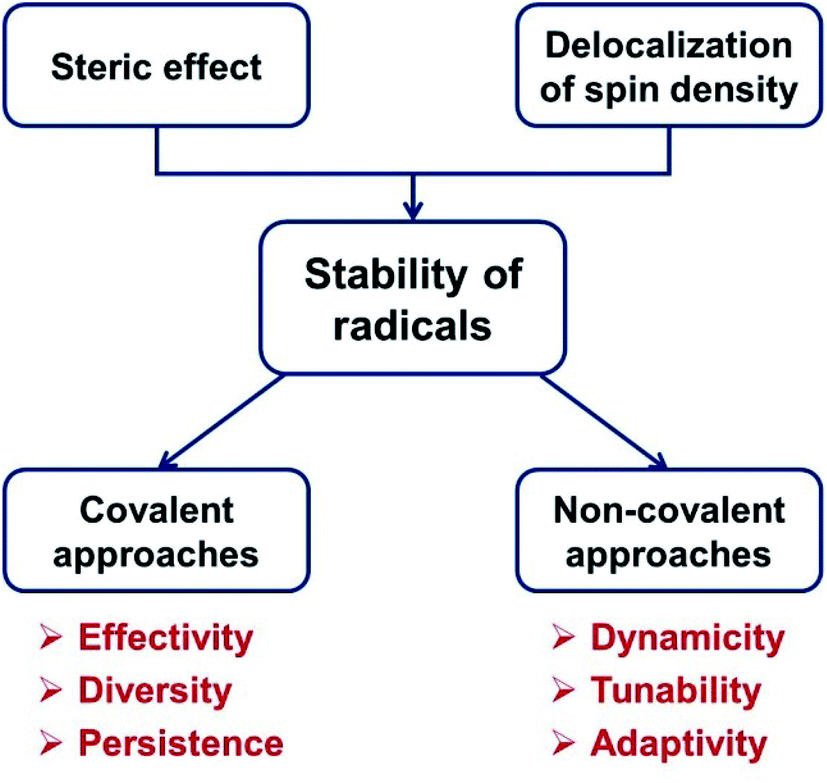 | ||
| Scheme 1 Covalent and non-covalent approaches to tune the stability of organic radicals. | ||
2. Tuning the activity of organic radicals by covalent approaches
In this section, we will introduce several methods to modulate the stability of organic radicals by covalent approaches, including steric protection and modulating the delocalization of spin density. In general, steric protection is inclined to provide kinetic stability to organic radicals, but thermodynamic stability can also be obtained through steric protection. Modulating the delocalization of spin density is usually described as an electronic effect, which mainly provides thermodynamic stability to radicals. The three major approaches to modulate the delocalization of spin density are the modulation of π-conjugates, incorporation of polar substituents and the fabrication of heteroatom based radicals. However, different methods in modulating the stability of radicals are often combined in practice. Therefore, we will introduce some examples, in which some respects are highlighted to illustrate the design of modulating the stability of organic radicals.2.1 Steric protection with bulky substituents
Under most conditions, organic radicals are unstable species because of their highly reactive single electrons. They suffer from side reactions such as dimerization, recombination, electron transfer, and so on. To stabilize an organic radical, steric protection is one of the most effective and widely used approaches. It is the most direct and natural idea that steric protection can prevent the interaction of organic radicals with others, thus inhibiting their intermolecular side reactions. In fact, through steric protection, organic radicals can be greatly stabilized and even isolated.Many examples of stable radicals are stabilized by steric protection. In the famous stable radical 2,2,6,6-tetramethylpiperidinyloxyl, known as TEMPO, four methyl groups serve as steric hindrance groups, which keep the TEMPO radicals from contacting with each other. Moreover, the stability of the triphenylmethyl radical, the first synthesized organic radical in history, is largely attributed to steric protection as well. The three surrounding phenyl groups, twisted by around 30° in a propeller conformation, prevent the contact of the spin center with other molecules.27 When the ortho hydrogens on phenyl groups are replaced by heavier elements, for example, chlorine, the steric protection can be further enhanced.28 In the case of a perchlorinated triphenylmethyl radical, the strong steric hindrance increases the twisted angle to about 50° and makes it extremely stable and unreactive unless under very harsh conditions.
Combining the stability with the special properties of organic radicals, materials with charming functions are produced. Li et al. reported the use of an open-shell molecule, (4-N-carbazolyl-2,6-dichlorophenyl)bis(2,4,6-trichlorophenyl)-methyl radical (TTM-1Cz), as an emitter to build organic light emitting diodes29 (OLEDs) (Fig. 1). Because organic radicals are doublet molecules, the radiative decay from the lowest singly unoccupied molecular orbital (SUMO) to the singly occupied molecular orbital (SOMO) is always spin-allowed, and thus the upper limit of inner quantum efficiency can reach 100%. With six chlorine atoms around the triphenylmethyl radical center, the TTM-1Cz radical was stable enough to withstand oxygen and light, and thus could be utilized in OLED devices. By a similar strategy, OLEDs with a maximum external quantum efficiency of 27% at a wavelength of 710 nm were achieved, which was the highest value for deep-red and near infrared LEDs.30 In the field of OLEDs, commonly radicals are not favored species because of their lack of stability. Steric protected stable radicals provide a new avenue to fabricate OLEDs with 100% internal quantum efficiency.
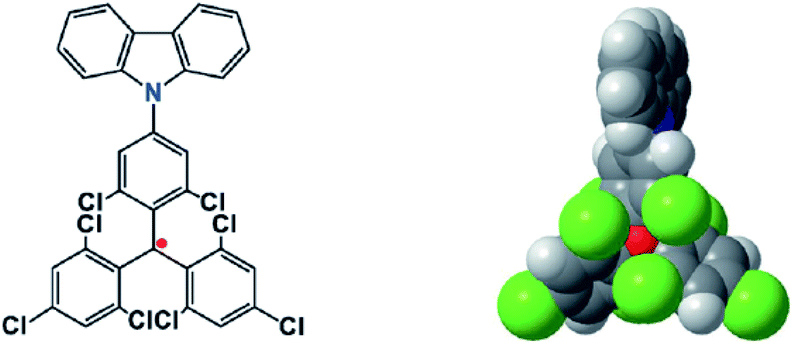 | ||
| Fig. 1 The chemical structure and configuration of a TTM-1Cz radical stabilized with ortho chlorine atoms. Reproduced from ref. 29 with permission from Wiley-VCH, Copyright 2015. | ||
Besides optoelectronic properties, stable radicals with steric protection can also be utilized in the field of energy storage. With the great redox reversibility of stable radicals like TEMPO, organic radical batteries possess long cycle lives and service time. In 2002, Nakahara et al. first reported the construction of a battery based on TEMPO modified polymethacrylate and provided a detailed standard model.31,32 Schubert et al. reported the use of polymeric TEMPO and polymeric viologen, which took advantage of their safety and low-cost, in the construction of a redox-flow battery.33,34
As a brief summary, steric protection is an effective and universal approach to stabilize organic radicals. For different kinds of radicals, neutral or ionized, carbon-based or heteroatom-based, and localized or delocalized, the incorporation of bulky substituents provides a protection effect on them. It should be noted that steric protection stabilized radicals benefit from the inhibition of their interactions with other molecules. However, interactions with other molecules are sometimes necessary for the applications of organic radicals. Moreover, although organic radicals are usually reactive species that need to be stabilized, sometimes radicals still need to be activated. Therefore, methods without the use of bulky substituents and capable of tuning the activity of organic radicals are developed.
2.2 Tuning the delocalization of spin density
Commonly, organic radicals have a strong tendency to form σ-dimers or conduct electron transfer. Therefore, the delocalization of spin density becomes a conventional approach to stabilize radicals. In this section, three main approaches to tune the delocalization of spin density will be presented, including the extension of π-conjugates, the introduction of polar substituents and the fabrication of heteroatom based radicals. | ||
| Fig. 2 The resonance structures of the phenalenyl radical. | ||
The extension of π-conjugates is a powerful tool to stabilize highly reactive radicals. For example, Osuka et al. stabilized the trimethylenemethane (TMM) diradical by fusing the TMM segment with three Ni(II) meso-triarylporphyrins40 (Fig. 3). The TMM diradical is the simplest non-Kekulé non-disjoint molecule with a triplet ground state (ΔEST = +16.1 kcal mol−1) and is extremely reactive. After the introduction of porphyrin moieties, the diradical possessed extraordinary stability, which could be stored for months in the solid state and stand heating at 80 °C in 1,2-dichlorobenzene in air for 10 h.
 | ||
| Fig. 3 The stable trimethylenemethane triplet diradical fused with trimeric porphyrin segments (Ar = 3,5-di-tert-butylphenyl). Reproduced from ref. 40 with permission from Wiley-VCH, Copyright 2018. | ||
It should be pointed that the extension of π-conjugates does not always stabilize radicals. For example, the phthalimide N-oxyl radical (PINO) is not as stable as TEMPO. The “instability” of PINO makes it quite different from TEMPO in the reactivity, because of which PINO is widely used in organic synthesis.41 It is believed that the high reactivity of PINO is due to the effect of the carbonyl groups, suggesting that different substituents will have considerable influence on the stability of radicals. So the effects of substituents will be discussed in next section.
For radical ions, the effect of substituents, especially charged substituents, would be quite significant on the stability of radicals.43,44 In general, the incorporation of oppositely charged substituents will stabilize the radical. In contrast, identically charged substituents will activate the radical and in most cases, lead to difficulty in the generation of the radical. Würthner et al. reported a perylene-3,4:9,10-tetracarboxylic acid bisimide (PBI) radical anion stabilized by the incorporation of a positively charged imidazolium substituent45 (Fig. 4). It is the first example of a zwitterionic PBI radical with remarkable stability which can be isolated and even fully characterized by X-ray diffraction under ambient conditions. Comparing the single crystal structural features of the zwitterionic PBI radical with the non-radical product, it was indicated that the zwitterionic PBI radical was stabilized by the electronic effect of the positively charged imidazolium group rather than steric protection.
 | ||
| Fig. 4 Imidazolium cation stabilized PBI radical anion and its single crystal structure. Reproduced from ref. 45 with permission from Wiley-VCH, Copyright 2015. | ||
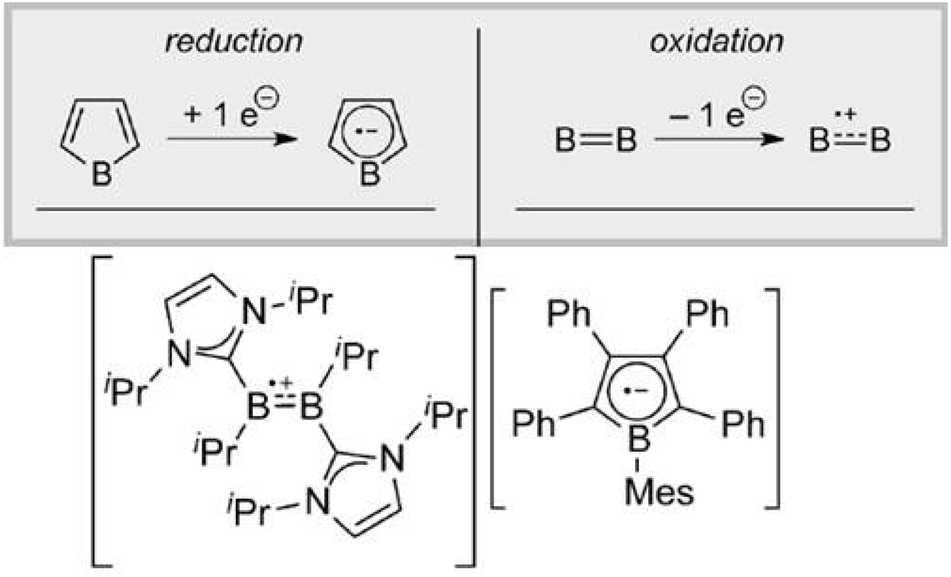 | ||
| Fig. 5 Boron-centered stable radical anion/radical cation pair. Reproduced from ref. 48 with permission from Wiley-VCH, Copyright 2015. | ||
To summarize this part, covalent approaches are powerful tools to modulate the stability of organic radicals. Organic radicals are usually unstable species, and therefore almost all the studies are focused on the stabilization of organic radicals. Both kinetic and thermodynamic stabilization of radicals can be achieved by covalent synthesis, including steric protection and the delocalization of spin density. Radicals can become air-stable and water-stable and are able to be purified with silica columns and characterized by single crystal X-ray diffraction. However, the stability of covalently modified radicals is fixed, meaning that their stability and reactivity will remain the same in a chemical system. If dynamicity can be introduced in modulating the stability of radicals, a tunable stability can be achieved, and thus the radicals are able to respond to external stimuli. To this end, non-covalent approaches to tune the stability of radicals are developed.
3. Tuning the activity of organic radicals by non-covalent approaches
As discussed above, the covalent methods to modulate the stability of radicals are highly effective; however, the irreversibility of the modulation “freezes” the stability of radicals. In contrast, non-covalent approaches can benefit from their dynamic nature to achieve tunable stability which may lead to stimuli-responsive and adaptive properties and so on.In some special cases where covalent steric protection cannot be applicable, non-covalent approaches provide a new path to tune the stability of organic radicals. In general, non-covalent approaches share the same principle with covalent approaches. They involve steric protection by host–guest chemistry and the tuning of the delocalization of spin densities by different non-covalent interactions.
Non-covalent approaches take advantage of their dynamicity. However, achieving effective tuning of radicals' stability through non-covalent interaction is the key problem. There are many approaches that are developed to achieve effective tuning of the radicals' stability, such as size-fitting, cooperative effect, the formation of well-confined organized structures and so on. In this section, different interactions to tune the stability of radicals will be introduced and the approaches to realize effective tuning will be mentioned with examples as well.
3.1 Steric protection by host–guest chemistry
The steric protection from covalently synthesized bulky substituents can be considered as “intramolecular steric protection”, while the chemistry of “intermolecular steric protection” can be correspondingly achieved by host–guest chemistry. The host–guest chemistry can provide specific non-covalent molecular recognition and a characteristic microenvironment. As mentioned above, the steric protection aims to prevent the contact of organic radicals with other molecules, thus inhibiting their dimerization and other side reactions with other molecules, especially molecular oxygen. Host–guest chemistry can function in a similar way to protect and stabilize organic radicals. It is a well-developed strategy to utilize host molecules for encapsulating organic radicals to stabilize them.In 1991, Turro et al. reported benzylic radicals stabilized in cyclodextrin49 (Fig. 6a). The solid state host–guest complex of α, α′-dimethyldibenzyl-ketone and cyclodextrin was fabricated by precipitation from saturated cyclodextrin–water solutions containing the guest. Through the photolysis of the solid state host–guest complex, benzylic radicals were generated. According to electron spin resonance (ESR), the transient benzylic radicals could remain 3 days in the absence of oxygen after inclusion of cyclodextrin in the solid state. In solution, benzylic radicals would form σ-dimers immediately. However, the inclusion of cyclodextrin strongly prevented them from coming into contact with each other, thus stabilizing the benzylic radicals.
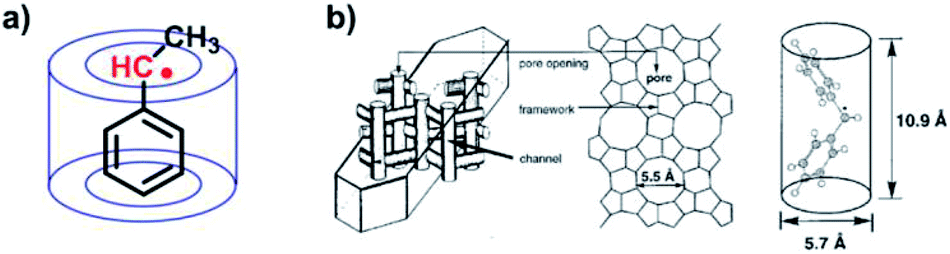 | ||
| Fig. 6 (a) Cyclodextrin stabilized benzylic radicals. (b) LZ-105 zeolite stabilized diphenylmethyl radicals. Reproduced from ref. 51 with permission from the American Chemical Society, Copyright 2000. | ||
In addition to macrocycle molecules, various kinds of hosts, such as micelles, liquid crystals, Nafion and zeolites can also stabilize radicals by steric protection.21,50 In 2000, Turro et al. reported the stabilization of diphenylmethyl radicals in zeolites51 (Fig. 6b). The unstable diphenylmethyl radicals became persistent radicals inside the channel of LZ-105 zeolite. This reminds us the great power of size-fitting. To protect radicals, it is necessary to “grab” the guest radical tightly. As shown in Fig. 6b, the diphenylmethyl radicals were precisely fitting the size of the LZ-105 channel, for which the encapsulated radicals were largely stabilized. Notably, these two examples were both carried out in the solid state. The solid state is a great arena to lock the exclusion and exchange of guest radicals; therefore contact between the radical and other molecules and side reactions are suppressed. In other words, restricting the exclusion and exchange of radicals helps to achieve strong stabilization through steric protection.
Sometimes, topological structures can be utilized to restrict the exchange of guests efficiently. For example, Anderson et al. reported the fabrication of a cucurbituril oligoaniline rotaxane to stabilize oligoaniline radical cations52 (Fig. 7). After the encapsulation of cucurbit[7]uril, the yield of oligoaniline radical cations significantly increased. In this case, steric protection was not the only reason for the stabilization, but the formation of a mechanically locked rotaxane indeed provided strong protection on the radical cation. Furthermore, the inclusion of cucurbit[7]uril could be utilized to stabilize the polyaniline radical cation by the fabrication of polymeric (pseudo)rotaxanes.53
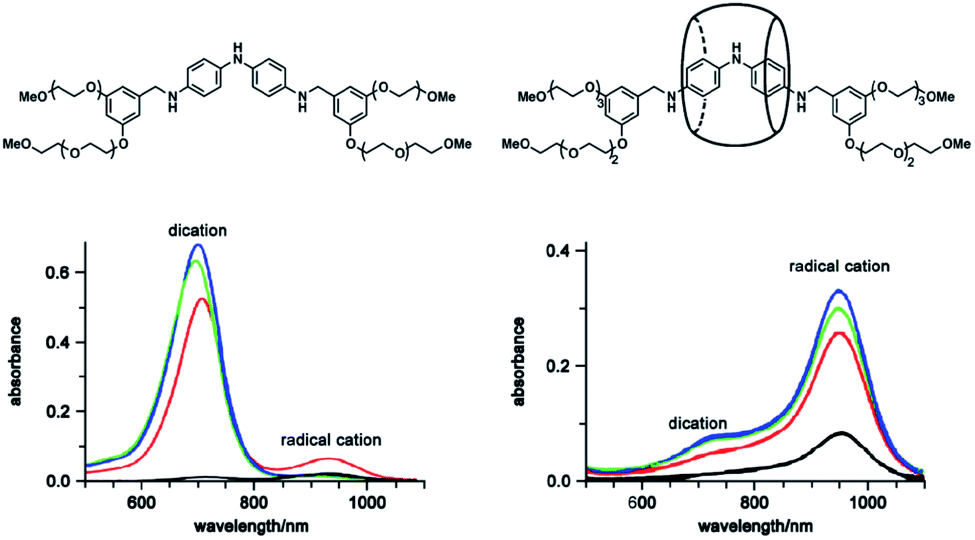 | ||
| Fig. 7 Structures of the dumbbell oligoaniline and the cucurbituril oligoaniline rotaxane and UV-Vis spectra of their radical cation. Reproduced from ref. 52 with permission from the American Chemical Society, Copyright 2007. | ||
Supramolecular steric protection can provide another approach to stabilize radicals under some special conditions, where conventional steric substituents are not applicable. Most recently, Tagmatarchis et al. reported an exciting study of a long-lived azafullerenyl radical stabilized by supramolecular shielding with [10]cycloparaphenylene ([10]CPP)54 (Fig. 8). The unpaired electron of the azafullerenyl radical C59N˙ was strongly localized next to the nitrogen atom, leading to a strong tendency to form a σ-dimer, (C59N)2. However, conventional steric hindrance groups could not stabilize the radical due to the concave fullerene geometry. After the inclusion of [10]CPP, the reactive radical center was inhibited, thus stabilizing the C59N˙;. With formation of the host–guest complex C59N˙⊂[10]CPP, the yield of radicals increased by 300 fold and the transient C59N˙ could hold a half-life time as long as 100 min. This work gives an excellent description of the concept of non-covalent synthesis and its utilization when covalent synthesis does not work. In addition, it also exhibits the potential of supramolecular approaches in overcoming disfavored geometry.
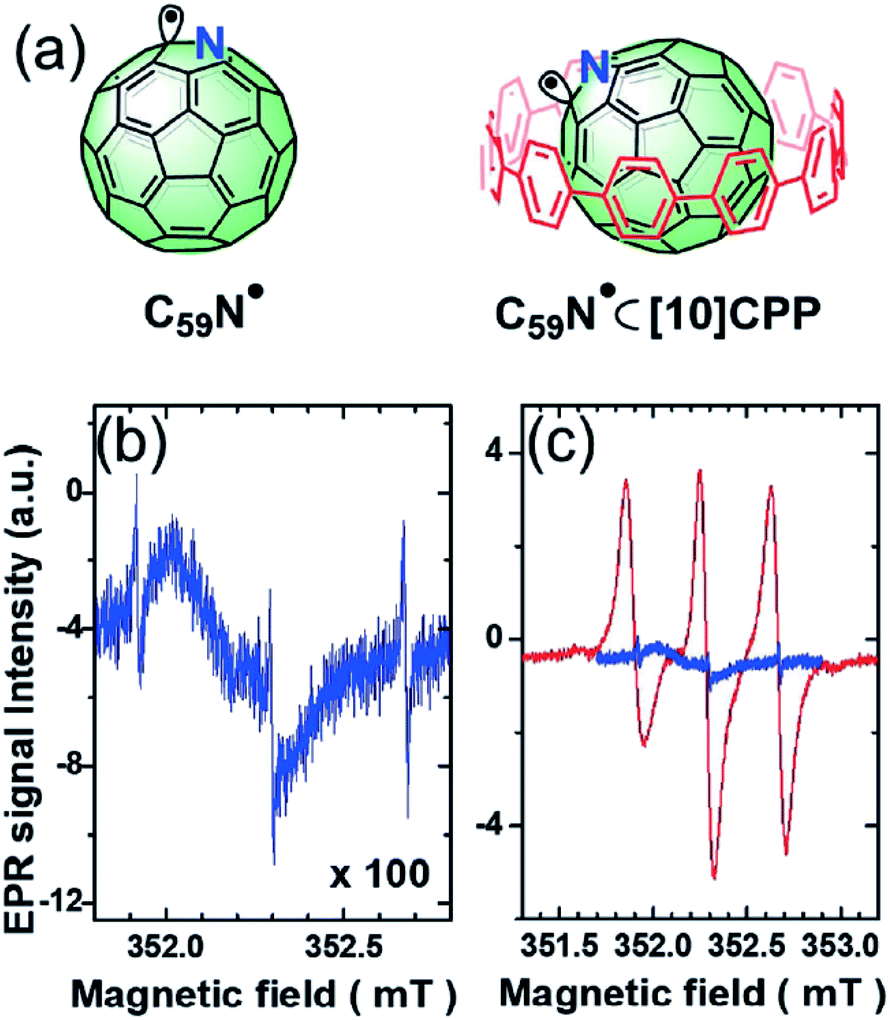 | ||
| Fig. 8 (a) The structures of C59N˙ and C59N˙⊂[10]CPP. (b) The ESR signal of C59N˙. (c) The comparison of the ESR signal of C59N˙ and C59N˙⊂[10]CPP. Reproduced from ref. 54 with permission from Wiley-VCH, Copyright 2019. | ||
Besides the encapsulation of organic radicals as guest molecules, the fabrication of ordered supramolecular structures based on organic radials can also provide steric protection to the radicals. Yin et al. reported the use of a perylenediimide (PDI) derivative and Zr-cluster to fabricate a metal–organic framework Zr–PDI55,56 (Fig. 9). After the loading of triethylamine vapor and irradiation at 455 nm, the PDI moiety was reduced to its radical anion PDI˙−. The metal–organic framework Zr–PDI˙− exhibited outstanding stability under exposure of air. Zr–PDI˙− could remain almost unaffected for over a month under ambient conditions. Therefore, Zr–PDI˙− showed efficient near-infrared photothermal conversion with an efficiency of 52.3% at 808 nm. This work describes an elegant approach to stabilize radicals by the utilization of radicals as building blocks to fabricate a well-defined supramolecular structure. In this way, the density of radicals in the supramolecular structure can be increased, which helps to improve the performance of the functional material based on organic radicals.
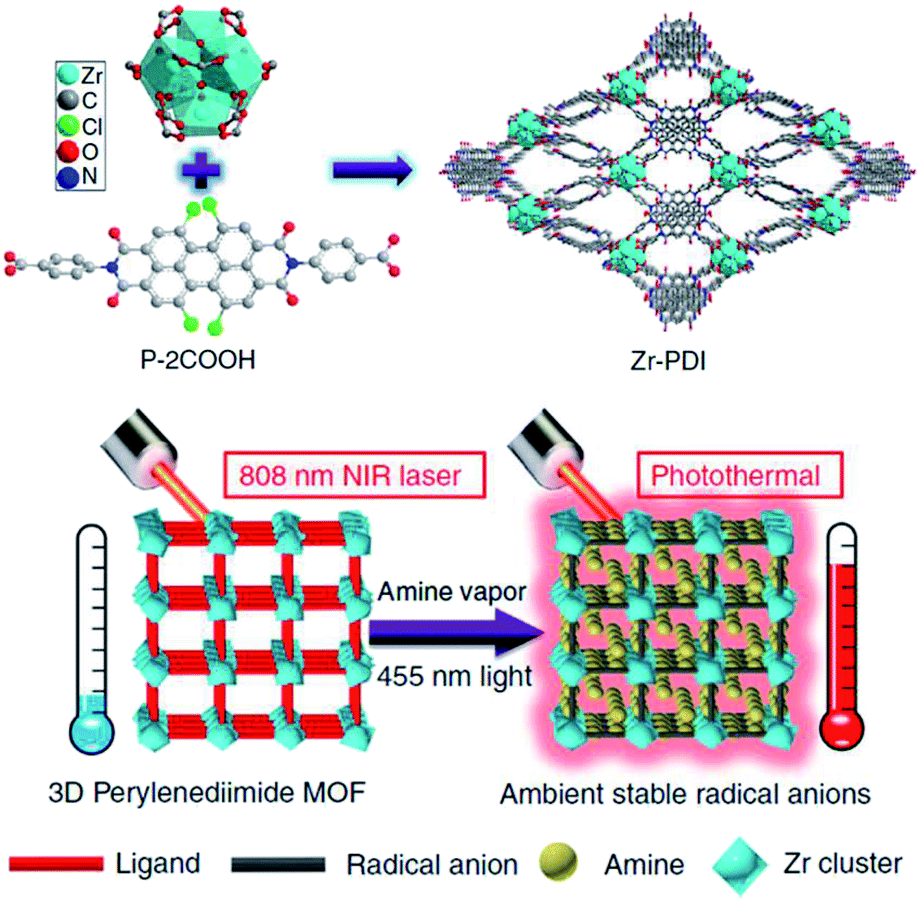 | ||
| Fig. 9 The fabrication of Zr–PDI and Zr–PDI˙− and their photothermal conversion abilities at 808 nm. Reproduced from ref. 55 with permission from Nature Publishing Group, Copyright 2019. | ||
3.2 Tuning the delocalization of spin density by non-covalent approaches
Besides the steric protection, non-covalent interactions can thermodynamically tune the stability of radicals and affect their redox properties. In this section, non-covalent interactions that are used in tuning the stability of radicals, such as hydrogen bonds, coordination bonds, electrostatic interaction and π–π interaction, will be introduced.Synthetic receptors have been designed to reproduce the hydrogen bonding patterns present in flavoenzymes to deepen the understanding of complex enzymes at a molecular/atomic level. Rotello et al. reported a family of diaminopyridine receptors to stabilize flavin radical anions58 (Fig. 10). The receptors could form triple hydrogen bonds with flavin, mimicking the natural hydrogen binding modes. The cyclic voltammetry studies of all the flavin–receptor complexes showed much less negative reduction potential than flavin. The anodic shift of reduction potential indicated the stabilization of flavin radical anions. The highest binding constant of this series of receptors is 537 M−1, accompanied by a 155 mV redox potential shift (E1/2). This work is a good example to show that complexation through hydrogen bonding can stabilize radical anions.
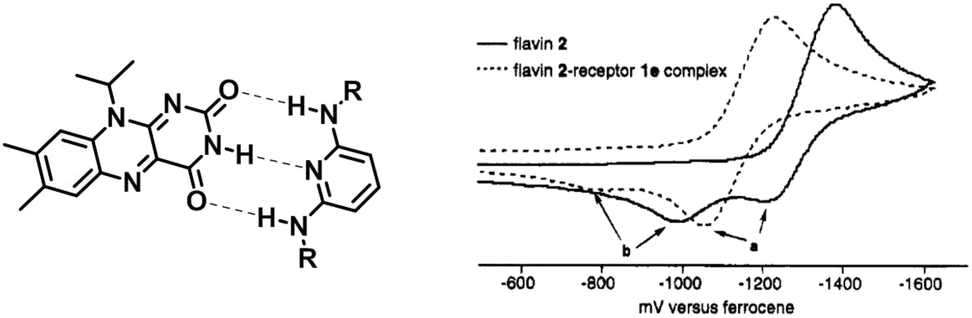 | ||
| Fig. 10 Triple hydrogen bond stabilized flavin radical anion. Reproduced from ref. 58 with permission from the American Chemical Society, Copyright 1995. | ||
The bond energy of a single hydrogen bond in solution is, however, not high enough to effectively tuning the stability of radicals. Cooperativity is a versatile and effective strategy to enhance the binding force of hydrogen bonding. Multiple hydrogen bonding can provide molecular recognition with high affinity and stability. A well-known example is a ureidopyrimidinone (UPy) unit bearing a quadruple hydrogen bonding array.59 On account of the high cooperativity, the binding constant of quadruple hydrogen bonding can be as high as 107 M−1 in chloroform, and the UPy dimer shows good tolerance to various ambient environments. Cooperativity can also promote the binding between a hydrogen bonding receptor and radical anion, and hence remarkable stabilization can be envisioned. Flood et al. designed a cyanostar macrocycle to stabilize tetrazine radical anions60 (Fig. 11). The cyanostar possesses a positive cavity in the middle, surrounded by 10 low-acidity C–H hydrogen bond donors. The tetrazine anion was produced by addition of 4 equiv. of cobaltocene. Cyanostars formed sandwich complexes with tetrazine radical anions in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which was determined by X-ray crystal structure analysis. Within the binding pocket, the lifetime of the radical anions was prolonged from 2 h to over 20 days in solution. If PF6− was added to competitively eject the radical anion from the macrocycle, the tetrazine radical anion MPTz˙− decomposed completely within 130 min, indicating the essential role of the macrocycles in the radical stabilization. This strategy may be broadened to other radical anions and unstable anions, demonstrating the universality of the encapsulation and hydrogen-bonding for radical anion stabilization. It is unusual that in this case, the stabilization effect provided by neutral species has exceeded the stabilization conferred by Cu+, a metal cation. This should be largely due to the simultaneous impact of 20 weak C–H hydrogen bonds, illustrating how cooperativity makes the effect of weak interactions outweigh strong interactions.
:1 ratio, which was determined by X-ray crystal structure analysis. Within the binding pocket, the lifetime of the radical anions was prolonged from 2 h to over 20 days in solution. If PF6− was added to competitively eject the radical anion from the macrocycle, the tetrazine radical anion MPTz˙− decomposed completely within 130 min, indicating the essential role of the macrocycles in the radical stabilization. This strategy may be broadened to other radical anions and unstable anions, demonstrating the universality of the encapsulation and hydrogen-bonding for radical anion stabilization. It is unusual that in this case, the stabilization effect provided by neutral species has exceeded the stabilization conferred by Cu+, a metal cation. This should be largely due to the simultaneous impact of 20 weak C–H hydrogen bonds, illustrating how cooperativity makes the effect of weak interactions outweigh strong interactions.
 | ||
| Fig. 11 The tetrazine radical anion stabilized by the 20 C–H hydrogen bonds with a cyanostar. Reproduced from ref. 60 with permission from the American Chemical Society, Copyright 2016. | ||
When encapsulated in protein, the excess binding energy can be harnessed to stabilize an otherwise inaccessible radical in an ambient environment. DeGrado et al. designed a metalloprotein that could stabilize the semiquinone radical anion SQ˙62 (Fig. 12). Due Ferri (DF) proteins were designed as model systems. One variant of the single-stranded form of DF-type proteins, 2A3H-DFsc (referred to as DFsc), bound two Zn(II) to form [DFsc-Zn(II)2], forming a well-structured four-helix bundle. In the presence of DFsc-Zn(II)2, an equimolar mixture of Q/QH2 was converted to SQ˙, which was characterized by the appearance of a new broad band that spanned 740–850 nm. The EPR signal further confirmed the formation of SQ˙. The control experiment showed that the apo DFsc alone could not induce the formation of SQ˙, while Zn(II) alone only provided a yield of 2% SQ˙. The reduction potential of [DFsc-Zn(II)2]–SQ˙ is ∼400 mV less than the reported reduction potential of free SQ˙ in solution, indicating strong stabilization by both the Zn coordination and the protein environment. Molecular dynamics simulation and QM/MM optimization demonstrated that the stabilization came from two factors: the coordination of SQ˙ to unsaturated Zn(II) and the hydrophobic environment around the SQ˙. These studies lead to a better comprehension about how metalloproteins stabilize an organic radical.
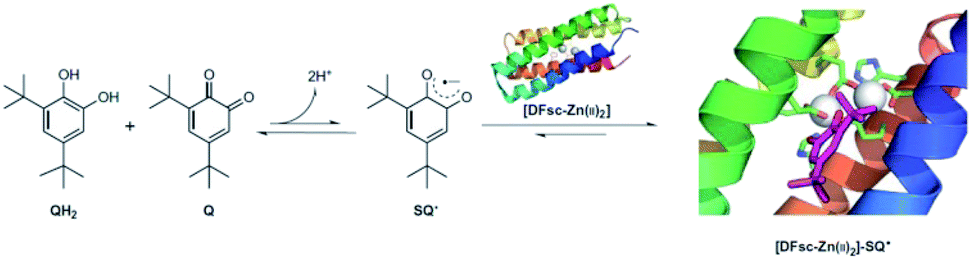 | ||
| Fig. 12 Semiquinone radical anion stabilized by the coordination interaction of Zn(II) in designed Due Ferri proteins. Reproduced from ref. 62 with permission from Nature Publishing Group, Copyright 2016. | ||
It is widely reported in the literature that the properties of radical ions are related to their counter ions. However, uncharged species with a strong dipole can also strongly influence the stability of radical ions. We reported a naphthalenediimide derivative radical anion (NDI˙−) stabilized by the electrostatic interaction of the carbonyl-fringed portals of cucurbit[7]uril63,64 (Fig. 13). As shown in Fig. 13, the cucurbit[7]urils were located on each side of NDI. With the electrostatic interaction of the carbonyl groups, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy of NDI were decreased by about 0.47 eV, leading to the increase of the reduction potential of NDI. Therefore, both the yield and life-time of NDI˙− generated from the photo-induced electron transfer process can be increased significantly. Such a strategy can be extended to other radical anions, for example, PDI˙−.65,66 An et al. reported the use of supramolecularly stabilized PDI˙− in photosensitized initiation of polymerization and photoinduced electron transfer reversible addition-fragmentation chain transfer (PET-RAFT) polymerization to synthesize polymers with an ultrahigh molecular weight in aqueous solution.67,68
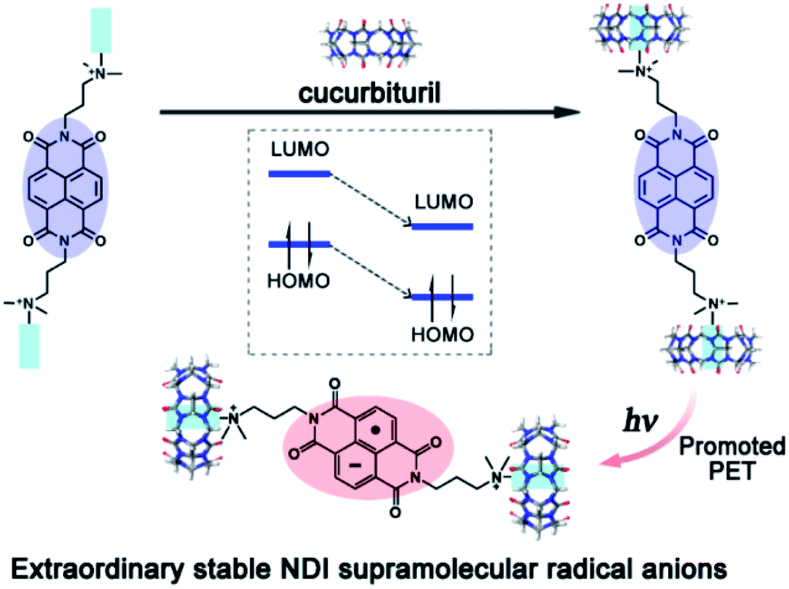 | ||
| Fig. 13 NDI˙− stabilized by the electrostatic effect of cucurbit[7]uril. Reproduced from ref. 63 with permission from Royal Society of Chemistry, Copyright 2015. | ||
In contrast, for radical cations, the effect of the electrostatic interaction of cucurbituril on the stability of radicals can be reversed. We reported that the radical cation of a derivative of 1,4-diketopyrrolo-[3,4-c]-pyrroles (DPP˙+) could be activated by cucurbiturils69,70 (Fig. 14). DPP˙+ served as the key intermediate of Fenton oxidation. The electrostatic interaction of cucurbit[7]uril would increase the spin density of DPP˙+ and increase its SOMO energy, thus improving the reactivity of DPP˙+, leading to the acceleration of Fenton oxidation. As mentioned before, the strength of electrostatic interaction is dependent on the distance between two dipoles. Therefore, in the folded conformation host–guest complex of DPP˙+ and cucurbit[8]uril with a decreased distance between the radical cation and carbonyl groups, the electrostatic interaction was further strengthened. The SOMO energy of DPP˙+ was increased by 1.06 eV and the Fenton oxidation was accelerated by 112 fold after the introduction of cucurbit[8]uril. These results show the great ability of electrostatic interaction in tuning the stability of radical ions. Though cucurbiturils are not charged species, their effect on radicals can be comparable to that of covalent interactions. The key point is the focus of dipoles. The rigid structure of cucurbiturils combines the dipoles of carbonyl groups together, and the formation of a well-defined supramolecular structure brings the radical cation and the carbonyl groups together. Combining those factors, an extraordinarily stabilized radical anion and activated radical cation are fabricated.
 | ||
| Fig. 14 DPP˙+ activated with cucurbit[7]uril and its further activation with cucurbit[8]uril. Reproduced from ref. 70 with permission from Royal Society of Chemistry, Copyright 2018. | ||
π–π interaction can stabilize aromatic radicals in the π-stack through the delocalization of spin density. Giuseppone et al. reported a triarylamine radical cation stabilized in its hierarchically self-assembled nanowire with triarylamine-based building blocks73 (Fig. 15). The radical cation was generated from the photo-oxidation of white light irradiation.
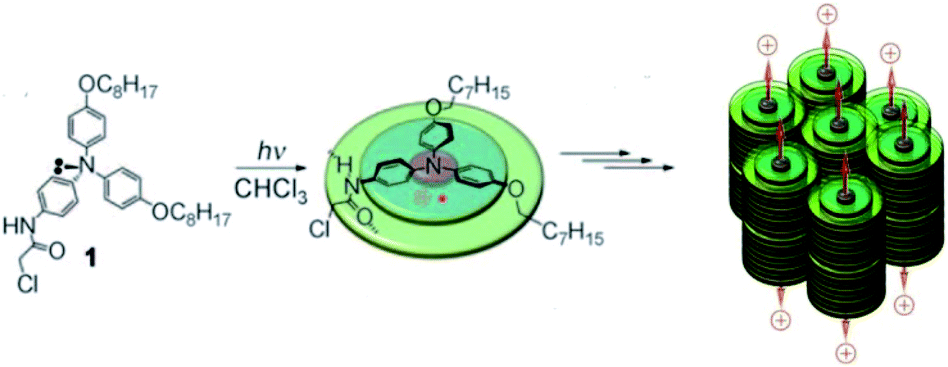 | ||
| Fig. 15 The triarylamine radical cation stabilized in its hierarchically self-assembled nanowire with triarylamine-based building blocks. Reproduced from ref. 73 with permission from Wiley-VCH, Copyright 2010. | ||
The decay of radical cations reached a plateau at a concentration of 6 radicals per 1000 triarylamine molecules and the remaining radical cation would last for over one week. After the destruction of the π-stack under heating, the signal of radical cations vanished in 2 h. Interestingly, the total disappearance of the aromatic 1H-NMR signals was observed with only 0.6% radical yield, which indicated the delocalization of spin density in the self-assembly. Then, the delocalization was further confirmed by a high-resolution magic angle spinning 1H-NMR experiment. On average, the spin of radical cations was delocalized on over 160 triarylamine molecules and that was responsible for the unusual stability of the radical cation. The radical cation was stabilized by the π–π interaction, and conversely, its generation could feed back to stabilize the π-stacks. The generation of radical cations would initiate the self-assembly of triarylamine molecules thus leading to the formation of nanowires. The radical stabilized nanowires benefited from strong stabilization and hole conduction properties, which could display metallic conduction properties. This work provides new insights into the mutual promotion between the stabilized radical cation and the highly organized supramolecular structure, by which their further applications in functional soft materials are expected.74,75
The charge transfer interaction between an electro-rich donor and electro-deficient acceptor is another interaction that is usually applied to tune the stability of radicals for its considerable strength.76 Sessler et al. reported a stable radical pair generated by spontaneous electron transfer between an electro-rich host and electro-deficient guest77 (Fig. 16). A tetrathiafulvalene calix[4]pyrrole (TTF-C4P) donor could form a charge transfer complex with a bisimidazolium quinone (BIQ2+) guest acceptor with the addition of specific anions. The strong charge transfer interaction led to spontaneous electron transfer to form a stable radical paired host–guest complex whose structure was confirmed by single crystal X-ray diffraction. This work demonstrated the ingenious utilization of dynamicity of supramolecular chemistry to develop a stimuli-responsive system. On the one hand, the stabilization of the radical pair could be modulated with specific anions. Only with anions like chloride, bromide and methylsulfate could the host calix[4]pyrrole transfer from a 1,3-alternate to the cone conformation, which favored the formation of a charge transfer complex. On the other hand, the introduction of a cationic competitive guest could also break the radical pair and further caused the back transfer of the single electron. The dual responsive properties to anions and cations give a perfect example of supramolecular tuning of the stability of radicals, including their generation and decay.
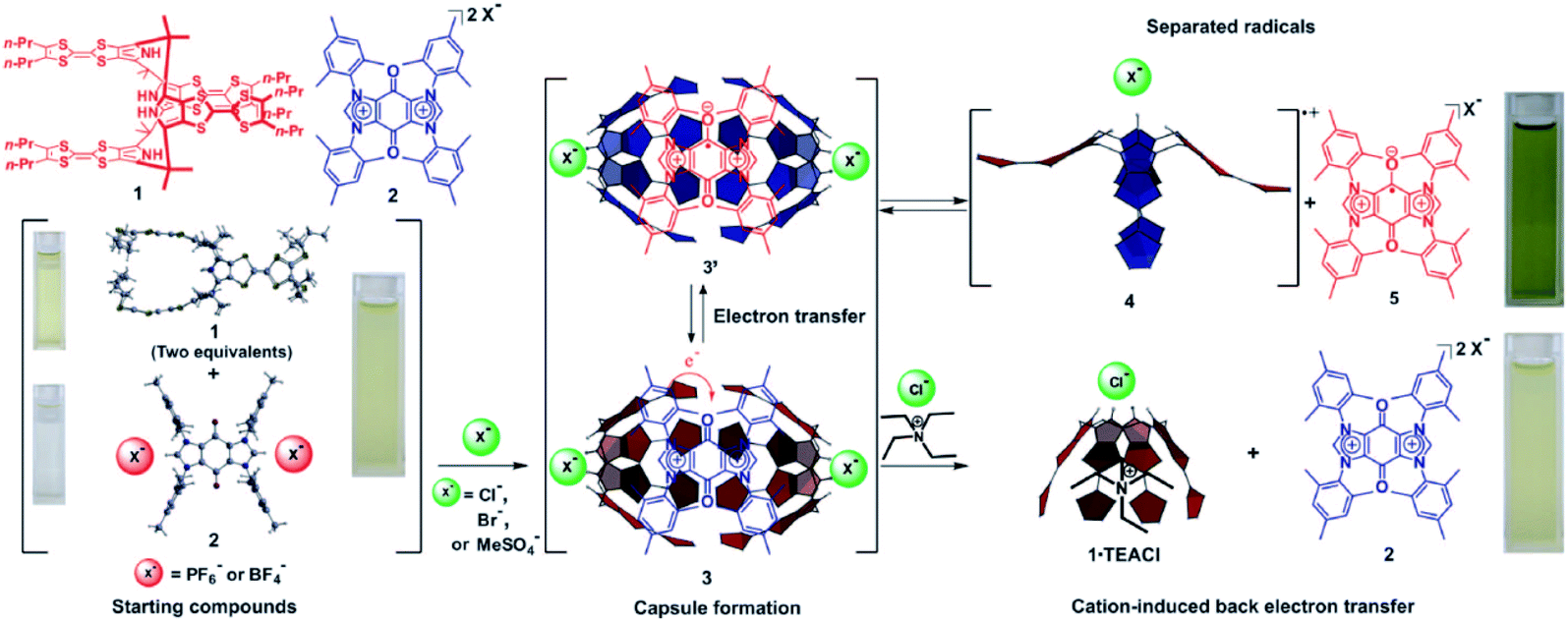 | ||
| Fig. 16 The chemical structures of the electro-rich host and the electro-deficient guest and their charge transfer to form a stable radical pair. Reproduced from ref. 77 with permission from American Association for the Advancement of Science, Copyright 2010. | ||
The intermolecular spin–spin interaction is of great importance in tuning the activity of radicals. Many great studies were based on the formation of radical dimer or radical based assemblies. In 2002, Kim et al. pioneered the early studies about the dimerization of methyl viologen radical cations (MV˙+) in the cavity of cucurbit[8]uril in solution.78 In 2004, they reported the stabilized dimer of tetrathiafulvalene radical cations (TTF˙+) in the cavity of cucurbit[8]uril.79 Stoddart et al. studied the supramolecular stabilization of TTF radical dimers in detail by the synthesis of [3]catenanes80 (Fig. 17). The two TTF formed a dimer in the [3]catenanes. With stepwise oxidation, it was transferred into a mixed-valence complex (TTF)2˙+, and then into a radical dimer (TTF˙+)2. Both of (TTF)2˙+ and (TTF˙+)2 were stabilized in the [3]catenanes to form air-stable radicals. Furthermore, the single crystal structures of the [3]catenanes containing (TTF)2, (TTF)2˙+ and (TTF˙+)2 are shown in Fig. 17. In all of these complexes, two TTF groups were placed face to face but with different distances. Among them, (TTF)2 was placed in a slipped-stacked arrangement. In the (TTF)2 containing [3]catenane, TTF groups were kept 3.68 Å away from each other with the π–π interaction among them, which meant a very little electronic interaction between them. When one TTF group was oxidized to TTF˙+, the distance was decreased to 3.56 Å by the charge transfer interaction between the electro-rich TTF and electro-deficient TTF˙+. After its further oxidation, the intermolecular spin–spin interaction pulled the two TTF˙+ together with a distance of 3.42 Å against the coulombic repulsion between the charged species. These data presented an intuitionistic description on the strength of π–π interaction between homogeneous aromatic systems, charge transfer interaction and spin–spin interaction, and moreover, their capability in tuning the stability of radicals. By the rational use of spin–spin interaction, air-stable and water-stable radical crystals with multi-redox responsive properties are successfully prepared.81,82 Besides, the spin–spin interaction can be also used in template synthesis with organic radicals as the template molecule.83,84
 | ||
| Fig. 17 The chemical structure of the [3]catenanes and their stepwise oxidation. The single crystal structure of (TTF)2, (TTF)2˙+ and (TTF˙+)2 containing [3]catenanes. Reproduced from ref. 80 with permission from Nature Publishing Group, Copyright 2010. | ||
The intermolecular spin–spin interaction cannot only stabilize the radical dimer, which is not commonly observed in the solution phase, but also create novel topological and optical properties. Supramolecular systems with unique functions can be achieved in combination of the tunable stability of radicals and the supramolecular topological change with the formation of radical dimers. Recently, we reported a light powered dissipative supramolecular polymerization system based on a MV˙+ dimer stabilized in the cavity of cucurbit[8]uril85 (Fig. 18). Upon the input of light power, MV end groups underwent photoreduction and formed a stabilized MV˙+ dimer in cucurbit[8]uril, which served as a linker to form supramolecular polymers. Without the light, the oxidation of MV˙+ led to the decay of supramolecular polymers. In this work, the stabilization of the radical dimer built up the supramolecular linker, and thus ensured the formation and the kinetic stability of supramolecular polymers. Nevertheless, the stable MV˙+ dimer exhibited satisfactory redox reversibility. Both of these factors support the dissipative supramolecular polymerization in a far-from-equilibrium state. In addition to the linear supramolecular polymer, various topological structures can be constructed driven by the stacking of radicals. For example, Li et al. reported the fabrication and stabilization of supramolecular macrocycles and two-dimensional and three-dimensional supramolecular organic frameworks by intermolecular spin–spin interactions.86–89
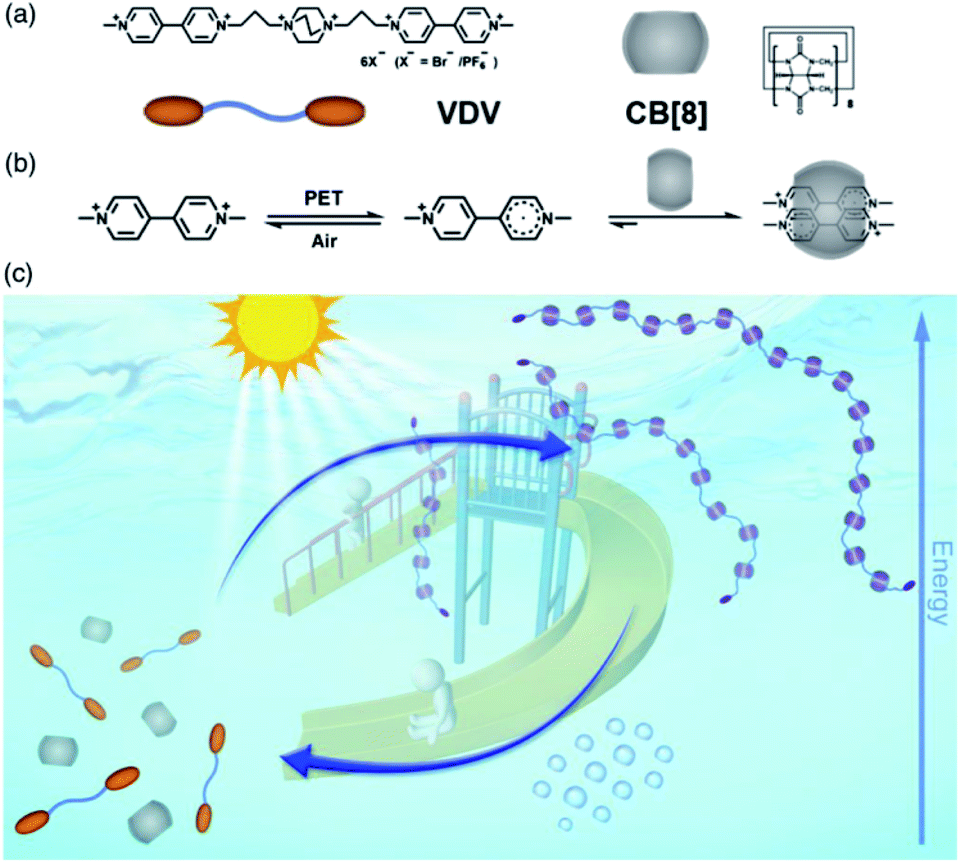 | ||
| Fig. 18 Light powered dissipative supramolecular polymerization system based on a MV˙+ dimer stabilized in cucurbit[8]uril. Reproduced from ref. 85 with permission from Chinese Chemical Society, Copyright 2019. | ||
The formation of radical dimers may lead to a narrower band gap than the radical itself, and the narrower band gap will further lead to absorption at longer wavelengths. We reported the utilization of an N,N′-dimethylated dipyridiniumthiazolo[5,4-d]thiazole radical dimer stabilized in the cavity of cucurbit[8]uril to achieve high-efficiency NIR-II photothermal conversion and therapy90 (Fig. 19). NIR-II light (1000–1350 nm) exhibits a large penetration depth and maximum permissible exposure and is considered to be suitable for the photothermal therapy of cancer.91 The radical dimer showed strong absorption in the NIR-II region with a molar absorption coefficient ε of 3.93 × 104 L mol−1 cm−1 and efficient photothermal conversion with an efficiency of about 54.6%. The improved stability helped its reversible photothermal conversion under irradiation and further application in photothermal therapy. The supramolecular radical dimer exhibited strong inhibition on HegG2 cancer cell growth under 1064 nm irradiation even penetrating through chicken breast tissue. This work opens a new door for the fabrication of functional materials by tailor-made assembly of organic radicals, which may provide improved stability and the creation of new functions.
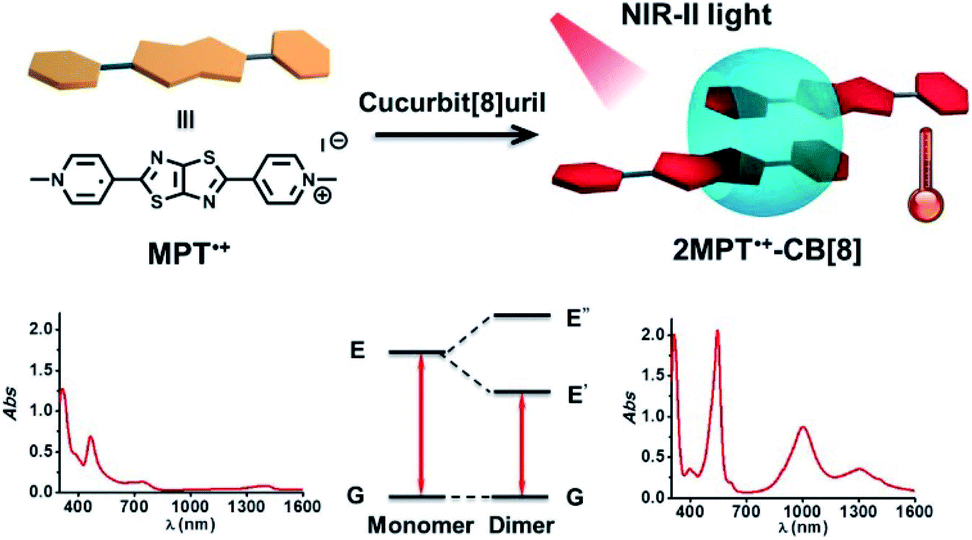 | ||
| Fig. 19 Supramolecular radical dimer with improved stability for NIR-II photothermal conversion and therapy. Reproduced from ref. 90 with permission from Wiley-VCH, Copyright 2019. | ||
4. Conclusions and outlook
In summary, we have reviewed covalent and non-covalent approaches to tune the stability of organic radicals, including steric protection and the delocalization of spin density. Both covalent and non-covalent approaches can tune the stability of organic radicals, towards their application in radical based functional materials and the promotion of radical mediated reactions. Covalent strategies can tune the activity of organic radicals in a large range and even achieve thermal stable radicals, while supramolecular strategies can tune the activity of organic radicals dynamically and reversibly.Although covalent and non-covalent approaches for tuning the activity of organic radicals have made significant progress, they still need to be further developed. There are many kinds of supramolecular hosts which may be used for tuning the stability of organic radicals. Depending on the nature and size of organic radicals, supramolecular hosts with a suitable size of cavity and electronic effect should be chosen. In addition, various non-covalent interactions could be employed, and combined interaction of non-covalent interactions may bring wonders out of our expectations.
The unique open-shell structures of radicals bring about extensive applications in functional materials, such as spin probes, magnetic materials, optoelectronic materials and biomedical materials. The function of materials originates from the single electron of radicals in this regard, and thus the persistence of radicals is necessary. Another kind of radical material is based on its redox properties, such as energy materials and conductive and semiconductive materials. The performances of these materials are closely related to the stability of radicals, especially redox potential and redox reversibility. For various radical based functional materials, the appropriate utilization of covalent approaches may guarantee the persistence of radicals. The introduction of dynamicity and reversibility by non-covalent approaches can push the development of functional radical materials with stimuli-responsiveness, switchability and adaptivity. Sometimes, the supramolecular arrangement of organic radicals can create new functions in long-range electron/hole transport, novel topological structures, the intermolecular coupling of molecular orbitals and others.
Radical mediated reactions usually possess low activation energy, and thus the product selectivity is considerably dependent on the reaction path of radical intermediates because of their high reactivity. Through supramolecular chemistry, the preorganization and molecular orientation can be controlled, leading to controlled product selectivity. Considering that many reactions involve the use of radicals as intermediates, there is plenty of room to be explored in this regard. This strategy can be further extended to other reactive intermediates, such as excited state molecules, reactive carbon ions and so on. We may enrich this field by supramolecular intermediate chemistry, allowing for controlling the activity of different intermediates for controlling not only the path of organic reactions but also their selectivity and chirality of the products.
Another future trend of this field may be tuning of radical's activity by interplay between covalent and non-covalent strategies. To combine covalent approaches for high stability of radicals with non-covalent approaches for dynamicity, both effective and dynamic tuning could be achieved. Moreover, dynamic covalent bonding may be an option for realization of effective and dynamic tuning in one pot. In a word, this is an open field that can only be limited by our imagination.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21821001).Notes and references
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- M. J. Schmidt, J. Borbas, M. Drescher and D. Summerer, J. Am. Chem. Soc., 2014, 136, 1238–1241 CrossRef CAS.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS.
- K. Nakahara, K. Oyaizu and H. Nishide, Chem. Lett., 2011, 40, 222–227 CrossRef CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie and B. W. Boudouris, Science, 2018, 359, 1391–1395 CrossRef CAS PubMed.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS.
- Q.-S. Gu, Z.-L. Li and X.-Y. Liu, Acc. Chem. Res., 2019 DOI:10.1021/acs.accounts.9b00381.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- H. Jiang and A. Studer, CCS Chem., 2019, 1, 38–49 CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Feng, P. Cheng, W. Yan, M. Boronat, X. Li, J.-H. Su, J. Wang, Y. Li, A. Corma, R. Xu and J. Yu, Science, 2016, 351, 1188–1191 CrossRef CAS.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS PubMed.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS.
- H. Tomioka, E. Iwamoto, H. Itakura and K. Hirai, Nature, 2001, 412, 626–628 CrossRef CAS PubMed.
- T. Hirano, W. Li, L. Abrams, P. J. Krusic, M. F. Ottaviani and N. J. Turro, J. Org. Chem., 2000, 65, 1319–1330 CrossRef CAS PubMed.
- C.-H. Tung, L.-Z. Wu, L.-P. Zhang and B. Chen, Acc. Chem. Res., 2003, 36, 39–47 CrossRef CAS PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- Y. Wang, M. Frasconi and J. F. Stoddart, ACS Cent. Sci., 2017, 3, 927–935 CrossRef CAS PubMed.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS PubMed.
- Y. Jiao, B. Tang, Y. Zhang, J.-F. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6077–6081 CrossRef CAS.
- D. Shimizu and A. Osuka, Chem. Sci., 2018, 9, 1408–1423 RSC.
- P. Andersen and B. Klewe, Acta Chem. Scand., 1967, 21, 2599–2607 CrossRef CAS.
- J. Veciana, J. Carilla, C. Miravitlles and E. Molins, J. Chem. Soc., Chem. Commun., 1987, 812–814 RSC.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef CAS PubMed.
- B. P. Sogo, M. Nakazaki and M. Calvin, J. Chem. Phys., 1957, 26, 1343–1345 CrossRef.
- D. H. Reid, Tetrahedron, 1958, 3, 339–352 CrossRef CAS.
- J. R. Bolton, J. Chem. Phys., 1967, 46, 408–409 CrossRef CAS.
- R. Mondal, C. Tönshoff, D. Khon, D. C. Neckers and H. F. Bettinger, J. Am. Chem. Soc., 2009, 131, 14281–14289 CrossRef CAS PubMed.
- Y.-Y. Lin, D. J. Gundlach, S. F. Nelson and T. N. Jackson, IEEE Trans. Electron Devices, 1997, 44, 1325–1331 CrossRef CAS.
- K. Kato, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 9491–9494 CrossRef CAS PubMed.
- R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS.
- Y. Kim and E. Lee, Chem.–Eur. J., 2018, 24, 19110–19121 CrossRef CAS PubMed.
- S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663–1667 RSC.
- D. Schmidt, D. Bialas and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 3611–3614 CrossRef CAS PubMed.
- P. P. Power, Chem. Rev., 2003, 103, 789–809 CrossRef CAS PubMed.
- P. J. Davidson, A. Hudson, M. F. Lappert and P. W. Lednor, J. Chem. Soc., Chem. Commun., 1973, 829–830 RSC.
- P. Bissinger, H. Braunschweig, A. Damme, C. Hörl, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2015, 54, 359–362 CrossRef CAS PubMed.
- V. P. Rao, M. B. Zimmt and N. J. Turro, J. Photochem. Photobiol., A, 1991, 60, 355–360 CrossRef CAS.
- H. García and H. D. Roth, Chem. Rev., 2002, 102, 3947–4007 CrossRef PubMed.
- S. Jockusch, T. Hirano, Z. Liu and N. J. Turro, J. Phys. Chem. B, 2000, 104, 1212–1216 CrossRef CAS.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384–12385 CrossRef CAS.
- Y. Liu, J. Shi, Y. Chen and C.-F. Ke, Angew. Chem., Int. Ed., 2008, 47, 7293–7296 CrossRef CAS PubMed.
- A. Stergiou, J. Rio, J. H. Griwatz, D. Arčon, H. A. Wegner, C. P. Ewels and N. Tagmatarchis, Angew. Chem., Int. Ed., 2019, 58, 17745–17750 CrossRef CAS PubMed.
- B. Lü, Y. Chen, P. Li, B. Wan, K. Müllen and M. Yin, Nat. Commun., 2019, 10, 767 CrossRef PubMed.
- C. Ji, W. Cheng, Q. Yuan, K. Müllen and M. Yin, Acc. Chem. Res., 2019, 52, 2266–2277 CrossRef CAS PubMed.
- S. Ghisla and V. Massey, Biochem. J., 1986, 239, 1–12 CrossRef CAS PubMed.
- E. Breinlinger, A. Niemz and V. M. Rotello, J. Am. Chem. Soc., 1995, 117, 5379–5380 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- C. R. Benson, E. M. Fatila, S. Lee, M. G. Marzo, M. Pink, M. B. Mills, K. E. Preuss and A. H. Flood, J. Am. Chem. Soc., 2016, 138, 15057–15065 CrossRef CAS PubMed.
- W. Kaim, Coord. Chem. Rev., 1987, 76, 187–235 CrossRef CAS.
- G. Ulas, T. Lemmin, Y. Wu, G. T. Gassner and W. F. DeGrado, Nat. Chem., 2016, 8, 354–359 CrossRef CAS PubMed.
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342–3346 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975–3980 RSC.
- Y. Yang, P. He, Y. Wang, H. Bai, S. Wang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16239–16242 CrossRef CAS PubMed.
- Y. Yang and Z. An, Polym. Chem., 2019, 10, 2801–2811 RSC.
- L. Shen, Q. Lu, A. Zhu, X. Lv and Z. An, ACS Macro Lett., 2017, 6, 625–631 CrossRef CAS.
- Y. Jiao, W.-L. Li, J.-F. Xu, G. Wang, J. Li, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8933–8937 CrossRef CAS.
- B. Tang, W.-L. Li, Y. Jiao, J.-B. Lu, J.-F. Xu, Z. Wang, J. Li and X. Zhang, Chem. Sci., 2018, 9, 5015–5020 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, CCS Chem., 2019, 1, 181–196 Search PubMed.
- E. Moulin, F. Niess, M. Maaloum, E. Buhler, I. Nyrkova and N. Giuseppone, Angew. Chem., Int. Ed., 2010, 49, 6974–6978 CrossRef CAS PubMed.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS.
- Y. Jiao, J.-F. Xu, Z. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22635–22640 CrossRef CAS PubMed.
- J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hudnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324–1327 CrossRef CAS PubMed.
- W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828–1829 RSC.
- A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Chem. Commun., 2004, 806–807 RSC.
- J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard III, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870–879 CrossRef CAS PubMed.
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS.
- M. T. Nguyen, D. P. Ferris, C. Pezzato, Y. Wang and J. F. Stoddart, Chem, 2018, 4, 2329–2344 CAS.
- H. Li, A. C. Fahrenbach, S. K. Dey, S. Basu, A. Trabolsi, Z. Zhu, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8260–8265 CrossRef CAS PubMed.
- Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC.
- Z. Yin, G. Song, Y. Jiao, P. Zheng, J.-F. Xu and X. Zhang, CCS Chem., 2019, 1, 335–342 CrossRef.
- W.-K. Wang, Y.-Y. Chen, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Chem.–Asian J., 2014, 9, 1039–1044 CrossRef CAS PubMed.
- L. Zhang, T.-Y. Zhou, J. Tian, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 4715–4721 RSC.
- C. Zhou, J. Tian, J.-L. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 341–345 RSC.
- J. Tian, Y.-D. Ding, T.-Y. Zhou, K.-D. Zhang, X. Zhao, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Chem.–Eur. J., 2014, 20, 575–584 CrossRef CAS PubMed.
- B. Tang, W.-L. Li, Y. Chang, B. Yuan, Y. Wu, M.-T. Zhang, J.-F. Xu, J. Li and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15526–15531 CrossRef CAS PubMed.
- Y. Cao, J.-H. Dou, N.-J. Zhao, S. Zhang, Y.-Q. Zheng, J.-P. Zhang, J.-Y. Wang, J. Pei and Y. Wang, Chem. Mater., 2017, 29, 718–725 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |