
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePot and time economies in the total synthesis of Corey lactone†
Nariyoshi
Umekubo
,
Yurina
Suga
and
Yujiro
Hayashi
 *
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: yujiro.hayashi.b7@tohoku.ac.jp
First published on 23rd December 2019
Abstract
The Corey lactone is a highly versatile intermediate for the synthesis of a variety of prostaglandin hormones that natively control a multitude of important physiological processes. Starting from commercially available compounds, we herein disclose a time-economical, one-pot enantioselective preparation of the Corey lactone by virtue of a new diphenylprolinol silyl ether-mediated domino Michael/Michael reaction to afford the substituted cyclopentanone core in a formal (3 + 2) cycloadditive fashion. More broadly, this work advances the on-demand, gram-scale synthesis of high-value targets involving chemically orthogonal transformations, whereby distinct reactions of acids, bases, organometalics, reductants and oxidants can be carried out in a single reaction vessel in a sequential fashion.
Introduction
The efficient synthesis of target molecules is a key aim in synthetic organic chemistry. For an ideal synthesis,1 chemists consider several economic factors. Such considerations include but are not limited to atom economy,2 step economy,3 and redox economy.4 In addition to these, we have proposed the concept of pot economy.5 Today one-pot operations are being advanced as effective ways to not only form key structural motifs in a single-pot sequence with stereoselectivity, but also to carry out a growing number of chemically distinct transformations that adopt apparently incompatible reagents such as strong and weak acids, bases, organometallics, Lewis acids, oxidants and reductants. Not only this, but one-pot operations circumvent several purification steps through strategic in situ quenching events, thereby minimizing chemical waste generation, and saving time. Thus, one-pot operations are not only efficient, but also tend to be more environmentally acceptable. Based on such advances in contemporary synthetic methods, we have accomplished the one-pot syntheses6 of (−)-oseltamivir, ABT341, and baclofen, the three-pot synthesis of PGE1 methyl ester, and the five-pot synthesis of estradiol methyl ether.The synthesis of molecules within the shortest possible time is also important, as can be inferred from the popular expression that “time is money”. A rapid synthesis reduces the cost of production and enables the possibility of on-demand synthesis. Hence, we have emphasized the importance of time economy in the synthesis of molecules, as recently reported in our 60 minute total synthesis of (−)-oseltamivir.6a
Prostaglandins (PGs) are known to act as local hormones with the ability to control a multitude of important physiological properties. Some of their derivatives also serve as useful medicines.7 Given the biological importance of PGs and their scarce availability from natural sources, the scientific community has invested a great deal of effort and ingenuity into their efficient synthesis.8 Corey's synthesis via the Corey lactone is a landmark achievement, which greatly facilitated the science of PGs.9 Even today, new synthetic routes using radical methods to access PGF2α from the Corey lactone have been developed by Baran.10 A three-component coupling method to access PGs was also reported by Noyori11 and organocatalysts have been applied to the synthesis of PGs by Aggarwal12 and our group.6d,13
Given the synthetic significance of the Corey lactone, many synthetic protocols has emerged for the synthesis sequel to the seminal synthetic precedent by Corey.9 In general, the reported synthetic methods usually involves several reaction steps together with a considerable amount of time.14 Stork's synthesis involves 7 steps, from cis-cyclopentene-1,4-diol, and it features a radical cyclization-trapping strategy.14i Woodward reported an innovative 11 step racemic synthesis starting from cis-cyclohexane-1,3,5-triol via rearrangement.14b The chiral Corey lactone was prepared from the chiral starting materials,14h,14p resolution14n and by using diastereoselective Diels–Alder reaction using a chiral auxiliary.9e,14r As for the asymmetric catalytic synthesis, Corey is also a pioneer, and his group elegantly synthesized the Corey lactone via an asymmetric Diels–Alder reaction that involved a chiral Lewis acid catalyst (Scheme 1).9 While chiral dirhodium mediated C–H insertion of diazoacetate was reported by Doyle for the synthesis of the key intermediate of the Corey lactone,14s Chen recently reported a kinetic resolution in the asymmetric catalytic Baeyer–Villiger oxidation, although only a half of the materials was utilized.14u In spite of the great progress in asymmetric catalytic reactions, it is still difficult to synthesize the Corey lactone via an asymmetric catalytic method. Although Corey's synthesis still remains a practical and excellent approach, it requires 10 steps, and the total reaction time is over 48 hours, excluding the required purification time. Thus, a more practicable and efficient asymmetric catalytic synthesis of this highly valued target within a short total reaction time and in a single vessel starting from commercially available compounds remains a prominent synthetic challenge. In this paper, we describe a practical and high-yielding synthesis of the Corey lactone by using a newly optimized organocatalyst-mediated asymmetric catalytic (3 + 2) cycloaddition reaction as a prelude to a reductive, oxidative sequence of events.
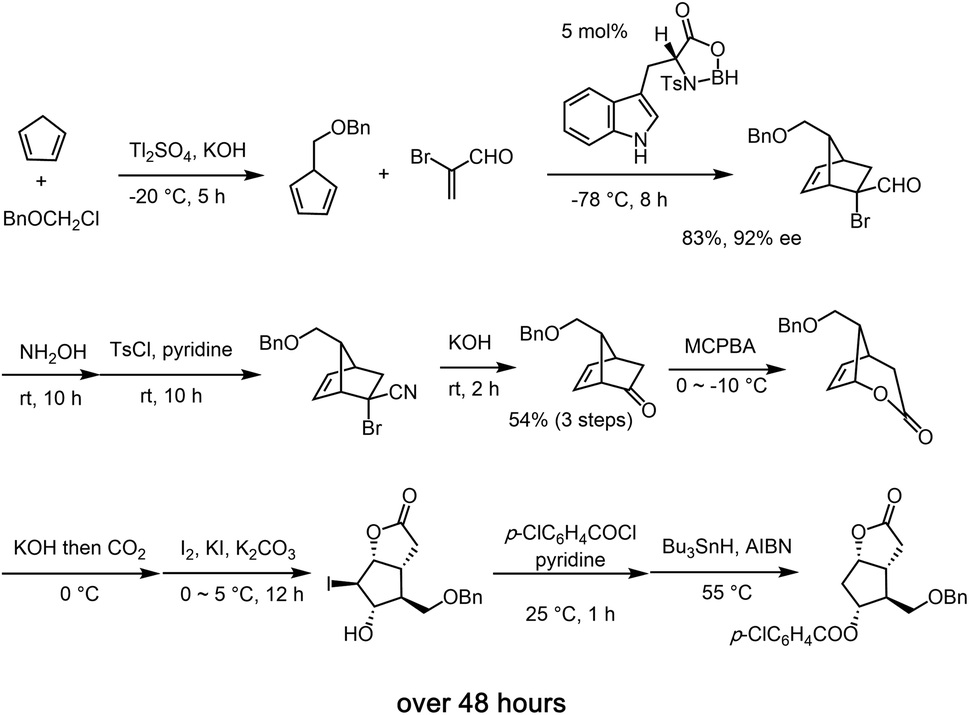 | ||
| Scheme 1 Corey's synthetic route of the Corey lactone. | ||
Results and discussion
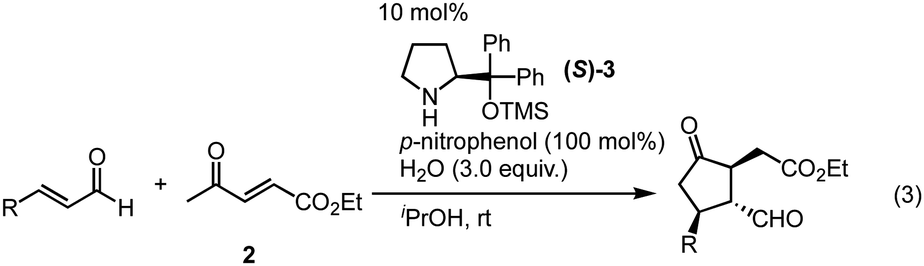
Previously, we have reported the asymmetric Michael reaction of cyclohexanone and α,β-enal catalyzed by two amine catalysts namely; (S)-diphenylprolinol silyl ether15 and 4-hydroxyproline (eqn (1)).16 Based on this precedent Michael reaction of a ketone as the Michael donor, we designed a domino17 Michael/Michael reaction of 3-(dimethylphenylsilyl)propenal (1)18 and ethyl 4-oxo-2-pentenoate (2). There are two possible pathways after the first Michael reaction (Scheme 2): the first Michael reaction would afford an enamine, which might react with the α,β-unsaturated ester moiety via a 5-exo mode to afford 419 or it might also react with the α,β-unsaturated ketone moiety via a 6-endo mode to afford 5.20 After several investigations, the desired cyclopentanone derivative 4 was found to be obtained without any observed formation of the cyclohexanone 5 (eqn (2)).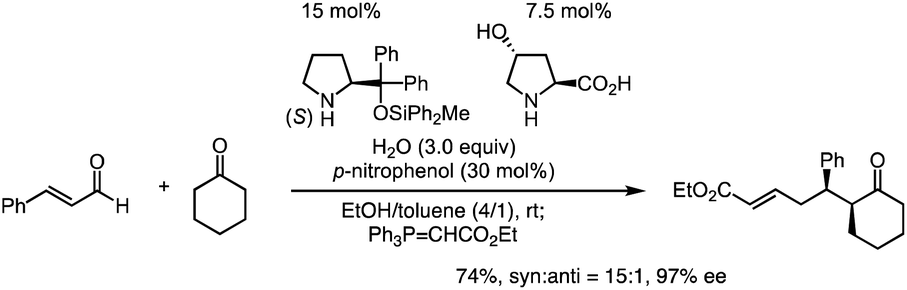 | (1) |
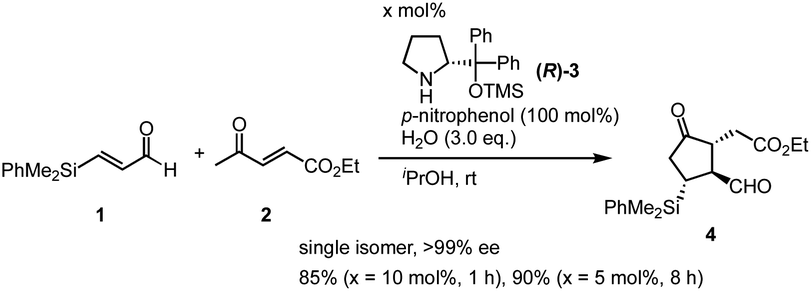 | (2) |
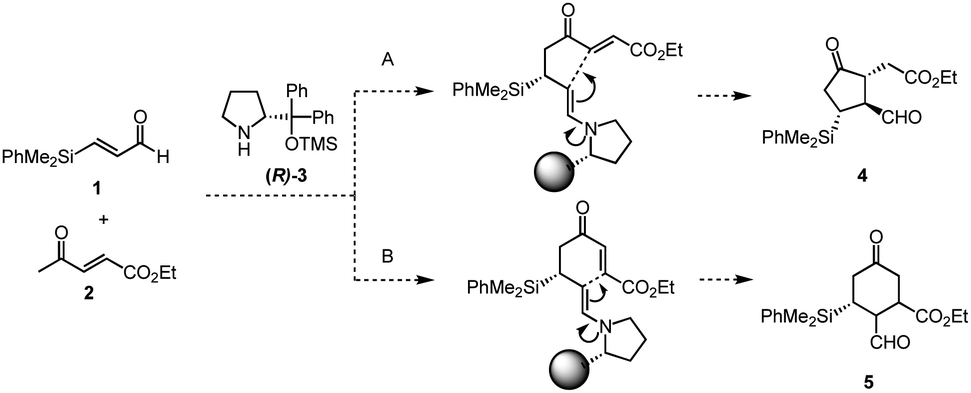 | ||
| Scheme 2 The possible reaction paths of A and B. | ||
We also found that two amine catalysts are not necessary and that the domino Michael/Michael reaction proceeded in the presence of (R)-diphenylprolinol silyl ether (R)-3 (10 mol%) and p-nitrophenol16 in i-PrOH to afford cyclopentanone 4 with good yields in nearly enantiomerically pure form as a single isomer within 1 hour. The reaction also proceeded in the presence of 5 mol% of the catalyst within 8 hours without decreasing optical purity. It should be noted that the three contiguous stereogenic centers are completely controlled, and that the stereochemistry perfectly matches that of PGs. Given that the first formal (3 + 2) cycloaddition reaction21 proceeded efficiently to give the cyclopentanone core of Corey lactone, we further investigated the one-pot synthesis of Corey lactone.
To this end, the rest of the transformations involve (1) reduction of aldehyde, (2) stereoselective reduction of ketone, (3) lactonization, and (4) Tamao–Fleming oxidation22 for the transformation of the C–Si bond into a C–O bond. It is a challenge to carry out these transformations in a single pot because they involve several unrelated reactions such as reduction and oxidation, under different reaction conditions such as acidic or basic media; thus, the residual reagents might also interfere with the subsequent reactions. After an intensive screening of the reaction conditions, a rapid one-pot sequential synthesis was eventually accomplished (Scheme 3). After the first domino reaction, both aldehyde and ketone are reduced by LiAlH(t-BuO)3 within 15 minutes at 60 °C to afford 6. Notably the ketone was reduced with excellent diastereoselectivity by a bulky reducing reagent, and the reaction time was only 15 minutes at an elevated temperature (60 °C) without any decrease in the diastereoselectivity. The addition of HBF4 quenched the reaction and generated dihydroxy ester, which immediately cyclized to afford lactone 7 under the acidic conditions. In the course of THF removal under reduced pressure (80 °C for 15 minutes), concurrent conversion of the Si–Ph bond into a Si–F bond proceeded to provide 8. Although this conversion requires several hours in general, we accidentally found that the reaction could be completed within 15 minutes under condensed conditions during the evaporation process for the solvent swap. After neutralization of the reaction mixture with K2CO3, the reaction with H2O2 and KF for 1 hour at 40 °C afforded the Corey lactone 9. NMR spectroscopic analysis of the crude material indicated that 9 was obtained in good quality (see ESI†) and it was confirmed that 9 could be easily purified by facile column chromatography. All operations could be conducted in a single vessel, and the total yield of the gram-scale synthesis using 2 (1.4 g) was 50%; the total reaction time was 152 minutes. All starting materials were available from commercial sources.
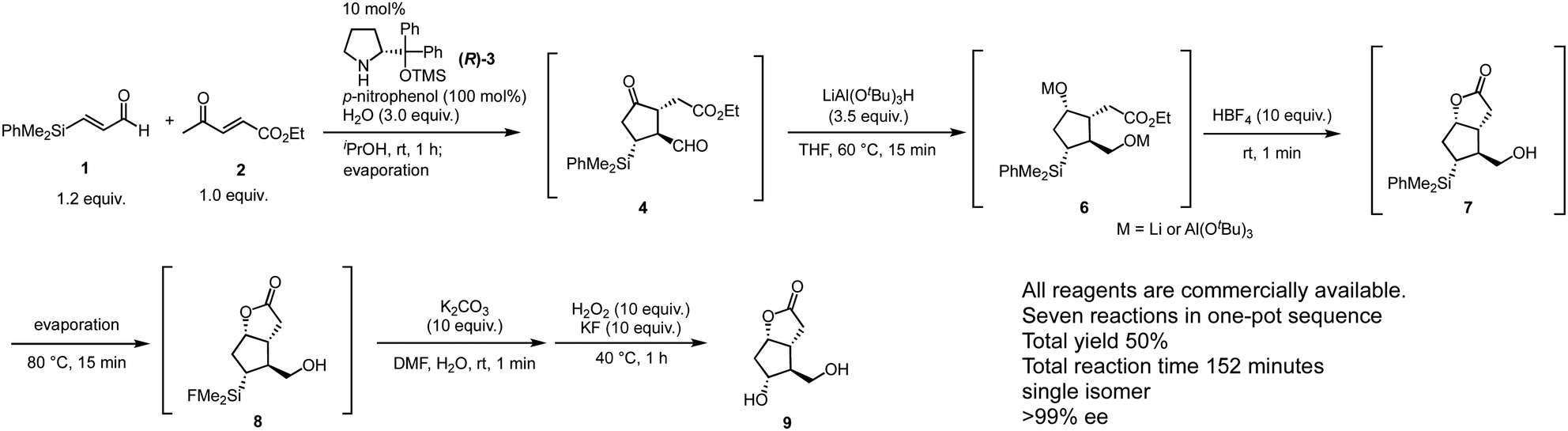 | ||
| Scheme 3 One-pot synthesis of the Corey lactone 9. | ||
This one-pot operation involves one domino reaction composed of two reaction steps: (1) asymmetric Michael reaction of ketone and α,β-enal and (2) successive intra molecular Michael reaction; and five subsequent reaction steps composed of (3) reduction of aldehyde, (4) stereoselective reduction of ketone, (5) lactone formation, (6) conversion of the Si–Ph bond into a Si–F bond, and (7) conversion of the C–SiMe2F bond into a C–OH bond. The total yield of 50% indicates that each reaction step proceeds in 90% yield (0.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 = 0.48). It should be noted that HBF4 played four roles in this process, namely (1) conversion of the O–Li or O–Al bond into an O–H bond after reduction, (2) decomposition of the excess LiAlH(t-BuO)3, (3) promotion of the formation of lactone 7, and (4) transformation of the Si–Ph bond into a Si–F bond. The ability to employ a single reagent to effect various roles in a multistep reaction constitutes an efficient feature of a one-pot process.5 It should also be mentioned that 7, which can be isolated, is a useful intermediate because the selective protection of the two hydroxy groups in 9 can be initiated at this stage.
7 = 0.48). It should be noted that HBF4 played four roles in this process, namely (1) conversion of the O–Li or O–Al bond into an O–H bond after reduction, (2) decomposition of the excess LiAlH(t-BuO)3, (3) promotion of the formation of lactone 7, and (4) transformation of the Si–Ph bond into a Si–F bond. The ability to employ a single reagent to effect various roles in a multistep reaction constitutes an efficient feature of a one-pot process.5 It should also be mentioned that 7, which can be isolated, is a useful intermediate because the selective protection of the two hydroxy groups in 9 can be initiated at this stage.
We then investigated the generality of the key domino reaction, namely the formal (3 + 2) cycloaddition reaction of α,β-unsaturated aldehyde and ethyl 4-oxo-2-pentenoate (2). The reaction of cinnamaldehyde was first examined under the best reaction conditions of 3-(dimethylphenylsilyl)propenal (1) using (S)-catalyst (S)-3, but the reaction was slow and afforded the product in 23% yield after 24 hours together with recovery of the starting materials. After re-optimization of the reaction conditions, it was observed that the amount of the reagent dramatically affected the yield. By using two equivalents of cinnamaldehyde in the presence of p-nitrophenol (100 mol%), the reaction proceeded at room temperature to afford the product as a single isomer in excellent yield (98%) in nearly optically pure form (>99% ee). With this good result obtained, the generality of the reaction was investigated (Table 1). For the β-substituent of α,β-unsaturated aldehyde, not only phenyl and 2-naphthyl but also electron-rich aromatics such as p-tolyl and p-methoxyphenyl substituents were suitable. Electron-deficient aromatics such as p-fluoro, p-chloro, and p-bromo phenyls, as well as m-bromo and o-bromo phenyls were all efficient aryl groups, affording the corresponding substituted cyclopentanone as a single isomer in nearly optically pure form. Heteroaromatic furyl was also a suitable substituent. It should be noted that, in all cases, excellent diastereo- and enantioselectivities were realized, and synthetically useful chiral substituted cyclopentanones were synthesized in nearly enantiomerically pure forms. In spite of the successful results in Table 1, β-alkyl substituted α,β-unsaturated aldehyde did not afford the product.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time [h] | Yieldb [%] | Drc | eed [%] |
a Unless otherwise shown, reactions were performed by employing α,β-unsaturated aldehyde (0.15 mmol), 2 (0.30 mmol), organocatalyst (S)-3 (0.015 mmol), water (0.45 mmol) and p-nitrophenol (0.15 mmol) in i-PrOH (75 μL) at room temperature for the indicated time.
b Isolated yield.
c Diastereomer ratio (dr) was determined by 1H-NMR analysis of a crude mixture.
d Enantiomeric excess (ee) of the products, as determined by HPLC analysis over a chiral solid phase after conversion to α,β-unsaturated ester by the treatment with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et. See ESI for details. CHCO2Et. See ESI for details.
|
|||||
| 1 | Phenyl | 10 | 98 | >98:2 |
>99 |
| 2 | 2-Naphthyl | 6 | 84 | >98:2 |
>99 |
| 3 | p-MeC6H4 | 12 | 91 | >98:2 |
>99 |
| 4 | p-MeOC6H4 | 15 | 88 | >98:2 |
>99 |
| 5 | p-FC6H4 | 6 | 81 | >98:2 |
>99 |
| 6 | p-ClC6H4 | 5 | 85 | >98:2 |
>99 |
| 7 | p-BrC6H4 | 4 | 83 | >98:2 |
>99 |
| 8 | m-BrC6H4 | 4 | 74 | >98:2 |
>99 |
| 9 | o-BrC6H4 | 7 | 82 | >98:2 |
96 |
| 10 | 2-Furyl | 24 | 80 | >98:2 |
96 |
Conclusions
In summary, a pot- and time-economical enantioselective total synthesis of the Corey lactone was accomplished in a single reaction vessel within 152 minutes from commercially available reagents. This work represents the shortest and fastest synthesis of a nearly enantiomerically pure Corey lactone with excellent yield. The key reaction is a diphenylprolinol silyl ether-mediated domino Michael/Michael reaction of α,β-enal and ethyl 4-oxo-2-pentenoate (2) to afford trisubstituted cyclopentanone as a single isomer with excellent enantioselectivity; three contiguous stereogenic centers are completely controlled with the desired configuration. A sequential, rapid total synthesis was then realized through the judicious selection of rapid reactions and versatile reagents allowing for a one-pot reductive–lactonization-oxidative sequence. Given the key organocatalyst-mediated formal asymmetric (3 + 2) cycloaddition reaction displayed wide generality, the highly functionalized cyclopentanone products constitute useful chiral synthetic intermediates not only for PGs but also for other natural products and drug targets. Importantly, this efficient synthesis illustrates that highly valued targets can now be prepared rapidly, on-demand through the strategic and sequential use of acids, bases, organometalics, reductants and oxidants in a single reaction vessel, a feat previously thought unreachable for a complex multi-stepped synthesis.5,6Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to dedicate this manuscript to Prof. E. J. Corey. This work was supported by JSPS KAKENHI grant number JP18H04641 in Hybrid Catalysis for Enabling Molecular Syn-thesis on Demand, JP18H04380 in Middle Molecular Strategy, and JP19H05630.Notes and references
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- (a) P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505 CrossRef CAS; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed; (c) P. A. Wender, Nat. Prod. Rep., 2014, 31, 433 RSC.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- (a) Y. Hayashi and S. Ogasawara, Org. Lett., 2016, 18, 3426 CrossRef CAS PubMed; (b) H. Ishikawa, M. Honma and Y. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 2824 CrossRef CAS PubMed; (c) Y. Hayashi, D. Sakamoto and D. Okamura, Org. Lett., 2016, 18, 4 CrossRef CAS PubMed; (d) Y. Hayashi and S. Umemiya, Angew. Chem., Int. Ed., 2013, 52, 3450 CrossRef CAS PubMed; (e) Y. Hayashi, S. Koshino, K. Ojima and E. Kwon, Angew. Chem., Int. Ed., 2017, 56, 11812 CrossRef CAS PubMed.
- C. D. Funk, Science, 2001, 294, 1871 CrossRef CAS PubMed.
- (a) S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286 CrossRef CAS PubMed; (b) H. Peng and F.-E. Chen, Org. Biomol. Chem., 2017, 15, 6281 RSC.
- (a) E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1995 Search PubMed; (b) E. J. Corey, N. M. Wesinshenker, T. K. Schaaf and W. Huber, J. Am. Chem. Soc., 1969, 91, 5675 CrossRef CAS PubMed; (c) E. J. Corey, T. K. Schaaf, W. Huber, U. Koelliker and N. M. Wesinshenker, J. Am. Chem. Soc., 1970, 92, 397 CrossRef CAS PubMed; (d) E. J. Corey, S. M. Albonico, U. Koelliker, T. K. Schaaf and R. K. Varma, J. Am. Chem. Soc., 1971, 93, 1490 CrossRef CAS PubMed; (e) E. J. Corey and H. E. Ensley, J. Am. Chem. Soc., 1975, 97, 6908 CrossRef CAS PubMed; (f) E. J. Corey and T. P. Loh, J. Am. Chem. Soc., 1991, 113, 8966 CrossRef CAS.
- J. T. Edwards, R. R. Merchant, K. S. MaClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed.
- R. Noyori and M. Suzuki, Angew. Chem., Int. Ed. Engl., 1984, 23, 847 CrossRef.
- (a) G. Coulthard, W. Erb and V. A. Aggarwal, Nature, 2012, 489, 278 CrossRef CAS PubMed; (b) S. Prévost, K. Thai, N. Schützenmeister, G. Coulthard, W. Erb and V. K. Aggarwal, Org. Lett., 2015, 17, 504 CrossRef PubMed; (c) H. Baars, M. Classen and V. K. Aggarwal, Org. Lett., 2017, 19, 6008 CrossRef CAS PubMed; (d) A. Pelšs, N. Gandhamsetty, J. R. Smith, D. Mailhol, M. Silvi, A. Watson, I. Perez-Powell, S. Prévost, N. Schützenmeister, P. Moore and V. K. Aggarwal, Chem.–Eur. J., 2018, 24, 9542 CrossRef PubMed.
- (a) S. Umemiya, D. Sakamoto, G. Kawauchi and Y. Hayashi, Org. Lett., 2017, 19, 1112 CrossRef CAS PubMed; (b) G. Kawauchi, S. Umemiya, T. Taniguchi, K. Monde and Y. Hayashi, Chem.–Eur. J., 2018, 24, 8409 CrossRef CAS PubMed.
- (a) E. J. Corey, Z. Arnold and J. Hutton, Tetrahedron Lett., 1970, 11, 307 CrossRef; (b) R. B. Woodward, J. Gosteli, I. Ernest, R. J. Friary, G. Nestler, H. Raman, R. Stitrin, C. Suter and J. K. Whitesell, J. Am. Chem. Soc., 1973, 95, 6853 CrossRef CAS PubMed; (c) J. S. Bindra, A. Grodski, T. K. Schaaf and E. J. Corey, J. Am. Chem. Soc., 1973, 95, 7522 CrossRef CAS PubMed; (d) D. Brewster, M. Myers, J. Ormerod, P. Otter, A. C. B. Smith, M. E. Spinner and S. Turner, J. Chem. Soc., Perkin Trans. 1, 1973, 2796 RSC; (e) G. Jones, R. A. Raphael and S. Wright, J. Chem. Soc., Perkin Trans. 1, 1974, 1676 RSC; (f) I. Tömösközi, L. Gruber, K. Go, I. Székely and V. Simonidesz, Tetrahedron Lett., 1976, 17, 4639 CrossRef; (g) S. Takano, N. Kubodera and K. Ogasawara, J. Org. Chem., 1977, 42, 786 CrossRef CAS PubMed; (h) F. Johson, K. G. Paul, D. Favara, R. Ciabatti and U. Guzzi, J. Am. Chem. Soc., 1982, 104, 2190 CrossRef; (i) G. Stork, P. M. Sher and H.-L. Chen, J. Am. Chem. Soc., 1986, 108, 6384 CrossRef CAS; (j) P. G. Baraldi, A. Barco, S. Benetti, G. P. Pollini, D. Simoni and V. Zanirato, Tetrahedron, 1987, 43, 4669 CrossRef CAS; (k) T. V. RajanBabu, J. Org. Chem., 1988, 53, 4522 CrossRef CAS; (l) K. Miyaji, Y. Ohara, Y. Takahashi, T. Tsuruda and K. Arai, Tetrahedron Lett., 1991, 32, 4557 CrossRef CAS; (m) J.-P. Vionnet and P. Renaud, Helv. Chim. Acta, 1994, 77, 1781 CrossRef CAS; (n) M. Node, D. Nakamura, K. Nishide and T. Inoue, Heterocycles, 1997, 46, 535 CrossRef CAS; (o) T. Yakura, S. Yamada, M. Azuma, A. Ueki and M. Ikeda, Synthesis, 1998, 973 CrossRef CAS; (p) M. Sannigrahi, D. L. Mayhew and D. L. J. Clive, J. Org. Chem., 1999, 64, 2776 CrossRef CAS PubMed; (q) E. Marot-ta, P. Righi and G. Rosini, Org. Lett., 2000, 2, 4145 CrossRef CAS PubMed; (r) D. Depre, L.-Y. Chen and L. Ghosez, Tetrahedron, 2003, 59, 6797 CrossRef CAS; (s) M. P. Doyle and A. J. Catino, Tetrahedron: Asymmetry, 2003, 14, 925 CrossRef CAS; (t) B. Augustyns, N. Maulide and I. E. Marko, Tetrahedron Lett., 2005, 46, 3895 CrossRef CAS; (u) K. Zhu, S. Hu, M. Liu, H. Peng and F.-E. Chen, Angew. Chem., Int. Ed., 2019, 58, 9923 CrossRef CAS PubMed.
- (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed; (b) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS PubMed.
- Y. Hayashi and N. Umekubo, Angew. Chem., Int. Ed., 2018, 57, 1958 CrossRef CAS PubMed.
- Domino reactions, see; (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (b) L. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef CAS PubMed; (c) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed; (d) B.-C. Hong, A. Raja and V. M. Sheth, Synthesis, 2015, 47, 3257 CrossRef CAS.
- P. Bolze, G. Dickmeiss and K. A. Jørgensen, Org. Lett., 2008, 10, 3753 CrossRef CAS PubMed , CAS number: 119480-92-1.
- Ma reported the diphenylprolinol silyl ether mediated (3 + 2) cycloaddition of α,β-enal and a highly activated three carbon component, 1,1,4-tri(ethoxycarbonyl)-3-hydroxy-1,3-butadiene, in which a β-keto ester is the nucleophilic moiety, and a highly electron deficient alkene activated by a ketone and two esters is the electrophilic moiety. A. Ma and D. Ma, Org. Lett., 2010, 12, 3634 CrossRef CAS PubMed.
- (a) D.-Q. Xu, A.-B. Xia, S.-P. Luo, J. Tang, S. Zhang, J.-R. Jiang and Z.-Y. Xu, Angew. Chem., Int. Ed., 2009, 48, 3821 CrossRef CAS PubMed; (b) L.-Y. Wu, G. Benvivenni, M. Mancinelli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7196 CrossRef CAS PubMed; (c) X. Feng, Z. Zhou, R. Zhou, Q.-Q. Zhou, L. Dong and Y.-C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (d) L. Dell'Amico, G. Rassu, V. Zambrano, A. Sartori, C. Curti, L. Battistini, G. Pelosi, G. Casiraghi and F. Zanardi, J. Am. Chem. Soc., 2014, 136, 11107 CrossRef PubMed.
- Selected examples of the organocatalyst mediated (3 + 2) cycloaddition reaction for the formation of cyclopentane skeleton, see; (a) D. Enders, C. Wang and J. W. Bats, Angew. Chem., Int. Ed., 2008, 47, 7539 CrossRef CAS PubMed; (b) B. Tan, P. J. Chua, X. Zeng, M. Lu and G. Zhong, Org. Lett., 2008, 10, 3489 CrossRef CAS PubMed; (c) B.-C. Hong, N. S. Dange, C.-S. Hsu and J.-H. Liao, Org. Lett., 2010, 12, 4812 CrossRef CAS PubMed; (d) W. Raimondi, G. Lettieri, J.-P. Dulcére, D. Bonne and J. Rodriguez, Chem. Commun., 2010, 46, 7247 RSC; (e) M. Laugeois, S. Ponra, V. Ratovelomanana-Vidal, V. Michelet and M. R. Vitale, Chem. Commun., 2016, 52, 5332 RSC; (f) K. S. Halskov, L. Næsborg, F. Tur and K. A. Jørgensen, Org. Lett., 2016, 18, 2220 CrossRef CAS PubMed; (g) J. Blom, A. Vidal-Albalat, J. Jørgensen, C. L. Barløse, K. S. Jessen, M. V. Iversen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2017, 56, 11831 CrossRef CAS PubMed.
- (a) K. Tamao, M. Akita and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS; (b) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05824a |
| This journal is © The Royal Society of Chemistry 2020 |