
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy†
Philippe
Schwaller
 *a,
Riccardo
Petraglia
a,
Valerio
Zullo
b,
Vishnu H.
Nair
a,
Rico Andreas
Haeuselmann
a,
Riccardo
Pisoni
a,
Costas
Bekas
a,
Anna
Iuliano
b and
Teodoro
Laino
a
*a,
Riccardo
Petraglia
a,
Valerio
Zullo
b,
Vishnu H.
Nair
a,
Rico Andreas
Haeuselmann
a,
Riccardo
Pisoni
a,
Costas
Bekas
a,
Anna
Iuliano
b and
Teodoro
Laino
a
aIBM Research GmbH, Zurich, Switzerland. E-mail: phs@zurich.ibm.com
bDepartment of Chemistry and Industrial Chemistry, University of Pisa, Pisa, Italy
First published on 3rd March 2020
Abstract
We present an extension of our Molecular Transformer model combined with a hyper-graph exploration strategy for automatic retrosynthesis route planning without human intervention. The single-step retrosynthetic model sets a new state of the art for predicting reactants as well as reagents, solvents and catalysts for each retrosynthetic step. We introduce four metrics (coverage, class diversity, round-trip accuracy and Jensen–Shannon divergence) to evaluate the single-step retrosynthetic models, using the forward prediction and a reaction classification model always based on the transformer architecture. The hypergraph is constructed on the fly, and the nodes are filtered and further expanded based on a Bayesian-like probability. We critically assessed the end-to-end framework with several retrosynthesis examples from literature and academic exams. Overall, the frameworks have an excellent performance with few weaknesses related to the training data. The use of the introduced metrics opens up the possibility to optimize entire retrosynthetic frameworks by focusing on the performance of the single-step model only.
1 Introduction
The field of organic chemistry has been continuously evolving, moving its attention from the synthesis of complex natural products to the understanding of molecular functions and activities.1–3 These advancements were made possible thanks to the vast chemical knowledge and intuition of human experts, acquired over several decades of practice. Among the different tasks involved, the design of efficient synthetic routes for a given target (retrosynthesis) is arguably one of the most complex problems. Key reasons include the need to identify a cascade of disconnections schemes, suitable building blocks and functional group protection strategies. Therefore, it is not surprising that computers have been employed since the 1960s,4 giving rise to several computer-aided retrosynthetic tools.Rule-based or similarity-based methods have been the most successful approach implemented in computer programs for many years. While they suggest very effective5,6 pathways to molecules of interest, these methods do not strictly learn chemistry from data but rather encode synthon generation rules. The main drawback of rule-based systems is the need for laborious manual encoding, which prevents scaling with increasing data set sizes. Moreover, the complexity in assessing the logical consistency among all existing rules and the new ones increases with the number of codified rules and may sooner or later reach a level where the problem becomes intractable.
1.1 The dawn of AI-driven chemistry
While human chemical knowledge will keep fueling the organic chemistry research in the years to come, a careful analysis of current trends5,7–20 and the application of basic extrapolation principles undeniably shows that there are growing expectations on the use of Artificial Intelligence (AI) architectures to mimic human chemical intuition and to provide research assistant services to all bench chemists worldwide.Concurrently to rule-based systems, a wide range of AI approaches have been reported for retrosynthetic analysis,9,12 prediction of reaction outcomes21–26 and optimization of reaction conditions.27 All these AI models superseded rule-based methods in their potential of mimicking the human brain by learning chemistry from large data sets without human intervention.
This extensive production of AI models for organic chemistry was made possible by the availability of public data.28,29 However, the noise contained in this data generated by the text-mining extraction process heavily holds back their potential. In fact, while rule-based systems30 demonstrated, through wet-lab experiments, the capability to design target molecules with less purification steps and hence, leading to savings in time and cost,31 the AI approaches6,9,12,16,32–38 still have a long way to go.
Among the different AI approaches39 those treating chemical reaction prediction as natural language (NL) problems40 are becoming increasingly popular. They are currently state of the art in the forward reaction prediction realm, scoring an undefeated accuracy of more than 90%.22 In the NL framework, chemical reactions are encoded as sentences using reaction SMILES41 and the forward- or retro-reaction prediction is cast as a translation problem, using different types of neural machine translation architectures. One of the most significant advantages of representing synthetic chemistry as a language is the inherent scalability for larger data sets, as it avoids important caveats such as the need for humans to assign reaction centers.6,30 The Molecular Transformer architecture42 is currently the most popular approach to treat chemistry as a language. Its trained models fuel the cloud-based IBM RXN43 for Chemistry platform.
1.2 Transformer-based retrosynthesis: current status
Inspired by the success of the Molecular Transformer22,42,43 for forward reaction prediction, a few retrosynthetic models based on the same architecture were reported shortly after.32,33,35–37 Zheng et al.32 proposed a template-free self-corrected retrosynthesis predictor built on the Transformer architecture. The model achieves 43.7% top-1 accuracy on a small standardized (50k reactions) data set.44 They were able to reduce the initial number of invalid candidate precursors from 12.1% to 0.7% using a coupled neural network-based syntax checker. Previous work reported less than 0.5% of invalid candidates in forward reaction prediction,22 without the need of any additional syntax checker. Karpov et al.33 described a Transformer model for retrosynthetic reaction predictions trained on the same data set.44 They were able to successfully predict the reactants with a top-1 accuracy of 42.7%. Lin et al.35 combined a Monte-Carlo tree search, previously introduced for retrosynthesis in the ground-breaking work by Segler et al.,12 with a single retrosynthetic step Transformer architecture for predicting multi-step reactions. In a single-step setting, the model described by Lin et al.35 achieved a top-1 prediction accuracy of over 43.1% and 54.1% when trained on the same small data set44 and a ten times larger collection, respectively. Duan et al.37 increased the batch size and the training time for their Transformer model and were able to achieve a top-1 accuracy of 54.1% on the 50k USPTO data set.44 Later on, the same architecture was reported to have a top-1 accuracy of 43.8%,36 in line with the three previous transformer-based approaches32,33,35 but significantly lower than the accuracy previously reported by Duan et al.37 Interestingly, the transformer model was also trained on a proprietary data set,36 including only reactions with two reactants with a Tanimoto similarity distribution peaked at 0.75, characteristic of an excessive degree of similarity (roughly two times higher than the USPTO). Despite the high reported top-1 accuracy using the proprietary training and testing set, it is questionable how a model that overfits a particular ensemble of identical chemical transformations could be used in practice. Recently, a graph enhanced transformer model45 and a mixture model46 were proposed, achieving a top-1 accuracy of 44.9% and more diverse reactant suggestions, respectively, with no substantial improvements over previous works.Except for the work of Lin et al.,35 all transformer-based retrosynthetic approaches were limited to a single step only. None of the previously reported works attempts the concurrent predictions of reagents, catalysts and solvent conditions but only reactants.
In this work, we present an extension of our Molecular Transformer architecture combined with a hyper-graph exploration strategy to design retrosynthetic pathways without human intervention. Compared to all other existing works using AI, we predict reactants as well as reagents for each retrosynthetic step, which significantly increases the difficulty of prediction.47 Throughout the article, we will refer to reactants and reagents (e.g. solvents and catalysts) as precursors (see Fig. 1). We criticize the use of the confidence level intrinsic to the retrosynthetic model (top-N accuracy) and introduce new metrics (coverage, class diversity, round-trip accuracy and Jensen–Shannon divergence) to evaluate the single-step retrosynthetic model, using the corresponding forward prediction and a reaction classification model. This provides a general assessment of each retrosynthetic step capturing the essential aspects a model should have to perform similarly to human experts in retrosynthetic analysis.
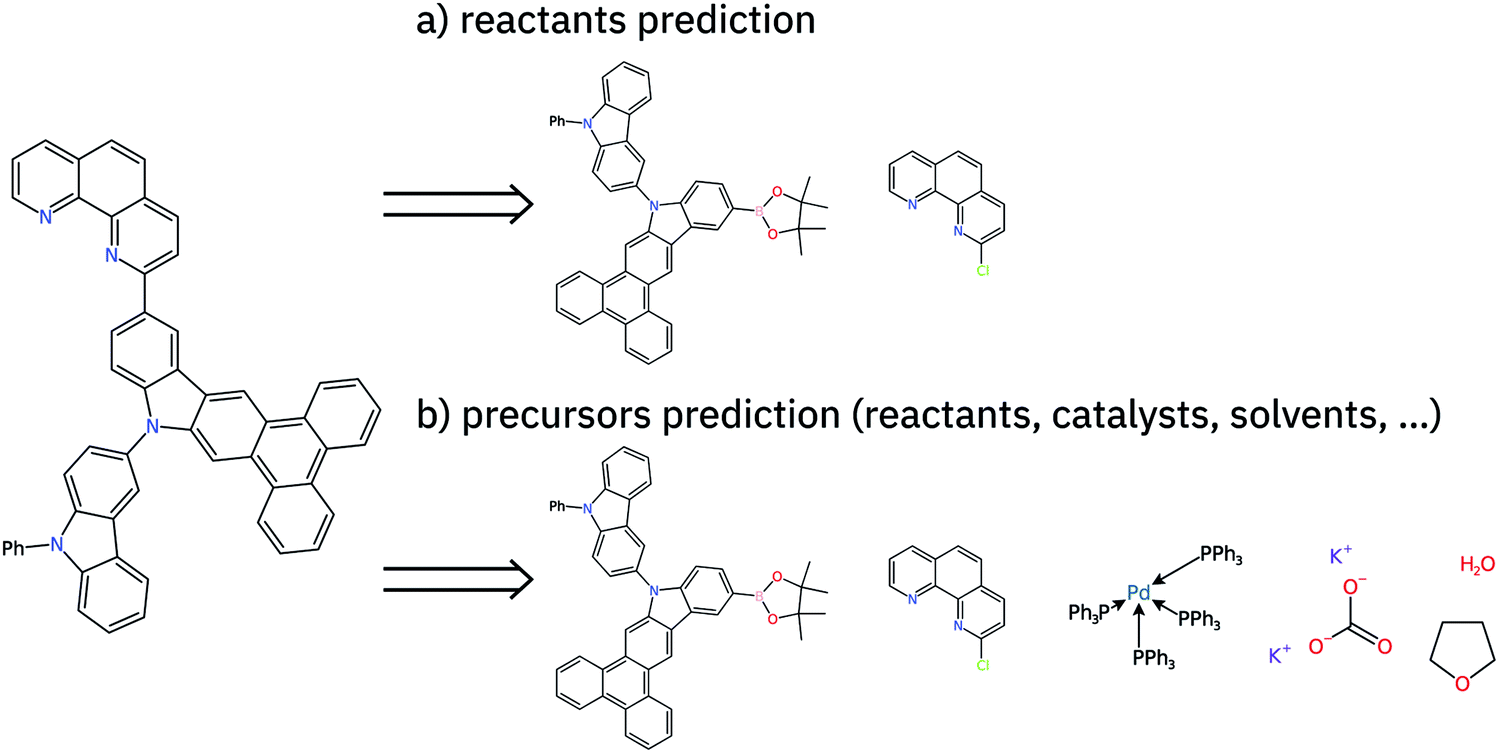 | ||
| Fig. 1 Retrosynthesis step suggestion for 13-(1,10-phenanthrolin-2-yl)-10-(9-phenyl-9H-carbazol-3-yl)-10H-phenanthro[9,10-b]carbazole using a chloro-Suzuki coupling reaction. In (a) only the reactants are predicted. In (b) all the precursors are predicted, which increases the overall difficulty of the single-step prediction task. While for (a) two molecules consisting of a total of 68 atoms are predicted, the target of (b) are six molecules consisting of 157 atoms. | ||
The optimal synthetic pathway is found through a beam search on the hyper-graph of the possible disconnection strategies. The hyper-graph is constructed on the fly, and the nodes are filtered and subject to further expansion based on a Bayesian-like probability that makes use of the forward prediction likelihood and the SCScore48 to prioritize synthetic steps. This strategy allows circumventing potential selectivity traps, penalizing non-selective reactions and precursors with higher complexity than the targets and leads to termination when commercially available building blocks are identified. We relate the quality of the retrosynthetic tree to the likelihood distributions of the forward prediction model and suggest the use of the Jensen–Shannon divergence to characterize the similarity of the distributions. This holistic analysis provides first the time a way to improve the quality of multi-step retrosynthetic tools systematically.
Finally, we critically assessed the entire AI framework by reviewing several retrosynthetic problems, some of them from literature data and others from academic exams. We show that reaching high performance on a subset of metrics for single-step retrosynthetic prediction is not beneficial in a multi-step framework. We also demonstrate that the use of all newly defined metrics provides an evaluation of end-to-end solutions, thereby focusing only on the quality of the single-step prediction model. The trained models and the entire architecture is freely available online.43 The potential of the presented technology is high, augmenting the skills of less experienced chemists but also enabling chemists to design and protect the intellectual property of non-obvious synthetic routes for given targets.
2 Methods
2.1 Evaluation metrics for single-step retrosynthetic models
The evaluation of retrosynthetic routes is a task for human experts. Unfortunately, every evaluation is tedious and difficult to scale to a large number of examples. Therefore, it is challenging to generate statistically relevant results for more than a few different model settings. By using an analogy with human experts, we propose to use a forward prediction model12,49 and a reaction classification model to assess the quality50 of the retrosynthetic predictions. The forward prediction model estimates the likelihood of the forward reaction of a single-step retrosynthesis and the classification model provides its corresponding class. Model scores have already been used as an alternative to human annotators to evaluate generative adversarial networks.51 In our context, we define a retrosynthetic prediction as valid if the suggested set of precursors leads to the original product when processed by the forward chemical reaction prediction model (see Fig. 2). More detail about the forward prediction and the reaction classification model can be found in the ESI.† Here we introduce four new metrics (round-trip accuracy, coverage, class diversity and the Jensen–Shannon divergence) to thoroughly evaluate retrosynthetic models.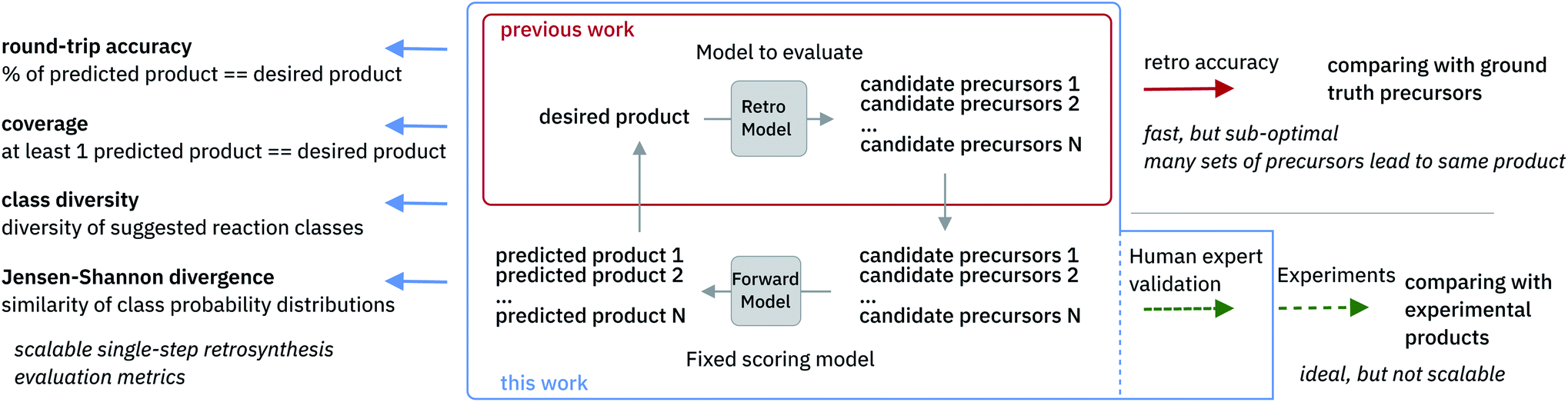 | ||
| Fig. 2 Overview of single-step retrosynthesis evaluation metrics. | ||
The round-trip accuracy quantifies what percentage of the retrosynthetic suggestions is valid. This metric is an crucial evaluation as it is desirable to have as many valid suggestions as possible. This metric is highly dependent on the number of beams, as generating more outcomes through the use of a beam search might lead to a smaller percentage of valid suggestions due to lower quality suggestions in case of a higher number of beams.
The coverage quantifies the number of target molecules for which the retrosynthetic model produces at least one valid precursors suggestion. With this metric, one wants to prevent rewarding models that produce many valid precursors for only a few reactions. Such a behavior could result in a relatively high round trip accuracy but would result in a small coverage. A retrosynthetic model should be able to produce valid suggestions for a wide variety of target molecules.
The class diversity is complementary to the coverage, as instead of relating to targets it counts the number of diverse reaction superclasses predicted by the retrosynthetic model, upon classification. A single-step retrosynthetic model should predict a wide diversity of disconnection strategies, which means generating precursors leading to the same product, with the corresponding reactions belonging to different reaction classes. Allowing a multitude of different disconnection strategies is beneficial for an optimal route search and essential, precisely when the target molecule contains multiple functional groups.
Finally, the Jensen–Shannon divergence, which is used to measure the similarity between the likelihood distributions of the suggested reactions belonging to the 12 different reaction superclasses i above a threshold of 0.5, is calculated as follows:
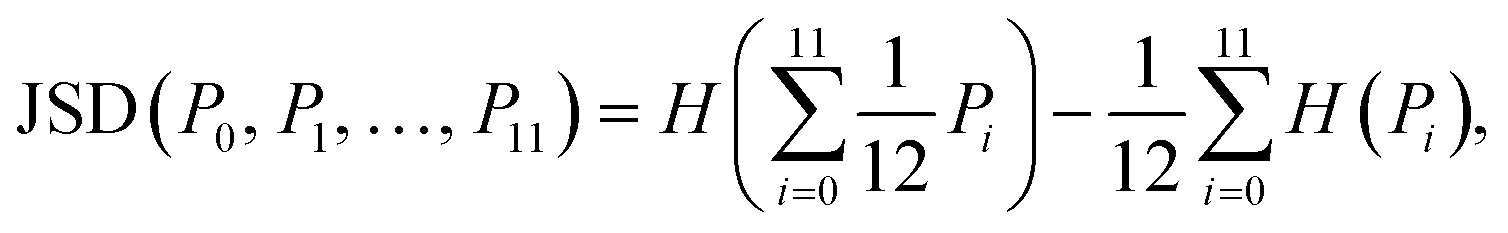 | (1) |
To compute the Jensen–Shannon divergence, we split the single-step retrosynthetic reactions into superclasses and use the likelihoods predicted by the forward model to build a likelihood density function for each class, which are then normalized by the entropy function in the Jensen–Shannon divergence equation. This metric is crucial to assess the quality of a sequence of retrosynthetic steps. Having a model with a dissimilar likelihood distribution would be equivalent to having a human expert favor a few specific reaction classes over others. This would result in an introduction of bias favoring those classes with dominant likelihood distributions. While it is desirable to have a peaked distribution, as this is an evident sign of the model learning from the data, it is also desirable to have all the likelihood distributions equally peaked, with none of them exercising more influence than the others during the construction of a large number of retrosynthetic trees. The inverse of the Jensen–Shannon divergence (1/JSD) is a measure of the similarity of the likelihood distributions among the different superclasses and we use this parameter as an effective metric to guarantee uniform likelihood distributions among all possible predicted reaction classes. Uneven distributions are directly connected to the nature of the training data set. All these four metrics have been critically designed and assessed with the help of human domain experts. Their combined use paves the way for a systematic improvement of entire retrosynthetic frameworks, by adequately tuning data sets that optimize the different single-step performance indicators in a multi-objective fashion. An example of the metrics is made available online.52
Additionally, we use the open-source chemoinformatics software RDKit53 to evaluate the percentage of syntactically valid predicted molecules (grammatically correct SMILES).
2.2 Hyper-graph exploration
A retrosynthetic tree is equivalent to a directed acyclic hyper-graph, a mathematical object composed of hyper-arcs (A) that link nodes (N). The main difference compared to a typical graph is that a hyper-arc can link multiple nodes, similar to what happens in a retrosynthesis: if a node represents a target molecule, the hyper-arcs connecting to different nodes represent all possible reactions involving those corresponding molecules. Hyper-arcs have an intrinsic direction defining whether the reaction is forward or retro (see Fig. 3).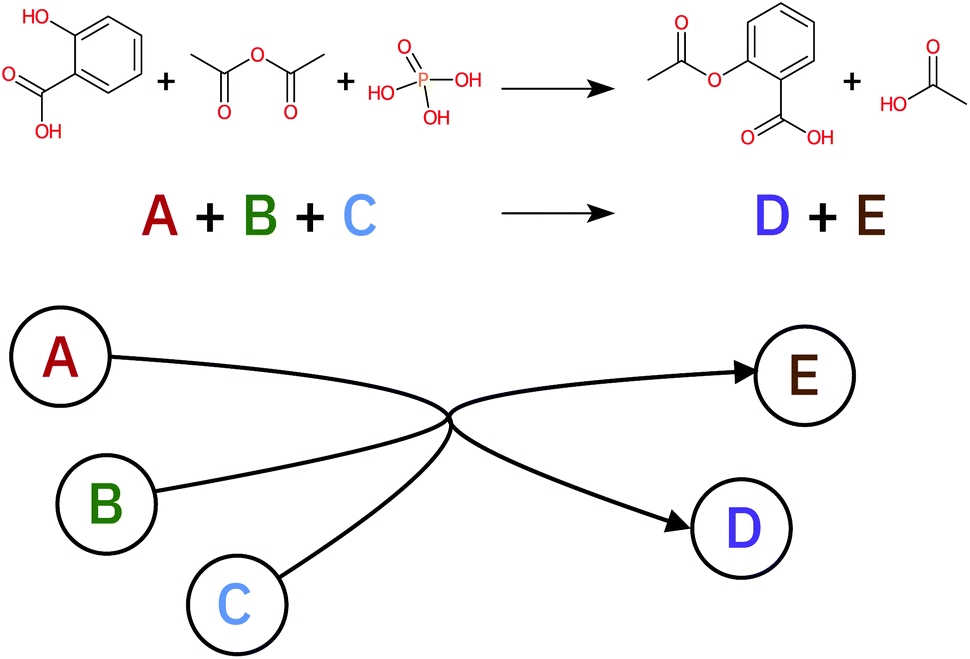 | ||
| Fig. 3 A generic reaction (top of the picture) can be represented as a hyper-graph. Each molecule involved in the reaction becomes a node in the hyper-graph while the hyper-arc, connecting the reactants and reagents to the product, represents the reaction arrow. | ||
A retrosynthetic route needs to be free of any loops, i.e. acyclic. This requirement renders the retrosynthetic route a hyper-tree,54 in which the root is the target molecule and the leaves are the commercially available starting materials (see Fig. 4). We use the database provided by eMolecules55 to determine if a molecule is available or not.
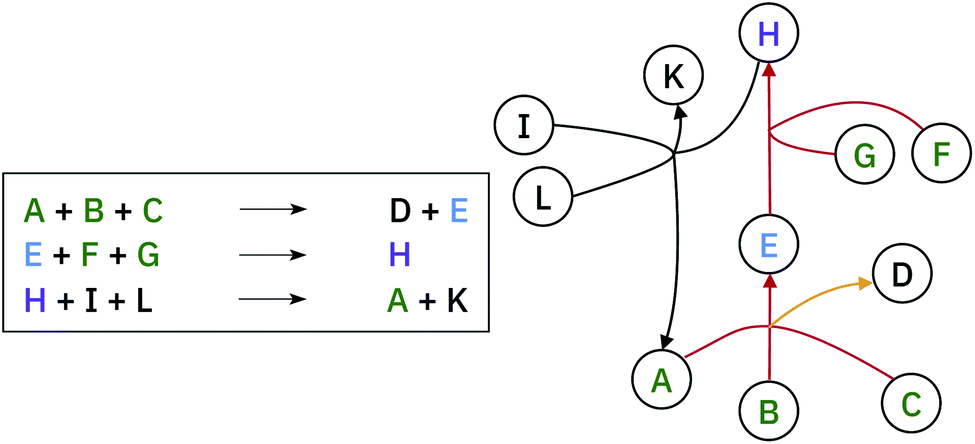 | ||
| Fig. 4 Example of hyper-graph complexity. The molecule H is the target (purple label). The red lines represent the synthetic path from commercially available precursors (highlighted in green) to the target molecule. The yellow line, does not affect the retrosynthesis of H, neither does the last reaction with black lines. | ||
In cases where the hyper-graph of the entire chemical space is available, an exhaustive search may reveal all the possible synthetic pathways leading to a target molecule from defined starting materials. Instead, here we build the hyper-tree on the fly: only the nodes and arcs expanding in the direction of the most meaningful retrosynthesis are calculated and added to the existing tree. The retrosynthesis exploration uses a SCScore48-based Bayesian-like probability to decide the direction along which the graph is expanded, driving the tree towards more simple precursors. In Fig. 5, we show a schematic representation of the multi-step retrosynthetic workflow. Given a target molecule, we use a single-step retrosynthetic model to generate a certain number of possible disconnections (i.e. precursors set). We canonicalize the predicted reaction smiles and determine their reaction class. We compute the SCScore as well as the reaction likelihood with the forward prediction model on the corresponding inchified entry. In order to discourage the use of non-selective reactions, we filter the single-step retrosynthetic predictions by using a threshold on the reaction likelihood returned by the forward model. The likelihood and SCScore of the filtered predictions are combined to compute a probability score to rank all the options. In case all the predicted precursors are commercially available the retrosynthetic analysis provides that option as a possible solution and the exploration of that tree branch is considered complete. If not, we repeat the entire cycle using the precursors as initial target molecules until we reach either commercially available molecules or the maximum number of specified retrosynthesis steps. The single-step forward and retrosynthetic predictive models, as well as the multi-step framework, do not contain explicitly encoded chemical knowledge: the only chemical knowledge embedded is the one learned from the data during the training processes. The algorithmic details and the path scoring function are detailed in the ESI.†
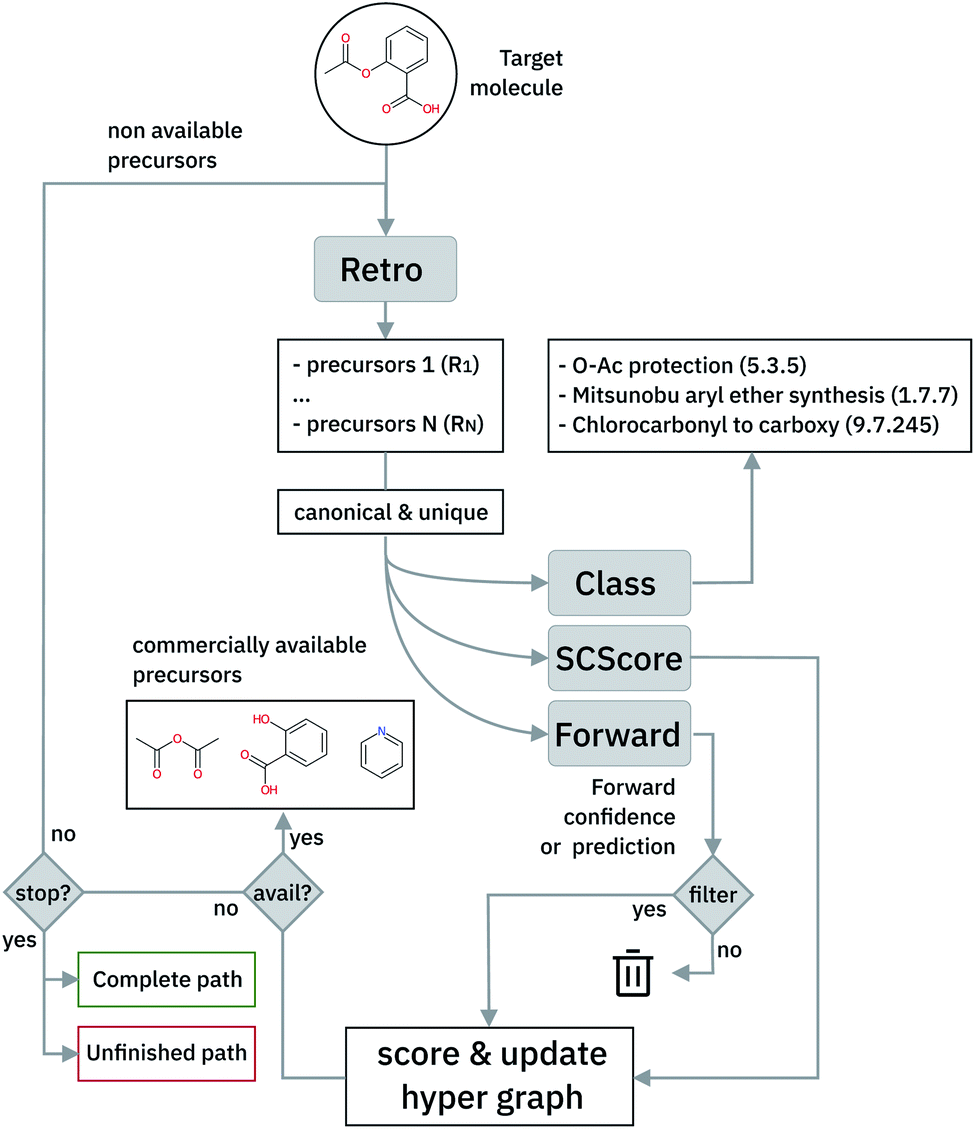 | ||
| Fig. 5 Schematic of the multi-step retrosynthetic workflow. | ||
3 Results and discussion
3.1 Single-step retrosynthesis
The top-N accuracy score is the preferred method to evaluate the quality of single-step predictive models. While this is entirely justified for the evaluation of forward reaction prediction, its usage in the context of single-step retrosynthetic models is misleading, as recently suggested also by Thakkar et al.38 Top-N accuracy means that the ground truth precursors were found within the first N suggestions of the retrosynthetic model. In contrast to forward prediction models, a target molecule rarely originates from one set of precursors only. Often the presence of different functional groups allows a multitude of possible disconnection strategies to exist, leading to different sets of reactants, as well as possible solvents and catalysts.The analysis of the USPTO stereo data set, derived from the text-mined open-source reaction data set by Lowe,28,29 and of the Pistachio data set,56 shows that 6% of the products, and 14% respectively, have at least two different sets of precursors. While these numbers only reflect the organic chemistry represented in each data set, the total number of possible disconnections is undoubtedly larger. Considering the limited size of existing data sets, it is evident that, in the context of retrosynthesis, the top-N accuracy rewards the ability of a model to retrieve expected answers from a data set more than that to predict chemically meaningful precursors. Therefore, a top-N comparison with the ground truth is not an adequate metric for assessing retrosynthetic models.
Here, we dispute the previous use of top-N accuracy in single-step retrosynthetic models6,9,12,16,32–37 and propose four new different metrics (round-trip accuracy, coverage, class diversity and Jensen–Shannon divergence,57 see Section 2.1) for their evaluation.
During the development phase, we trained different retrosynthetic transformer-based models with two different data sets, one fully based on open-source data (stereo) and one on based commercially available data from Pistachio (pistachio). In some cases, the data set was inchified58 (labelled with _i). Table 1 shows the results for the retrosynthetic models, evaluated using a fixed forward prediction model (pistachio_i) on two validation sets (stereo and pistachio). The coverage represents the percentage of desired products for which at least one valid precursor set was suggested. It was slightly better for stereo but above 90% for all the model combinations, which is an important requirement to guarantee the possibility to always offer at least one disconnection strategy. Likewise, the class diversity, which is an average of how many different reaction classes are predicted in a single retrosynthetic step, was comparable for both models with slightly better performance for the pistachio model. The round-trip accuracy, which is the percentage of precursor sets leading to the initial target when evaluated with the forward model, was better for stereo than for pistachio. Despite the stereo retrosynthetic model performed better than the pistachio model in terms of round-trip accuracy and coverage, the synthesis routes generated with this model were of lower quality and often characterized by a sequence of illogical protection/deprotection steps as determined by the human expert assessment (last column in Table 1). This apparent paradox became clear when we analyzed in detail how humans approach the problem of retrosynthesis.
| Model Retro | Forw. | Test data | RT [%] | Cov. [%] | CD |
|
ismi [%] | hu. ev. |
|---|---|---|---|---|---|---|---|---|
| ste_i | pist_i | ste | 81.2 | 95.1 | 1.8 | 16.5 | 0.5 | − |
| ste_i | pist_i | pist | 79.1 | 93.8 | 1.8 | 20.6 | 1.1 | − |
| pist_i | pist_i | pist | 74.9 | 95.3 | 2.1 | 22.0 | 0.5 | + |
| pist | pist_i | pist | 71.1 | 92.6 | 2.1 | 27.2 | 0.6 | ++ |
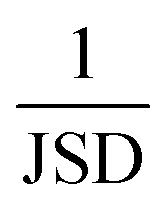
Solving retrosynthetic problems requires a careful analysis of which ones among multiple precursors could lead to the desired product more efficiently, as seen in Fig. 6 for 5-bromo-2-methoxypyridine. Humans address this issue by mentally listing and analyzing all possible disconnection sites and retaining only the options, for which the corresponding precursors are thought to produce the target molecule most selectively.
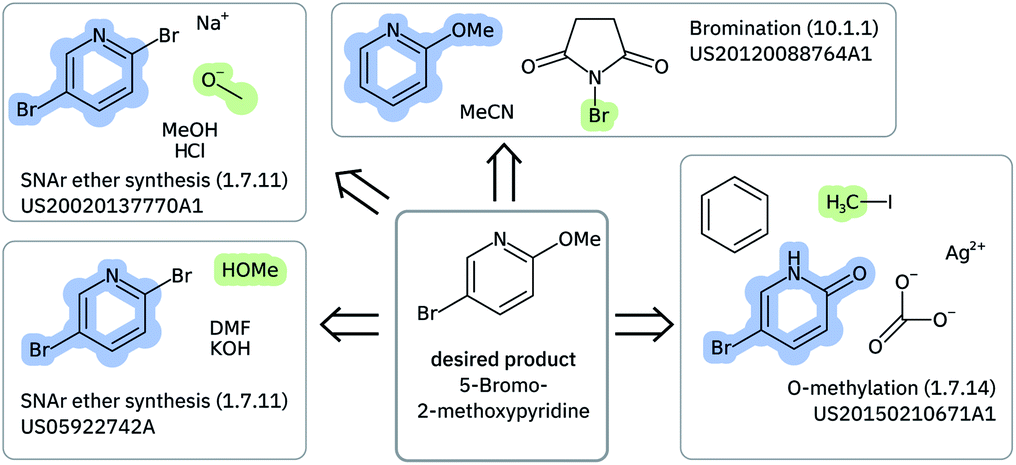 | ||
| Fig. 6 Highlighting a few of the precursors and reactions leading to 5-bromo-2-methoxypyridine that are found in the US Patents data set. The molecules were depicted with CDK.59 | ||
For an expert, it is not sufficient to always find at least one disconnection site (coverage) and be sure that the corresponding precursors will selectively lead to the original target (round-trip accuracy). It is necessary to generate a diverse sample of disconnection strategies to cope with competitive functional group reactivity (class diversity). Moreover, most important, every disconnection class needs to have a similar probability distribution to all the other classes (Jensen–Shannon divergence, JSD). Continuing the parallelism with human experts, if one was exposed to the same reaction classes for many years, the use of those familiar schemes in the route planning would appear more frequently, leading to strongly biased retrosynthesis. Therefore, it is essential to reduce any bias in single-step retrosynthetic models to a minimum.
To evaluate the bias of single-step models, we use the JSD of the likelihood distributions for the prediction divided in different reaction superclasses, which we report in Table 1 as 1/JSD. The larger this number, the more similar the likelihood distributions of the reactions belonging to different classes are and hence, the less dominant (lower bias) individual reaction classes are in the multi-step synthesis. In Fig. 7, we show the likelihood distributions for the different models in Table 1. Except for the resolution class, all of the distributions show a peak close to 1.0, which clearly shows that the model learned how to predict the reaction in those classes. The resolution class is instead relatively flat as a consequence of the poor data quality/quantity for stereochemical reactions both in the stereo and pistachio data set. Interestingly, one can see that for the stereo model the likelihood distributions of the deprotection, reduction and oxidation reactions are different (and generally more peaked) from all other distributions generated with the same model. This statistical imbalance favors those reaction classes and explains the occurrence of illogical loops of protection/deprotection or oxidation/reduction strategies. While peaked distributions are desirable, as this is a consequence of the model learning to predict disconnection strategies in a precise class, the dissimilarity (JSD) between the twelve probability distributions reflects an intrinsic bias, likely due to unbalanced data sets. Among the few models reported, the pistachio model was found to have the best similarity (1/JSD) score and is the one analyzed in the subsequent part of the manuscript and made available online.
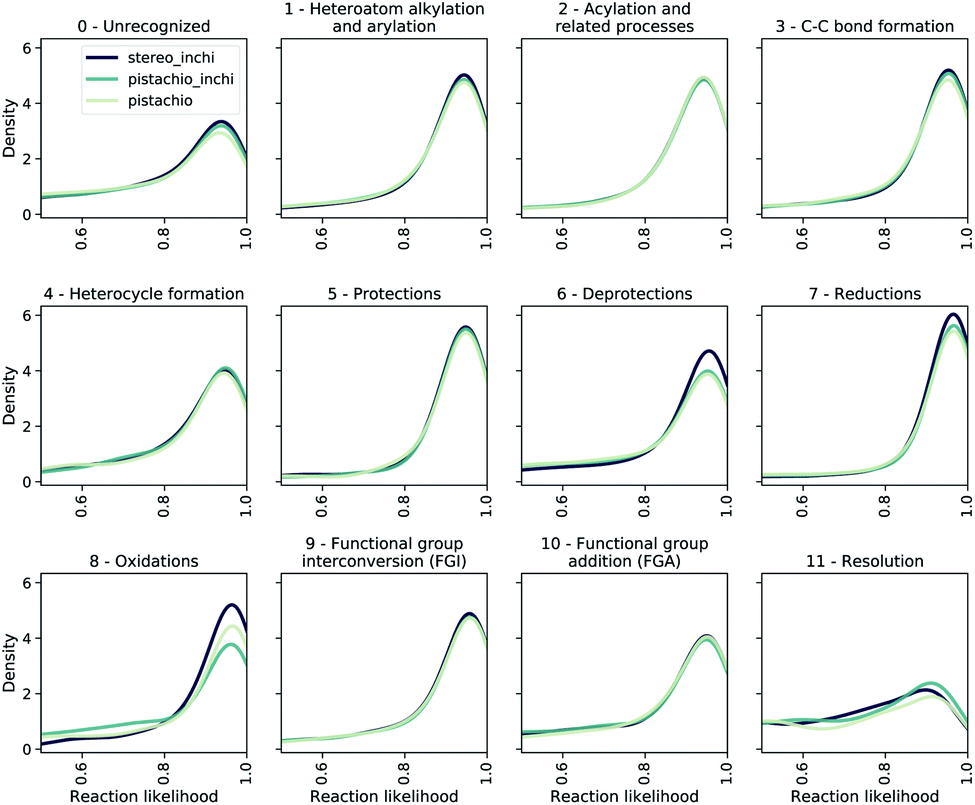 | ||
| Fig. 7 The likelihood distributions predicted by a forward model (pistachio_i) for the reactions suggested by different retro models. We show the likelihood range between 0.5 and 1.0. | ||
The class diversity and similarity scores require the identification of the reaction class for each prediction. We used a transformer-based reaction classification model, as described in.50 In Fig. 8, we report the ground truth classified by the NameRXN60 tool, the class distribution predicted by our classification model on the ground truth reactions and finally, the class distributions predicted for the reactions suggested by the retrosynthesis models (see Table 1). We observe that the classifications made by our class prediction model are in agreement with the ones of NameRXN60 and match them with an accuracy of 93.8%. The distributions of the single-step retrosynthetic models resemble the original one with the number of unrecognized reactions nearly halved. All of the models learned to predict more recognizable reactions, even for products, for which there was an unrecognized reaction in the ground truth.
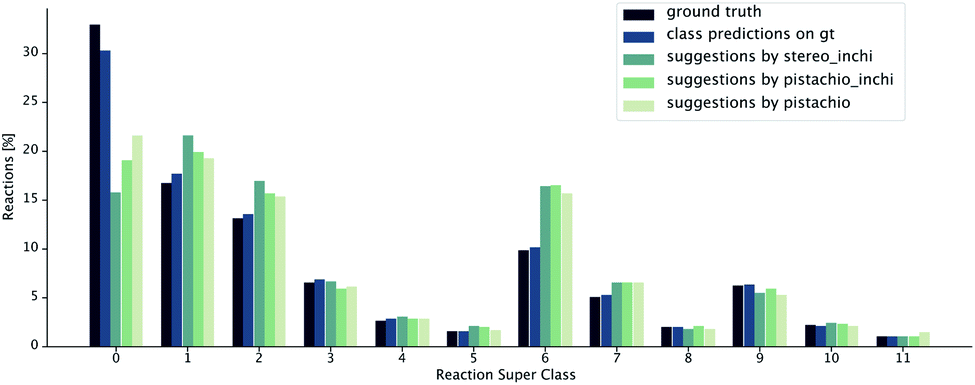 | ||
| Fig. 8 Distribution of reaction superclasses for the ground truth,60 the predicted superclasses for the ground truth reactions and the predicted superclasses for the reactions suggested by the different retrosynthesis models. | ||
3.2 A holistic evaluation of the pathway prediction
An evaluation of the model was carried out through performing the retrosynthesis of the compounds reported in Fig. 9. Some of these are known compounds, for which the synthesis is reported in the literature (1, 2, 5, 7, 8), others (3, 4, 6, 9) are unknown structures, which are similar to structures reported in the organic synthesis textbooks. The latter are challenging targets for students and useful to test the capability of the model to solve entirely new synthetic problems. For the first group, the evaluation of the model could be made by comparing the proposed retrosynthetic analysis with the known synthetic pathway. For the second group, a critical evaluation of the proposed retrosynthesis, which takes into account the level of chemo-, regio-, and stereoselectivity for every retrosynthetic step was performed. The parameters used for each retrosynthesis are reported in the ESI.† In some cases, the default values were changed to increase the hyper-graph exploration and yield better results. As an output, the model generates several retrosynthetic sequences for each compound, each one with a different confidence level. Because the model predicts not only reactants but also reagents, solvents and catalysts, there are several sequences with similar confidence level and identical disconnection strategies and differing only by the suggested reaction solvents in a few steps. Therefore, we report only one of the similar sequences in the ESI.†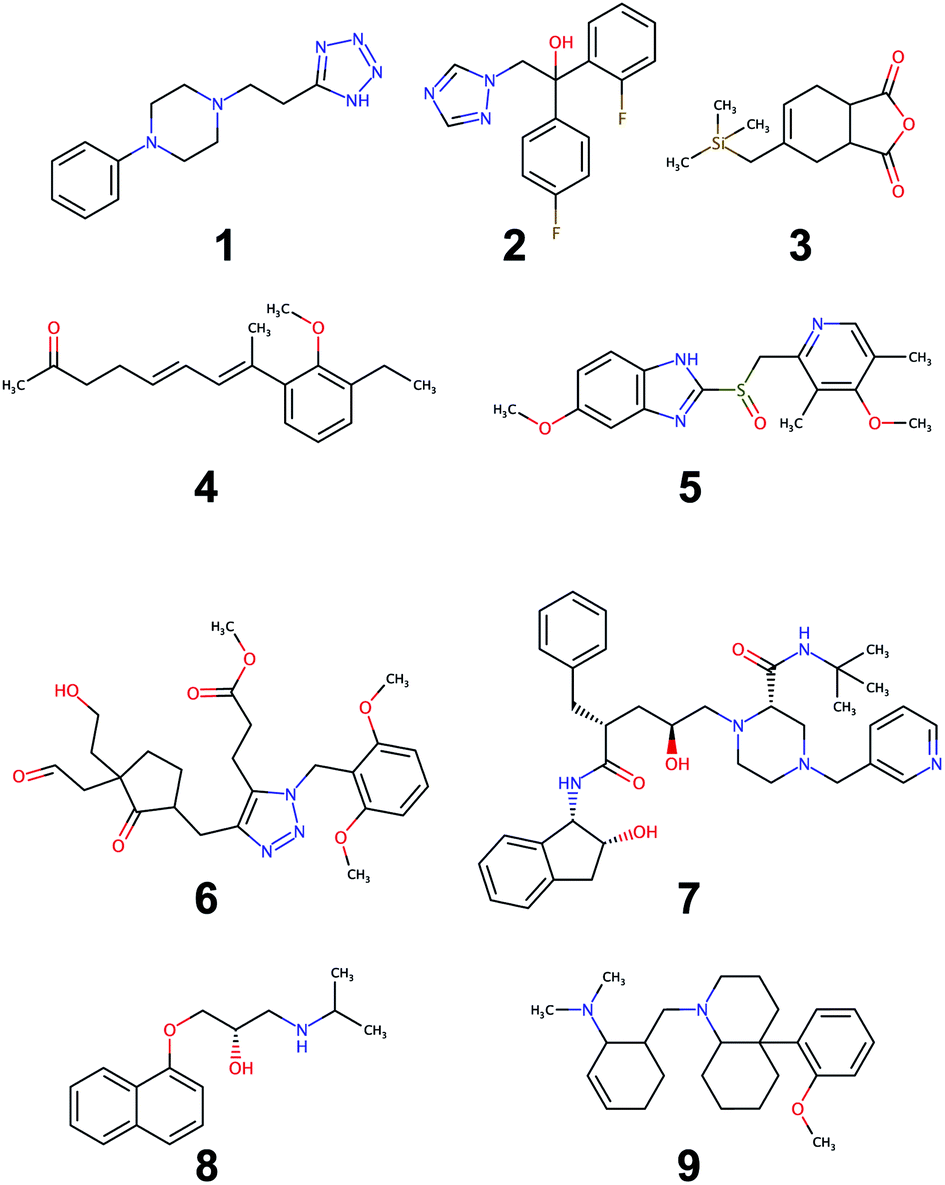 | ||
| Fig. 9 Set of molecules used to assess the quality of retrosynthesis. | ||
All of the retrosynthetic routes generated for compounds 1, 2 and 3 fulfill the criteria of chemoselectivity. The highest confidence sequence (called “sequence 0”) of 1 corresponds to the reported synthesis of the product61 and starts from the commercially available acrylonitrile. The other two sequences (17 and 22) use synthetic equivalents of acrylonitrile and also show its preparation. For compound 2, the highest confidence retrosynthetic sequence (sequence 0) does not correspond to the synthetic pathway reported in the literature, where the key step is the opening of an epoxide ring. Two other sequences (5 and 23) report this step, and one of them (sequence 5) corresponds to the literature synthesis.62 The retrosynthetic sequence for compound 3 provides a Diels–Alder reaction as the first disconnection strategy and proposes a correct retrosynthetic path for the synthesis of the diene from available precursors. A straightforward retrosynthetic sequence was also found in the case of compound 4, where the diene moiety was disconnected by two olefination reactions and the sequence uses structurally simple compounds as starting material. It may be debatable whether the two olefinations through a Horner–Wadsworth–Emmons reaction, can really be stereoselective towards the E-configurated alkenes or whether the reduction of the conjugate aldehyde by NaBH4 can be completely chemoselective towards the formation of the allylic alcohol. Only experimental work can solve this puzzle and give the correct answer.
The retrosynthesis of racemic omeprazole 5 returned a sequence consisting of one step only because the model finds in its library of available compounds the sulfide precursor of the final sulfoxide. When repeating the retrosynthesis using benzene as starting molecule in conjunction with a restricted set of available compounds, we obtained a more complete retrosynthetic sequence with some steps in common with the reported one.63 However, although all of the steps fulfill the chemoselectivity requirement, the sequence is characterized by some avoidable protection–deprotection steps. This sequence nicely reflects the bias present in the likelihood distributions of the different superclasses for the chosen model. Although the single-step retrosynthetic model has the best Jensen–Shannon divergence among all of the trained models, there is still room for improvements that we will explore in the future. A higher similarity across the likelihood distributions will prevent the occurrence of illogical protection–deprotection, esterification/saponification steps.
Besides, the reported sequence for 5 lists a compound not present in the restricted set of available molecules as starting material. A “de novo” retrosynthesis of this compound solved the problem. The retrosynthetic sequence of the structurally complex compound 6 was possible only with wider settings allowing a more extensive hyper-graph exploration. The result was a retrosynthetic route starting from simple precursors: notably, the sequence also showed the synthesis of the triazole ring through a Huisgen cycloaddition. However, we recognized the occurrence of some chemoselectivity problems in step 6, when the enolate of the ketone is generated in the presence of an acetate group, used as protection of the alcohol. This problem could be avoided by using a different protecting group for the alcohol. By contrast, the alkylation of the ketone enolate by means of a benzyl bromide bearing an enolizable ester group in the structure appears less problematic, due to the high reactivity of the bromide. The retrosynthesis of the chiral stereodefined compound indinavir, 7, completed in one step, through finding a very complex precursor in the set of available molecules. Sequences of lower confidence resulted in more retrosynthetic steps, disconnecting the molecule as in the reported synthesis64 but stopped at the stereodefined epoxide, with no further disconnection paths available. However, when the retrosynthesis was performed on the same racemic molecule, a chemoselective retrosynthetic pathway was found, disconnecting the epoxide and starting from simple precursors. Similarly, for the other optically active compound, propranolol, 8, which was disconnected according to the published synthetic pathway65 only when the retrosynthesis was performed on the racemic compound. The problem experienced with stereodefined molecules reflects the poor likelihood distribution of the resolution superclass in Fig. 7. Because all current USPTO derived data sets (stereo and pistachio) have particularly noisy stereochemical data we decided to retain only few entries in order to avoid jeopardizing the overall quality. With a limited number of stereochemical examples available in the training set, the model was not able to learn reactions belonging to the resolution class, failing to provide disconnection options for stereodefined centers.
The retrosynthesis of the last molecule, 9, succeeded only with intensive hypergraph exploration settings. However, the retrosynthetic sequence is tediously long, with several avoidable esterification–saponification steps. Similar to 5, the bias in the likelihood distributions is the one reason for this peculiar behavior. In addition, a non-symmetric allyl bromide was chosen as precursor of the corresponding tertiary amine: this choice entails a regioselectivity problem, given that the allyl bromide can undergo nucleophilic displacement not only at the ipso position, giving rise to the correct product, but also at the allylic position, resulting in the formation of the regioisomeric amine. Lastly, the model was unable to find a retrosynthetic path for one complex building block, which was not found in the available molecule set. However, a slight modification of the structure of this intermediate enabled a correct retrosynthetic path to be found, which could also be easily applied to the original problem, starting from 1,3-cyclohexanedione instead of cyclohexanone. Although the targets in examples 1, 5 and 8 are found in the training set of the single-step retrosynthesis model, the multistep synthesis prediction system selected alternative reactions, which are not present in the training data. For the nine compounds, the predicted retrosynthetic paths show for every step the reactants and almost always also reaction solvents. All the proposed reagents match the ones usually reported for similar transformations. We also made a comparison of our retrosynthetic architecture with previous work,6,12 using the same compounds for the assessments (see ESI†). The model performed well on the majority of these compounds, showing problems in the case of stereodefined compounds as in the previous examples. Retrosynthetic paths were easily obtained only for their racemic structure. The proposed retrosyntheses in some cases are similar to those reported6 while, for some compounds12 they are different but still chemoselective. Only in a few cases, the model failed to find a retrosynthesis.
4 Conclusion
In this work, we presented an extension of our Molecular Transformer architecture combined with a hyper-graph exploration strategy to design retrosynthesis without human intervention. We introduce a single-step retrosynthetic model predicting reactants as well as reagents for the first time. We also introduce four new metrics (coverage, class diversity, round-trip accuracy and Jensen–Shannon divergence) to provide a thorough evaluation of the single-step retrosynthetic model. The optimal synthetic pathway is found through a beam search on the hyper-graph of the possible disconnection strategies and allows to circumvent potential selectivity traps. The hypergraph is constructed on the fly, and the nodes are filtered, and further expanded based on a Bayesian-like probability score until commercially available building blocks are identified. We assessed the entire framework by reviewing several retrosynthetic problems to highlight strengths and weaknesses. As confirmed by the statistical analysis, the entire framework performs very well for a broad class of disconnections. An intrinsic bias towards a few classes (reduction/oxidation/esterification/saponification) may lead, in some cases, to illogical disconnection strategies that are a peculiar fingerprint of the current learning process. Also, an insufficient ability to handle stereochemical reactions is the result of the poor quality training data set that covers only a few examples in the resolution class. The use of the four new metrics, combined with the critical analysis of the current model, provides a well defined strategy to optimize the retrosynthetic framework by focusing exclusively on the performance of the single-step retrosynthetic model without the need to manually review the quality of entire retrosynthetic routes. A key role in this strategy will be the construction of statistically relevant training data sets to improve the confidence of the model in different types of reaction classes and disconnections.Conflicts of interest
There are no conflicts to declare.References
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- Y. Ai, N. Ye, Q. Wang, K. Yahata and Y. Kishi, Angew. Chem., 2017, 129, 10931–10935 CrossRef.
- X. Liu, X. Li, Y. Chen, Y. Hu and Y. Kishi, J. Am. Chem. Soc., 2012, 134, 6136–6139 CrossRef CAS PubMed.
- E. J. Corey, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465 CrossRef.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed.
- J. S. Schreck, C. W. Coley and K. J. M. Bishop, ACS Cent. Sci., 2019, 5, 970–981 CrossRef CAS PubMed.
- I. A. Watson, J. Wang and C. A. Nicolaou, J. Cheminf., 2019, 11, 1 Search PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed.
- R. Fagerberg, C. Flamm, R. Kianian, D. Merkle and P. F. Stadler, J. Cheminf., 2018, 10, 19 Search PubMed.
- D. Lowe, Nature, 2018, 555, 592–593 CrossRef CAS PubMed.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- F. Feng, L. Lai and J. Pei, Front. Chem., 2018, 6, 199 CrossRef PubMed.
- J. Savage, A. Kishimoto, B. Buesser, E. Diaz-Aviles and C. Alzate, Chemical Reactant Recommendation Using a Network of Organic Chemistry, ACM, New York, USA, 2017 Search PubMed.
- M. H. S. Segler and M. P. Waller, Chemistry, 2017, 23, 5966–5971 CrossRef CAS PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- A. Masoumi, M. Soutchanski and A. Marrella, th International Workshop on Semantic Web Applications and Tools for Life Sciences SWATLS, 2013 Search PubMed.
- J. Law, Z. Zsoldos, A. Simon, D. Reid, Y. Liu, S. Y. Khew, A. P. Johnson, S. Major, R. A. Wade and H. Y. Ando, J. Chem. Inf. Model., 2009, 49, 593–602 CrossRef CAS PubMed.
- M. H. Todd, Chem. Soc. Rev., 2005, 34(3), 247–266 RSC.
- C. W. Coley, D. A. Thomas, J. A. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers and H. Gao, et al. , Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- P. Schwaller, T. Gaudin, D. LÃąnyi, C. Bekas and T. Laino, Chem. Sci., 2018, 9, 6091–6098 RSC.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS PubMed.
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed.
- M. H. S. Segler and M. P. Waller, Chemistry, 2017, 23, 6118–6128 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS PubMed.
- C. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- D. M. Lowe, Ph.D. thesis, University of Cambridge, 2012.
- D. Lowe, Chemical reactions from US patents (1976-Sep2016), 2017, http://https://figshare.com/articles/Chemical_reactions_from_US_patents.
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, Nat. Chem., 2009, 1, 31–36 CrossRef CAS PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. SzymkuÄĞ, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. SieredziÅĎska, M. Mrksich, S. L. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- S. Zheng, J. Rao, Z. Zhang, J. Xu and Y. Yang, arXiv preprint arXiv:1907.01356, 2019.
- P. Karpov, G. Godin and I. V. Tetko, International Conference on Artificial Neural Networks, 2019, pp. 817–830 Search PubMed.
- X. Liu, P. Li and S. Song, bioRxiv, 2019, 677849 Search PubMed.
- K. Lin, Y. Xu, J. Pei and L. Lai, arXiv preprint arXiv:1906.02308, 2019.
- A. A. Lee, Q. Yang, V. Sresht, P. Bolgar, X. Hou, J. L. Klug-McLeod and C. R. Butler, Chem. Commun., 2019, 55, 12152–12155 RSC.
- H. Duan, L. Wang, C. Zhang and J. Li, arXiv preprint arXiv:1908.00727, 2019.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. Bjerrum, Chem. Sci., 2020, 11, 154–168 RSC.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 1, 1–16 Search PubMed.
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Angew. Chem., Int. Ed., 2014, 53, 8108–8112 CrossRef CAS PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- Molecular Transformer, https://github.com/pschwllr/MolecularTransformer, accessed Jul 29, 2019 Search PubMed.
- IBM RXN for Chemistry, https://rxn.res.ibm.com, accessed Oct 10, 2019 Search PubMed.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- Anonymous, Submitted to International Conference on Learning Representations, 2020 Search PubMed.
- B. Chen, T. Shen, T. S. Jaakkola and R. Barzilay, arXiv preprint arXiv:1910.09688, 2019.
- R.-R. Griffiths, P. Schwaller, et al., chemrxiv preprint, DOI:10.26434/chemrxiv.7366973.v1, 2018.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed.
- H. Satoh and K. Funatsu, J. Chem. Inf. Comput. Sci., 1995, 35, 34–44 CrossRef CAS.
- P. Schwaller, A. Vaucher, V. H. Nair and T. Laino, chemrxiv preprint, DOI:10.26434/chemrxiv.9897365.v1, 2019.
- T. Salimans, I. Goodfellow, W. Zaremba, V. Cheung, A. Radford and X. Chen, Advances in neural information processing systems, 2016, pp. 2234–2242 Search PubMed.
- Retrosynthesis metrics example, https://gist.github.com/pschwllr/fb795e5384817c8b79089902bf4d0920, accessed Feb 03, 2020 Search PubMed.
- G. Landrum, P. Tosco, B. Kelley, S. Riniker, P. Gedeck, N. Schneider, R. Vianello, A. Dalke, R. R. Schmidt, B. Cole, A. Savelyev, S. Turk, M. Swain, A. Vaucher, D. Nealschneider, M. WÃşjcikowski, A. Pahl, J.-P. Ebejer, F. Berenger, A. Stretton, J. L. Varjo, N. O'Boyle, D. Cosgrove, P. Fuller, J. H. Jensen, G. Sforna, D. Gavid, K. Leswing, S. Leung and J. van Santen, rdkit/rdkit: 2019_03_4 (Q1 2019) Release, 2019, DOI:10.5281/zenodo.3366468.
- J. Nieminen and M. Peltola, Appl. Math. Lett., 1999, 12, 35–38 CrossRef.
- eMolecules, https://www.emolecules.com, accessed Oct 29, 2019 Search PubMed.
- Nextmove Software Pistachio, http://www.nextmovesoftware.com/pistachio.html, accessed Jul 29, 2019 Search PubMed.
- J. Lin, IEEE Trans. Inf. Theor., 1991, 37, 145–151 CrossRef.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, J. Cheminf., 2015, 7, 23 Search PubMed.
- E. L. Willighagen, J. W. Mayfield, J. Alvarsson, A. Berg, L. Carlsson, N. Jeliazkova, S. Kuhn, T. Pluskal, M. Rojas-Chertó and O. Spjuth, et al. , J. Cheminf., 2017, 9, 33 Search PubMed.
- Nextmove Software NameRXN, http://www.nextmovesoftware.com/namerxn.html, accessed Jul 29, 2019 Search PubMed.
- D. Lednicer and L. A. Mitscher, The organic chemistry of drug synthesis, Wiley, New York, 1980, vol. 2 Search PubMed.
- P. A. Worthington, in Synthesis and Fungicidal Activity of Triazole Tertiary Alcohols, 1987, ch. 27, pp. 302–317 Search PubMed.
- H. Cotton, T. Elebring, M. Larsson, L. Li, H. SÃűrensen and S. von Unge, Tetrahedron: Asymmetry, 2000, 11, 3819–3825 CrossRef CAS.
- J. F. Larrow, E. Roberts, T. R. Verhoeven, K. M. Ryan, C. H. Sennayake, P. J. Reider and E. N. Jacobsen, Org. Synth., 1999, 76, 46 CrossRef CAS.
- A. F. Crowther and L. H. Smith, J. Med. Chem., 1968, 11, 1009–1013 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Model and data description, detailed hyper-graph expansion algorithm, predicted retrosynthetic pathways. See DOI: 10.1039/c9sc05704h |
| This journal is © The Royal Society of Chemistry 2020 |