
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-triggered self-amplifying degradable polyurethanes with the ability to translate local stimulation to continuous long-range degradation†
Yanhua
Xu
a,
Samya
Sen
 b,
Qiong
Wu
a,
Xujia
Zhong
a,
Randy H.
Ewoldt
b and
Steven C.
Zimmerman
*a
b,
Qiong
Wu
a,
Xujia
Zhong
a,
Randy H.
Ewoldt
b and
Steven C.
Zimmerman
*a
aDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: sczimmer@illinois.edu
bDepartment of Mechanical Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 3rd March 2020
Abstract
A new type of base-triggered self-amplifying degradable polyurethane is reported that degrades under mild conditions, with the release of increasing amounts of amine product leading to self-amplified degradation. The polymer incorporates a base-sensitive Fmoc-derivative into every repeating unit to enable highly sensitive amine amplified degradation. A sigmoidal degradation curve for the linear polymer was observed consistent with a self-amplifying degradation mechanism. An analogous cross-linked polyurethane gel was prepared and also found to undergo amplified breakdown. In this case, a trace amount of localized base initiates the degradation, which in turn propagates through the material in an amplified manner. The results demonstrate the potential utility of these new generation polyurethanes in enhanced disposability and as stimuli responsive materials.
Introduction
“Smart” polymers that degrade in response to external triggers have found applications in many fields, including drug delivery, transient electronics, encapsulation and sensing.1–6 More recently, chain-shattering degradable polymers and self-immolative degradable polymers have attracted considerable attention because their backbones can be completely degraded into small fragments with high sensitivity to different types of triggers including pH, light, and redox agents.7–16 However, both types of polymers require a stoichiometric amount of triggering agent and degradation rates are constant at best. Some self-immolative polymers suffer from slow or incomplete breakdown because side reactions occur as the degradation proceeds down the polymer backbone. An alternative approach uses autocatalytic degradation chemistry wherein a specific catalytic trigger causes chain cleavage and generation of additional triggers for acceleration of the degradation.17–21Phillips and coworkers reported that ROMP polymers with appropriate pendant chains could exhibit dramatic changes in macroscopic properties through amplified, self-propagating side-chain reactions.22,23 In particular, a global switch in hydrophobicity and a change in the optical properties of a film occurred with local stimulation. In an effort to develop polymeric materials that might degrade with accelerated rate profiles and inspired by acid amplifier small molecules, we recently reported poly(3-iodopropyl)acetals that breakdown liberating HI.24 In essence such polymers carry the seeds of their own destruction,25 with liberated acid catalyzing further cleavage in an autocatalytic loop. It is important to determine the generality of the autocatalytic polymer degradation strategy by developing breakdown pathways using other triggers such as base, light and redox agents. Herein we report a new polyurethane that undergoes self-amplified degradation mediated by base and further show that in analogous gels, a small localized addition of base leads to rapid long-range breakdown (Fig. 1).
 | ||
| Fig. 1 Design of base-triggered self-amplifying degradable polyurethanes that have higher sensitivity to catalytic amount of base and exhibit an autocatalytic degradation mechanism. | ||
Results and discussion
As recently noted, base-degradable polymers are underdeveloped relative to acid-degradable polymers.26,27 In designing auto-catalytic base-degradable polyurethanes, the base amplifiers reported by Ichimura and others were considered.28–32 Within this class of small molecules, the Fmoc protected carbamate offered a convenient aromatic scaffold for functionalization and the potential for conventional polyurethane synthesis. The actual polyurethanes studied, 1 and 1c, were prepared in six steps as shown in Scheme 1. Functionalization of the fluorene ring was achieved through a Friedel–Crafts acylation after protection of the alcohol group with acetic anhydride, thus affording 4 or 5. Acidic deprotection and reduction with BH3·THF produced intermediate 6 and 7, which were further converted to diol monomer 8 and 9, respectively, by selectively reducing the benzylic alcohol group with Et3SiH. Traditional polycondensation was performed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of diol monomer and hexylmethylene diisocyanate to afford polymer 1 and 1c.
:1 ratio of diol monomer and hexylmethylene diisocyanate to afford polymer 1 and 1c.
 | ||
| Scheme 1 Reagents and conditions: (a) acetic anhydride, pyridine, DCM, 25 °C; (b) succinic anhydride, AlCl3, DCM, 0 °C to 25 °C (56% for 4 and 59% for 5); (c) 18% HCl, acetone, reflux; (d) BH3·THF, THF, 25 °C (32% for 6 and 34% for 7); (e) Et3SiH, BF3·OEt2, 0 °C (30% for 8 and 25% for 9); (f) hexamethylene diisocyanate, DBTDL, NMP, 25 °C (75% for 1 and 70% for 1c). | ||
As illustrated in Scheme 2, the addition of base can abstract the weakly acidic fluorenyl methine proton on the polymer 1 backbone, followed by E1cB elimination and decarboxylation to generate a dibenzofulvene and stoichiometric amine that can catalyse additional cleavage reactions before or after addition to the dibenzofulvene unit. The two polyurethanes 1 and 1c are identical structurally except that control polymer 1c is unable to undergo base-triggered degradation because the additional methylene group prevents the E1cB elimination from occurring. Both 1 and 1c were characterized by gel permeation chromatography (GPC) with DMF as the eluent (Fig. S6 and S7†). Polymer 1 has a Mn = 22 kDa (Ð = 2.1) and control polymer 1c has Mn = 11 kDa (Ð = 2.6). The 1H NMR was consistent with the expected structure of 1 (Fig. S3†) and 1c (Fig. S4†).
 | ||
| Scheme 2 Degradation mechanism of base-triggered self-amplifying degradable polyurethane. | ||
Thermal gravimetric analysis (TGA) of polymer 1 and polymer 1c revealed the onset of thermal degradation to occur around 120 °C and 280 °C respectively (Fig. S25 and S26†) and the onset thermal temperature at 120 °C of polymer 1 correlates well to what Simeunovic and his coworkers reported.33 The Tg of polymer 1 and 1c were determined to be 61 °C and 45 °C, respectively, the latter value measured by differential scanning calorimetry (DSC) (Fig. S27†).
Several bases were found to trigger the autocatalytic degradation of 1 (Fig. S11†), with hexylamine chosen for further study because its basicity and steric hindrance is most similar to the amplified amine species. Thus, the base-triggered degradation of polymers 1 and 1c in DMF solution was initiated by the addition of hexylamine and monitored by gel permeation chromatography (GPC). When 1 was exposed to 5 mol% hexylamine (per repeat unit), it showed a progressive and significant decrease in molecular weight over a 12 h period. As seen in Fig. 2a, the reduction in polymer size over time is nonlinear. Thus, the retention time of the 1 shifts only 1 min during the first 2 h but between 6 h and 9 h significantly broadens and shifts to longer times. In contrast, under the same conditions, the GPC of polymer 1c remained unchanged over 24 h (Fig. S8†).
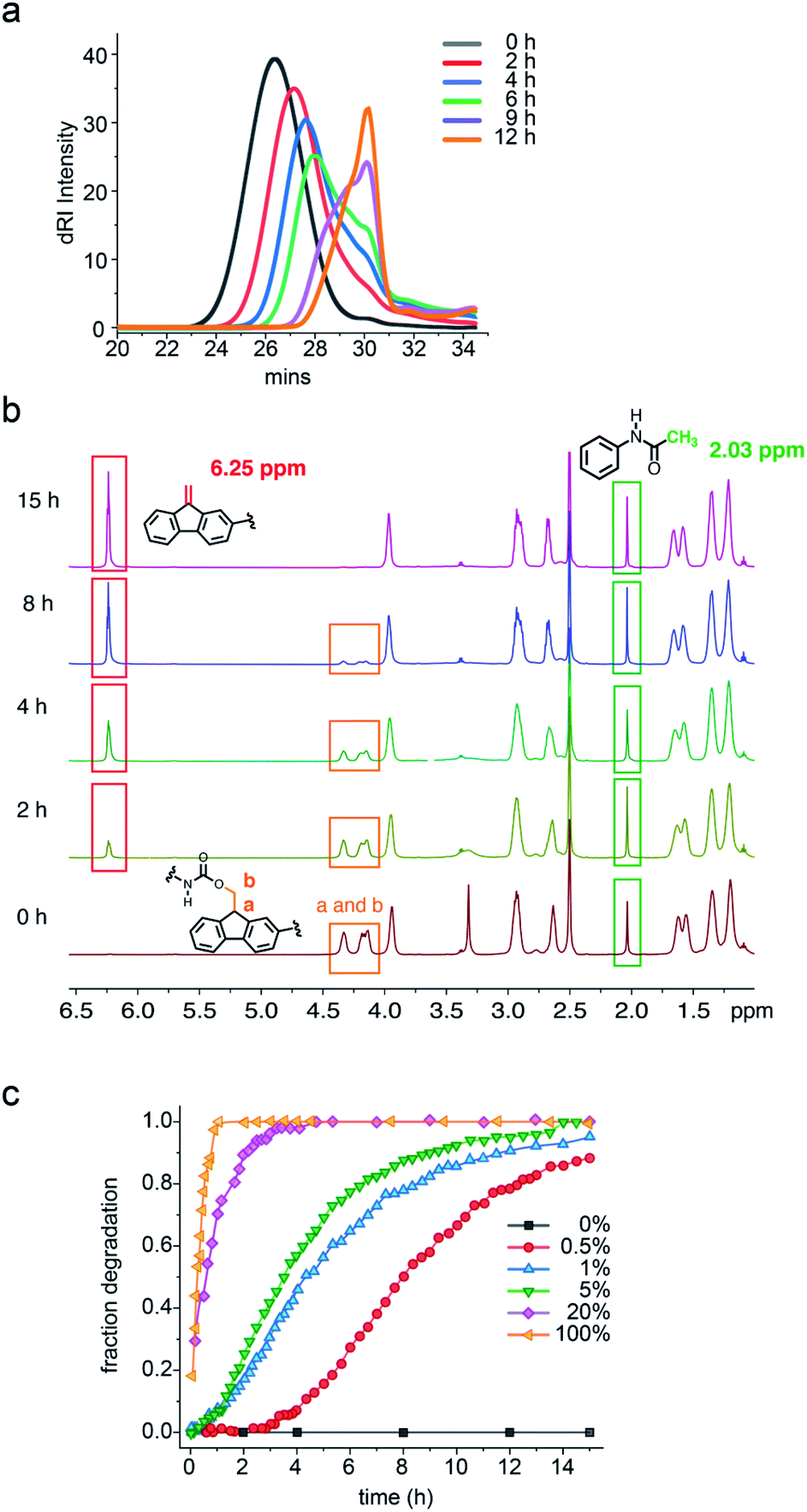 | ||
| Fig. 2 (a) GPC of 1 over time after addition of 5 mol% hexylamine (per repeat unit). (b) Time-dependent 1H NMR spectra of 1 in DMSO-d6 with 5 mol% hexylamine as trigger and acetamide as internal standard. (c) 1H NMR degradation of 1 over time with different hexylamine concentrations. Orange: 100 mol%, purple: 20 mol%, green: 5 mol%, blue: 1 mol%, red: 0.5 mol%, gray: 0 mol%. Fraction degradation calculated from ratio of signals at δ 6.25 or 4.33 ppm (alkene product or a/b protons of starting polymer) and internal standard at 2.03 ppm. Lines connecting the points are to guide the eyes. | ||
1H NMR was used to monitor the molecular details of the degradation of polymers 1 and 1c in the presence of hexylamine in DMSO-d6 solution. Consistent with the GPC study, no change in the NMR of 1c was observed over 24 h with 5 mol% hexylamine (Fig. S10†). In the case of 1, addition of 5 mol% of hexylamine led to the simultaneous disappearance of the methine and methylene protons labelled a and b at δ 4.33 and 4.16 ppm, respectively and the appearance of alkene protons at δ 6.25 ppm from the dibenzofulvene elimination product (Fig. 2b and S9†).
To determine how the concentration of the base trigger affects the rate of the degradation, quantitative 1H NMR-monitored kinetics were carried out in the presence of 0.5 mol%, 1 mol%, 5 mol%, 20 mol% and 100 mol% hexylamine. As seen in Fig. 2c, a stoichiometric amount of hexylamine induced complete polymer degradation at room temperature within 1 h. The rate profile and time for complete degradation correlated with the amount of base trigger. Thus, with no added base the polymer was stable, whereas for 0.5 mol%, 1 mol%, 5 mol%, 20 mol% and 100 mol% hexylamine the degradation reached 90% at ca. 15 h, 12 h, 10 h, 2 h, and 47 min, respectively. Most exciting was the observation that the three lowest concentration hexylamine experiments (0.5 to 5 mol%) exhibited obvious induction periods and sigmoidal conversion curves indicative of autocatalytic degradation.
Additional support for the autocatalytic, base amplification mechanism came from fitting the degradation data of polymer 1 to an autocatalytic kinetic model (eqn (S3)†).23,24,34–36 In this model, rate constants k1 and k2 separately represent the nonautocatalytic and autocatalytic, amine-accelerated rate constants (see ESI† for details). Consistent with the mechanism shown in Scheme 2, fitting the sigmoidal curves seen in Fig. 2c, led to k2 values that were quite close and k2c0 values that are larger than the k1 values (Table 1). The latter is especially true for the 0.5 mol% hexylamine run, in which the k2c0 (6.7 × 10−3 min−1) is 30 times larger than k1 (2.1 × 10−4 min−1). This larger k2c0 value is characteristic of an autocatalytic reaction (Table 1 and Fig. S14–S16†).
| Trigger | k 1 (min−1) | k 2 (M−1 min−1) | k 2 c 0 (min−1) |
|---|---|---|---|
| a Rate constants k1 and k2 are defined in text, c0 is initial concentration of degradable group. Concentration (M) is [Fmoc]. See ESI for additional details of the kinetic fit. The 1 mol% run was repeated and values found within 20%, establishing reproducibility. | |||
| 0.5% | 2.1 × 10−4 | 0.28 | 6.7 × 10−3 |
| 1% | 1.4 × 10−3 | 0.19 | 4.3 × 10−3 |
| 5% | 1.8 × 10−3 | 0.23 | 5.6 × 10−3 |
To further characterize the degradation of 1, liquid chromatography coupled mass spectrometry (LC-MS) was utilized to identify the major degradation products, and further indicate the chemical structure of the polymer repeating units. Analysis of the degradation products from polymer 1 through LC-MS revealed two major peaks, degradation product 1 with higher intensity appearing at 6.4 min (m/z = 393.4) and degradation product 2 with a lower intensity appearing at 9.3 min (m/z = 669.4) (Fig. S12†). These products are consistent with two types of repeating units in 1 (Fig. 3) and degradation product 1 demonstrates the ability of polymer 1 to form amine products via Fmoc deprotection.
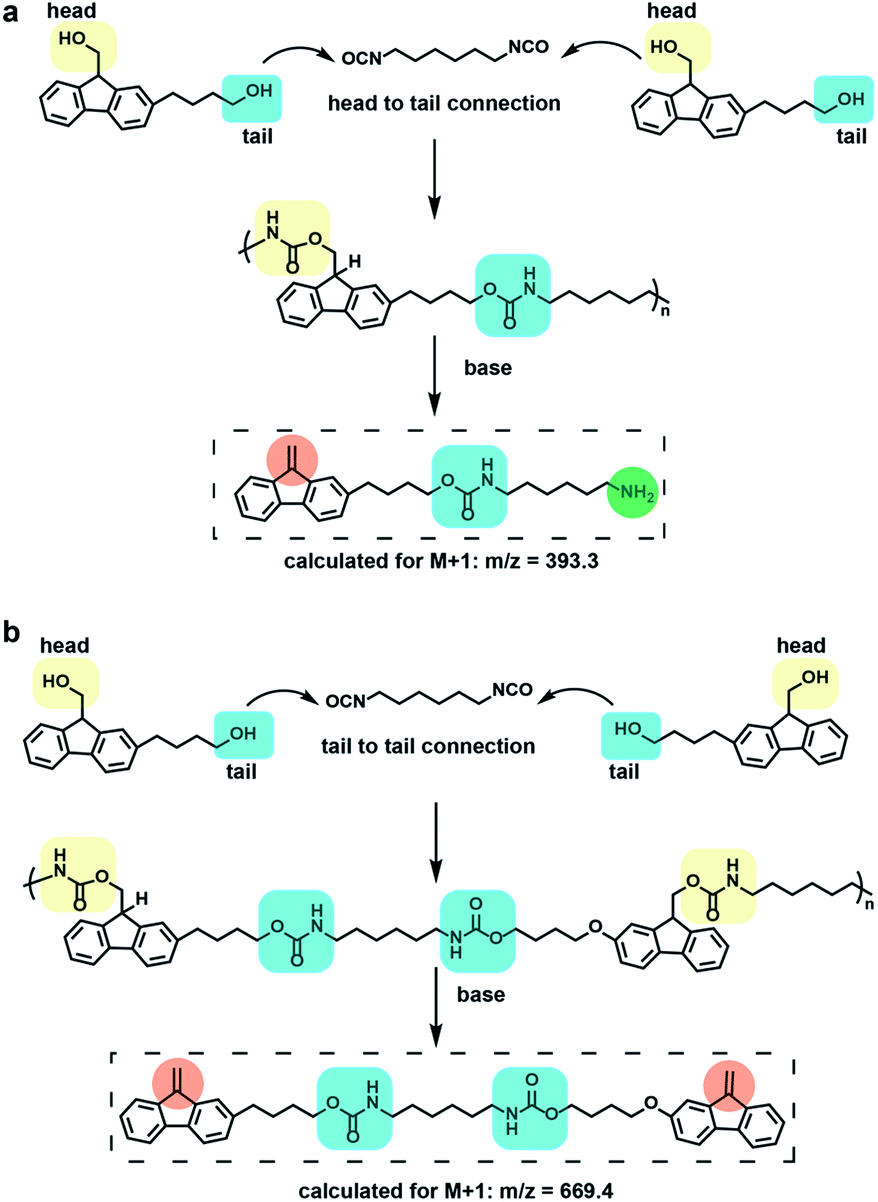 | ||
| Fig. 3 (a) Major repeating unit formed by head to tail connection and its corresponding degradation product observed by LC-MS. (b) Minor repeating units formed by tail to tail connection and its corresponding degradation product observed by LC-MS. | ||
Polyurethanes are important and widely used polymeric materials commonly found in plastics, adhesives and coatings.37,38 Unlike polymer 1, these materials are usually prepared from a polyol that produces cross-linking. The combination of cross-linking and the stability of the urethane linkage makes polyurethanes highly durable but also limits their end-of-life breakdown. To examine whether the base-amplified degradation might be applicable to bulk materials, triol 6 was prepared (see ESI†) and polymerized with hexamethylene diisocyanate and dibutyltindilaurate (DBTDL) as catalyst in N-methylpyrrolidone (NMP) with bromothymol blue present to visualize the gel and provide a pH indicator (Fig. 4b and S17†). The polymerization was performed at room temperature in a circular Teflon mold for 24 h to give a polyurethane film of 11 with a 500 μm thickness.
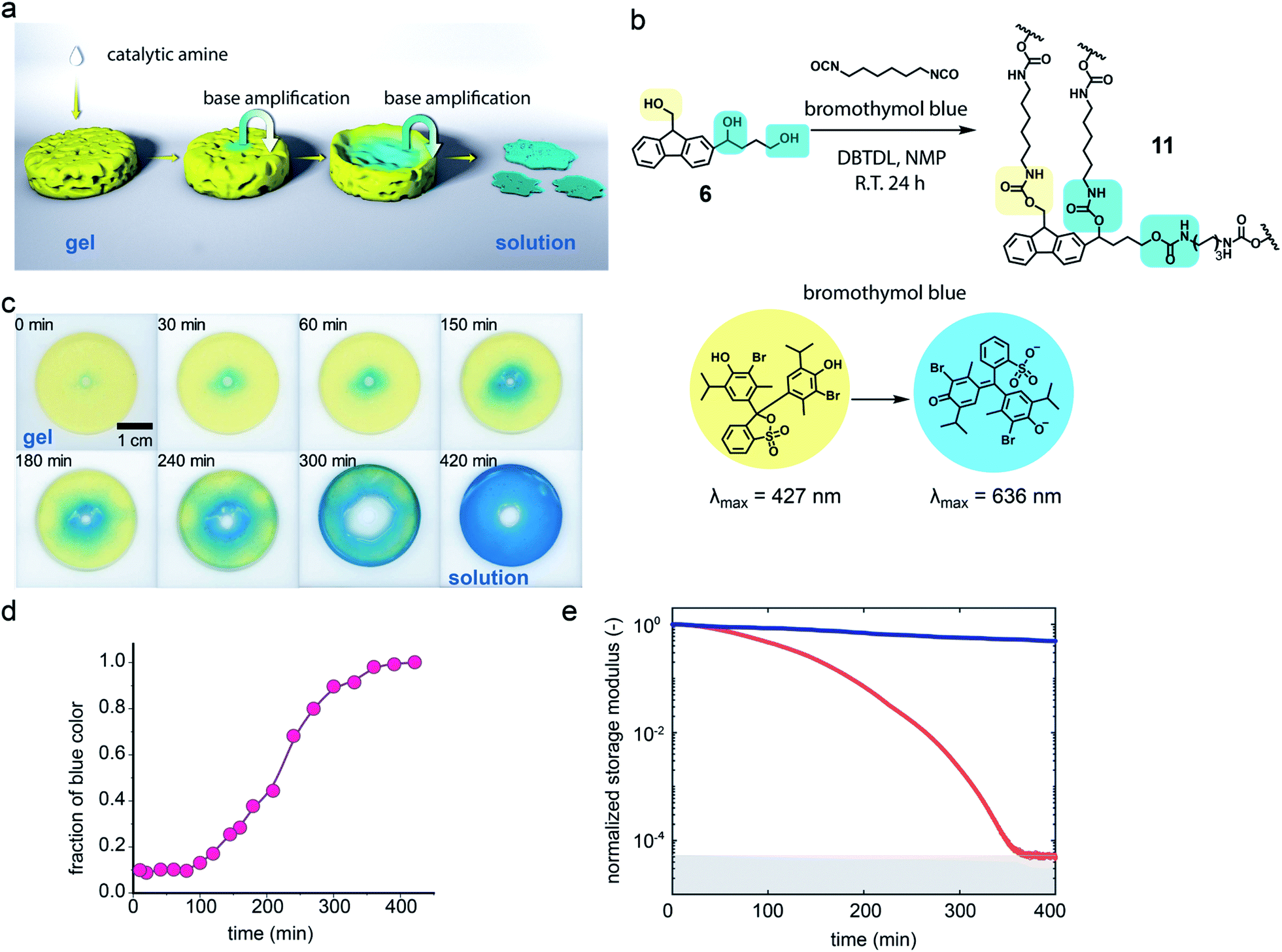 | ||
Fig. 4 (a) Representation of local base-stimulation triggering long range self-amplifying macroscopic degradation. (b) Synthesis of base-triggered self-amplifying degradable polymeric network 11 with bromothymol blue pH indicator. (c) Photographs of radial degradation of 11 as a disk-shaped polymeric film. (d) Quantification of loss of yellow area versus time plot. Smooth curve fit added to guide the eye. (e) Rheological study of degradation of polymeric film ( and and  curve with and without addition of hexylamine, respectively). curve with and without addition of hexylamine, respectively). | ||
To characterize the polymer film, it was immersed in additional NMP which induced significant swelling, but did not dissolve the gel. This observation is consistent with a cross-linked gel. To demonstrate the urethane network, the polymer film was dried under high vacuum and characterized by attenuated total reflection infrared spectroscopy (ATR-FTIR). The absorption peaks at 1694 cm−1 and 1252 cm−1 were assigned to the urethane structure and the absorption peak at 3326 cm−1 was assigned to unreacted hydroxyl groups in the polymer network (Fig. S18†).39
Degradation study of the polymer film was performed with film being swelled by NMP solution. In the degradation study, the centre of the polymer film changed from yellow to blue after 2 μL of a 180 mM hexylamine NMP solution was added in the centre and photographs were acquired over time (Fig. 4c). It was observed that the degradation area kept increasing, producing a deep blue colour, suggesting the formation of increasing numbers of terminal amino groups with conversion of the bromothymol blue pH indicator to its blue coloured ring open form. Quantification of the degradation area using the blue colour change for the polymer film was assisted by Image-Pro Plus (Fig. 4d). An increase in degradation area also simulates a sigmoidal curve, with a nonlinear increase from 10% at 100 min to 90% at 300 min, which is consistent with an autocatalytic degradation process for the crosslinked gel. The complete degradation of the polymer film required 420 min and over this period the yellow solid film became a blue solution (see ESI Video†). Six prominent degradation products were observed and characterised by high resolution electrospray ionization mass spectrometry (HR-ESI-MS, see Fig. S19†). Three of the six degradation products must contain a urethane structure at the benzylic position, consistent with the proposed cross-linked structure. Several of the degradation products have m/z values indicative of dibenzofulvene units or amino groups and this observation is consistent with the proposed Fmoc degradation mechanism.
The degradation process was also monitored by rheology. The storage modulus of the gel was measured and no major rheological change was observed from the polymeric network without addition of the base trigger (blue curve, Fig. 4e and S23†). However, the bulk polymeric network underwent a rapid decrease in storage modulus from about 5300 Pa to nearly 0 Pa upon addition of a very small amount of a dilute hexylamine solution in NMP at room temperature (red curve, Fig. 4e and S23†). In this case autocatalytic equations (eqn (S4) and (S5)†) that relate the storage modulus to degradation time were utilized to quantify the gel breakdown kinetics as described in more detail in the ESI.† In particular, these equations relate the storage modulus decrease to the concentration of crosslinks, thus enabling inference of apparent chemical rate constants. The fitting of triplicate runs (Fig. S24†) gave k2c0 = 15.9 ± 5.3 min−1, which is much larger than the k1 = 2.1 × 10−3 ± 1.1 × 10−3 min−1. These observations are consistent with an autocatalytic degradation process.
Conclusion
In conclusion, we developed a new type of self-amplifying degradable polymer with self-accelerating degradation properties using the well-developed base-sensitive Fmoc protecting group used in peptide synthesis. The incorporation of Fmoc in every repeating unit provides extremely sensitive polymeric materials with a small amount of base leading to rapid and amplified degradation. The base amplification process may be useful in applications where rapid production of an amine base is desirable. The crosslinked gel provides a rare example where a tiny local stimulation generates long range, rapid macroscopic degradation. In principle such a degradation might propagate over very large distances. Our current efforts are focused on generalizing this self-amplified degradation process to other kinds of triggers such as light, ions, and ROX agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by the National Science Foundation (NSF CHE-1709718) and the National Institutes of Health (R01 AR058361).Notes and references
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071–16087 CrossRef CAS PubMed.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- E. R. Gillies and J. M. Fréchet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS PubMed.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- Y. Zhang, Q. Yin, L. Yin, L. Ma, L. Tang and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 6435–6439 CrossRef CAS PubMed.
- Y. Zhang, L. Ma, X. Deng and J. Cheng, Polym. Chem., 2013, 4, 224–228 RSC.
- K. Cai, J. Yen, Q. Yin, Y. Liu, Z. Song, S. Lezmi, Y. Zhang, X. Yang, W. G. Helferich and J. Cheng, Biomater. Sci., 2015, 3, 1061–1065 RSC.
- S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2017, 139, 10002–10008 CrossRef CAS PubMed.
- A. P. Esser-Kahn, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2010, 132, 10266–10268 CrossRef CAS PubMed.
- M. G. Olah, J. S. Robbins, M. S. Baker and S. T. Phillips, Macromolecules, 2013, 46, 5924–5928 CrossRef CAS.
- K. Yeung, H. Kim, H. Mohapatra and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 5324–5327 CrossRef CAS PubMed.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- N. Fomina, C. McFearin, M. Sermsakdi, O. Edigin and A. Almutairi, J. Am. Chem. Soc., 2010, 132, 9540–9542 CrossRef CAS PubMed.
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed.
- X. Sun, S. D. Dahlhauser and E. V. Anslyn, J. Am. Chem. Soc., 2017, 139, 4635–4638 CrossRef CAS PubMed.
- J.-A. Gu, V. Mani and S.-T. Huang, Analyst, 2015, 140, 346–352 RSC.
- M. S. Baker and S. T. Phillips, J. Am. Chem. Soc., 2011, 133, 5170–5173 CrossRef CAS PubMed.
- X. Sun, D. Shabat, S. T. Phillips and E. V. Anslyn, J. Phys. Org. Chem., 2018, 31, e3827 CrossRef PubMed.
- X. Sun and E. V. Anslyn, Angew. Chem., Int. Ed., 2017, 56, 9522–9526 CrossRef CAS PubMed.
- H. Kim, M. S. Baker and S. T. Phillips, Chem. Sci., 2015, 6, 3388–3392 RSC.
- H. Mohapatra, H. Kim and S. T. Phillips, J. Am. Chem. Soc., 2015, 137, 12498–12501 CrossRef CAS PubMed.
- K. A. Miller, E. G. Morado, S. R. Samanta, B. A. Walker, A. Z. Nelson, S. Sen, D. T. Tran, D. J. Whitaker, R. H. Ewoldt, P. V. Braun and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 2838–2842 CrossRef CAS PubMed.
- Anon, Nature, 2019, 566, 157 Search PubMed.
- C. M. Possanza Casey and J. S. Moore, ACS Macro Lett., 2016, 5, 1257–1260 CrossRef CAS.
- H.-C. Wang, Y. Zhang, C. M. Possanza, S. C. Zimmerman, J. Cheng, J. S. Moore, K. Harris and J. S. Katz, ACS Appl. Mater. Interfaces, 2015, 7, 6369–6382 CrossRef CAS PubMed.
- K. Arimitsu, M. Miyamoto and K. Ichimura, Angew. Chem., Int. Ed., 2000, 39, 3425–3428 CrossRef CAS PubMed.
- H. Mohapatra, K. M. Schmid and S. T. Phillips, Chem. Commun., 2012, 48, 3018–3020 RSC.
- K. Arimitsu and K. Ichimura, J. Mater. Chem., 2004, 14, 336–343 RSC.
- K. Arimitsu, K. Tomota, S. Fuse, K. Kudo and M. Furutani, RSC Adv., 2016, 6, 38388–38390 RSC.
- K. Arimitsu, H. Kitamura, R. Mizuochi and M. Furutani, Chem. Lett., 2015, 44, 309–311 CrossRef CAS.
- S. Höck, R. Marti, R. Riedl and M. Simeunovic, Chimia, 2010, 64, 200–202 CrossRef PubMed.
- F. Mata-Perez and J. F. Perez-Benito, J. Chem. Educ., 1987, 64, 925–927 CrossRef CAS.
- O. P. Lee, H. Lopez Hernandez and J. S. Moore, ACS Macro Lett., 2015, 4, 665–668 CrossRef CAS.
- J. F. Perez-Benito, J. Phys. Chem. A, 2011, 115, 9876–9885 CrossRef CAS PubMed.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- F. E. Golling, R. Pires, A. Hecking, J. Weikard, F. Richter, K. Danielmeier and D. Dijkstra, Polym. Int., 2019, 68, 848–855 CrossRef CAS.
- H. Hao, J. Shao, Y. Deng, S. He, F. Luo, Y. Wu, J. Li, H. Tan, J. Li and Q. Fu, Biomater. Sci., 2016, 4, 1682–1690 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06582b |
| This journal is © The Royal Society of Chemistry 2020 |