
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stable covalent organic framework for photocatalytic carbon dioxide reduction†
Zhiwei
Fu
 a,
Xiaoyan
Wang
a,
Adrian M.
Gardner
b,
Xue
Wang
ac,
Samantha Y.
Chong
a,
Gaia
Neri
b,
Alexander J.
Cowan
b,
Lunjie
Liu
a,
Xiaobo
Li
a,
Anastasia
Vogel
a,
Rob
Clowes
a,
Matthew
Bilton
d,
Linjiang
Chen
*ac,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*ac
a,
Xiaoyan
Wang
a,
Adrian M.
Gardner
b,
Xue
Wang
ac,
Samantha Y.
Chong
a,
Gaia
Neri
b,
Alexander J.
Cowan
b,
Lunjie
Liu
a,
Xiaobo
Li
a,
Anastasia
Vogel
a,
Rob
Clowes
a,
Matthew
Bilton
d,
Linjiang
Chen
*ac,
Reiner Sebastian
Sprick
*a and
Andrew I.
Cooper
*ac
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool L7 3NY, UK. E-mail: ssprick@liverpool.ac.uk; aicooper@liverpool.ac.uk; lchen@liverpool.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, Chadwick Building, Peach Street, Liverpool L69 7ZF, UK
cLeverhulme Research Centre for Functional Materials Design, Materials Innovation Factory and Department of Chemistry, University of Liverpool, Oxford Street, Liverpool L7 3NY, UK
dImaging Centre at Liverpool, University of Liverpool, Liverpool L69 3GL, UK
First published on 21st November 2019
Abstract
Photocatalytic conversion of CO2 into fuels is an important challenge for clean energy research and has attracted considerable interest. Here we show that tethering molecular catalysts—a rhenium complex, [Re(bpy)(CO)3Cl]—together in the form of a crystalline covalent organic framework (COF) affords a heterogeneous photocatalyst with a strong visible light absorption, a high CO2 binding affinity, and ultimately an improved catalytic performance over its homogeneous Re counterpart. The COF incorporates bipyridine sites, allowing for ligation of the Re complex, into a fully π-conjugated backbone that is chemically robust and promotes light-harvesting. A maximum rate of 1040 μmol g−1 h−1 for CO production with 81% selectivity was measured. CO production rates were further increased up to 1400 μmol g−1 h−1, with an improved selectivity of 86%, when a photosensitizer was added. Addition of platinum resulted in production of syngas, hence, the co-formation of H2 and CO, the chemical composition of which could be adjusted by varying the ratio of COF to platinum. An amorphous analog of the COF showed significantly lower CO production rates, suggesting that crystallinity of the COF is beneficial to its photocatalytic performance in CO2 reduction.
Introduction
There is now little doubt that the human production of CO2 has contributed to climate change, which will affect life on earth.1 One potential approach to reducing CO2 emissions is its conversion into value-added products using solar energy.2,3 A range of inorganic semiconductors has been investigated as photocatalysts for CO2 reduction, such as TiO2,4 ZrO2,5 In2O3,6 CdS,7 or ZnGa2O4.8 Unfortunately, many inorganic photocatalysts either have unsuitably aligned conduction/valence band positions or relatively large band gaps, hence limiting visible light absorption. By contrast, the band gap in organic semiconductors can be tuned readily through the incorporation of a diverse range of monomers.9–11 In recent years, porous organic materials such as carbon nitrides,12–14 conjugated microporous polymers (CMPs),15,16 covalent triazine-based frameworks (CTFs)17 and hyper-crosslinked polymers (HCPs)18 have been studied for photocatalytic CO2 reduction. Those organic materials are typically amorphous; by contrast, covalent organic frameworks (COFs) can combine porosity with crystallinity.19–23 COFs have been investigated as photocatalysts for water splitting,24,25 and for electrocatalytic CO2 reduction.26,27 These materials also have potential for direct photocatalytic CO2 reduction: for example, an azine-based COF, N3-COF, was shown to exhibit gas phase photocatalytic CO2 reduction.28 Likewise, a 2D imine triazine-COF loaded with rhenium29 and a β-ketoenamine-linked COF decorated with both nickel and a light-absorbing dye30 were studied for the same reaction. All of these COFs have limited effective conjugation lengths in the 2D plane of the framework because they are based on imine, azine, or β-ketoenamine-linkers. This results in blue-shifted absorption on-sets, which limit the ability of the materials to absorb visible light.31,32Here, we used Knoevenagel condensation (Fig. 1a) such that olefins become the COF linkers.31,33,34 Our aim was to increase the conjugation length in the framework and hence, perhaps, to improve the performance of these materials for CO2 reduction. The cyanovinyl-groups as a result of the Knoevenagel condensation have been shown to be beneficial for CO2 uptake35 which might increase the efficiency of CO2 reduction. The COF was loaded with [Re(CO)5Cl] giving a heterogeneous analogue of the well-studied homogeneous catalyst [Re(bpy)(CO)3Cl] with enhanced stability.36
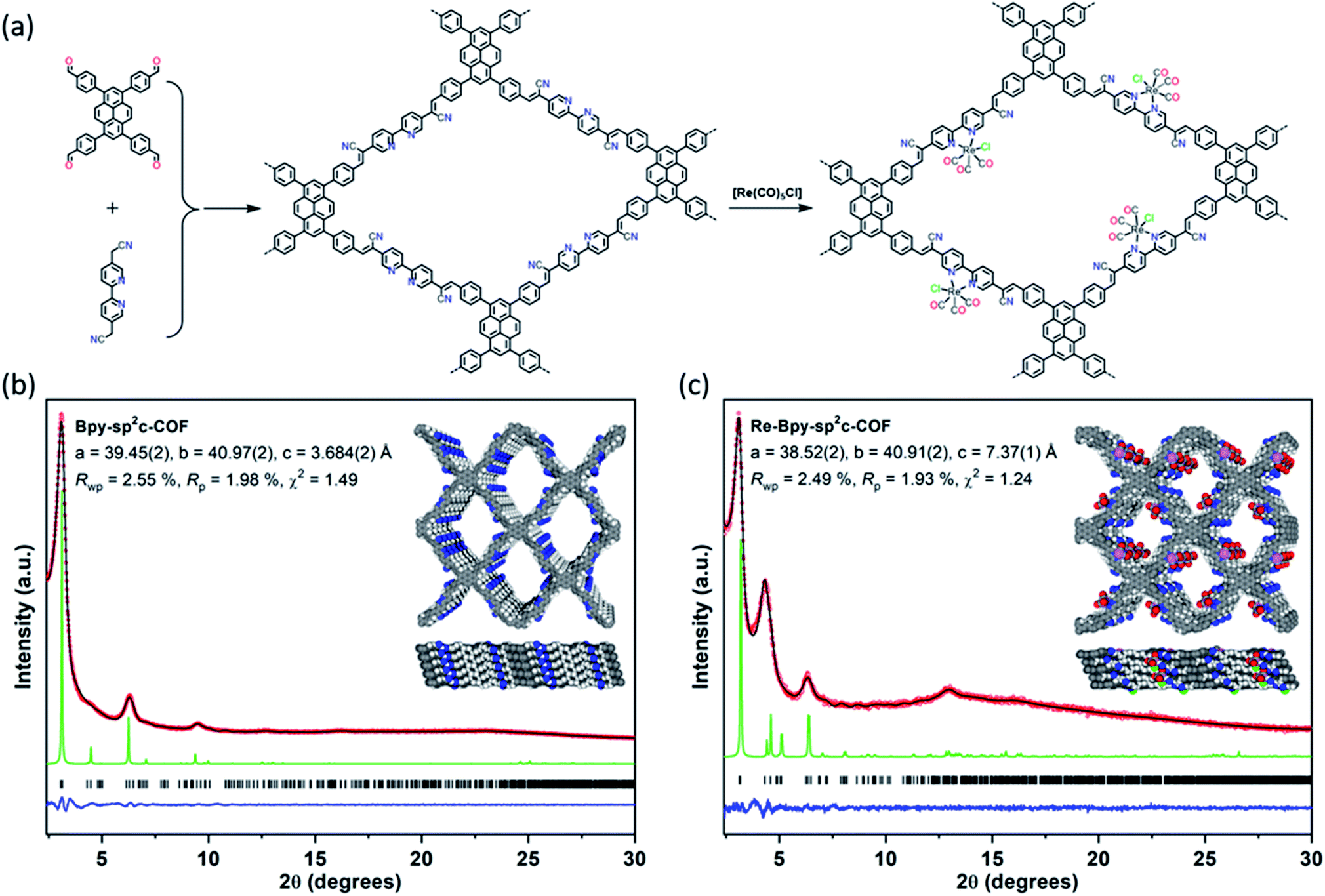 | ||
Fig. 1 (a) Synthesis of Bpy-sp2c-COF and Re-Bpy-sp2c-COF. Conditions for Bpy-sp2c-COF: KOH (4 M) 1,2-dichlorobenzene and 1-butanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture), 120 °C, 72 hours; (b) and (c) PXRD patterns of Bpy-sp2c-COF (b) and Re-Bpy-sp2c-COF (c) obtained experimentally (red circles), simulated from the eclipsed AA-stacking mode (green), profiles calculated from Le Bail fitting (black) and residual (blue). Reflection positions are shown by tick marks. Structural models for Bpy-sp2c-COF (b) and Re-Bpy-sp2c-COF (c) with eclipsed AA stacking patterns, shown parallel to the pore channel along the crystallographic c axis (top) and parallel to the layers (bottom). :1 mixture), 120 °C, 72 hours; (b) and (c) PXRD patterns of Bpy-sp2c-COF (b) and Re-Bpy-sp2c-COF (c) obtained experimentally (red circles), simulated from the eclipsed AA-stacking mode (green), profiles calculated from Le Bail fitting (black) and residual (blue). Reflection positions are shown by tick marks. Structural models for Bpy-sp2c-COF (b) and Re-Bpy-sp2c-COF (c) with eclipsed AA stacking patterns, shown parallel to the pore channel along the crystallographic c axis (top) and parallel to the layers (bottom). | ||
Results and discussion
We synthesized a two-dimensional (2D) sp2c-COF via the Knoevenagel condensation of 1,3,6,8-tetrakis(4-formylphenyl)pyrene (TFPPy) and 5,5′-bis(cyanomethyl)-2,2′-bipyridine in 1,2-dichlorobenzene and 1-butanol at 120 °C (Fig. 1a). The powder X-ray diffraction (PXRD) pattern of Bpy-sp2c-COF (Fig. 1b and S9†) is in good agreement with the profile predicted for the eclipsed (AA) stacked structure (Fig. 1b, inside). Diffraction peaks are observed at 3.1°, 4.5°, 6.2°, and 9.5°, corresponding to the (110), (200), (220), and (330) reflections, suggesting that Bpy-sp2c-COF has uniform 1D diamond-shaped pores.Nitrogen sorption experiments were performed at 77 K and the Brunauer–Emmett–Teller surface area (SABET) for Bpy-sp2c-COF was calculated to be 432 m2 g−1. This SABET is lower than that predicted for the atomistic model of a perfectly crystalline structure (2041 m2 g−1), but this is commonly observed for sp2c-COFs which typically have surface areas ranging from 322 m2 g−1 for sp2c-COF-2 (ref. 37) up to 692 m2 g−1 for sp2c-COF.31
The pore size distribution profile shows a narrow pore size distribution with a pore width of 2.4 nm (Fig. 2a, inset curve), further supporting an AA stacking sequence that is predicted to have a pore size of 2.4 nm. Fourier-transform infrared (FT-IR) spectroscopy shows a distinct peak at 2217 cm−1 relating to a –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N vibration band, indicating the formation of Bpy-sp2c-COF (Fig. 2c).
N vibration band, indicating the formation of Bpy-sp2c-COF (Fig. 2c).
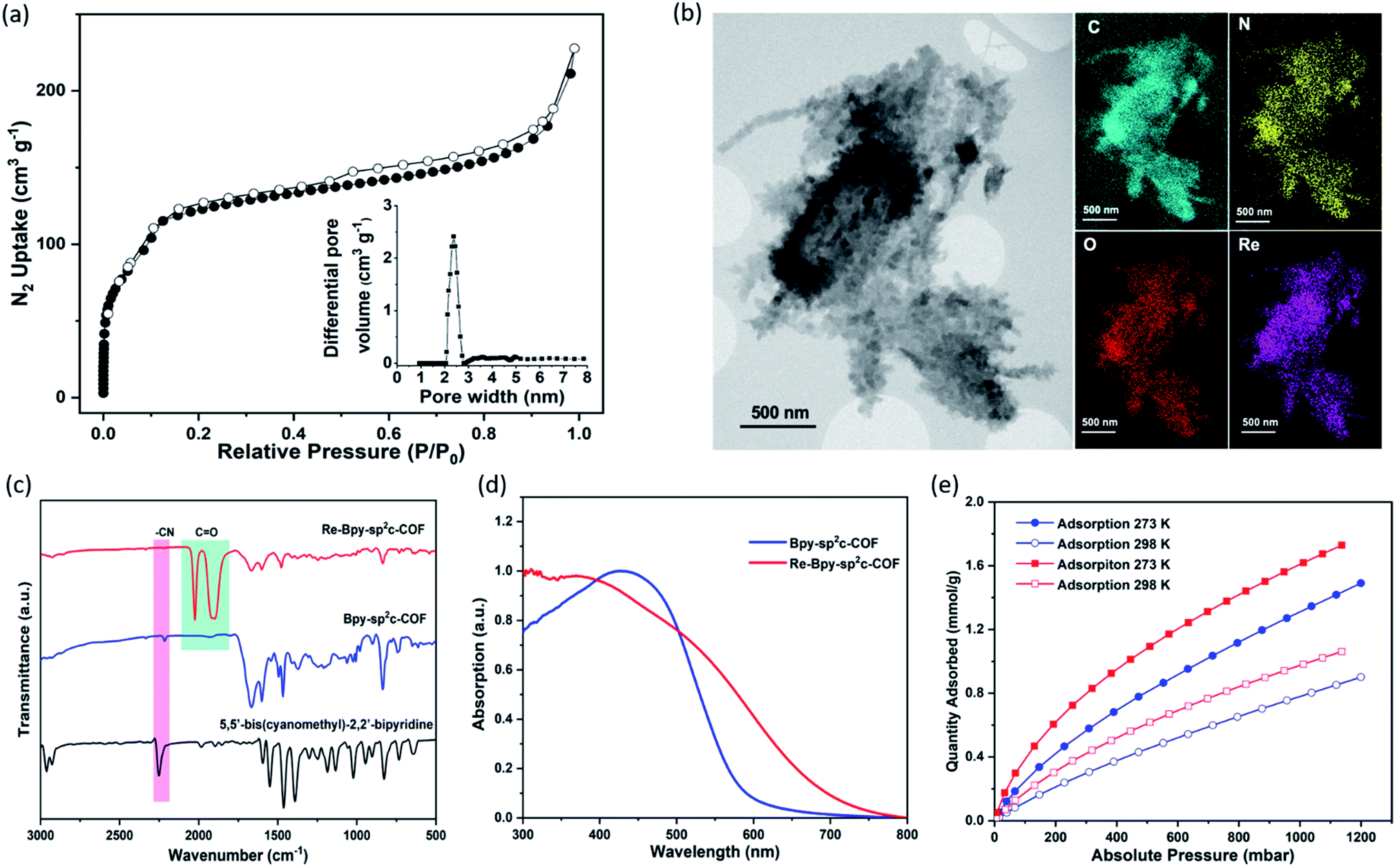 | ||
| Fig. 2 (a) N2 Adsorption (filled dots) and desorption (open dots) isotherm profiles of Bpy-sp2c-COF measured at 77 K. Inset: profile of the calculated pore size distribution of Bpy-sp2c-COF; (b) STEM images and EDX mapping of Re-Bpy-sp2c-COF; (c) FT-IR Spectra of Bpy-sp2c-COF, Re-Bpy-sp2c-COF and 5,5′-bis(cyanomethyl)-2,2′-bipyridine; (d) Solid-state reflectance UV-vis spectra; (e) CO2 adsorption isotherms of Bpy-sp2c-COF (blue) and Re-Bpy-sp2c-COF (red). | ||
Re-complexes for photocatalytic CO2 reduction were first reported in 1983,38 and they are well studied owing to their high efficiency and selectivity for CO formation.39,40 Here, we used the bipyridine sites in Bpy-sp2c-COF to ligate [Re(CO)5Cl] to give Re-Bpy-sp2c-COF. The PXRD pattern of the Re-incorporated COF (Fig. 1c and S10†) shows peaks at 3.1°, 4.5°, 6.2°, 12.9°, corresponding to (110), (200), (220), (−221) reflections predicted for the Re-loaded, AA-stacked model (Fig. 1c).
The BET surface area (SABET) for Re-Bpy-sp2c-COF was calculated to be 323 m2 g−1 (Fig. S38†). Scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDX) mapping images (Fig. 2b) show a uniform distribution of C, N, O, and Re in Re-Bpy-sp2c-COF, further suggesting that the Re moiety has been incorporated throughout the structure of COFs. Inductively coupled plasma-optical emission spectrometry (ICP-OES) measurements show that 18 wt% of Re has been incorporated into the material. This ratio corresponds to ligation of half of the bipyridine sites by the Re complex, which was also the ratio in the atomistic model built to represent Re-Bpy-sp2c-COF (Fig. 1c). The FT-IR spectrum for the Re-modified COF (Fig. 2c) shows new peaks at 1900 cm−1, 1917 cm−1, 2024 cm−1, corresponding to the CO-stretching bands of the incorporated [Re(CO)3Cl] complex.29,36 UV-visible diffuse reflectance spectra (Fig. 2d) show a red-shift of the absorption edge from 589 nm to 694 nm for Re-Bpy-sp2c-COF compared to Bpy-sp2c-COF. Finally, X-ray photoelectron spectra for Re 4f, Cl 2p and N 2s regions look very similar for Re-Bpy-sp2c-COF compared to the molecular catalyst [Re(bpy)(CO)3Cl] indicating that complexation of Re is similar in both cases (Fig. S40†).
We next studied the CO2 uptake for this material up to 1200 mbar at both 273 and 298 K. Re-Bpy-sp2c-COF adsorbs 1.7 mmol g−1 CO2 at 273 K and 1.1 mmol g−1 at 298 K (Fig. 2e). Re-Bpy-sp2c-COF has a high isosteric heat of adsorption (31 kJ mol−1), showing that the COF has good affinity toward CO2 (Fig. S19†).
Photocatalytic CO2 reduction experiments were conducted in a quartz flask under 1 atmosphere CO2 in a mixture of acetonitrile (MeCN) and triethanolamine (TEOA) in 30:1 ratio and under visible light illumination (λ > 420 nm, 300 W Xe light source). TEOA acts as the sacrificial electron donor and proton source, while MeCN is used to disperse the catalyst. Over a total of 17.5 hours irradiation under visible light (Fig. S16†), Re-Bpy-sp2c-COF produced CO with a rate of 1040 μmol g−1 h−1 and 81% selectivity over H2, which equals to a TON of 18.7 for CO, outperforming its homogeneous counterpart under the same conditions, which is deactivated after 3 hours with a TON of 10.3 (Fig. S16†).
An apparent quantum yield (AQY) of 0.5% was measured at 420 nm for CO production. The small amount of H2 possibly originates from competing proton reduction of water traces in the TEOA or oxidative dehydrogenation of TEOA.13 In the absence of the Re-complex, Bpy-sp2c-COF only generated trace amounts of CO while H2 was not detected. No other liquid phase products, i.e. HCOOH and methanol, were observed.
The combination of CO2 conversion rate and CO/H2 selectivity of Re-Bpy-sp2c-COF compares favorably with other reported COFs (see Table S3†). For example, a rhenium modified 2D imine triazine-COF produced around 750 μmol g−1 h−1 CO with 98% selectivity,29 and a β-ketoenamine-linked COF modified with nickel, plus the use of an additional dye, gave a CO production rate of 811 μmol g−1 h−1 CO with 96% selectivity.30 These comparisons should be made with caution, though, since photocatalytic rates also depend strongly on the precise experimental set-up that is used.41
Control experiments were carried out to confirm that the source of the CO generated is indeed a photocatalytic process (Table S2†). Under an argon atmosphere in absence of CO2, Re-Bpy-sp2c-COF generated 14.9 μmol g−1 h−1 CO and 285.3 μmol g−1 h−1 H2. The small amount of CO produced is possibly a result of decomposition of organic residues during photocatalysis1 or decomposition of TEOA as an ineffective side-reaction.13 No gas production was observed in the dark or in the absence of hole scavenger. Experiments with isotopically labelled 13CO2 resulted in the formation of 13CO, strongly suggesting that CO2 was the source of the produced CO (Fig. 3a).
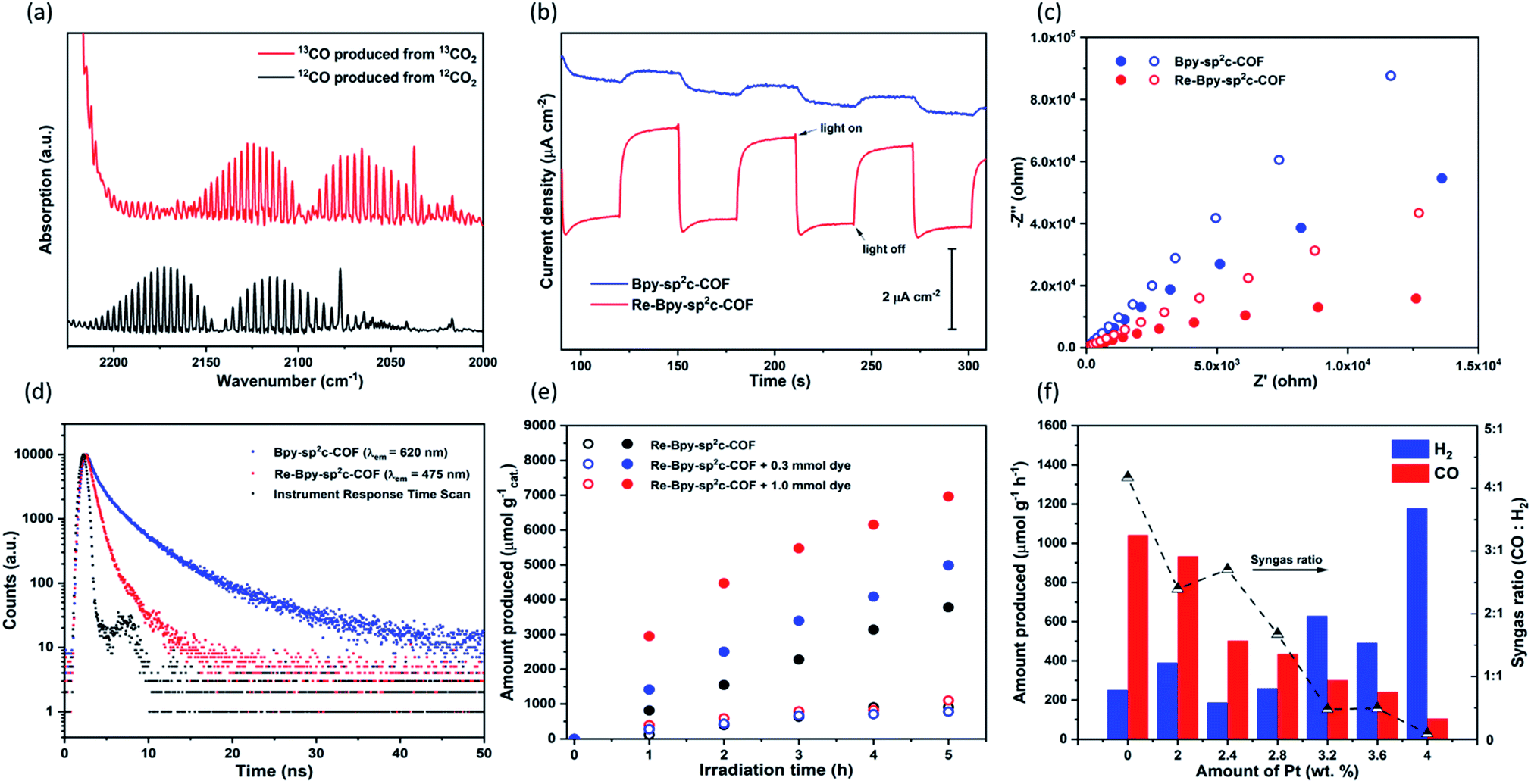 | ||
| Fig. 3 (a) FT-IR Spectra of 13CO produced in the photoreduction of 13CO2 (b) transient photocurrent response at 0.5 V vs. RHE under intermittent light irradiation for Bpy-sp2c-COF and Re-Bpy-sp2c-COF; (c) Nyquist plots of Bpy-sp2c-COF (blue) and Re-Bpy-sp2c-COF (red) at a voltage of 0.5 V vs. RHE under dark (open dots) and light irradiation (closed dots); (d) time-correlated single-photon counting experiments for Bpy-sp2c-COF and Re-Bpy-sp2c-COF in MeCN/TEOA (30/1) solution purged with CO2 (λexc = 405 nm); (e) CO (closed dots) and H2 (open dots) production using visible light (λ > 420 nm, 300 W Xe light source) for Re-Bpy-sp2c-COF and Re-Bpy-sp2c-COF with dye (1 mg catalyst or 1 mg catalyst with 0.3 mmol or 1.0 mmol dye in 5 mL solvent with ratio of MeCN/TEOA = 30/1); (f) photocatalytic syngas generation of Pt modified Re-Bpy-sp2c-COF under visible light irradiation (λ > 420 nm, 300 W Xe light source). | ||
Re-Bpy-sp2c-COF appears to be stable under the photocatalysis conditions, evident from the post-illumination FT-IR spectra (Fig. S7†) of the sample after 17.5 hours of continuous visible light illumination (λ > 420 nm, 300 W Xe light source). The material also retains most of its crystallinity and PXRD patterns show that some order is retained after photolysis for 17.5 hours (Fig. S11†). This shows that the material has very good stability compared to other previous reports.29,30 When the run was extended to a total of 50 hours a further loss of crystallinity is observed (Fig. S43 and S44†) along a loss of activity, highlighting that stability is still one of the important challenges in the field. Nevertheless, it seems that in making a heterogeneous analogue of [Re(bpy)(CO)3Cl] an increase in stability is observed (Fig. S43†), possibly by preventing the formation of the dimer of the Re-complex36 which can occur with Re(bpy)CO3Cl in solution, or by the shielding of the Re-centre within the COF structure from photodecomposition side reactions.42
Photoelectrochemical experiments were conducted using FTO glass as a photocathode in 0.1 M Na2SO4 solution (Fig. 3b). All samples were tested at a constant voltage of 0.5 V vs. reversible hydrogen electrode (RHE). The photocurrent of Re-Bpy-sp2c-COF photocathode was about 2 μA cm−1, which was more than four times higher than a Re-free Bpy-sp2c-COF photoanode. Additionally, Nyquist plots (Fig. 3c) showed the arc radii for Bpy-sp2c-COF and Re-Bpy-sp2c-COF under irradiation were smaller than those in dark, verifying that charge carriers were generated in Bpy-sp2c-COF and Re-Bpy-sp2c-COF under irradiation. The Nyquist plots of Re-Bpy-sp2c-COF under irradiation have smaller semicircles than those of Bpy-sp2c-COF. Both measurements taken together show that the Re bearing material acts as a better photo-electro catalyst indicating that the material is better at separating and transferring charges, which is also in line with computational predictions (vide infra).
We then went on to use emission spectroscopy to study the mechanism of the photocatalysis for the Re loaded Bpy-sp2c-COF. The photocatalyst Bpy-sp2c-COF in acetonitrile suspension shows the presence of two emissive states, with λmax at 475 and 640 nm. The excitation spectrum shows the 640 nm emission arises from a broad range of absorption bands in the UV/vis spectrum (from 300 to 500 nm), in contrast the sharp emission band centred at 475 nm is a result of excitation into a single band at 390 nm (Fig. S20†). Time-correlated single-photon counting (TCSPC) measurements show the lifetime of the 640 nm emissive state of Bpy-sp2c-COF is insensitive to the TEOA scavenger (Table S1 and Fig. S21†) and the emission yield is also unchanged (Fig. S20 and S21†). In contrast the 475 nm emission lifetime (2.48 ns to 0.72 ns) and yield is very sensitive to the presence of the TEOA electron donor (Fig. S20 and S21†), indicating that reductive quenching of this excited state can occur. Previous studies on closely related sp2c pyrene COFs have also reported the presence of two emissive states for COF samples.37 Therein, emission at 640 nm was attributed to the presence of a delocalised excited state across both pyrene and the sp2-carbon backbone on the basis of the significantly red-shifting of the emission when compared to that typically measured for excimer state of pyrene systems alone (ca. 480 nm). Interestingly following exfoliation of the COF, a second emission at ca. 468 nm was observed, proposed to be due to exfoliated COF where the removal of the π–π stacking force allows twisting of the structure and a loss of conjugation across the backbone. Here, the samples are sonicated prior to use and a similar assignment is also proposed.
Addition of the Re catalytic site to the COF framework leads to a marked change in the measured photophysical behaviour. With Re-Bpy-sp2c-COF a single emissive state (λmax = 475 nm), proposed to be due exfoliated COF material remains. Such an assignment is in-line with the noted insensitivity of emission at this wavelength to the presence of the Re centre as the loss of conjugation of the exfoliated structure may be expected to prevent efficient electron or energy transfer from the COF framework to the Re centre. Significantly the delocalised COF excited 640 nm emissive state of Bpy-sp2c-COF is completely absent in the Re-Bpy-sp2c-COF (Fig. S22†). The quenching of the emission by the Re centre indicates possible electron transfer from the COF backbone to the catalytically active Re complex. The assignment of the sensitisation of the catalytic centre following photon absorption by the COF framework is supported by the DFT calculations below and the good agreement between the wavelength dependent CO measurement (Fig. S35†) and the excitation spectrum of the 640 nm emission of the Bpy-sp2c-COF sample (Fig. S20†).
To further explore the photophysics of the system we have also carried out transient absorption (TA) spectroscopic studies on both Re-Bpy-sp2c-COF and Bpy-sp2c-COF (Fig. 4). Following excitation at 400 nm, 800 μW (5 kHz) of Bpy-sp2c-COF we observe complex TA spectra with broad negative bands between 450 to ca. 700 nm that formed within 0.5 ps. There is minimal absorption by the ground state of Bpy-sp2c-COF (Fig. 2d) at wavelengths longer than 600 nm. Therefore, the negative signal is proposed to be the overlap of stimulated emission from both the conjugated Bpy-sp2c-COF structure (λmax = 640 nm) and the exfoliated Bpy-sp2c-COF (λmax = 475 nm), overlapped with the ground state bleach, giving rise to the complex shape. The complex nature of the bleach makes determining accurate kinetics challenging, therefore we use t50% (the time taken for the bleach to decrease by 50%) as a rough measure of the lifetime of the photogenerated excited state. For Bpy-sp2c-COF at 550 nm, t50% = ca. 5 ps (Fig. S46†). Within 0.5 ps a photoinduced absorption (PIA) is present at 770 nm, which decays within 10 ps to form a new PIA centred at 700 nm that decays over the course of the experiment to leave only a small PIA by 3 ns.
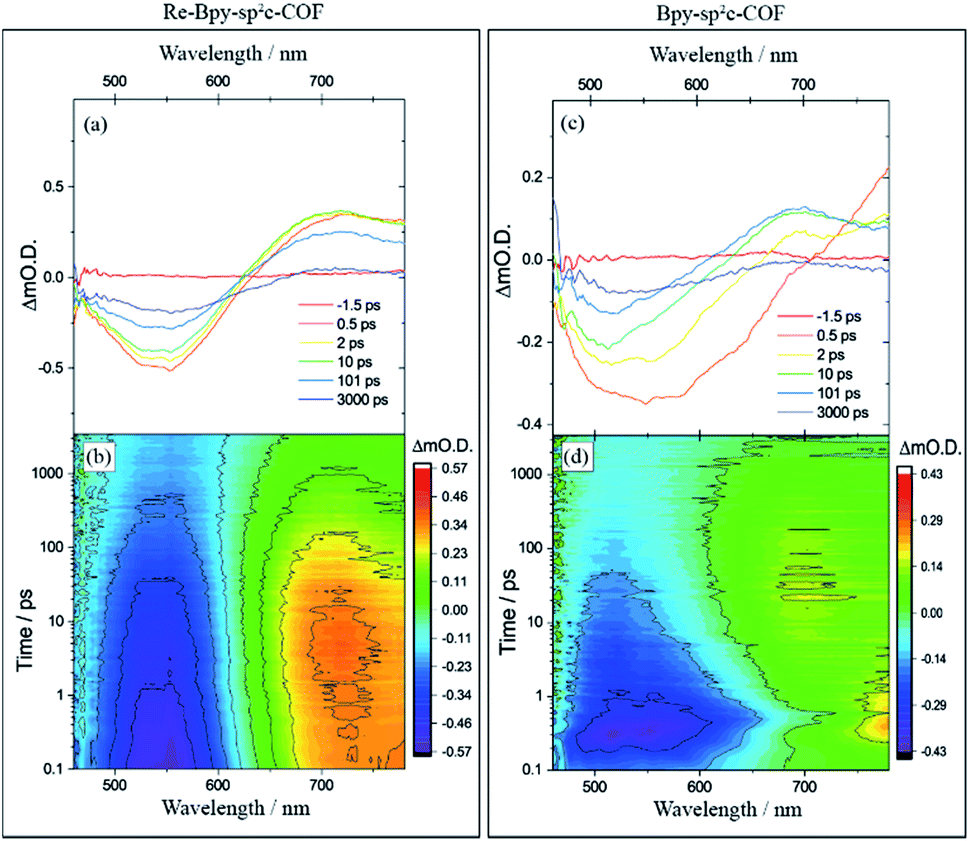 | ||
| Fig. 4 Transient absorption spectra of (a) Re-Bpy-sp2c-COF and (c) Bpy-sp2c-COF at pump-probe time delays chosen to highlight the changing nature of the excited electronic states probed, and the complete transient absorption surface probed (b) Re-Bpy-sp2c-COF and (d) Bpy-sp2c-COF. All spectra are recorded in CH3CN following 400 nm excitation. | ||
Re-Bpy-sp2c-COF shows a simpler TA spectrum following 400 nm excitation (Fig. 4). A negative band is formed, centred at 540 nm, again assigned to a combination of ground state bleaching and stimulated emission from the exfoliated COF framework (λmax = 475 nm; Fig. S23†). It is notable that this negative feature is substantially narrower than that observed for Bpy-sp2c-COF and for Re-Bpy-sp2c-COF at 550 nm, t50% = ca. 200 ps, significantly longer than observed in the absence of the Re. A PIA centred at ca. 770 nm is again observed to be formed within 0.5 ps, with a blue shift of this initially formed PIA observed within the first 5 ps, forming a band centred at ∼720 nm. This state continues to decay over the course of the time-delays probed, as the negative band assigned to ground state bleaching and stimulated emission, recovers. Although no direct spectral fingerprint is observed for the formation of the reduced Re centre by TA spectroscopy in the UV/vis spectral region, it is clear from the simplification of the TA spectra, combined with the greatly increased lifetime of the ground state bleach, that the presence of the Re centre within the COF leads to the formation of a long-lived charge separated (non-emissive) state.
Density functional theory (DFT) and time-dependent (TD) DFT calculations—performed on representative molecular models Bpy-sp2c(L) and Re-Bpy-sp2c(L) of Bpy-sp2c-COF and Re-Bpy-sp2c-COF, respectively—show that the electron affinity (EA) and the ionization potential (IP) of both COFs straddle the reduction potential of CO2 to CO, as well as the proton reduction potential, and the oxidation potential of TEOA (Fig. S47 and S48†). This provides a thermodynamic explanation for the ability of Re-Bpy-sp2c-COF to drive CO2 reduction to CO, in the presence of the sacrificial agent TEOA. Relative energy levels of the dye and the molecular COF models confirm that it is thermodynamically allowed for excited electrons on the dye to be transferred to Re-Bpy-sp2c-COF (Fig. S48†), in line with the dye-sensitization effects observed experimentally.
TD-CAM-B3LYP calculations predict that the lowest-energy, excited electronic state (S1) for both Bpy-sp2c(L) and Re-Bpy-sp2c(L) corresponds to the LUMO ← HOMO transition, with a strong oscillator strength (Table S4†). Electron distributions of the excited-state frontier orbitals show that for both Bpy-sp2c(L) and Re-Bpy-sp2c(L) the HOMO orbital is predominantly located on the pyrene unit of the COF, with the LUMO orbital mainly located on the bipyridine unit (with or without ligated Re complex; Fig. S49 and S50†). Analyses of excited-state, inter-fragment charge transfer between the building units of the COFs indicate that appreciable amounts of electrons are transferred from the pyrene fragment to the bipyridine fragment (Table S5†), with a sizable electron–hole distance as measured by the charge centroids of the orbitals involved (Δr in Table S4†). Our computational results clearly support that there is electron transfer from the COF backbone to the catalytically active Re complex upon electronic excitation.
The CO2 reduction mechanism of the COF compared to the homogenous catalysts is therefore different. Here we propose pyrene excitation to a bipyridine based LUMO. In contrast, in solution excitation upon irradiation forms a metal to bipyridine excited state (3MLCT) which is then quenched by an electron donor.43
Crystallinity25 and accessible surface area44 have been shown to be important factors for the photocatalytic activity of organic photocatalysts. To probe whether these factors also influence the performance of the COF in photocatalytic CO2 reduction, we synthesized an amorphous analogue by using 1,4-dioxane instead of a 1,2-dichlorobenzene/1-butanol mixture under otherwise exactly the same experimental conditions, Re-Bpy-sp2c-P (PXRD, see Fig. S12†), which shows low CO2 uptakes and BET surface area (Fig. S19 and S39†). Despite having comparable FT-IR, UV-visible and PL spectra (Fig. S8, S15 and S24†), the amorphous polymer showed significantly lower activity for CO2 reduction (Table S2†) after being loaded with Re with a TON of 2.3 after 12 hours compared to 12.9 for Re-Bpy-sp2c-COF. This highlights that morphological properties, such as crystallinity and porosity, are important in these materials.
We showed previously that COFs that have accessible pores can potentially act as a host for dyes giving rise to increased photocatalytic activity for hydrogen production.25 Here, we used (Ir[dF(CF3)ppy]2(dtbpy))PF6 (ppy = 2-phenylpyridine, tbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl) in conjunction with Re-Bpy-sp2c-COF to further enhance the photocatalytic performances. Different amounts of the dye were used, and the CO production rates were enhanced by 32% and 84% compared to the unsensitized COF when using 0.3 mmol and 1.0 mmol of the dye, respectively, with 1 mg COF over 5 hours (Fig. 3e). The H2 production rates were unaffected. The highest CO production rates were 1400 μmol h−1 g−1, with a selectivity of 86% for CO, over 5 hours from Re-Bpy-sp2c-COF loaded with 1.0 mmol dye. It appears to be an electron transfer mechanism between the dye and the COF via oxidative quenching as suggested by emission quenching experiments (Fig. S32†).
Finally, we explored Re-Bpy-sp2c-COF loaded with additional in situ photodeposited colloidal Pt as a photocatalyst for syngas production, hence, simultaneous evolution of CO and H2. Syngas is used in chemical industry on large scale for processes, such as Fischer–Tropsch, and control of the ratio is important. The production of syngas with tunable ratio of CO and H2 has been reported for electrocatalysts45,46 and inorganic photocatalysts.47,48 By adding different amounts of Pt, Re-Bpy-sp2c-COF could produce high rates of CO-rich or H2-rich mixtures ranging from approximately 4:1 to 1:10 for CO:H2 (Fig. 3f).
Conclusions
In conclusion, we have synthesized a new porous, crystalline bipyridine-containing sp2c-COF, which was post-synthetically modified with a rhenium complex to enhance the photocatalytic CO2 reduction performance. Re-Bpy-sp2c-COF achieved a CO production rate of 1040 μmol g−1 h−1 with 81% selectivity over H2 over 17.5 h illumination. This performance was enhanced over 5 hours by up to 84% by dye-sensitization, giving a CO production rate of 1400 μmol h−1 g−1 and a CO/H2 selectivity of 86%. Based on a range of experimental and computational techniques it appears that Re-Bpy-sp2c-COF operates by a markedly different mechanism compared to the homogeneous catalyst [Re(bpy)(CO)3Cl] which the Re-Bpy-sp2c-COF also outperforms in terms of stability. Crystallinity and porosity seem to be important in these materials since an amorphous, low-porosity analogue showed almost no photocatalytic activity.Author contributions
A. I. C. and Z. F. conceived the project. Z. F. synthesized and characterized the materials and performed photocatalysis experiments. L. C. conceived the modelling strategy and L. C. and X. W. performed the calculations. S. Y. C. carried out PXRD analyses. R. S. S. performed the TCSPC experiments. G. N. and A. J. C. carried out isotopic labelling experiments. L. L. performed photoelectrochemical measurements. A. M. G. carried out TA measurements and analysed the results with A. J. C. R. C. and X. L. configured the photocatalysis platform, including setup and methods for GC. R. C., X-Y. W. and L. C. interpreted the gas sorption isotherms. M. B. and X-Y. W. performed STEM imaging. A. I. C., R. S. S. and L. C. co-supervised the project. A. V. provided expertise and feedback. Data was interpreted by all authors and the manuscript was prepared by A. I. C., R. S. S., L. C., and Z. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC) (EP/N004884/1, EP/P034497/1, EP/S017623/1), Newton Fund (EP/R003580/1), and the Leverhulme Trust via the Leverhulme Research Centre for Functional Materials Design. Z. F., X. W. and L. L. thank the China Scholarship Council for a PhD studentship. We thank Dr Jesum A. Fernandes for help with XPS measurements and Dr John Ward for useful discussions.Notes and references
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Chem. Commun., 1997, 9, 841–842 RSC.
- Y. X. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y. L. Men, F. F. Cao, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400–6404 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS.
- S. Guo, H. Zhang, Y. Chen, Z. Liu, B. Yu, Y. Zhao, Z. Yang, B. Han and Z. Liu, ACS Catal., 2018, 8, 4576–4581 CrossRef CAS.
- H. P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T. X. Wang, A. Thomas and B. H. Han, ACS Catal., 2019, 10, 3959–3968 CrossRef.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem.–Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- S. Wang, M. Xu, T. Peng, C. Zhang, T. Li, I. Hussain, J. Wang and B. Tan, Nat. Commun., 2019, 10, 676 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- S. J. Lyle, P. J. Waller and O. M. Yaghi, Trends Chem., 2019, 1, 172–184 CrossRef.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- Y. Fu, X. Zhu, L. Huang, X. Zhang, F. Zhang and W. Zhu, Appl. Catal., B, 2018, 239, 46–51 CrossRef CAS.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- X. Zhuang, W. Zhao, F. Zhang, Y. Cao, F. Liu, S. Bi and X. Feng, Polym. Chem., 2016, 7, 4176–4181 RSC.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- A. Yassin, M. Trunk, F. Czerny, P. Fayon, A. Trewin, J. Schmidt and A. Thomas, Adv. Funct. Mater., 2017, 17, 1700233 CrossRef.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- E. Jin, J. Li, K. Geng, Q. Jiang, H. Xu, Q. Xu and D. Jiang, Nat. Commun., 2018, 9, 4143 CrossRef PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Chem. Commun., 1983, 536–538 RSC.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed.
- M. Schwarze, D. Stellmach, M. Schröder, K. Kailasam, R. Reske, A. Thomas and R. Schomäcker, Phys. Chem. Chem. Phys., 2013, 15, 3466–3472 RSC.
- E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374–7376 RSC.
- T. W. Schneider, M. Z. Ertem, J. T. Muckerman and A. M. Angeles-Boza, ACS Catal., 2016, 6, 5473–5481 CrossRef CAS.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- M. B. Ross, Y. Li, P. De Luna, D. Kim, E. H. Sargent and P. Yang, Joule, 2019, 3, 257–264 CrossRef CAS.
- M. B. Ross, C. T. Dinh, Y. Li, D. Kim, P. De Luna, E. H. Sargent and P. Yang, J. Am. Chem. Soc., 2017, 139, 9359–9363 CrossRef CAS PubMed.
- J. S. Lee, D. Il Won, W. J. Jung, H. J. Son, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2017, 56, 976–980 CrossRef CAS PubMed.
- J.-C. Hu, M.-X. Gui, W. Xia, J. Wu, Y.-N. Zhou, N. Feng, J. Xiao, H. Liu, C.-H. Tung, L.-Z. Wu and F. Wang, J. Mater. Chem. A, 2019, 7, 10475–10482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03800k |
| This journal is © The Royal Society of Chemistry 2020 |