
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction of a dihydroboryl cation to a boryl anion and its air-stable, neutral hydroboryl radical through hydrogen shuttling†
Stephan
Hagspiel
ab,
Merle
Arrowsmith
 ab,
Felipe
Fantuzzi
abc,
Alexander
Hermann
ab,
Valerie
Paprocki
ab,
Regina
Drescher
ab,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
ab,
Felipe
Fantuzzi
abc,
Alexander
Hermann
ab,
Valerie
Paprocki
ab,
Regina
Drescher
ab,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cInstitut für Physikalische und Theoretische Chemie, Julius-Maximilians-Universität Würzburg, Emil-Fischer-Straße 42, 97074 Würzburg, Germany
First published on 25th November 2019
Abstract
The addition of Lewis bases to a cyclic (alkyl)(amino)carbene (CAAC)-supported dihydroboron triflate yields the mixed doubly base-stabilised dihydroboryl cations [(CAAC)BH2L]+. Of these, [(CAAC)2BH2]OTf (OTf = triflate) underwent facile two-electron reduction with KC8 owing to a 1,2-hydride migration from boron to the carbene carbon to yield a stable hydroboryl anion. One-electron oxidation of the latter yielded the first neutral hydroboryl radical, which is bench-stable in the solid state.
Introduction
Cyclic (alkyl)(amino)carbenes (CAACs) have become the ligands of choice for the stabilisation of many main group compounds in low oxidation states owing to their excellent σ-donor and π-acceptor properties derived from a relatively high-lying HOMO and low-lying LUMO.1–4 In the field of low-valent mononuclear boron chemistry, they have been successfully employed to synthesise unusual boron(II) species such as boryl radicals ([(CAAC)BXY]˙; X, Y = anionic ligands, e.g.I, Fig. 1a),5–10 boryl radical cations ([(CAAC)LBY]˙+, L = Lewis donor)10–13 and boryl anions ([(CAAC)BXY]−, e.g.II),14–17 as well as boron(I) species such as borylenes ((CAAC)LBX, e.g.III, and (CAAC)BNR2).6–8,11–13,16,18–20 In all these compounds, the accumulation of negative charge on the low-valent boron centre is stabilised through π backbonding to the CAAC ligand(s) (Fig. 1a), making many of them surprisingly stable under inert conditions.1–4 Recently, transient dicoordinate (CAAC)-stabilised borylenes have drawn particular attention as compounds capable of activating and catenating N2,21–25 the latter reaction being unprecedented even in transition metal chemistry.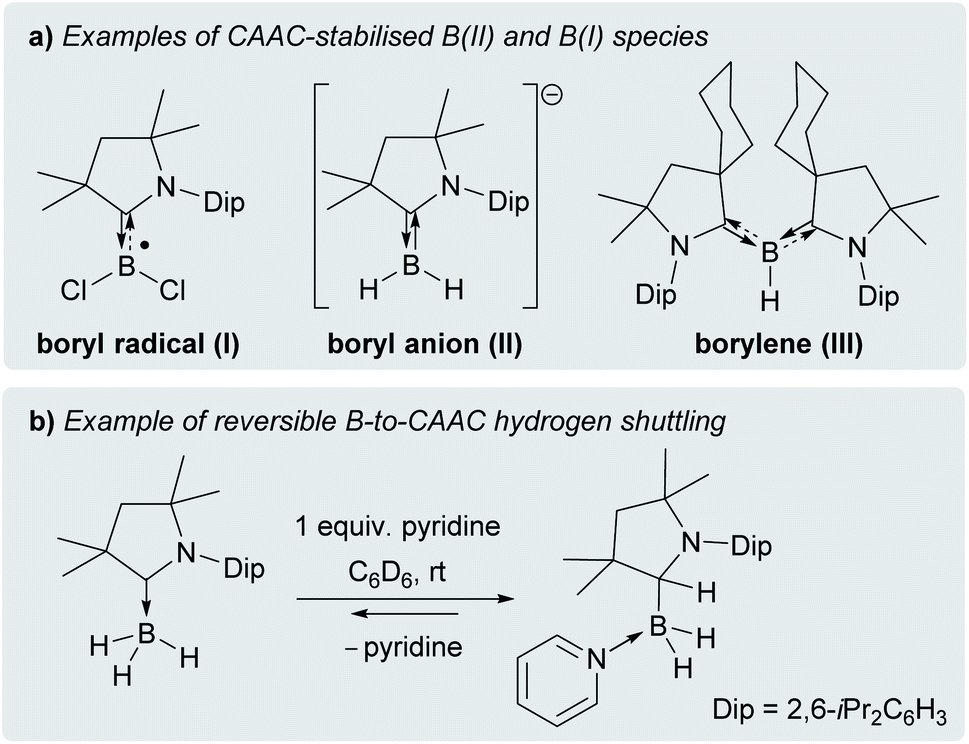 | ||
| Fig. 1 (a) Selected examples of CAAC-stabilised B(II) and B(I) species; (b) example of reversible Lewis-base-induced B-to-CAAC hydrogen shuttling. | ||
Furthermore, CAACs have been shown to activate element-hydrogen σ bonds, including H–H, N–H, P–H, Si–H and B–H by addition to their nucleophilic carbene carbon.3,4 In CAAC-supported hydroboron compounds, the B–H bond activation process can be reversible (Fig. 1b)14,26,27 and is favoured by electron-donating ligands at boron,8,26–30 thereby affording additional stabilisation for electron-rich lower oxidation state species through facile hydrogen shuttling. In this contribution we combine the excellent σ-donating/π-accepting and B–H bond activating properties of CAACs to synthesise and isolate a solvent-free alkyl(hydro)boryl anion, and selectively oxidise it to the corresponding radical, which is surprisingly air-stable in the solid state.
Results and discussion
Following a procedure by Bertrand and co-workers,12 methyl trifluoromethanesulfonate (MeOTf) was employed to abstract a hydride from (CAACMe)BH3 (CAACMe = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene). The resulting triflate derivative 1 was treated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with a series of Lewis bases in benzene to generate the bis(base)-stabilised boronium cations [(CAACMe)BH2L]OTf (2-L,L = CAACMe, IMeMe = 1,3-dimethylimidazol-2-ylidene, PMe3, Scheme 1a), all presenting a characteristic upfield 11B NMR BH2 triplet in the −22 to −30 ppm region.§ In the case of the 4-dimethylaminopyridine (DMAP) derivative, 2-DMAP (δ11B = −10.6 ppm, broad), the synthesis had to be carried out in THF as treatment of 1 with one equivalent of DMAP in benzene resulted in the formation of the bis(DMAP) adduct 3-DMAP (δ11B = 4.2 ppm, Scheme 1b), in which the second DMAP equivalent has promoted a typical 1,2-migration of one hydrogen atom from boron to the CAACMe ligand.26 The solid-state structure of 3-DMAP (Fig. 2) evidences the binding of the DMAP residues and the migration of H1 to C1, which is now sp3-hybridised (B1–C1 1.619(4), C1–N1 1.490(3) Å). In contrast, the binding of a second equivalent of pyridine to 2-Pyr (δ11B = −9.3 ppm, broad) was found to be reversible: even in neat pyridine only ca. 75% conversion to 3-Pyr (δ11B = 6.9 ppm) was observed. The use of 4,4′-bipyridine as a base led to the formation of the 4,4′-bipyridine-bridged bis(boronium) species 4-Bipy (δ11B = −8.6 ppm, broad, Scheme 1c). Attempts to synthesise the derivative 2-thf in THF resulted in ring-opening polymerisation of the solvent within two days at room temperature.
:1 ratio with a series of Lewis bases in benzene to generate the bis(base)-stabilised boronium cations [(CAACMe)BH2L]OTf (2-L,L = CAACMe, IMeMe = 1,3-dimethylimidazol-2-ylidene, PMe3, Scheme 1a), all presenting a characteristic upfield 11B NMR BH2 triplet in the −22 to −30 ppm region.§ In the case of the 4-dimethylaminopyridine (DMAP) derivative, 2-DMAP (δ11B = −10.6 ppm, broad), the synthesis had to be carried out in THF as treatment of 1 with one equivalent of DMAP in benzene resulted in the formation of the bis(DMAP) adduct 3-DMAP (δ11B = 4.2 ppm, Scheme 1b), in which the second DMAP equivalent has promoted a typical 1,2-migration of one hydrogen atom from boron to the CAACMe ligand.26 The solid-state structure of 3-DMAP (Fig. 2) evidences the binding of the DMAP residues and the migration of H1 to C1, which is now sp3-hybridised (B1–C1 1.619(4), C1–N1 1.490(3) Å). In contrast, the binding of a second equivalent of pyridine to 2-Pyr (δ11B = −9.3 ppm, broad) was found to be reversible: even in neat pyridine only ca. 75% conversion to 3-Pyr (δ11B = 6.9 ppm) was observed. The use of 4,4′-bipyridine as a base led to the formation of the 4,4′-bipyridine-bridged bis(boronium) species 4-Bipy (δ11B = −8.6 ppm, broad, Scheme 1c). Attempts to synthesise the derivative 2-thf in THF resulted in ring-opening polymerisation of the solvent within two days at room temperature.
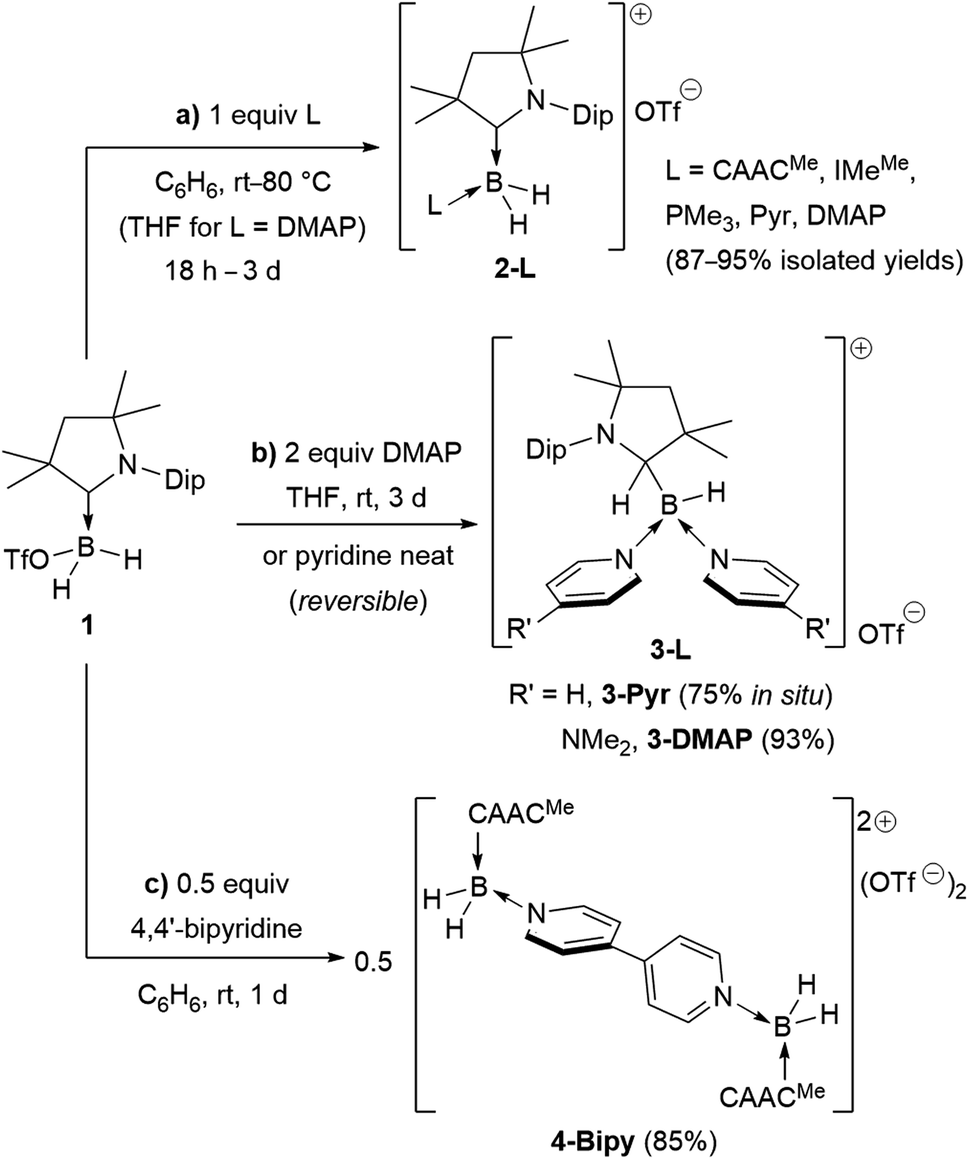 | ||
| Scheme 1 Syntheses of bis- and tris(base)-stabilised boronium cations (a) 2-L, (b) 3-L and (c) 4-L. Isolated yields in brackets. IMeMe = 1,3,4,5-tetramethylimidazol-2-ylidene, Pyr = pyridine, DMAP = 4-dimethylaminopyridine. | ||
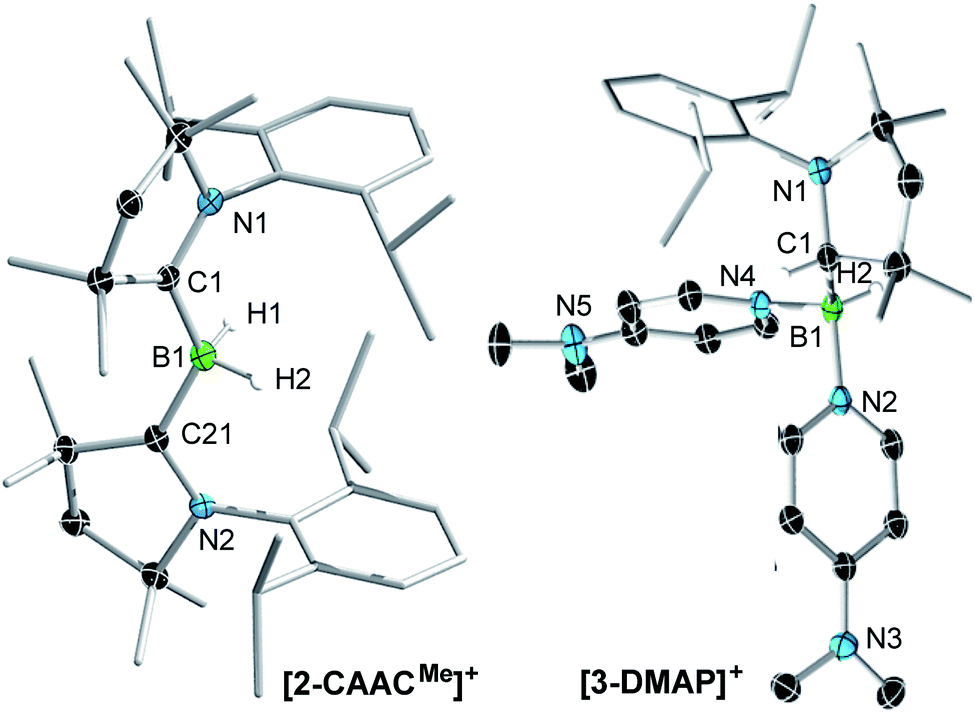 | ||
| Fig. 2 Crystallographically derived molecular structures of the 2-CAACMe (one of the two crystallographically distinct cations present in the asymmetric unit) and 3-DMAP cations. Atomic displacement ellipsoids are set at 50% probability. Ellipsoids of CH3 and iPr groups, triflate counteranion and hydrogen atoms omitted for clarity except for boron-bound hydrides.‡ Selected bond lengths (Å) for 2-CAACMe: B1–C1 1.597(7), B1–C21 1.607(7), B1–H1 1.11(6), B1–H2 1.16(6), C1–N1 1.316(6), C21–N2 1.310(6); for 3-DMAP B1–C1 1.619(4), B1–N2 1.585(3), B1–N4 1.597(3), B1–H2 1.10(2), C1–N1 1.490(3). | ||
Attempts to reduce 2-L, 3-L and 4-L under various conditions all resulted in unselective reactions, except for 2-CAACMe, which was readily reduced with excess KC8 to the red-coloured (alkyl)hydroboryl anion 5 by 1,2-migration of one hydrogen atom from boron to CAACMe (Scheme 2a). The 11B NMR spectrum of 5 shows a single broad resonance at 16.7 ppm, significantly downfield-shifted from that of other CAAC-stabilised boryl anions, which range from δ11B = −4.7 ppm for [(CAACMe)BH2]− to δ11B = −17.9 ppm for [(CAACCy)B(CN)2]−,14–17 likely because of the electron-withdrawing nature of the aminoalkyl substituent CAACMeH. The 1H{11B} NMR spectrum shows a BH doublet at 1.90 ppm (3J = 6.6 Hz), coupling to the BCH resonance of the CAACMeH ligand at 4.38 ppm, as well as two sets of unsymmetrical CAACMe ligand resonances. An X-ray crystallographic analysis revealed a monomeric structure with a trigonal-planar boron atom (Σ∠B1 359(1)°), in which the potassium cation bound to the BH hydride (K1⋯H2 2.53(3) Å) is encapsulated by the ligand sphere through η6–π interactions with the Dip (=2,6-diisopropylphenyl) substituents of the CAACMe and CAACMeH ligands (Fig. 3). The B1–C1A bond length of 1.439(11) Å is significantly shorter than in the 2-CAACMe precursor (B–Cavg. 1.69 Å, Fig. 2) and typical of a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. This is indicative of strong π backdonation from the lone pair of the boryl anion to the π-accepting CAACMe ligand, as found in all CAAC-stabilised boryl anions.6,14–17 According to DFT calculations carried out at the ωB97XD/6-31+G* level of theory, the HOMO of 5 possesses π-bonding character between B1 and C1A, with a nodal plane located at the C1A-N1′ bond region (Fig. 4). As in 3-DMAP, a 1,2-hydride shift has occurred and C1B is now sp3-hybridised (B1–C1B 1.633(9), N1–C1B 1.520(8) Å). The presence of the hydrogen atom at boron was further confirmed by a solid-state infrared absorption at 2329 cm−1, corresponding to the B–H stretching mode. The computed B–H stretching mode of 2352 cm−1 at ωB97XD/6-31+G* agrees well with the experimental value.
C double bond. This is indicative of strong π backdonation from the lone pair of the boryl anion to the π-accepting CAACMe ligand, as found in all CAAC-stabilised boryl anions.6,14–17 According to DFT calculations carried out at the ωB97XD/6-31+G* level of theory, the HOMO of 5 possesses π-bonding character between B1 and C1A, with a nodal plane located at the C1A-N1′ bond region (Fig. 4). As in 3-DMAP, a 1,2-hydride shift has occurred and C1B is now sp3-hybridised (B1–C1B 1.633(9), N1–C1B 1.520(8) Å). The presence of the hydrogen atom at boron was further confirmed by a solid-state infrared absorption at 2329 cm−1, corresponding to the B–H stretching mode. The computed B–H stretching mode of 2352 cm−1 at ωB97XD/6-31+G* agrees well with the experimental value.
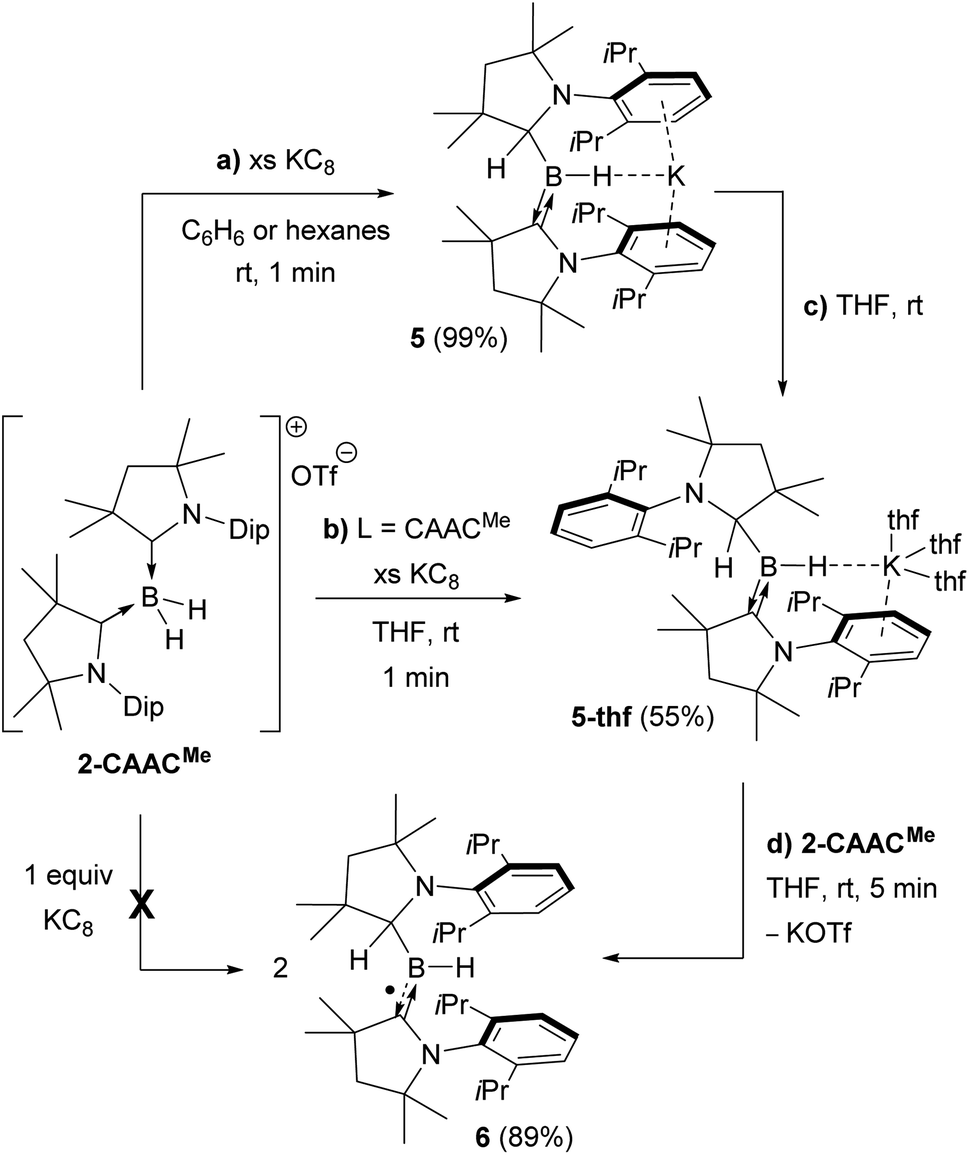 | ||
| Scheme 2 Reduction of 2-CAACMe to boryl anions (a) 5 and (b)–(c) 5-thf, and subsequent comproportionation to (d) boryl radical 6. | ||
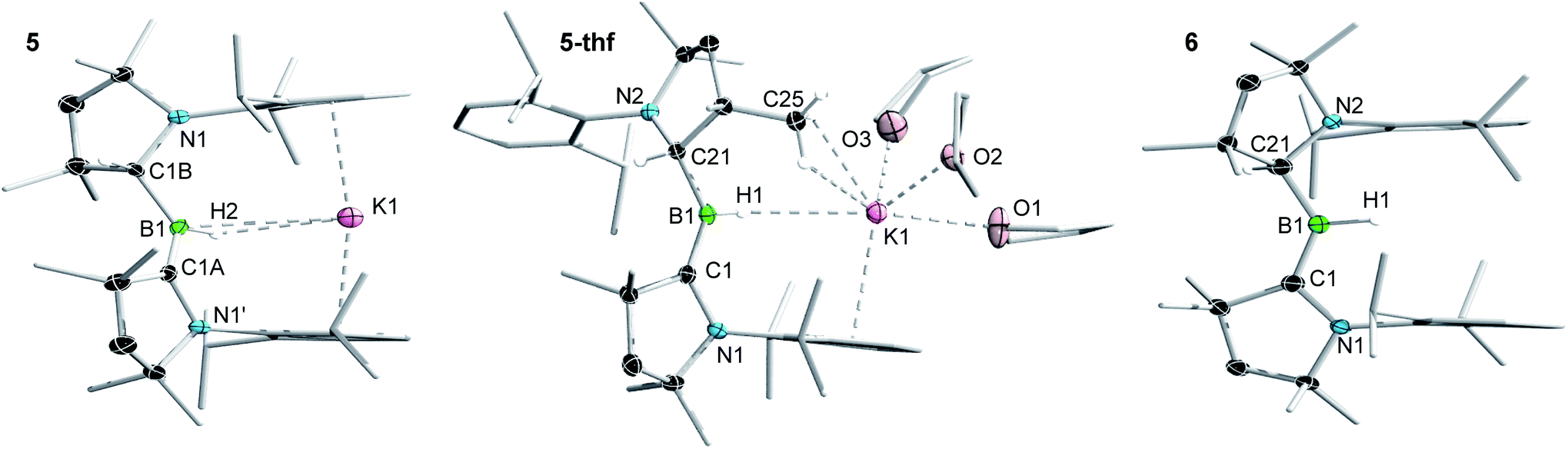 | ||
| Fig. 3 Crystallographically derived molecular structures of 5, 5-thf and 6. Atomic displacement ellipsoids are set at 50% probability. Ellipsoids of CH2, CH3 and iPr groups and hydrogen atoms omitted for clarity except for boron-bound hydrides.‡ Selected bond lengths (Å) and angles (°) for 5: B1–C1A 1.439(11), B1–C1B 1.633(9), B1–H2 1.14(3), C1A-N1′ 1.450(7), C1B–N1 1.520(8), K1⋯H1 2.53(3), K1⋯B1 3.141(4), K1⋯centroid 2.91, Σ∠B1 359.4(12), Σ∠C1A 359.7(5), B1–H2–K1 111.8(12); for 5-thf: B1–C1 1.452(2), B1–C21 1.620(2), B1–H1 1.159(17), C1–N1 1.4601(18), C21–N2 1.5076(19), K1⋯H1 2.653(16), K1⋯B1 3.599(2), K1⋯centroid 2.95, K1⋯C25 3.2933(17), Σ∠B1 359.9(1), Σ∠C1 359.9(1), B1–H1–K1 138(1); for 6: B1–C1 1.5174(18), B1–C21 1.5817(18), B1–H1 1.142(18), C1–N1 1.3777(15), C21–N2 1.4616(15), Σ∠B1 359.5(6), Σ∠C1 359.6(1). | ||
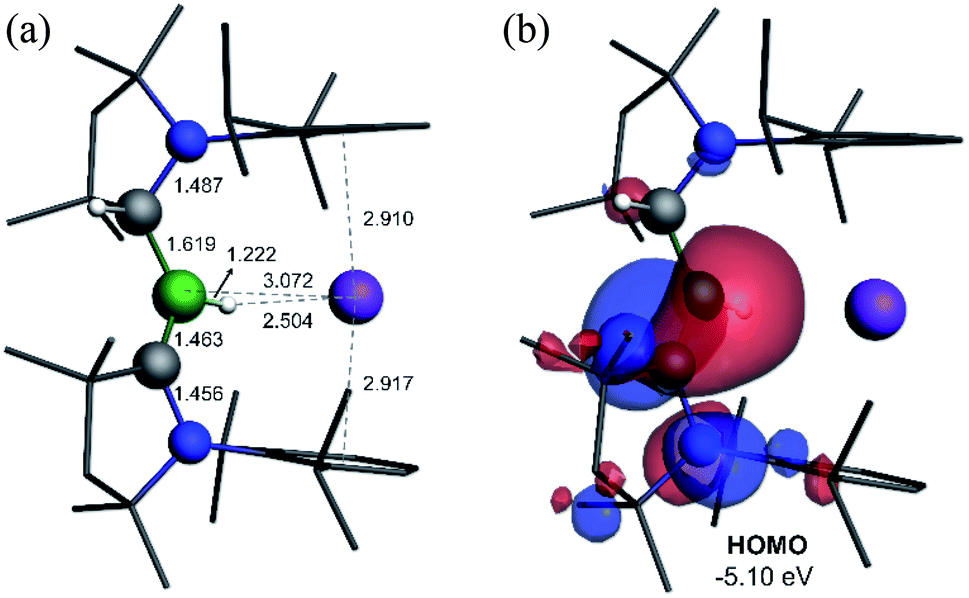 | ||
| Fig. 4 (a) Calculated structure of 5 at the ωB97XD/6-31+G* level of theory. (b) Plot of the HOMO of 5 (ωB97XD/6-311++G**). | ||
The reduction of 2-CAACMe in THF or the dissolution of 5 in THF both yielded the analogue 5-thf (Scheme 2b, c and Fig. 3), in which the hydride-bound potassium cation is η6–π-stabilised now only by the Dip substituent of the neutral CAACMe ligand, its coordination sphere being completed by three THF molecules and an agostic interaction with one of the vicinal methyl groups (C25) of the CAACMeH ligand. The bond lengths and angles of the boryl anion core change little compared to those of solvent-free 5, the major difference being the conformation of the pyrrolidine rings of CAACMeH and CAACMe, which flip so that the Dip substituents now point in opposite directions.
Cyclic voltammograms of 2-CAACMe and 5-thf in THF (0.1 M [nBu4N][PF6]) were essentially identical, showing a reversible redox event at E1/2 = −2.31 V and an irreversible oxidation around −0.90 V (relative to Fc/Fc+), suggesting that chemical oxidation of 5 to 6 should be possible. Indeed, the reaction of 5-thf with 2-CAACMe led to quantitative comproportionation to the boryl radical 6 (Scheme 2d). Attempts to generate 6 by the direct one-electron reduction of 2-CAACMe failed, resulting instead in incomplete consumption of 2-CAACMe and generating a mixture of 5 and 6. Radical 6 is deep purple in solution (λmax = 523 nm in the UV-vis spectrum) and 11B NMR-silent. In the solid state, however, isolated crystals of 6 are deep orange. X-ray diffraction analysis showed a structure very similar to 5 bar the potassium cation, with a trigonal planar B1 centre (Σ∠B1 359.5(6)°) and the Dip groups of the CAACMeH and CAACMe ligands both pointing in the same direction (Fig. 3). Unlike in 5 and 5-thf, the B1–C1 and C1–N1 bonds at the neutral CAACMe ligand (1.5174(18) and 1.4601(18) Å, respectively) are within the range typical of partial double bonds, as is typical for CAAC-stabilised boryl radicals due to the delocalisation of the unpaired electron over the N1–C1–B1 π framework.5–9,21,22,31–33
The IR spectrum of 6 shows a B–H stretching band at 2533 cm−1 (calc.: 2558 cm−1 at ωB97XD/6-31+G*), ca. 200 wavenumbers higher than that in 5, and 100 higher than in Bertrand's hydroborylene III (Fig. 1a, ν(B–H) = 2455 cm−1), suggesting a significant strengthening of the B–H bond in radical 6. The EPR spectrum of 6 displays a broad triplet from the hyperfine coupling to the 14N nucleus (a14N = 18.5 MHz, Fig. 5a). The simulated spectrum further provides hyperfine coupling parameters to the quadrupolar 11B nucleus (a11B = 9.7 MHz), which is responsible for the line-broadening, and the BH and CAACMeH1H nuclei (a1H = 13.6 and 4.8 MHz, respectively). The presence of two distinct couplings to these 1H nuclei suggests that the compound displays no fluxional B-to-CAAC hydrogen migration in solution.
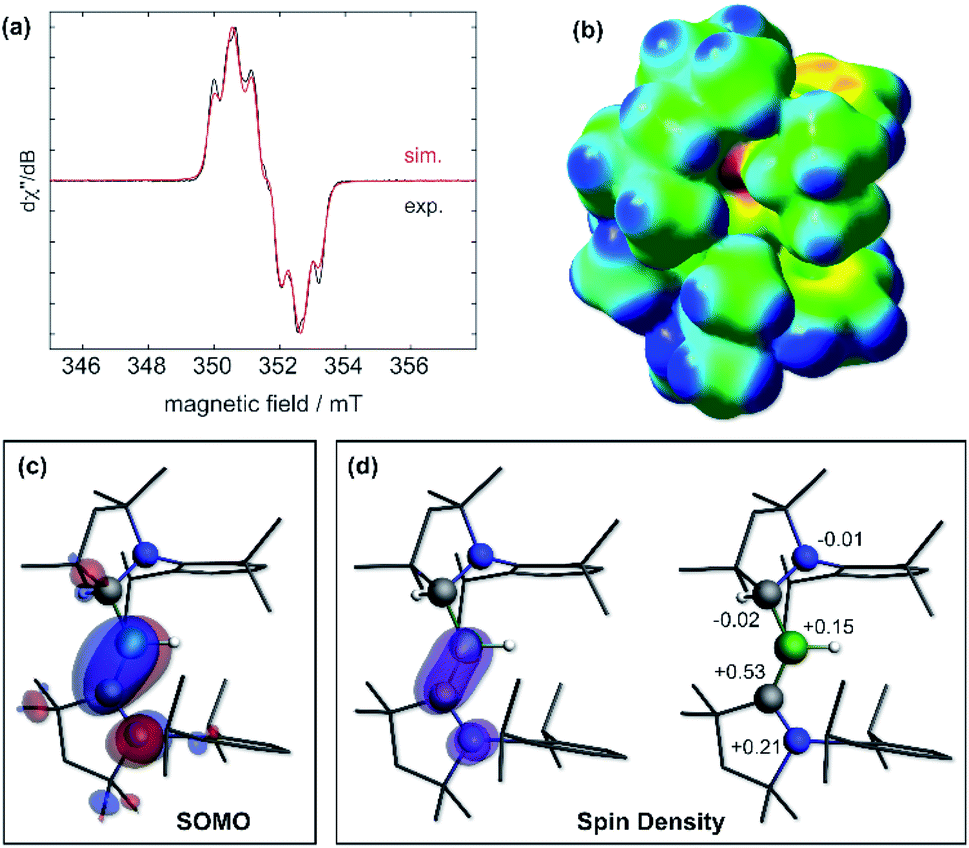 | ||
| Fig. 5 (a) Experimental (black solid line) and simulated (red line) continuous-wave X-band EPR spectra of 6 in hexane solution at rt. Simulation parameters: giso = 2.0027, a(11B) = 9.7 MHz, a(14N) = 18.5 MHz, a(1H(H1)) = 13.6 MHz and a(1H(H21)) = 4.8 MHz; (b) electrostatic potential (ESP) map of 6 at the ωB97XD/6-31+G* level of theory. ESP charges following the notation of Fig. 3: N2: −0.46, C21: −0.01, B1: +0.19, H1: −0.17, C1: −0.27, N1: −0.14. (c) Plot of the SOMO of 6 (surface isovalue: ± 0.03 [e a0−3]1/2). (d) Left: plot of the calculated spin density of 6 (surface isovalue: 0.005 [e a0−3]). Right: Mulliken atomic spin densities. | ||
Calculations show that the SOMO consists mainly of the B1–C1 π bond with some π-antibonding character on the C1–N1 bond (Fig. 5c). The calculated Mulliken atomic spin densities are 53% on C1, 21% on N1 and only 15% on B1, showing that the unpaired electron is mainly delocalised on the CAAC ligand (Fig. 5d), as already suggested by the much stronger EPR hyperfine coupling to N1 than B1 (vide supra). To our knowledge, 6 is the first example of a neutral, structurally characterised hydroboryl radical. Moreover, to our surprise, isolated crystals of 6 proved air-stable at room temperature over a period of one week, making this compound a rare example of an air-stable boron-centred radical. This is presumably owed to a combination of the high degree of spin delocalisation, the low spin density at boron and the very effective encapsulation of the B–H unit by the CAACMe and CAACMeH ligands as seen in the electrostatic potential map in Fig. 5b. The only other air-stable boron-based radical reported is a permethylated icosahedral borane [closo-B12(CH3)12]˙− radical anion, in which the unpaired electron is trapped and delocalised within the B12 cage.34
Reactions of the boryl anion 5 with a wide range of electrophiles including haloboranes, organohalides, heavier group 14 chlorides, as well as Zn(II), Cu(I) and Au(I) halides all resulted in quantitative oxidation of 5 to radical 6, and reduction of the corresponding electrophile. This contrasts with the boron nucleophile behaviour observed for CAAC-stabilised cyanoboryl anions.16,17 With elemental sulfur, double oxidation back to the 2-CAACMe cation was observed by NMR spectroscopic analysis (δ11B = −22.4 ppm, t, 1J11B–1H = 84.7 Hz), the counteranion presumably being a Sn2− polysulfide (7, Scheme 3a). The only nucleophilic reactivity observed was with methyl triflate, which yielded clean salt metathesis to the methylated trialkylborane 8 through migration of the second hydride to the remaining CAACMe ligand (δ11B = 93.9 ppm, Scheme 3b).
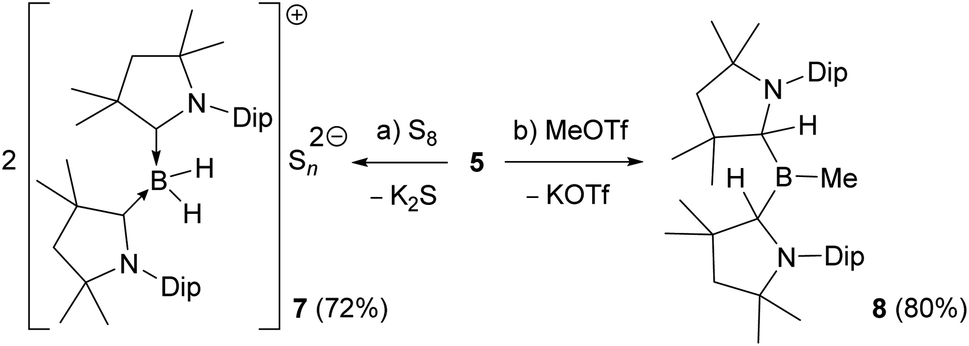 | ||
| Scheme 3 (a) Reducing and (b) nucleophilic reactivity of boryl anion 5. | ||
Conclusions
We have shown herein that the ability of CAACs to stabilise electron-rich boron centres and reversibly activate B–H bonds can be harnessed together to reduce a [L2BH2]+ cation to a [LRBH]− anion without the usual need for halide abstraction, thanks to B-to-CAAC hydrogen shuttling. This boryl anion reacts principally as a one-electron reducing agent to yield the neutral hydroboryl radical [LRBH]˙, the surprising stability of which is ensured by the unique stereoelectronic properties of the two encapsulating CAACMe ligands.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft for financial support. S. H. is grateful for a doctoral fellowship from the Studienstiftung des Deutschen Volkes. F. F. thanks the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and the Alexander von Humboldt (AvH) Foundation for a Capes-Humboldt postdoctoral fellowship.Notes and references
- S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727 RSC.
- U. S. D. Paul, M. J. Krahfuß and U. Radius, Chem. Unserer Zeit, 2018, 53, 212 CrossRef.
- M. Melaimi, R. J. M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346 CrossRef CAS.
- M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Härterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019, 10, 5095 RSC.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360 CrossRef CAS.
- J.-S. Huang, W.-H. Lee, C.-T. Shen, Y.-F. Lin, Y.-H. Liu, S.-M. Peng and C.-W. Chiu, Inorg. Chem., 2016, 55, 12427 CrossRef CAS PubMed.
- S. Kumar Sarkar, M. M. Siddiqui, S. Kundu, M. Ghosh, J. Kretsch, P. Stollberg, R. Herbst-Irmer, D. Stalke, C. Stückl, B. Schwederski, W. Kaim, S. Ghorai, E. D. Jemmis and H. W. Roesky, Dalton Trans., 2019, 48, 8551 RSC.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837 RSC.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed.
- M. Arrowsmith, J. D. Mattock, S. Hagspiel, I. Krummenacher, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15272 CrossRef CAS PubMed.
- M. Arrowsmith, J. D. Mattock, J. Böhnke, I. Krummenacher, A. Vargas and H. Braunschweig, Chem. Commun., 2018, 54, 4669 RSC.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, M. A. Celik, J. Erdmannsdörfer, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2017, 56, 11263 CrossRef CAS.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590 CrossRef CAS.
- J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368 CrossRef PubMed.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grüne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2106, 55, 14462 Search PubMed.
- M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282 CrossRef CAS PubMed.
- M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329 CrossRef PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231 CrossRef PubMed.
- C. Hering-Junghans, Angew. Chem., Int. Ed., 2108, 57, 6738 CrossRef.
- A. J. Ruddy, D. M. C. Ould, P. D. Newman and R. L. Melen, Dalton Trans., 2018, 47, 10377 RSC.
- D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066 RSC.
- M. Arrowsmith, J. Böhnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287 CrossRef CAS PubMed.
- S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem. –Eur. J., 2016, 22, 13032 CrossRef PubMed.
- M. R. Momeni, E. Rivard and A. Brown, Organometallics, 2013, 32, 6201 CrossRef CAS.
- G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444 CrossRef CAS PubMed.
- A. Deißenberger, E. Welz, R. Drescher, I. Krummenacher, R. D. Dewhurst, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 1842 CrossRef PubMed.
- J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. Röhr, R. Mitrić, B. Engels, F. Meyer and H. Braunschweig, Nat. Commun., 2018, 9, 1197 CrossRef PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, C. Claes, W. C. Ewing, I. Krummenacher, K. Lubitz and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 11271 CrossRef CAS PubMed.
- T. Peymann, C. B. Knobler and M. F. Hawthorne, Chem. Commun., 1999, 2039 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, EPR, UV-vis, IR, CV, X-ray crystallographic data and details of the computational analyses. CCDC 1956847–1956854. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05026d |
| ‡ The boron-bound hydrides of each structure were detected as residual electron density in the difference Fourier map and freely refined. |
| § The X-ray crystallographically-determined structures of 1, 2-Pyr and 2-DMAP can be found in the ESI, Fig. S55–S57.† |
| This journal is © The Royal Society of Chemistry 2020 |