DOI:
10.1039/C9RA10857B
(Paper)
RSC Adv., 2020,
10, 4218-4231
PLGA nanoparticle preparations by emulsification and nanoprecipitation techniques: effects of formulation parameters
Received
24th December 2019
, Accepted 13th January 2020
First published on 27th January 2020
Abstract
This study presents the influence of the primary formulation parameters on the formation of poly-DL-lactic-co-glycolic nanoparticles by the emulsification-solvent evaporation, and the nanoprecipitation techniques. In the emulsification-solvent evaporation technique, the polymer and tensoactive concentrations, the organic solvent fraction, and the sonication amplitude effects were analyzed. Similarly, in the nanoprecipitation technique the polymer and tensoactive concentrations, the organic solvent fraction and the injection speed were varied. Additionally, the agitation speed during solvent evaporation, the centrifugation speeds and the use of cryoprotectants in the freeze-drying process were analyzed. Nanoparticles were characterized by dynamic light scattering, laser Doppler electrophoresis, and scanning electron microscopy, and the results were evaluated by statistical analysis. Nanoparticle physicochemical characteristics can be adjusted by varying the formulation parameters to obtain specific sizes and stable nanoparticles. Also, by adjusting these parameters, the nanoparticle preparation processes have the potential to be tuned to yield nanoparticles with specific characteristics while maintaining reproducible results.
Introduction
Polymeric nanoparticle (PNP) preparation techniques are of great interest in biomedical research, in particular for drug delivery applications. The objective of PNPs in drug delivery applications is to achieve drug release in a controlled manner; to be biocompatible with tissues and cells; to reach higher intracellular uptake than free drugs; to improve the stability of active substances; and to be able to target specific tissues.1–3 An essential factor in the design of these systems is the size; the PNPs should be big enough to prevent fast incorporation into blood vessels but small enough to prevent elimination from the immune system.4 The PNP preparation techniques should be analyzed and adapted to each active substance to meet all these characteristics. Besides, it is essential to study the PNP preparation techniques to allow the production of reproducible large batches.5 The necessity to scale-up the PNP preparation process to the industrialized scale has been stated in earlier studies.6,7
Among the polymers used in the preparation of PNP, the most widely used is poly-DL-lactic-co-glycolic acid (PLGA). PLGA is successfully used in the research of drug delivery systems, because of its biodegradability and low systemic toxicity, it has also been approved by the US Food and Drug Administration (FDA) for medical applications.8–11 PLGA can be formulated as PNP using several different methods, such as emulsification-evaporation, nanoprecipitation or solvent displacement, solvent diffusion, and phase-inversion; with sizes ranging from 10 to 1000 nm.12,13 When encapsulating hydrophobic compounds, two of the most commonly used techniques are the emulsification-solvent evaporation technique and the nanoprecipitation technique.14–16 The emulsification technique is based in a mixture of a volatile non-water miscible solvent and an aqueous solution, which are emulsified by the application of high shear force. Then the volatile solvent is evaporated, forming in the process the PNP. This method is advantageous because it is nontoxic, rapid in reaction rate, and produces very small particles.17 Nevertheless, a disadvantage in the technique is the standardization for each specific drug, the high energy used in the process, which could affect the stability of certain drugs.18,19 On the other hand, in the nanoprecipitation method, the nanoparticles are formed in one step.20,21 Nanoprecipitation, involves the use of miscible solvents and its advantages include simplicity, good reproducibility, and low energy input.22,23 After the nanoparticle formulation with most techniques, some potentially toxic impurities such as organic solvents, surfactant excess, residual monomers, and large polymer aggregates must be eliminated. Some of the most common particle purification methods for laboratory scale include dialysis, gel filtration, evaporation under reduced pressure, and ultracentrifugation.24 Several works analyze the experimental parameters of different techniques for the encapsulation of specific drugs.25–34 The effect of some formulation parameters on the size of PLGA nanoparticles encapsulating bovine serum albumin, prepared by double emulsion solvent evaporation method, indicate that PLGA and PVA concentrations have a direct effect on the particle size.35 Halayqa and Domańska analyzed the effect of some parameters in the emulsion-evaporation technique to improve the efficiency of encapsulation and size in the preparation of PNP loaded with perphenazine and chlorpromazine hydrochloride.36 The influence of different experimental parameters on the incorporation efficiency of paclitaxel in the PLGA nanoparticles prepared by the interfacial deposition method was reported in the literature.37 Also, Derman et al., improve the encapsulation efficiency of caffeic acid phenethyl ester, using the oil in water (o/w) single emulsion solvent evaporation method, reporting high and sustained drug release.38 The same single emulsion technique was reported for PLGA nanoparticles preparations with simultaneously loaded vincristine sulfate and quercetin, by evaluating six independent parameters.39 The effects of different formulation parameters on particle size, zeta potential, drug loading efficiency and drug release of PLGA nanoparticles loaded with ciprofloxacin HCL and prepared by w/o/w emulsification solvent evaporation method is also reported in the literature.40 The effect of process and formulation parameters using nanoprecipitation on prednisolone loaded PLGA nanoparticles was studied.41 Also, Muthu et al., varied the concentration of PLGA and risperidone in the preparation of nanoparticles to improve the encapsulation efficiency.42 Govender et al. evaluated some formulation parameters to enhance the incorporation of a water-soluble drug (procaine hydrochloride) into PLGA nanoparticles.43 Madani et al. analyzed the effect of formulation parameters over the size of paclitaxel-loaded PLGA nanoparticles prepared by the emulsion and the precipitation methods.44 Other works have been focused not only in parameter studies but also in scale-up processes. Galindo-Rodríguez et al. made a comparative scale-up of three manufacturing processes; salting-out, emulsification–diffusion, and nanoprecipitation.45 Also, He et al. worked in the scalable fabrication of size-controlled chitosan nanoparticles for oral delivery of insulin.46 A comparison of the techniques of emulsion diffusion, solvent displacement, and double emulsion to prepare doxorubicin-loaded nanoparticles has also been reported in the literature.47 Sah et al. provided the fundamentals into emulsion solvent evaporation/extraction, salting-out, nanoprecipitation, membrane emulsification, microfluidic technology, and flow focusing.48 Mora-Huertas et al. describes the effect of the oil used in the recipes and preparation methods of nanoprecipitation and emulsification over the behavior of nanocapsules.49 Paliwal et al. made a review of the production methods of polymeric and lipid nanoparticles, their scale-up techniques, and commercialization challenges.50 Despite all the efforts in the encapsulation techniques of several actives into PLGA nanoparticles, is still essential to adapt these processes for the encapsulation of other different compounds. Therefore, this work aims to offer insights into the variable behaviors in the PLGA nanoparticles preparation processes to facilitate the encapsulation of other actives and for tune the nanoparticle-desired characteristics. Several factors that intervene in the PNP preparation processes were analyzed. In the emulsification-solvent evaporation technique, the polymer and tensoactive concentrations, the organic solvent fraction, and the sonication amplitude effects were analyzed. Similarly, in the nanoprecipitation technique were varied the polymer and tensoactive concentrations, the organic solvent fraction, and the injection speed. Additionally, the agitation speed during the solvent evaporation, the centrifugation speeds, and the use of cryoprotectants in the lyophilization process were studied. Nanoparticles were characterized by dynamic light scattering (DLS), laser Doppler electrophoresis, and scanning electron microscopy (SEM).
Materials and methods
Materials
PLGA acid terminated (50/50 DL-lactide/glycolide copolymer) with a molecular weight of 17 kg mol−1 (P17A), and PLGA (50/50 DL-lactide/glycolide copolymer) with a molecular weight of 153 kg mol−1 (P153) were received as gift sample from Corbion Purac, Gorinchem, The Netherlands. Polyvinyl alcohol (86–89% hydrolysis, low molecular weight, PVA) was obtained from Alfa Aesar, Ward Hill, Massachusetts, USA. Dichloromethane (DCM) was obtained from Fisher Scientific Inc., Fair Lawn, New Jersey, USA. Acetonitrile (AC) was obtained from Sigma Aldrich, Inc., St. Louis, MO, USA.
Preparation of PNP
PLGA nanoparticles were prepared by the single emulsification and the nanoprecipitation techniques.14,23,51 Fig. 1 illustrates the PNP preparation scheme for both techniques and the parameters evaluated within the study. Also, Table 1 presents the values of the parameters considered in each experiment; these parameters were analyzed individually while the rest of the conditions and procedures were maintained constant, as a control. The control conditions and procedure in the single emulsification technique are the following: an organic solution (DCM) containing 10 mg mL−1 of PLGA was added into an aqueous solution of 5% of PVA (with an organic solvent fraction of 0.167). The mixture was then emulsified for 1 min at 75% amplitude (90 μm) under an ice bath using a QSonica 500 sonicator (QSonica LLC, Newtown, Connecticut, USA). The control conditions and procedure in the nanoprecipitation technique are briefly described. An organic solution (AC) containing 10 mg mL−1 of PLGA was injected into an aqueous solution of 5% of PVA (with an organic solvent fraction of 0.167), using a NE-300 Just Infusion™ Syringe Pump (New Era Pump Systems Inc, Farmingdale, New York, USA). After both, the emulsification and the nanoprecipitation, the control parameters used in the solvent evaporation and purification are as follows: the organic solvent was evaporated under magnetic stirring at 400 rpm, at room temperature. Then, PNP were washed by three centrifugation cycles using a Sigma 3-30KS centrifuge (Sigma Laborzentrifugen GmbH Osterode am Harz, Germany) operated at 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 565 × g (20000 rpm) for 20 minutes, discarding supernatant and resuspending the pellet nanoparticles in deionized water.
565 × g (20000 rpm) for 20 minutes, discarding supernatant and resuspending the pellet nanoparticles in deionized water.
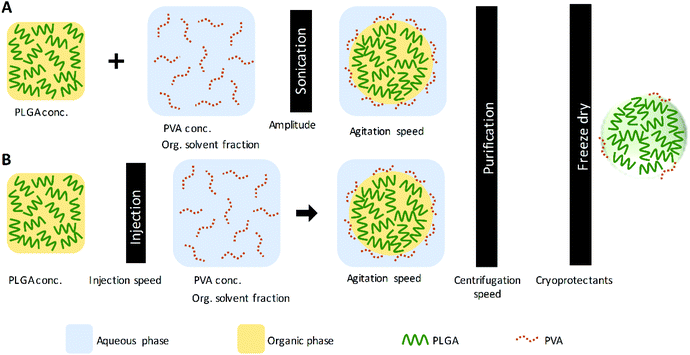 |
| | Fig. 1 Nanoparticle preparation scheme by (A) the single emulsification and (B) the nanoprecipitation techniques. | |
Table 1 Formulation parameters analyzed in the nanoparticle preparations by the single emulsification and the nanoprecipitation techniques
| Emulsification technique |
Nanoprecipitation technique |
| PLGA concentration (mg mL−1) |
5 |
10 |
15 |
PLGA concentration (mg mL−1) |
5 |
10 |
15 |
| PVA concentration (%) |
1 |
3 |
5 |
PVA concentration (mg mL−1) |
1 |
3 |
5 |
| Organic solvent fraction |
0.50 |
0.33 |
0.167 |
Organic solvent fraction |
0.50 |
0.33 |
0.167 |
| Sonication amplitude (%) |
25 |
50 |
75 |
Injection speed (mL min−1) |
0.6 |
1.2 |
2.4 |
| Speed of agitation in evaporation (rpm) |
200 |
300 |
400 |
Speed of agitation in evaporation (rpm) |
200 |
300 |
400 |
| General analysis |
| Purification [rpm (×g)] |
10000 (9391) |
15000 (21130) |
20000 (37565) |
| Cryoprotectant |
Sucrose |
Lactose |
Glucose |
A cryoprotectant solution (sucrose, glucose or lactose) was added to the PNP solution and then placed into a freezer at −80 °C. Finally, PNP were freeze-dried in lyophilizer freezone 2.5 Liter Benchtop (Labconco, Kansas City, Missouri, USA). Experiments were performed by triplicate.
Nanoparticle characterization
Nanoparticle size, size distribution, and zeta potentials were measured using a Zetasizer Nano ZS equipment (Malvern Instruments Ltd., Worcestershire, United Kingdom). Measurements of PNP sizes were performed by dynamic light scattering (DLS). Refraction index used in the analysis was 1.33 and water was used as the dispersant. Each sample was measured three times with 10 runs respectively for size analysis. Zeta potential of each sample was measured by duplicated with at least 10 runs at constant temperature (25 °C) by laser Doppler electrophoresis. Size averages and zeta potentials were obtained from three independent experiments. All the DLS sizes, PDI, and zeta potentials were measured after purification unless noted. Surface morphology of PNP was analyzed by scanning electron microscopy (SEM) through a field emission scanning electron microscopy (Hitachi S-4800 FE-SEM, Hitachi Corporation, Tokyo, Japan). Samples were prepared by placing a small number of lyophilized nanoparticles on a double-sided carbon tape, previously placed on a SEM stub. Compressed air was used to remove loose nanoparticles. The platinum coating was applied using with an Anatech Hummer 6.2 sputter system (Anatech USA, Hayward, California, USA). A total of 60 seconds under 10 mA under argon plasma were applied. A beam strength of 1.0 kV and a working distance in the range of 8–9 mm were used to visualize nanoparticles.
Data analysis
Statistical analysis for behavioral experiments was carried out on PNP size, polydispersity index and zeta potential measurements using R (ver. 3.0.1) with RStudio (ver. 1.2.1335). Results were analyzed by ANOVA followed by Tukey's HSD test (α = 0.05 was used, unless otherwise indicated) comparing different parameters such as PLGA concentration, PVA concentration, organic and aqueous phase ratio, sonication and agitation speed for nanoparticles prepared by emulsification. In the same manner, parameters studied for nanoparticles prepared by nanoprecipitation were PLGA concentration, PVA concentration, organic and aqueous phase ratio, and agitation speed. Also, the effect of the use of different cryoprotectants was considered in the statistical analysis. When the p-values were minor or equal than 0.05, the differences were considered statistically significant.
Results and discussion
PLGA nanoparticles were prepared using two of the most commonly used techniques to encapsulate hydrophobic compounds: the single emulsification solvent evaporation and the nanoprecipitation. The most critical parameters were investigated during the fabrication of PLGA nanoparticles to evaluate their effects on the mean particle size, polydispersity index (PDI) and zeta potential (ζ).
PLGA concentration effects on the size of the PNP
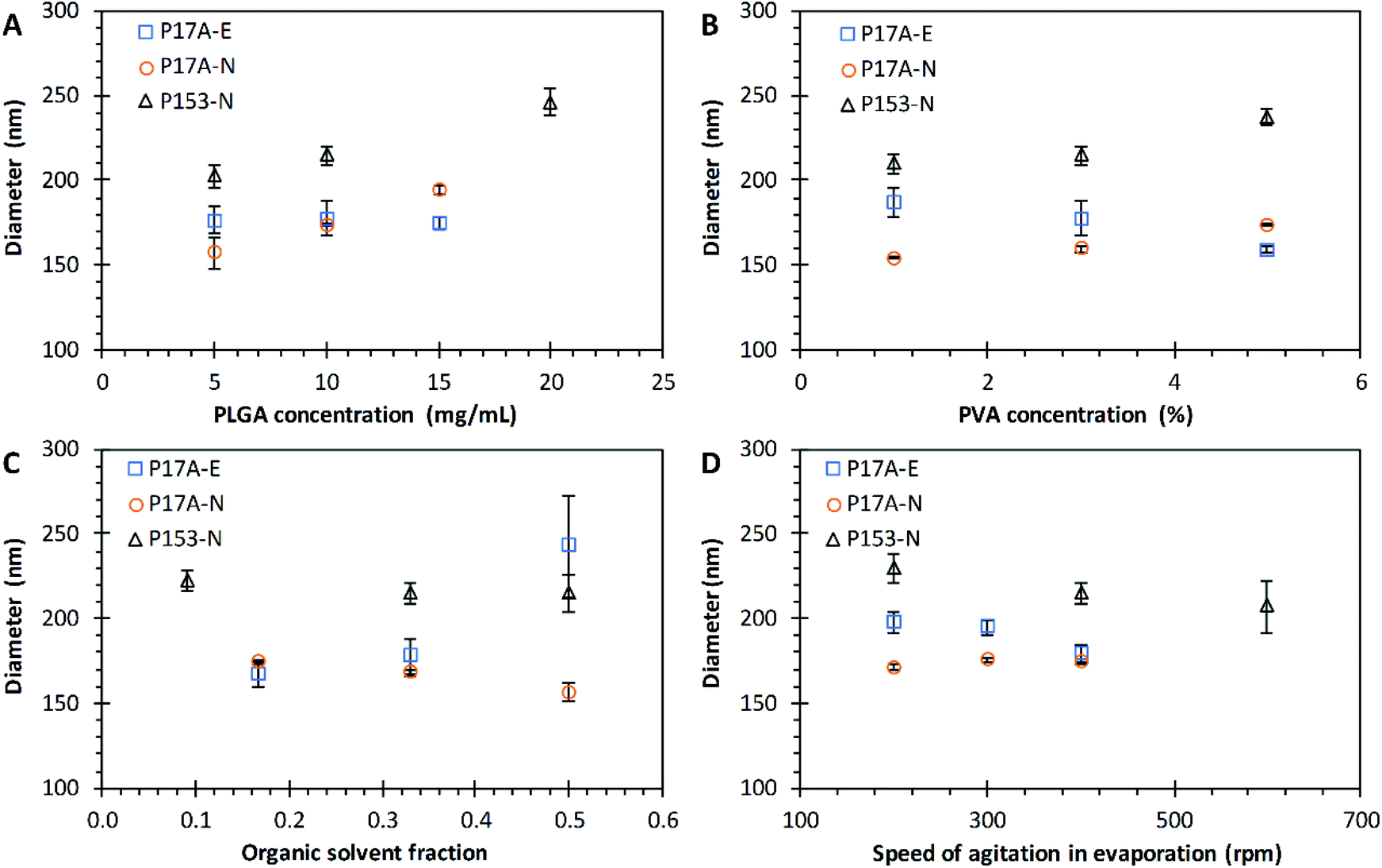
The effect of PLGA concentration over the diameter size of nanoparticles prepared by the emulsification and nanoprecipitation techniques are presented in Fig. 2A. Three concentrations of P17A were used in the nanoparticle preparations: 5 mg mL−1, 10 mg mL−1, and 15 mg mL−1. In this range of PLGA concentrations, no significant effect on the diameter size of PNP prepared by emulsification technique was observed, with diameters around 175 nm in all formulations (p > 0.05). Similarly, Xie et al., reported no significant effects of PLGA concentration over the diameter size in nanoparticles prepared by the double emulsification-evaporation solvent technique.52 In contrast, several authors report an increase in the particle size by increasing the polymer concentration.53–56 This difference is probably due to the PLGA concentration study range, the effect of the PLGA copolymers, lactic acid, and glycolic acid on the size of the PNP, and the molecular weight of the PLGA.57–59 In addition to molecular weight and co-polymer composition, PLGA can undergo several end-terminal modifications (such ester end-capping of its free carboxylic acid end-group) that affect the final physicochemical characteristics considerably.60 The end group of PLGA is a factor that affects the hydrophilicity of the polymer,61 PLGA with a final ester group is more hydrophobic than PLGA with a carboxyl group.62 For the nanoprecipitation technique, the same three concentrations of P17A than for emulsification technique were used. In this case, the size of nanoparticles increased proportionally to the concentration of P17A, obtaining nanoparticles with average sizes of 157.0 ± 9.0 nm, 174.0 ± 0.33 nm, and 194.5 ± 2.61 nm, respectively to P17A concentrations of 5 mg mL−1, 10 mg mL−1, and 15 mg mL−1; p = <0.05. A linear fit of experimental results correlates the PNP diameter size (D) with the concentration of P17A in the organic solvent (CP17A) following the equation D = 3.7511CP17A + 137.69, with an excellent coefficient of determination (R2 = 0.9971). This equation could help to tune the diameter size of nanoparticles of P17A prepared by nanoprecipitation. In addition, the size increment of PNP regarding the polymer concentration could be related to the phenomenon of super-saturation, which is important for the nucleation process, as discussed in the literature.63 Other authors explain that an increase in the viscosity of the organic phase means more polymer is contained in the drops formed during the nanoprecipitation process, leading to larger PNP.3,64 The nanoprecipitation technique was also employed to prepare nanoparticles with the polymer P153. The results were similar to the ones obtained by nanoprecipitation with P17A polymer; the size of the nanoparticles increase by increasing the concentration of P153 (CP153). The average diameter values were 202.5 ± 6.84 nm, 214.8 ± 5.60 nm, and 246.3 ± 7.51 nm, respectively to CP153 of 5 mg mL−1, 10 mg mL−1, and 20 mg mL; p = <0.05. A linear fit of this data results in the equation D = 2.952CP153 + 186.76, with an excellent R2 of 0.9968; showing that is possible to tune the diameter of P153 nanoparticles in function of CP153. These results indicate that the amount of PLGA plays an important role in the final size of PNP prepared by the nanoprecipitation technique. Also, the diameter size of PNP prepared by this technique could be tuned in function of the PLGA concentration used in the formulation. In contrast, the emulsification technique do not show a significant effect of the concentration of PLGA over the diameter size of PNP in the range studied.
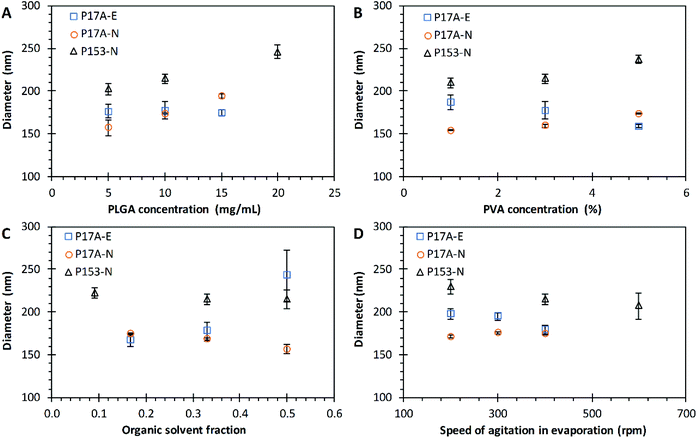 |
| | Fig. 2 Effect of varying formulation parameters on nanoparticle diameter size. (A) PLGA concentration; (B) PVA concentration; (C) organic solvent fraction; and (D) speed of agitation in evaporation. Preparations by the emulsification technique with PLGA of 17 kg mol−1, P17A-E ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N ( ), and PLGA of 153 kg mol−1, P153-N ( ). Data represent mean ± SD (n = 3). ). Data represent mean ± SD (n = 3). | |
PVA concentration effects on the size of the PNP
Another parameter considered during the PLGA nanoparticle preparations was concentration of PVA in the aqueous phase. Aqueous solutions of PVA of 1%, 3%, and 5% were used for the preparation of PNP in both techniques. The amount of surfactant plays an essential role in the emulsification-solvent evaporation process, by the protection of the droplets containing the PLGA against coalescence.65,66 Fig. 2B illustrates the effect of PVA concentration (CPVA) on the particle size for PNP prepared by both techniques. In the emulsification technique, the PNP sizes decreased as the CPVA increased, with average diameter values of 187.0 ± 8.5 nm, 177.7 ± 1.6 nm, and 159.1 ± 2.0 nm, correspondingly to CPVA of 1%, 3%, and 5%. However, this results are not statistically significant (p = 0.051), probably to the number of repetitions. Nevertheless, a trendline analysis of the experimental results indicate a fair linear fit with the equation D = −6.975CPVA + 195.53 (R2 = 0.9643). Miladi et al. found that the particle size decreases from 301 nm to 255 nm, when the PVA concentration increased from 0.25% to 1%, respectively.
They explained this result by the increased in the viscosity of the aqueous phase after the augment in the PVA concentration.67 Therefore, the decrease in the size of the PNP obtained in this work is probably due to the difference in the stability of the emulsion, formulated with different concentrations of PVA. Kejdušová et al. reported that an increase in the concentration of PVA guarantees a better stabilization of the system; hence, a decrease in the coalescence of the emulsion.66 Since PNP are forming from the emulsion droplets after solvent evaporation, its size depends on the size and stability of these droplets.68 Also, according to Sahoo et al. at concentrations lower than 2.5% w/v of PVA, this one exists as a single molecule in solution, and above this concentration, PVA exists as an aggregated form and has an enhanced surfactant activity. Besides, they found that the residual PVA associated with the nanoparticles increased by increasing the miscibility of the solvent with water.69 Therefore, droplets formed during emulsification would be more stable, resulting in smaller PNP. On the other hand, it was found that by using P17A in the nanoprecipitation technique, PNP sizes increased as the CPVA increases, with average diameter values of 154.5 ± 0.76 nm, 159.8 ± 1.85 nm, and 174.0 ± 0.33 nm, regarding to CPVA of 1%, 3%, and 5%, with statistically significant differences (p < 0.05). A fair linear fit of this data comes with the equation D = 4.8917CPVA + 148.09 (R2 = 0.9352). Similar results in the nanoparticle size when varying the CPVA were obtained by using P153 with the nanoprecipitation technique (Fig. 2B). The PNP sizes increased as CPVA increased (p < 0.05). In addition, a reasonable linear fit of the data could be observed, with the equation D = 6.835CPVA + 200.16 (R2 = 0.8803). These observed tendencies of PNP size increments by increasing the CPVA in the nanoprecipitation technique could be due to PVA deposited on the surface of the PNP, as reported in previous works.70 In agreement, Murakami et al., found that a certain amount of PVA remained adsorbed on the surface of the PLGA nanoparticles after the washing steps.71 Also, Badri et al. report similar results, they found that the augment of PVA, increased the particle size from 169 nm to 283 nm.22 Different tendencies in the size of nanoparticles were obtained between both techniques when the CPVA increased, finding that in the emulsification technique the size of PNP decreased, while in the nanoprecipitation technique the size of PNP increased. This difference could be because in the emulsification technique the stability of the emulsion is a critical step in the formation of the nanoparticle.72
Organic solvent fraction effects on the size of the PNP
The effect of organic solvent fraction (Fos) on the diameter size of the PNP prepared by the emulsification and nanoprecipitation techniques was evaluated. The polymer P17A was used in emulsification and nanoprecipitation; additionally, the polymer P153 was evaluated by the nanoprecipitation technique. The outcome of Fos over the diameter of PNP for both techniques is presented in Fig. 2C. An increase in the PNP diameter size with an increment of the Fos was observed in the emulsification technique (with P17A), with diameters of 167.5 ± 7.8 nm, 177.7 ± 10.6 nm, and 242.8 ± 30.0 nm, correspondingly to Fos of 0.167, 0.330, and 0.500, these results were statistically significant (p < 0.05). However, a poor linear fit of this data comes with the equation D = 227.25Fos + 120.48 (R2 = 0.8581). Other authors report similar results, for example, Habib et al., found that the size of the nanoparticles decreased when using acetone, with Fos values from 0.330 to 0.167.73 Also, a study with nanoparticles of mPEG-PLGA prepared by emulsion solvent evaporation reported that as the Fos increased, the system reduced the net shear stress due to a constant external energy input, which led to the increased size of the nanoparticles.74 The organic solvent fraction in the emulsification technique played an important role in the resulting PNP sizes. On the other hand, the evaluation of the Fos used in nanoprecipitation with P17A and P153 (Fig. 2C), shows that the size of the PNP decreased as Fos increased. The average diameter sizes values of PNP prepared with P17A were of 174.0 ± 0.3 nm, 167.8 ± 1.3 nm, and 156.6 ± 5.5 nm, respectively to the Fos of 0.167, 0.330, and 0.500; (p < 0.05). Also, a good linear fit of this data results in the equation D = −52.346Fos + 183.54 (R2 = 0.9786), showing that is possible to tune the diameter of P17A nanoparticles in function of Fos. Additionally, the average diameter sizes values of PNP prepared with P153 were 227 ± 6.1 nm, 214.8 ± 5.6 nm, and 214.6 ± 11.2 nm, correspondingly to Fos of 0.090, 0.167, and 0.500. Nevertheless, the statistical analysis did not show a significant difference between the range of Fos used (p > 0.05), possibly for the number of repetitions. The effect of the organic solvent fraction in the nanoprecipitation technique has been reported lately in the literature.75,76 Chaudhary et al. reported smaller cefixime loaded PLGA nanoparticles when the organic solvent fraction decreased, using a modified precipitation method; attributing this size reduction to the coalescence prevention.77 Also, de Oliveira et al. found that the mean particle size decreased when the organic solvent fraction decreased; if the size reduction was a result of the formation of a higher number of nucleation sites. This increase in the nucleation sites consequently leads to the generation of smaller particles.78 The nature of the organic solvent is also an important factor when manufacturing PNP via nanoprecipitation. The organic solvent should dissolve the polymer as well as miscible with water.79 Hence, other authors have studied the effect of the miscibility of the organic solvent in water. Cheng et al., report that an increase of water miscibility led to a decrease in the average docetaxel-loaded PEGylated PLGA nanoparticle size, which is presumably due to more efficient solvent diffusion and polymer dispersion into water.80 Also, Huang and Zhang report that solvents with a high diffusion coefficient favor the formation of smaller PNP with the narrower distribution.81 Therefore, it can be highlighted that the effect of the size of the PNP, depends not only on the range of study but also of the nature of the organic and aqueous phases used. Almoustafa et al. advises that choosing the solvent is the primary step in size tuning and encapsulation efficiency for the nanoprecipitation technique.20 Moreover, in the case of PNP with and encapsulated drug, the effect of the organic solvent fraction in particle size also depends on the solubility of the drug in the external phase.82
Speed of agitation effects on the size of the PNP
The effects of speed of agitation (Arpm) on the solvent evaporation over the diameter size of PNP prepared by the emulsification and nanoprecipitation techniques are presented in Fig. 2D. The PNP prepared by the emulsification technique with P17A were agitated at 200 rpm, 300 rpm, and 400 rpm, resulting in average diameter values of PNP of 198.2 ± 6.1 nm, 195.0 ± 4.1 nm, and 179.4 ± 5.2 nm, respectively. This results are statistically significant (p < 0.05); however only a reasonable linear fit of this data was found, resulting in the equation D = −0.0939Arpm + 219.06 (R2 = 0.8745). In analogous way, for the nanoprecipitation technique (P17A), the same three agitation speeds were evaluated, obtaining PNP with average diameter sizes of 171.4 ± 1.6 nm, 175.7 ± 1.7 nm and 174.0 ± 0.3 nm, respectively to speed of agitation of 200 rpm, 300 rpm, and 400 rpm; (p < 0.05). Despite the results are statistically significant, the diameter values were in a very close range; also, a bad linear fit of this data was found (not reported). Furthermore, in the same technique but using P153, the PNP diameter sizes resulted in 229.5 ± 8.3 nm, 214.8 ± 5.6 nm, and 207.0 ± 15.6 nm for 200 rpm, 300 rpm, and 600 rpm, respectively (Fig. 2D), with significant effects of the agitation (p < 0.05). A good linear fit of this data results in the equation D = −0.0562Arpm + 239.57, with R2 of 0.9705. Malkani et al., found that the agitation speed in the nanoprecipitation process had influence in the particle size of the celecoxib nanosuspension. This could be due to faster evaporation of the organic solvent, increasing the rate of drug precipitation and resulting in larger particles.83 Also, Lince et al., explain that the speed of agitation influences the nucleation speed of the polymer, where poor mixes result in slow nucleation speeds, generating larger particles; while proper mixing promotes the increase of the nucleation speed, leading to the formation of small particles.63 During the evaporation of the solvent, the surface hardening of the nanoparticles takes place.84 Therefore, the Arpm is an essential parameter in the formation of nanoparticles. In the range of speed of agitation studied for both techniques, emulsification and nanoprecipitation, the PNP size tend to decrease as the speed of agitation in the solvent evaporation increase.
PLGA concentration effects on the PDI of the PNP
The effects of PLGA concentration over the polydispersity index of PNP were evaluated in both techniques, emulsification and nanoprecipitation (Fig. 3A). The PLGA concentration did affect the PDI average value in the emulsification technique, with values of 0.140 ± 0.066, 0.110 ± 0.004, and 0.097 ± 0.024 when the P17A concentration increased from 5 mg mL−1, 10 mg mL−1, and 15 mg mL−1, respectively; in the three cases indicating a homogeneous size distribution. However, according to the statistical analysis, this differences are not significant (p > 0.05), probably indicating that a major number of repetitions are needed. Dangi and Shakya also found that the PDI was reduced by increasing the concentration of PLGA from 0.5 to 1.0.85 In addition, in this range of study, the P17A concentration have a significant effect on the PDI with the nanoprecipitation technique. The resulting values of PDI were 0.067 ± 0.005, 0.048 ± 0.002, and 0.041 ± 0.009 for P17A concentrations of 5 mg mL−1, 10 mg mL−1, and 15 mg mL−1, respectively (p < 0.05). These PDI were smaller than the ones obtained by the emulsification technique, indicating a narrow distribution size and the formation of uniform and homogeneous PNP for all formulations. Also, a fair linear fit comes with the equation PDI = −0.0026CP17A + 0.078 (R2 = 0.9297). The same effect of PDI over the nanoprecipitation technique was observed when P153 was used, obtaining a maximum value of PDI of 0.08, but with no statistical significances (p > 0.05). The results presented in the Fig. 3A, for both techniques, emulsification and nanoprecipitation, shown a slight trend to decrease the PDI values as increasing the concentration of PLGA.
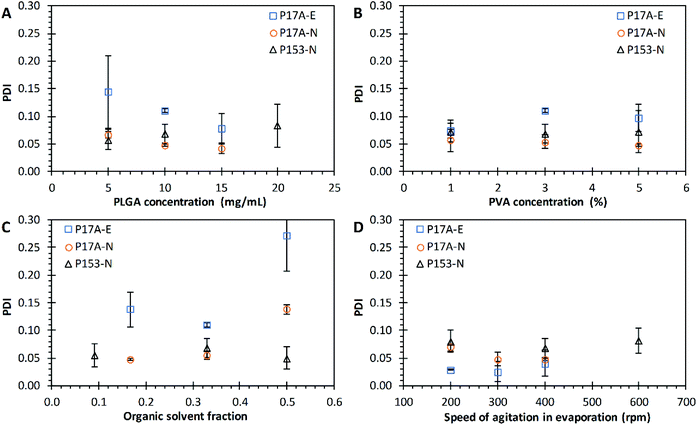 |
| | Fig. 3 Effect of varying formulation parameters on nanoparticle PDI. (A) PLGA concentration; (B) PVA concentration; (C) organic solvent fraction; and (D) speed of agitation in evaporation. Preparations by the emulsification technique with PLGA of 17 kg mol−1, P17A-E ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N ( ), and PLGA of 153 kg mol−1, P153-N (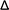 ). Data represent mean ± SD (n = 3). ). Data represent mean ± SD (n = 3). | |
PVA concentration effects on the PDI of the PNP
The effects of PVA concentration over the PDI of PNP were evaluated in both techniques, emulsification and nanoprecipitation, and are presented in Fig. 3B. The PNP average values of PDI obtained from the emulsification technique (P17A) were 0.074 ± 0.013, 0.111 ± 0.004, and 0.097 ± 0.002, corresponding to PVA concentrations of 1%, 3%, and 5%. However, the statistical analysis shows that an increase in PVA concentration does not show a significant effects on the final PDI (p > 0.05). Similarly, for the nanoprecipitation technique, with P17A and P153, the PDI values did not show variations in the range of PVA concentrations evaluated, obtaining PDI values around 0.05 (p > 0.05), and PDI values around 0.07 (p > 0.05), respectively. The solubility, viscosity, and surface tension of PVA vary in function of the temperature, concentration, % hydrolysis, and molecular weight of this polymer.86 In this context, several authors report that the degree of hydrolysis and the molecular weight of PVA have influence the PNP size, and the PNP polydispersity index.87–90
Organic solvent fraction effects on the PDI of the PNP
Fig. 3C presents the effect of Fos over the PDI values for the emulsification and the nanoprecipitation techniques. The resulting PDI values from the emulsification technique (P17A) were 0.138 ± 0.031, 0.111 ± 0.004, and 0.272 ± 0.064, corresponding to the Fos of 0.167, 0.330, and 0.500. These PDI values indicate that Fos affects directly to the monodispersity of the PNP (p < 0.05). Comparable, for the nanoprecipitation technique with P17A, the PDI values increased as the Fos increased, getting values of 0.048 ± 0.002, 0.056 ± 0.008, and 0.138 ± 0.008, for Fos of 0.167, 0.330, and 0.500, respectively (p < 0.05). In addition, when using P153 polymer and the nanoprecipitation technique, the average PDI values ranged between 0.05, and 0.07 for all the formulations, which indicates a narrow size distribution. Nevertheless, the statistical analysis of this data show no dependency of Fos with the PDI (p > 0.05). Despite all the values obtained show monodispersed PNP systems, the data obtained with both techniques and the two polymers were poorly fitted into a linear curve (data not shown).
Speed of agitation effects on the PDI of the PNP
The effects of Arpm on the solvent evaporation over the PDI of PNP prepared by the emulsification and nanoprecipitation techniques are presented in Fig. 3D. The PDI values obtained from the emulsification technique (P17A) were found between 0.03 and 0.04 for the range of Arpm studied, indicating a narrow size distribution, but with no significant differences (p > 0.05). Narayanan et al., reported that hyaluronidase loaded PLGA nanoparticles size is reduced due to shear stress, where PVA could participate stabilizing the reduced particles and preventing the formation of aggregates.91 The results of PDI obtained from the nanoprecipitation technique (P17A) also presented low PDI values, indicating narrow size distributions. In this case, the data resulted in significant differences (p < 0.05) but resulted in a poor fit to a linear behavior (data not shown). Similarly, the PDI values obtained with the nanoprecipitation technique, but using the polymer P153, shown narrow size distributions for all the Arpm studied, but with no significant differences (p > 0.05). Despite no trends were obtained with the Arpm over the PDI of PNP, is notable that all formulations presented narrow size distributions.
PLGA concentration effects on the ζ of the PNP
The effects of PLGA concentration over the ζ of PNP for both techniques were evaluated (Fig. 4A). The ζ values of PNP prepared by the emulsification technique ranged between −26.8 mV and −30.0 mV for all the P17A concentrations evaluated (p > 0.05). This negative charge was due to the presence of terminal carboxylic groups (–COOH) on the surface of nanoparticles.92,93 Similar results were obtained for the nanoprecipitation technique, with ζ values of PNP in the range of −20.8 mV and −23.4 mV for all the P17A concentrations evaluated (p < 0.05). Moreover, when using the same nanoprecipitation technique but with P153, the ζ values were in the range of −5.0 mV and −5.9 mV. The considerable difference in the ζ results between the formulations with P17A and P153 it is due to the polymers termination groups, which are acid terminated, and ester terminated, respectively. It should be mentioned that the anionic nature that PLGA gives to the nanoparticles also depends on its molecular weight and concentration.94,95
 |
| | Fig. 4 Effect of varying formulation parameters on nanoparticle zeta potential. (A) PLGA concentration; (B) PVA concentration; (C) organic solvent fraction; and (D) speed of agitation in evaporation. Preparations by the emulsification technique with PLGA of 17 kg mol−1, P17A-E ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N ( ), and PLGA of 153 kg mol−1, P153-N ( ). Data represent mean ± SD (n = 3). ). Data represent mean ± SD (n = 3). | |
PVA concentration effects on the ζ of the PNP
Fig. 5C presents the ζ of PNP obtained with different CPVA for both techniques, emulsification and nanoprecipitation. The ζ for the emulsification technique (P17A) shows an increase as the CPVA increase, with values of −29.4 ± 6.8 mV, −26.8 ± 5.0 mV, and −26.6 ± 5.5 mV, for CPVA of 1%, 3%, and 5%, respectively; however, these values do not present statistical differences (p > 0.05). Prabha and Labhasetwar reported that PVA attached to the surface nanoparticle affects the charge since the nanoparticles with the highest amount of PVA associated with the surface, presented a decrease in their anionic charge.96 In the case of the nanoprecipitation technique (P17A), the ζ decreased as the CPVA increased. The ζ values are −15.0 ± 1.0 mV, −18.3 ± 3.1 mV, and −23.4 ± 1.7 mV, corresponding to CPVA of 1%, 3%, and 5%. The influence of CPVA over the ζ is statistically significant, with a p < 0.05; also, a good linear fit could be obtained with the equation ζ = −2.1028CPVA − 12.566 (R2 = 0.9851). In addition, the ζ that results from the nanoprecipitation technique when the polymer P153 was used ranged from −3.7 mV to −6.0 mV (p > 0.05). As mentioned above, the differences in PNP ζ between the formulations with P17A and P153 are due to the terminations groups of PLGA.
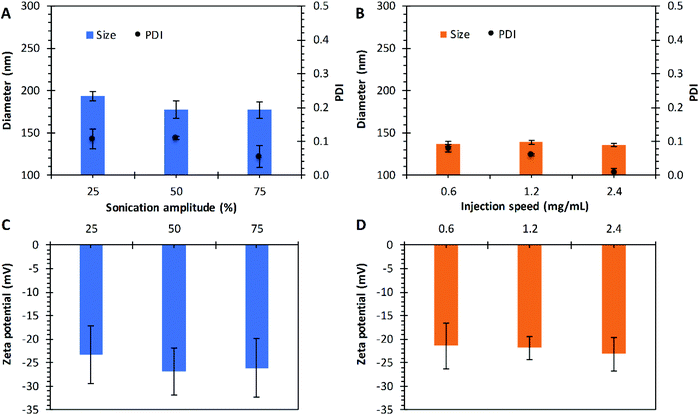 |
| | Fig. 5 Effect of sonication amplitude and injection speed on nanoparticles prepared by the emulsification and nanoprecipitation techniques, respectively. Diameter size and PDI as function of sonication amplitude (A), and the injection speed (B). Zeta potential as a function of the sonication amplitude (C), and the injection speed (D). Preparations made with PLGA of 17 kg mol−1. Data represent mean ± SD (n = 3). | |
Organic solvent fraction effects on the ζ of the PNP
The effects of Fos over the ζ of PNP for both techniques were evaluated and the results are presented in Fig. 4C. The ζ values obtained in the emulsification technique with P17A ranged between −26.5 mV and −31.0 mV for all the Fos evaluated (p > 0.05). Also, the ζ values with the same polymer but with the nanoprecipitation technique ranged between −16.2 mV and −23.4 mV for the same range of Fos (p < 0.05). Despite the statistical differences, no good linear fit was found with this data. Also, Fig. 4C shows that when the polymer P153 with the nanoprecipitation technique was used, the ζ values ranged between −4.4 mV and −5.9 mV (p > 0.05). The differences are consistent with the terminations groups of PLGA used in the preparations, as discussed in previous sections.
Speed of agitation effects on the ζ of the PNP
The effect of the Arpm in the evaporation of the organic solvent over the ζ of PNP was evaluated for both techniques; emulsification and nanoprecipitation, as presented in Fig. 4D. The ζ values obtained with the polymer P17A in the emulsification technique ranged between −22 mV and −25 mV for all the Arpm evaluated (p < 0.05). A fair linear fit was found with the equation D = −0.0169Arpm − 18.571 (R2 = 0.9353). The ζ results obtained with the nanoprecipitation technique and the P17A polymer ranged between −17.3 mV and −23.4 mV for all the Arpm evaluated (p < 0.05); however the results not present a good linear fit (results not shown). In the nanoprecipitation technique with P153, the ζ values were between −5.2 mV and −5.9 mV. These values were not significantly affected by the agitation (p > 0.05).
Sonicator amplitude and injection rate effects
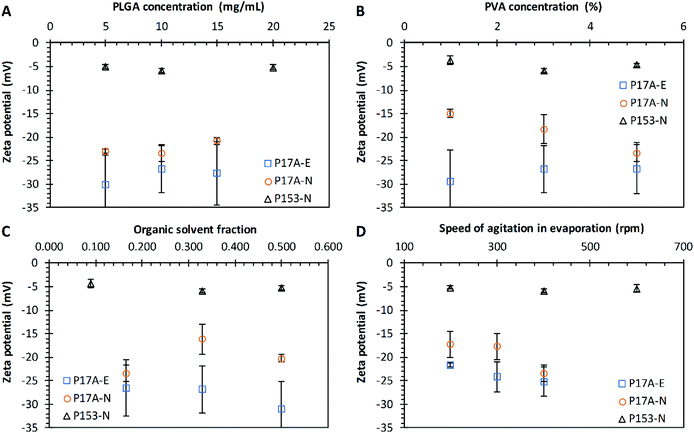
The sonication amplitude was evaluated in the emulsification technique with P17A; where three different sonication amplitudes were used: 25%, 50%, and 75% corresponding to 30 μm, 60 μm, and 90 μm of amplitude. The PNP sizes obtained at different amplitudes are presented in Fig. 5A. Particle with average diameters of 193.4 ± 5.4 nm, 177 ± 10.6 nm, and 177 ± 10.0 nm were obtained for amplitudes of 25%, 50%, and 75%, respectively. The average PDI values obtained for 25% and 50% amplitude were 0.11, and it decreases to 0.05 when sonication was set at 75%, although not statistical significance was found for the diameters (p > 0.05), probably due to the use of an ice bath and short sonication periods. Literature reports that by increasing the power and duration of sonication, a reduction in the mean diameter of the nanoparticles is obtained, and the particle population distribution could change from bimodal to unimodal.97,98 This effect can be attributed to the fact that the emulsion is carried out under high shear stress, which reduces the size of the emulsion droplets, correlating directly to the final size of the nanoparticles.99
In the nanoprecipitation technique with P17A, the injection speed was adjusted to 0.6 mL min−1, 1.2 mL min−1, and 2.4 mL min−1. The diameters obtained ranged between 135 nm and 139 nm, with PDI ranging between 0.01 and 0.08, as depicted in Fig. 5B. Therefore, no variation is appreciated within the range of study (p > 0.05). Reports in the literature suggest that the resulting nanoparticle size can be predicted by the diffusion coefficient of solvent in water with the presence of the polymer, and explain that the organic solvent injection speed only affects the rate of mass transport, but not the diffusion coefficient of the solvent.81 In the present study varying the sonication amplitude and injection rate, PLGA acid terminated was used in both techniques, emulsion, and nanoprecipitation. This comparison can be noticed by comparing the zeta potential of both methods where surface charge values ranged between −21 mV and −27 mV for both techniques (Fig. 5C and D).
Purification process
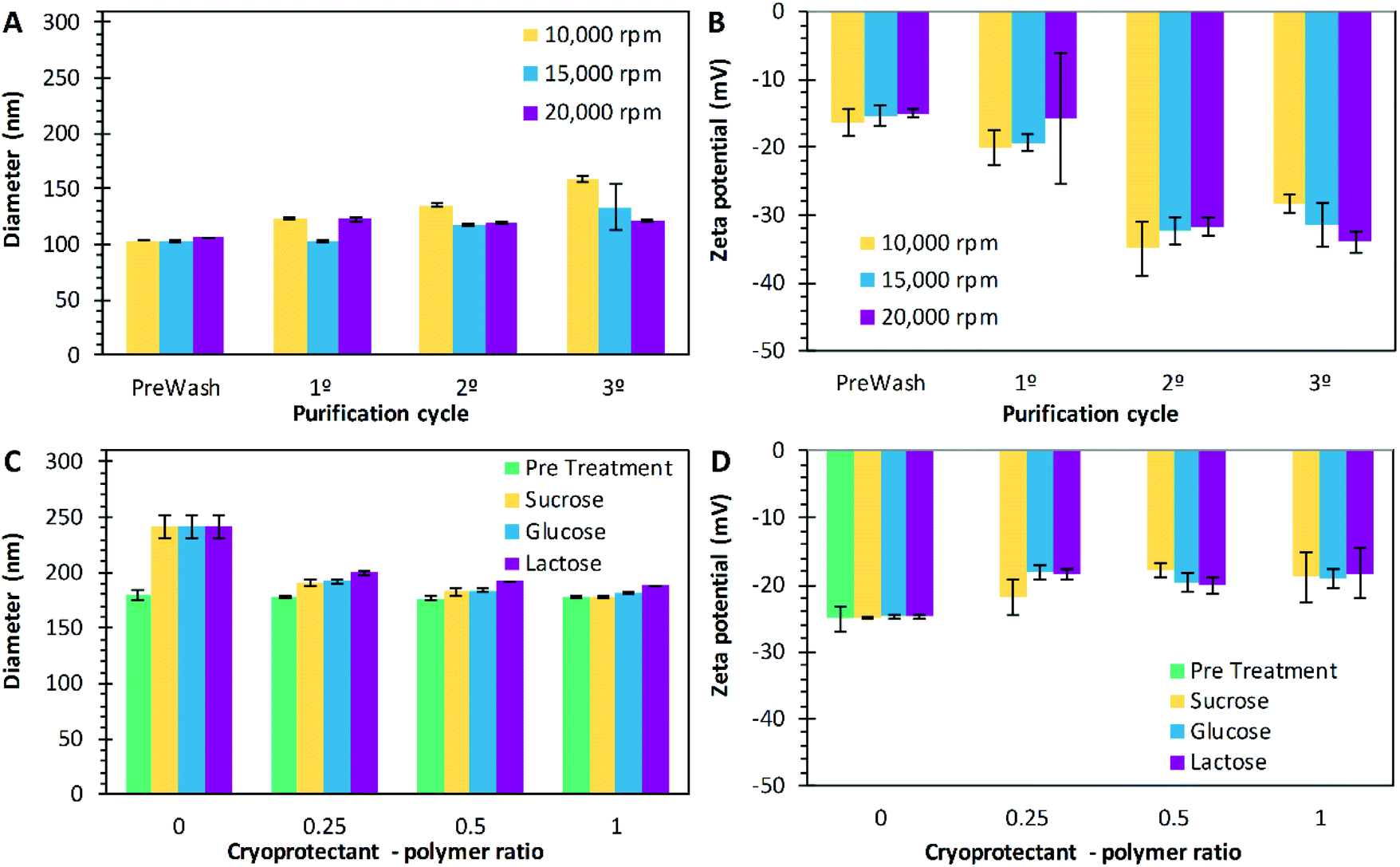
The nanoparticle purification step follows the solvent evaporation in both techniques (emulsification and nanoprecipitation), removing the excess of PVA. Several methods of purification can be used, but centrifugation is practical to perform on small scale experiments.100,101 In this work, three different centrifugation speeds were tested, 10000 rpm (9391 × g), 15000 rpm (21130 × g), and 20000 rpm (37565 × g). For this study, the particles were prepared by the technique of emulsification with the following conditions, 10 mg mL−1 of P17A in the organic phase, 5% of CPVA, 0.167 of Fos, 400 of Arpm, and 75% of sonication amplitude. These nanoparticles were used as a model to evaluate the purification process. In summary, an increase in the overall size of the nanoparticles was consistent across the centrifugation cycles used in the purification, where the PNP had an overall increase of 56 nm, 30 nm, and 16 nm, as the centrifugation speed increased to 10000 rpm, 15000 rpm, and 20000 rpm, respectively (Fig. 6A). Katas et al., explain that centrifugation could increase particle size by the compaction of the particles due to high-speed spinning and, therefore, forming aggregates.102 However, when the PNP were centrifuged, the PVA adsorbed on the surface could be washed more efficiently at 20000 rpm of centrifugation, thus explaining the drop in PNP size following this washing, compared with centrifugation at 10000 rpm, and 15000 rpm. Variations on the PDI through the purification process indicates the presence of a non-uniform population of PNP. For the experiments performed at 10000 rpm and 15000 rpm, the PDI values start at 0.15 on the prewash measurement, along with the process, they increased, and at the end, the PDI values were 0.22 and 0.27 respectively. However, when purification was performed at 20000 rpm, the PDI value decreased from 0.15 to 0.04, representing a monodisperse population of PNP. As mentioned above, when centrifugation speeds of 20000 rpm are used, the purification of the PNP becomes more efficient, decreasing the PDI of nanoparticles. On the other hand, zeta potential decreased in the progression of the purification cycles for all preparations (Fig. 6B). The values decreased from −16 mV to −28 mV corresponding to the prewash and the third cycle, respectively, when purifying at 10000 rpm. Similarly, values decreased from −15 mV to −31 mV from prewash to the third cycle, respectively, when purifying at 15000 rpm; and from −15 mV to −34 mV from prewash to the third cycle, respectively, when purifying at 20000 rpm (Fig. 5B). This effect could be attributed to the removal of the PVA layer on the surface of the nanoparticles as the purifying process is carrying out. By removing the PVA layer, the carboxyl groups from the PLGA become exposed and the surface charge is altered.103,104
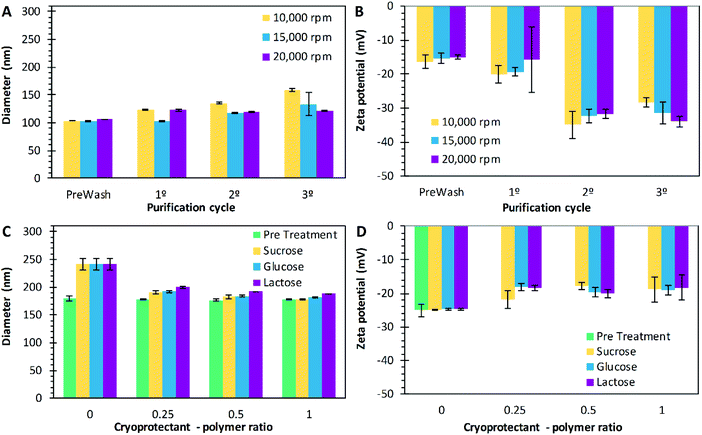 |
| | Fig. 6 Diameter size (A) and zeta potentials (B) of nanoparticles after the centrifugation cycles at 10000 rpm (9391 × g) ( ), 15000 rpm (21130 × g) ( ), 15000 rpm (21130 × g) ( ), and 20000 rpm (37565 × g) ( ), and 20000 rpm (37565 × g) ( ). Diameter size (C) and zeta potentials (D) of PLGA nanoparticles when different cryoprotectant – polymer ratios were used: pre-treatment ( ). Diameter size (C) and zeta potentials (D) of PLGA nanoparticles when different cryoprotectant – polymer ratios were used: pre-treatment ( ), sucrose ( ), sucrose ( ), glucose ( ), glucose ( ), and lactose ( ), and lactose ( ). Data represent mean ± SD (n = 3). ). Data represent mean ± SD (n = 3). | |
Cryoprotectants
The final part of the PNP preparation is freeze drying. During this stage, the use of cryoprotectants is commonly used to prevent nanoparticles from aggregation. Authors have used sugars such as trehalose, sucrose, lactose, glucose, and mannitol as cryoprotectants while freeze-drying nanoparticles.105,106 These sugars affect the glass transition temperature, and allow a high redispersion speed, and stabilization upon storage.107 Holzer et al. evaluated the effect of sucrose, trehalose, and mannitol on the storage stability of PNP.108 In the present work, the effect of sucrose, glucose, and lactose at different ratios (0:1, 0.25:1, 0.5:1 and 1:1) concerning initial mass of PLGA were evaluated. The diameter size, PDI and ζ of the PNP were measured before and after the lyophilization process. Before lyophilization, it was found that the sizes of the PNP were not affected by the addition of the cryoprotectants (p > 0.05); however, the ζ did change (Fig. 6D). For sucrose, from −25 mV to −18 mV, glucose changed from −25 mV to −22 mV and lactose from −25 mV to −19 mV. Rampino et al., report that the mechanism by which sugars protect particles resides in the interaction with the solute via hydrogen bonding.109 Therefore, the decrease of the zeta potential could be explained, by the coverage of the surface of the PNP. Besides, the PDI was maintained between 0.02 and 0.04 for all preparations, indicating a homogeneous size distribution. After lyophilization, the PNP were resuspended in water, and their size (Fig. 6C) and zeta potential (Fig. 6D) were measured again. When sucrose was added, PNP diameters of 190.4 nm, 183.3 nm, and, 178.3 nm were obtained for mass ratios of 0.25:1, 0.5:1, and 1:1, respectively; glucose had a similar result for the same ratios, with PNP sizes of 193 nm, 184.3 nm, and 182.1 nm, respectively. Similarly, as the mass ratio of lactose incremented, the size resulted in values of 200 nm, 193 nm, and 189 nm for mass ratios of 0.25:1, 0.5:1, and 1:1, respectively. According to the statistical analysis, these are all significant changes in size, with values of p limiting with zero, for α = 0.001, for the three cryoprotectants used. Also, when sucrose was used, the PDI values of 0.21, 0.11, 0.07, and 0.04 were obtained for the mass ratios of 0, 0.25, 0.5, and 1, respectively (p < 0.05). Similarly, for the glucose and the lactose, the PDI values decreased as the mass ratio of cryoprotectant increased, with values of p < 0.05. Tang and Shapiro reported smaller PNP average sizes with the addition of cryoprotectants, attributing the results to the ability of sugar additive to form a glassy amorphous matrix around the particles, preventing the particles from sticking together during the water removal.110
Also, Saez et al., discuss that the addition of cryoprotective agents makes the frozen mass behave more as fluid than a solid and it provides better mechanical protection of the PNP. Consequently, PNP aggregation or any alteration due to the pressure developed by the growth of crystals is avoided.111 Besides, all the zeta potential values decreased when the cryoprotectants were added (Fig. 6D), compared to the particles without cryoprotectants, with significant results for the use of sucrose (p = 0.004) and glucose (p = 0.044); but not significant for the use of lactose (p = 0.060).
Surface morphology
The morphological characteristics of dried PNP were determined by SEM analysis, showing a regular spherical shape and smooth surface. Fig. 7 shows the micrograph of PLGA nanoparticles prepared by emulsification technique with the following conditions: 10 mg mL−1 of PLGA in the organic phase, 5% of CPVA, Fos of 0.167, Arpm of 400 rpm, 75% of sonication amplitude and 20000 rpm of centrifugation speed. The histogram analysis of Fig. 7 (inset) obtained by the software Image J 1.8.0_112 (counting more than 200 particles) shown a diameter of 87 ± 40 nm. The diameter obtained from the micrograph is in concordance with the diameter sizes reported by DLS analysis (177.7 ± 10.6 nm); this by comparing those hydrodynamic diameters in solution to the diameters measured under drying conditions in SEM. Similar images could be obtained for all the preparations.
 |
| | Fig. 7 Scanning electron micrograph of PLGA nanoparticles. | |
Conclusions
A systematic study presenting the effects of the primary formulation parameters involved in the preparation of PLGA nanoparticles, via the emulsification technique and the nanoprecipitation technique is presented. The PLGA concentration, PVA concentration, organic solvent fraction, and agitation speed were evaluated in both techniques. Also, in the emulsion technique was evaluated the sonication amplitude; w in the nanoprecipitation technique, the organic phase injection velocity was studied. Additionally, the purification process, the use of cryoprotectants, and the surface morphology were studied. It was found that polymer concentration, as well as types of polymer-terminated chain, have significant influence regarding the physicochemical characteristics evaluated (size, PDI, and zeta potential). The particle size could be controlled by the PLGA concentration and PVA concentration in the nanoprecipitation technique. While in the emulsification technique, the sizes could be controlled by the organic solvent fraction. For both techniques, the use of high velocities of agitation in the evaporation of the solvents decreases the average diameter size. Also, by increasing the concentration of PLGA a slight decrease the PDI values can be achieved. Also, the ζ is not significantly affected by the variables explored; but the termination of PLGA polymer affects these values significantly. The centrifugation, lyophilization and the use of cryoprotectants are significative in the formulation process. The use of cryoprotectants, even in smaller mass ratios, help to maintain the size of nanoparticles after the lyophilization process. In summary, we conclude that uniform nanoparticles can be successfully prepared by adjusting the formulation parameters in both techniques. Moreover, physicochemical characteristics show to be suitable for therapeutic use, by obtaining uniform size, stable and reproducible nanoparticles. Also, the PNP preparation is sensitive to modifications in almost every step of the formulation, purification, and storage process. Therefore, by adjusting the parameters mentioned above, both techniques have the potential to be tuned to obtain any final nanoparticle desired characteristics, while maintaining reproducible results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Karol Yesenia Hernández-Giottonini, Rosalva Josefina Rodríguez-Córdova, and Omar Peñuñuri-Miranda thanks CONACYT for the graduate scholarships 302373, 281669, and 638788 respectively.
References
- B. Kumar, K. Jalodia, P. Kumar and H. K. Gautam, J. Drug Delivery Sci. Technol., 2017, 41, 260–268 CrossRef CAS.
- S. Patel, D. Singh, S. Srivastava, M. Singh, K. Shah, D. Chauahn and N. Chauhan, Adv. Pharmacol. Pharm., 2017, 5, 31–43 CAS.
- J. Ghitman, R. Stan and H. Iovu, UPB Scientific Bulletin, Series B: Chemistry and Materials Science, 2017, 79, 101–112 CAS.
- J. D. Robertson, L. Rizzello, M. Avila-Olias, J. Gaitzsch, C. Contini, M. S. Magón, S. A. Renshaw and G. Battaglia, Sci. Rep., 2016, 6, 1–9 CrossRef.
- K. M. El-say and H. S. El-sawy, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, H. Kim, J. M. Yoo, K. S. Lee, T. Kim, H. Shin, A. K. Sinha, S. G. Kwon, K. Kang, T. Hyeon and Y. E. Sung, J. Am. Chem. Soc., 2017, 139, 6669–6674 CrossRef CAS PubMed.
- P. L. Saldanha, V. Lesnyak and L. Manna, Nano Today, 2017, 12, 46–63 CrossRef CAS.
- C. Gutiérrez-Valenzuela, P. Guerrero-Germán, A. Tejeda-Mansir, R. Esquivel, R. Guzmán-Z and A. Lucero-Acuña, Appl. Sci., 2016, 6, 1–15 Search PubMed.
- M.-Y. Hsu, C.-H. Feng, Y.-W. Liu and S.-J. Liu, Appl. Sci., 2019, 9, 1077 CrossRef CAS.
- B. Colzani, L. Pandolfi, A. Hoti, P. A. A. Iovene, A. Natalello, S. Avvakumova, M. Colombo and D. Prosperi, Int. J. Nanomed., 2018, 13, 957–973 CrossRef CAS.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Güvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, Front. Pharmacol., 2018, 9, 1–19 CrossRef PubMed.
- R. Goyal, L. K. Macri, H. M. Kaplan and J. Kohn, J. Controlled Release, 2016, 240, 77–92 CrossRef CAS PubMed.
- X. Yang, Y. Chen, M. Wang, H. H. Zhang, X. Li and H. H. Zhang, Adv. Funct. Mater., 2016, 26, 8427–8434 CrossRef CAS.
- A. Lucero-Acuña, J. J. Jeffery, E. R. Abril, R. B. Nagle, R. Guzman, M. D. Pagel and E. J. Meuillet, Int. J. Nanomed., 2014, 9, 5653–5665 Search PubMed.
- S. Haque, B. J. Boyd, M. P. McIntosh, C. W. Pouton, L. M. Kaminskas and M. Whittaker, Curr. Nanosci., 2018, 14, 448–453 CrossRef CAS PubMed.
- A. Maaz, W. Abdelwahed, I. A. A. Tekko, S. Trefi, A. Maaz, W. Abdelwahed, I. A. Tekko and S. Tref, International Journal of Academic Scientific Research, 2015, 3, 1–12 Search PubMed.
- S. Mallakpour and V. Behranvand, eXPRESS Polym. Lett., 2016, 10, 895–913 CrossRef CAS.
- F. Masood, Mater. Sci. Eng., C, 2016, 60, 569–578 CrossRef CAS PubMed.
- J. Yang, S. Han, H. Zheng, H. Dong and J. Liu, Carbohydr. Polym., 2015, 123, 53–66 CrossRef CAS PubMed.
- H. A. Almoustafa, M. A. Alshawsh and Z. Chik, Int. J. Pharm., 2017, 533, 275–284 CrossRef CAS PubMed.
- S. Betala, M. M. Varma and K. Abbulu, J. Drug Delivery Ther., 2018, 8, 187–191 CrossRef CAS.
- W. Badri, K. Miladi, Q. A. Nazari, H. Fessi and A. Elaissari, Colloids Surf., A, 2017, 516, 238–244 CrossRef CAS.
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, 1–4 CrossRef.
- V. J. Pansare, D. Tien, P. Thoniyot and R. K. Prud'homme, J. Membr. Sci., 2017, 538, 41–49 CrossRef CAS.
- K. Nair, S. Vidyanand, J. James and G. S. Kumar, J. Appl. Polym. Sci., 2012, 124, 2030–2036 CrossRef CAS.
- C. Bouaoud, S. Xu, E. Mendes, J. G. J. L. Lebouille, H. E. A. De Braal and G. M. H. Meesters, J. Appl. Polym. Sci., 2016, 133, 1–11 CrossRef.
- H. Mahdavi, H. Mirzadeh, H. Hamishehkar, A. Jamshidi, A. Fakhari, J. Emami, A. R. Najafabadi, K. Gilani, M. Minaiyan, M. Najafi, M. Tajarod and A. Nokhodchi, J. Appl. Polym. Sci., 2010, 116, 528–534 CAS.
- S. A. Fahmy and W. Mamdouh, J. Appl. Polym. Sci., 2018, 46133, 1–9 Search PubMed.
- J. Tang, J.-Y. Chen, J. Liu, M. Luo, Y.-J. Wang, X. Wei, X. Gao, B. Wang, Y.-B. Liu, T. Yi, A.-P. Tong, X.-R. Song, Y.-M. Xie, Y. Zhao, M. Xiang, Y. Huang and Y. Zheng, Int. J. Pharm., 2012, 431, 210–221 CrossRef CAS PubMed.
- C. G. Keum, Y. W. Noh, J. S. Baek, J. H. Lim, C. J. Hwang, Y. G. Na, S. C. Shin and C. W. Cho, Int. J. Nanomed., 2011, 6, 2225–2234 CAS.
- S. A. Joshi, S. S. Chavhan and K. K. Sawant, Eur. J. Pharm. Biopharm., 2010, 76, 189–199 CrossRef CAS PubMed.
- B. Sahana, K. Santra, S. Basu and B. Mukherjee, Int. J. Nanomed., 2010, 5, 621–630 CAS.
- S. Jahan and A. Haddadi, Int. J. Nanomed., 2015, 10, 7371–7384 CAS.
- J. Vandervoort and A. Ludwig, Int. J. Pharm., 2002, 238, 77–92 CrossRef CAS.
- T. Adebileje, A. Valizadeh and A. Amani, J. Contemp. Med. Sci., 2017, 3, 306–312 CrossRef CAS.
- M. Halayqa, U. Doma and U. Domańska, Int. J. Mol. Sci., 2014, 15, 23909–23923 CrossRef PubMed.
- C. Fonseca, S. Simões and R. Gaspar, J. Controlled Release, 2002, 83, 273–286 CrossRef CAS PubMed.
- S. Derman, J. Nanomater., 2015, 2015, 341848 Search PubMed.
- X. Song, Y. Zhao, S. Hou, F. Xu, R. Zhao, J. He, Z. Cai, Y. Li and Q. Chen, Eur. J. Pharm. Biopharm., 2008, 69, 445–453 CrossRef CAS PubMed.
- K. Dillen, J. Vandervoort, G. Van Den
Mooter, L. Verheyden and A. Ludwig, Int. J. Pharm., 2004, 275, 171–187 CrossRef CAS.
- R. P. Darade, F. J. Sayyad and A. Patil, Indo Am. J. Pharm. Res., 2014, 4, 5111–5120 Search PubMed.
- M. S. Muthu, M. K. Rawat, A. Mishra and S. Singh, Nanomedicine, 2009, 5, 323–333 CrossRef CAS PubMed.
- T. Govender, S. Stolnik, M. C. Garnett, L. Illum and S. S. Davis, J. Controlled Release, 1999, 57, 171–185 CrossRef CAS.
- F. Madani, S. S. Esnaashari, B. Mujokoro, F. Dorkoosh, M. Khosravani and M. Adabi, Adv. Pharm. Bull., 2018, 8, 77–84 CrossRef CAS PubMed.
- S. A. Galindo-Rodríguez, F. Puel, S. Briançon, E. Allémann, E. Doelker and H. Fessi, Eur. J. Pharm. Sci., 2005, 25, 357–367 CrossRef PubMed.
- Z. He, H. Huang, J. L. Santos, Y. Chen, K. W. Leong, Y. Hu, L. Liu, H.-Q. Mao and H. Tian, Biomaterials, 2017, 130, 28–41 CrossRef CAS PubMed.
- S. Pieper and K. Langer, Mater. Today: Proc., 2017, 4, S188–S192 Search PubMed.
- E. Sah and H. Sah, J. Nanomater., 2015, 2015, 1–22 CrossRef.
- C. E. Mora-huertas, O. Garrigues, H. Fessi and A. Elaissari, Eur. J. Pharm. Biopharm., 2012, 80, 235–239 CrossRef CAS.
- R. Paliwal, R. J. Babu and S. Palakurthi, AAPS PharmSciTech, 2014, 15, 1527–1534 CrossRef CAS PubMed.
- A. Lucero-Acuña and R. Guzmán, Int. J. Pharm., 2015, 494, 249–257 CrossRef PubMed.
- S. Xie, S. Wang, B. Zhao, C. Han, M. Wang and W. Zhou, Colloids Surf., B, 2008, 67, 199–204 CrossRef CAS.
- C. E. Astete and C. M. Sabliov, J. Biomater. Sci., Polym. Ed., 2006, 17, 247–289 CrossRef CAS.
- H. Y. Kwon, J. Y. Lee, S. W. Choi, Y. Jang and J. H. Kim, Colloids Surf., A, 2001, 182, 123–130 CrossRef CAS.
- R. M. Mainardes and R. C. Evangelista, Int. J. Pharm., 2005, 290, 137–144 CrossRef CAS PubMed.
- M. C. Operti, D. Fecher, E. A. W. van Dinther, S. Grimm, R. Jaber, C. G. Figdor and O. Tagit, Int. J. Pharm., 2018, 550, 140–148 CrossRef CAS.
- C. Rodrigues de Azevedo, M. von Stosch, M. S. Costa, A. M. Ramos, M. M. Cardoso, F. Danhier, V. Préat and R. Oliveira, Int. J. Pharm., 2017, 532, 229–240 CrossRef CAS PubMed.
- A. Frank, S. K. Rath and S. S. Venkatraman, J. Controlled Release, 2005, 102, 333–344 CrossRef CAS PubMed.
- J. Herrmann and R. Bodmeier, J. Controlled Release, 1995, 36, 63–71 CrossRef CAS.
- R. P. Félix Lanao, A. M. Jonker, J. G. C. Wolke, J. A. Jansen, S. C. G. Van Hest and J. C. M. Leeuwenburgh, Tissue Eng., Part B, 2013, 19, 380–390 CrossRef.
- R. H. Ansary, M. B. Awang and M. M. Rahman, Trop. J. Pharm. Res., 2014, 13, 1179–1190 CrossRef.
- J. Panyam, D. Williams, A. Dash, D. Leslie-Pelecky and V. Labhasetwar, J. Pharm. Sci., 2004, 93, 1804–1814 CrossRef CAS.
- F. Lince, D. L. Marchisio and A. A. Barresi, J. Colloid Interface Sci., 2008, 322, 505–515 CrossRef CAS PubMed.
- X. Wu, Y. Chang, Y. Fu, L. Ren, J. Tong and J. Zhou, Starch/Staerke, 2016, 68, 258–263 CrossRef CAS.
- H. Rachmawati, Y. L. Yanda, A. Rahma and N. Mase, Sci. Pharm., 2016, 84, 191–202 CrossRef CAS PubMed.
- J. Vysloužil, P. Doležel, M. Kejdušová, E. Mašková, J. Mašek, R. Lukáč, V. Košťál, D. Vetchý and K. Dvořáčková, Acta Pharm., 2014, 64, 403–417 Search PubMed.
- K. Miladi, S. Sfar, H. Fessi and A. Elaissari, Ind. Crops Prod., 2015, 72, 24–33 CrossRef CAS.
- A. Lamprecht, N. Ubrich, M. H. Perez, C.-M. Lehr, M. Hoffman and P. Maincent, Int. J. Pharm., 2000, 196, 177–182 CrossRef CAS PubMed.
- S. K. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105–114 CrossRef CAS PubMed.
- E. Chiesa, R. Dorati, T. Modena, B. Conti and I. Genta, Int. J. Pharm., 2018, 536, 165–177 CrossRef CAS PubMed.
- H. Murakami, M. Kobayashi, H. Takeuchi and Y. Kawashima, Int. J. Pharm., 1999, 187, 143–152 CrossRef CAS PubMed.
- A. Maaz, W. Abdelwahed, I. A. Tekko and S. Trefi, International Journal of Academic Scientific Research, 2015, 3, 1–12 Search PubMed.
- S. M. Habib, A. S. Amr and I. M. Hamadneh, J. Am. Oil Chem. Soc., 2012, 695–703 CrossRef CAS.
- H. Wang, Y. Jia, W. Hu, H. Jiang, J. Zhang and L. Zhang, Pharmaceut. Dev. Technol., 2013, 18, 694–700 CrossRef CAS PubMed.
- S. A. Guhagarkar, V. C. Malshe and P. V Devarajan, AAPS PharmSciTech, 2009, 10, 695–703 CrossRef PubMed.
- S. Das, P. K. Suresh and R. Desmukh, Nanomedicine, 2010, 6, 318–323 CrossRef CAS PubMed.
- Sonam, H. Chaudhary and V. Kumar, Int. J. Biol. Macromol., 2014, 64, 99–105 CrossRef CAS PubMed.
- A. M. De Oliveira, E. Jäger, A. Jäger, P. Stepánek and F. C. Giacomelli, Colloids Surf., A, 2013, 436, 1092–1102 CrossRef CAS.
- T. Kantaria, T. Kantaria, S. Kobauri, M. Ksovreli, T. Kachlishvili, N. Kulikova, D. Tugushi and R. Katsarava, Appl. Sci., 2016, 6, 444 CrossRef.
- J. Cheng, B. A. Teply, I. Sherifi, J. Sung, G. Luther, F. X. Gu, E. Levy-Nissenbaum, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Biomaterials, 2007, 28, 869–876 CrossRef CAS PubMed.
- W. Huang and C. Zhang, Biotechnol. J., 2018, 13, 1–19 Search PubMed.
- N. Morsi, D. Ghorab, H. Refai and H. Teba, Int. J. Pharm., 2016, 506, 57–67 CrossRef CAS PubMed.
- A. Malkani, A. A. Date and D. Hegde, Drug Delivery Transl. Res., 2014, 365–376 CrossRef CAS PubMed.
- P. Hassanzadeh, I. Fullwood, S. Sothi and D. Aldulaimi, in Gastroenterology and Hepatology from Bed to Bench, 2011, vol. 4, pp. 63–69 Search PubMed.
- R. S. Dangi and S. Shakya, Int. J. Pharm. Life Sci., 2013, 4, 2810–2818 CAS.
- V. G. Kadajji and G. V. Betageri, Polymers, 2011, 3, 1972–2009 CrossRef CAS.
- H. Katas, E. Cevher and H. O. Alpar, Int. J. Pharm., 2009, 369, 144–154 CrossRef CAS PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- M. Azizi, F. Farahmandghavi, M. Joghataei, M. Zandi, M. Imani, M. Bakhtiary, F. A. Dorkoosh and F. Ghazizadeh, J. Polym. Res., 2013, 20, 1–14 CAS.
- D. Moinard-Chécot, Y. Chevalier, S. Briançon, L. Beney and H. Fessi, J. Colloid Interface Sci., 2008, 317, 458–468 CrossRef PubMed.
- K. Narayanan, V. M. Subrahmanyam and J. V. Rao, Enzyme Res., 2014, 2014, 162962 CAS.
- M. Nahar and N. K. Jain, Pharm. Res., 2009, 26, 2588–2598 CrossRef CAS PubMed.
- S. Dyawanapelly, U. Koli, V. Dharamdasani, R. Jain and P. Dandekar, Drug Delivery Transl. Res., 2016, 6, 365–379 CAS.
- R. Donno, A. Gennari, E. Lallana, J. M. Rios De La Rosa, R. D'Arcy, K. Treacher, K. Hill, M. Ashford and N. Tirelli, Int. J. Pharm., 2017, 534, 97–107 CrossRef CAS.
- D. H. Abdelkader, S. A. El-Gizawy, A. M. Faheem, P. A. McCarron and M. A. Osman, J. Drug Delivery Sci. Technol., 2018, 43, 160–171 CrossRef CAS.
- S. Prabha and V. Labhasetwar, Pharm. Res., 2004, 21, 354–364 CrossRef CAS PubMed.
- A. Budhian, S. J. Siegel and K. I. Winey, Int. J. Pharm., 2007, 336, 367–375 CrossRef CAS PubMed.
- N. Sharma, P. Madan and S. Lin, Asian J. Pharm. Sci., 2016, 11, 404–416 CrossRef.
- A. Tripathi, R. Gupta and S. A. Saraf, Int. J. PharmTech Res., 2010, 2, 2116–2123 CAS.
- C. Fornaguera and C. Solans, Int. J. Polym. Sci., 2018, 2018, 1–10 CrossRef.
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26, 1025–1058 CrossRef CAS PubMed.
- H. Katas, S. H. U. Chen, A. A. Osamuyimen, E. Cevher and H. O. Y. A. Alpar, J. Microencapsulation, 2008, 25, 541–548 CrossRef CAS PubMed.
- I. Takeuchi, S. Kobayashi, Y. Hida and K. Makino, Colloids Surf., B, 2017, 155, 35–40 CrossRef CAS PubMed.
- H. Murakami, Y. Kawashima, T. Niwa, T. Hino, H. Takeuchi and M. Kobayashi, Int. J. Pharm., 1997, 149, 43–49 CrossRef CAS.
- C. Pinto Reis, R. J. Neufeld, A. J. Ribeiro and F. Veiga, Nanomedicine, 2006, 2, 8–21 CrossRef PubMed.
- W. Abdelwahed, G. Degobert and H. Fessi, Int. J. Pharm., 2006, 324, 74–82 CrossRef CAS PubMed.
- P. Fonte, S. Soares, A. Costa, J. C. Andrade, V. Seabra, S. Reis and B. Sarmento, Biomatter, 2012, 2, 329–339 CrossRef PubMed.
- M. Holzer, V. Vogel, W. Mäntele, D. Schwartz, W. Haase and K. Langer, Eur. J. Pharm. Biopharm., 2009, 72, 428–437 CrossRef CAS PubMed.
- A. Rampino, M. Borgogna, P. Blasi, B. Bellich and A. Cesàro, Int. J. Pharm., 2013, 455, 219–228 CrossRef CAS PubMed.
- K. S. Tang, S. M. Hashmi and E. M. Shapiro, Nanotechnology, 2013, 24, 1–19 Search PubMed.
- A. Saez, M. Guzmán, J. Molpeceres and M. R. Aberturas, Eur. J. Pharm. Biopharm., 2000, 50, 379–387 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Patricia Guerrero-Germán
b,
Patricia Guerrero-Germán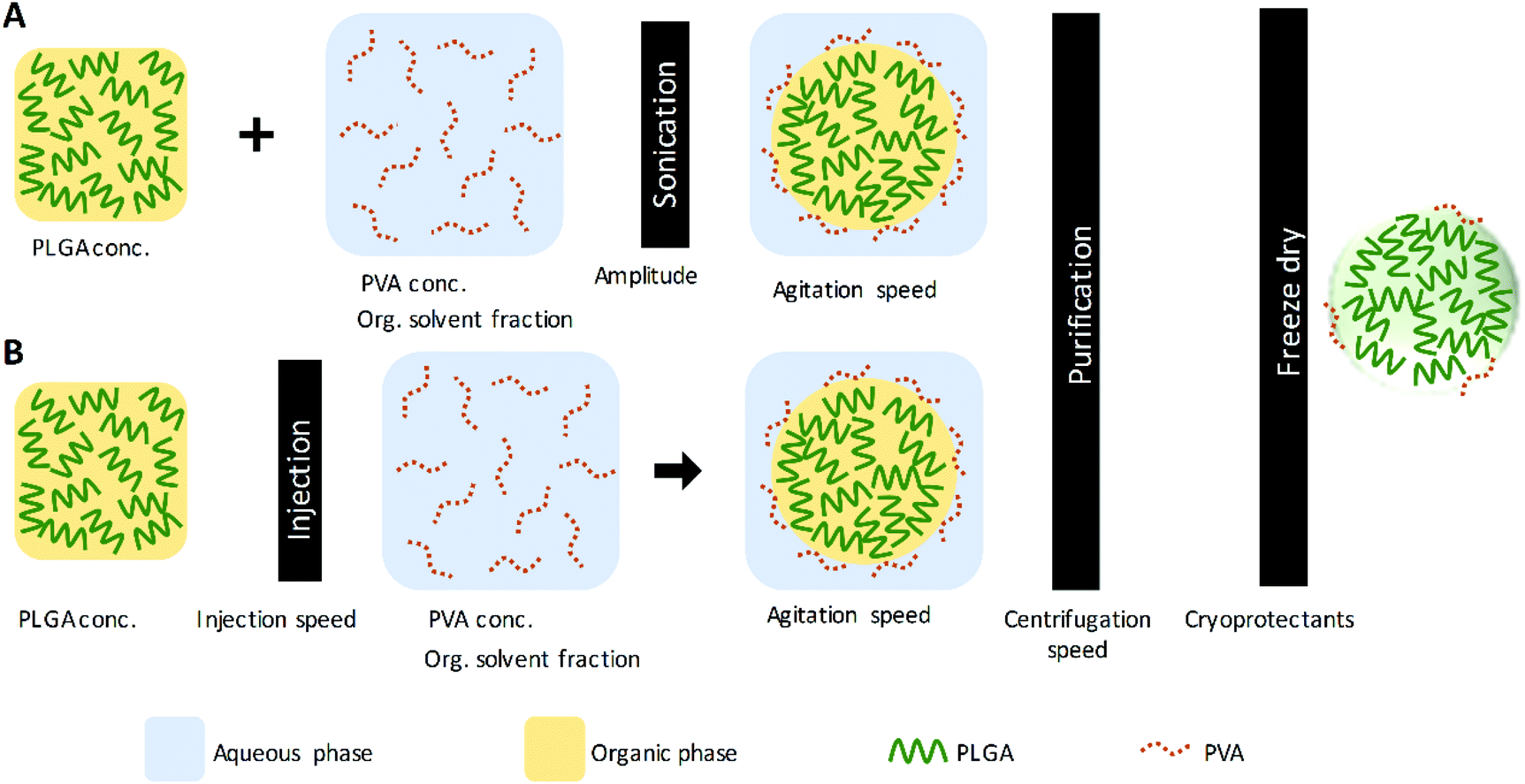
 ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N (
). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N (
), and PLGA of 153 kg mol−1, P153-N ( ). Data represent mean ± SD (n = 3).
). Data represent mean ± SD (n = 3).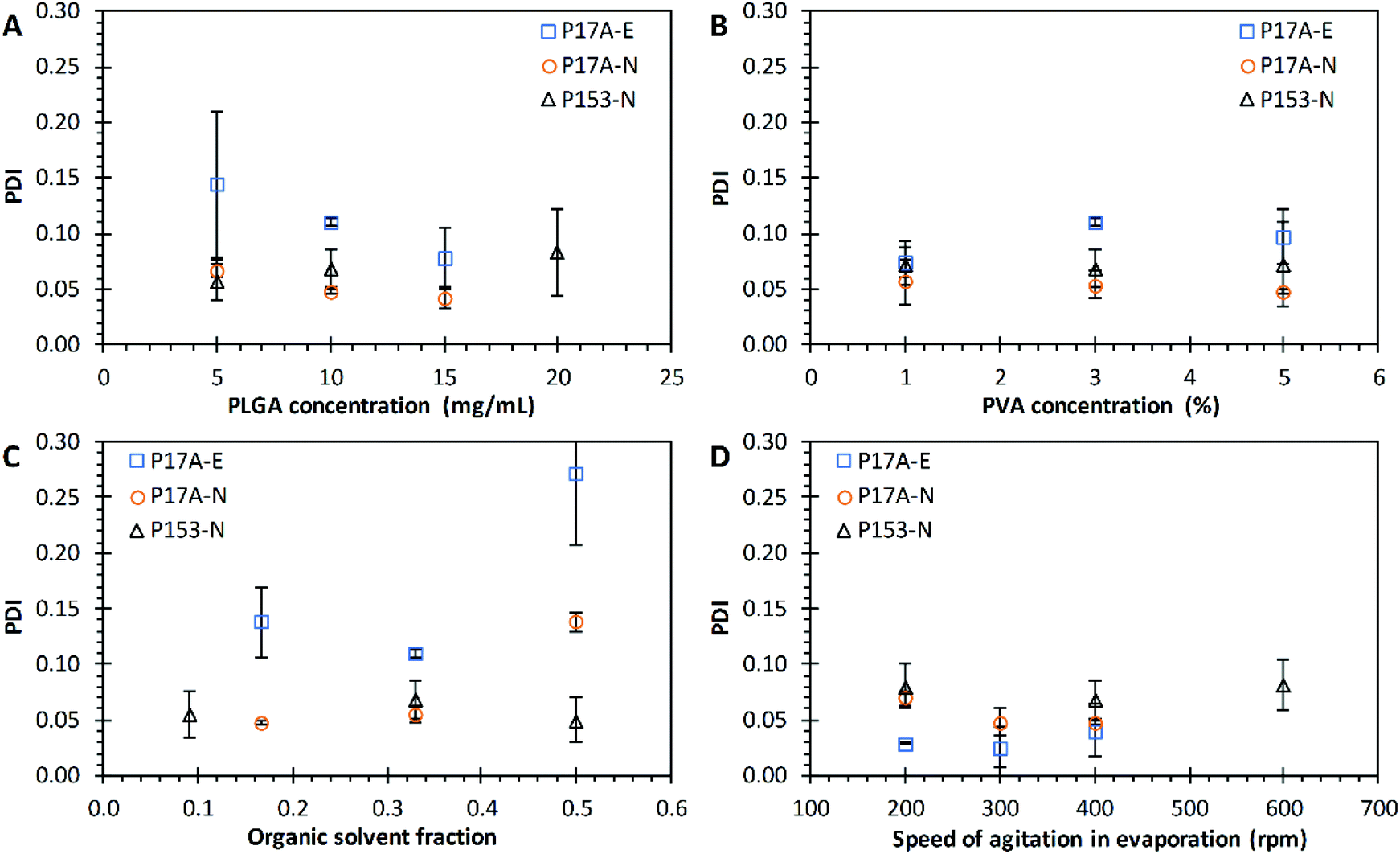
 ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N (
). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N (
), and PLGA of 153 kg mol−1, P153-N ( ). Data represent mean ± SD (n = 3).
). Data represent mean ± SD (n = 3).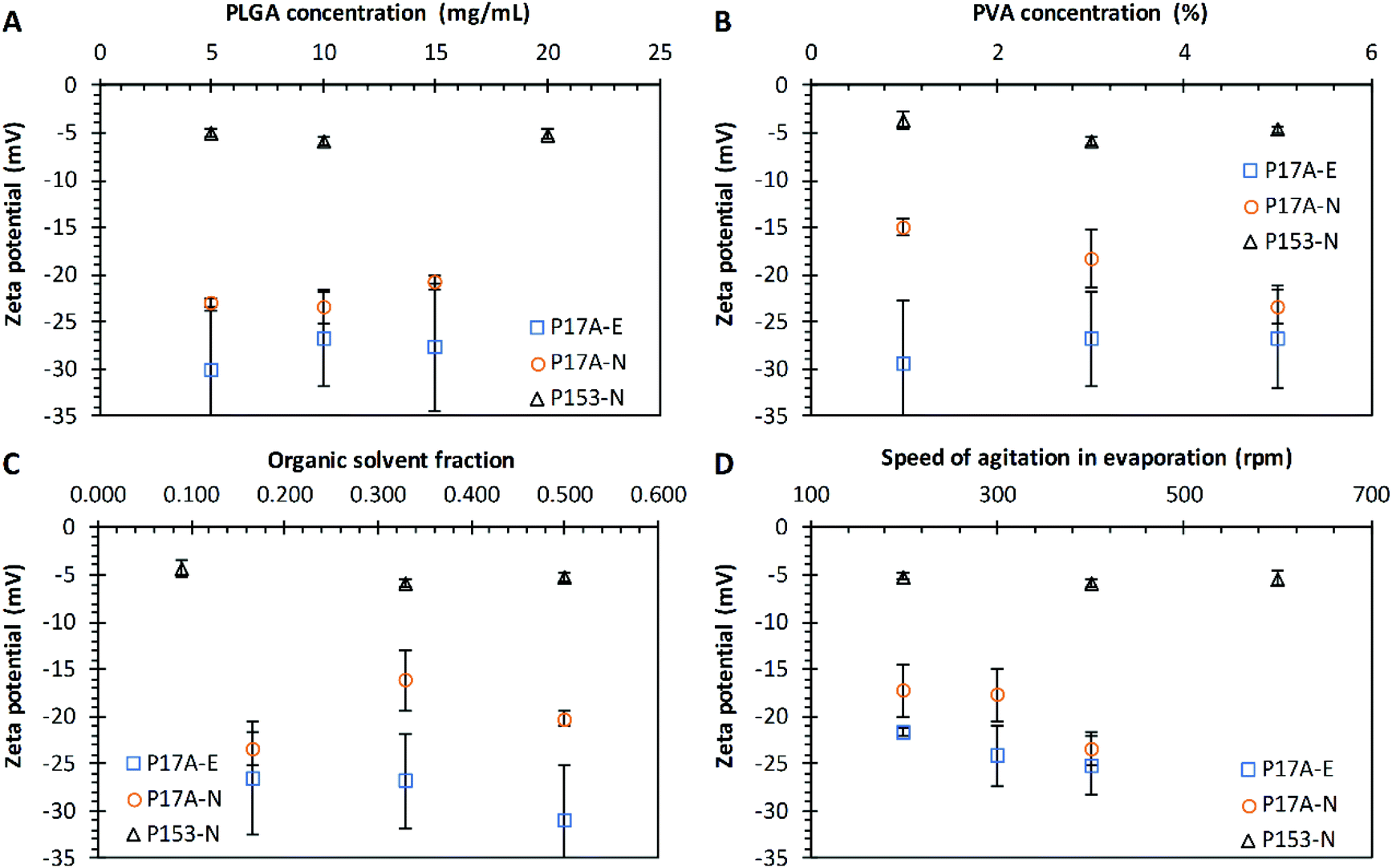
 ). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N (
). Preparations by the nanoprecipitation technique with PLGA of 17 kg mol−1, P17A-N ( ), and PLGA of 153 kg mol−1, P153-N (
), and PLGA of 153 kg mol−1, P153-N ( ). Data represent mean ± SD (n = 3).
). Data represent mean ± SD (n = 3).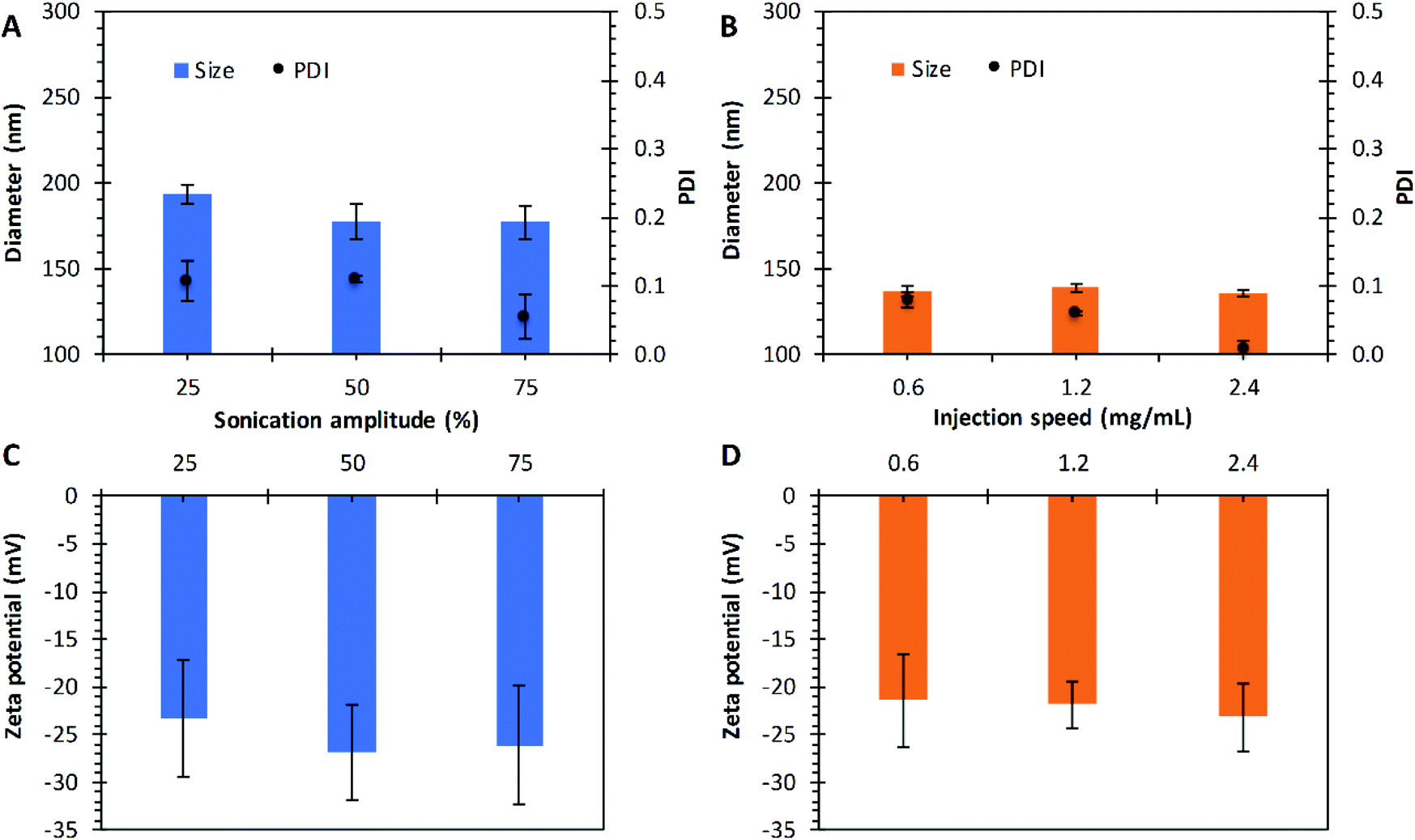
 ), 15
), 15 ), and 20
), and 20 ). Diameter size (C) and zeta potentials (D) of PLGA nanoparticles when different cryoprotectant – polymer ratios were used: pre-treatment (
). Diameter size (C) and zeta potentials (D) of PLGA nanoparticles when different cryoprotectant – polymer ratios were used: pre-treatment ( ), sucrose (
), sucrose ( ), glucose (
), glucose ( ), and lactose (
), and lactose ( ). Data represent mean ± SD (n = 3).
). Data represent mean ± SD (n = 3).