
Surface domain heterojunction on rutile TiO2 for highly efficient photocatalytic hydrogen evolution†
Kuo
Lin‡
,
Fang
Xiao‡
,
Ying
Xie
 ,
Kai
Pan
,
Lei
Wang
,
Wei
Zhou
* and
Honggang
Fu
*
,
Kai
Pan
,
Lei
Wang
,
Wei
Zhou
* and
Honggang
Fu
*
Key Laboratory of Functional Inorganic Material Chemistry, Ministry of Education of the People's Republic of China, Heilongjiang University, Harbin 150080, P. R. China. E-mail: zwchem@hotmail.com; fuhg@vip.sina.com
First published on 30th September 2020
Abstract
Compared with the highly active anatase TiO2, rutile TiO2 usually presents poor photocatalytic performance due to high electron–hole recombination. Herein, we propose a surface domain heterojunction (SDH) structure between adjacent micro-domains with and without chemisorbed chlorine on rutile TiO2, which utilizes the potential difference between these domains to form a built-in field that promotes charge separation. Single-crystal rutile TiO2 nanorods assembled into radial microspheres with SDHs were fabricated, and these exhibited excellent solar-driven photocatalytic hydrogen evolution, ∼8-fold higher than that of the pristine one. Experimental results and density functional theory calculations reveal that the exceptional photocatalytic performance can be attributed to the in situ formation of chemisorbed chlorine, which forms SDHs that separate electrons and holes efficiently and results in surface reconfiguration, exposing the tri-active sites, increasing the O-site active centers and enhancing the catalytic activity of the 4-coordinated (Ti4c) and 5-coordinated Ti sites (Ti5c). This SDH strategy can extend to other halogen elements and thus provides an universal approach for the rational design of high-efficiency TiO2 photocatalysts toward sustainable solar-fuel evolution.
New conceptsA new concept of surface domain heterojunction (SDH) structure has been developed for promoting charge separation. The SDH structure is formed between adjacent micro-domains with and without chemisorbed chlorine on the rutile TiO2 surface, which utilizes potential difference between diverse domains to form a built-in field that enhances spatial charge separation. The SDH contains various connected domains with different stoichiometry. If the band alignment of the two different domains is suitable, the photogenerated holes and electrons diffuse in opposite directions along the surface, resulting in the efficient inhibition of electron–hole recombination. This mechanism has been proposed based on experimental results and density functional theory calculations. SDHs indeed facilitate the separation of the photo-induced electron–hole pairs. This study provides a new way for the development of novel TiO2-based photocatalysts for highly efficient solar-energy conversion. |
Photocatalytic hydrogen fuel evolution is a sustainable and green solar-energy conversion technology.1–5 One of the most efficient and stable benchmark photocatalysts, titanium dioxide (TiO2) is of particular interest due to high chemical stability, low cost, and excellent photoelectrochemical characteristics.6–8 However, rutile TiO2 is the most thermodynamically stable catalyst and usually presents rapid photoexcited electron–hole recombination and exhibits low photocatalytic performance compared with the anatase phase, which can be ascribed to its direct bandgap semiconductor characteristic and nearly inert surface in the stoichiometric form.9–11 So far, numerous techniques, such as surface engineering, doping with metal or non-metal elements, controlling the morphology, adjusting the bandgap and tuning the junctions have been attempted to promote the charge separation efficiency and prolong the lifetime of the photogenerated electron–hole pairs.12–16 Among them, chlorine (Cl)-decorated TiO2 was found to promote charge carrier separation and improve photocatalytic hydrogen evolution obviously.17–19 Li and coworkers found that the addition of Cl favored the formation of Cl-involved intermediates, which could decrease the activation energy of water splitting, thus promoting hydrogen evolution efficiently.17 Rayalu and coworkers believed that adsorbed Cl acted as an internal electron donor that restrained the photogenerated holes, which enhanced charge separation and photocatalytic hydrogen production.18 The decoration of TiO2 with Cl indeed played a critical role in improving the photocatalytic hydrogen evolution activity, but the principle behind high-efficiency charge separation and photocatalytic hydrogen evolution is still ambiguous and controversial. The photocatalytic hydrogen evolution procedure includes the adsorption, hydrogen formation and desorption stages.20 Among these, desorption is vital for a continuous hydrogen evolution process because it is difficult for the subsequent adsorption without the efficient desorption of hydrogen. To date, it is unknown whether the Cl decorations on rutile TiO2 influence hydrogen desorption. The design and fabrication of Cl-decorated rutile TiO2 for highly efficient photocatalytic hydrogen evolution and understanding the nature of decorated Cl are still greatly challenging.
Considering the surface nature of materials, combining surface engineering and bandgap engineering to construct a photocatalyst surface with different stoichiometric domains and tunable electronic structures is significant. Inspired by Cl decoration and heterojunctions, the surface domain heterojunction (SDH) strategy has been introduced to promote the separation of the photogenerated electron–hole pairs in rutile TiO2, as depicted in Fig. 1a. The SDHs contain various connected domains with different stoichiometry. If the band alignment of the two different domains is suitable, the photogenerated holes and electrons will diffuse in opposite directions along the surface. To demonstrate this mechanism, first-principles calculations were carried out, and the computational details are summarized in Supplementary Information. Two rutile (110) facets with and without Cl-decorations were considered, as shown in Fig. 1c. After geometry optimization, the O atoms in the topmost layer of the pure rutile (110) surface relax only slightly outward, while an obvious reconstruction of the Cl-decorated surface happens, as illustrated in Movies S1 and S2 (ESI†). Such reconfiguration does not only lead to the stabilization of the surface but also change the electronic structure of Domain I. Fig. 1b clearly shows that the total density of states (TDOSs) of the facet shift to a relatively low position after Cl decoration. To align the band structures of the two facets according to the vacuum level, their work functions were calculated, as shown in Fig. S1 (ESI†). It was found that the Fermi energy of the Cl-decorated facet was ∼0.285 eV lower than that of rutile TiO2 (110). As a result, the photogenerated holes in the valence band (VB) of Domain I are injected into that of Domain II, accompanied by the injection of the photogenerated electrons from the conduction band (CB) of Domain II to that of Domain I (Fig. 1c). Our calculation confirmed that the type-II band alignment in SDH could facilitate the separation of the photogenerated electron–hole pairs. More importantly, the two close-contact domains take full advantage of the active sites and the surface space of the catalyst to participate in the oxidation and reduction processes, respectively. Therefore, we speculate that SDHs could favor charge separation and promote the photocatalytic hydrogen evolution performance.
 | ||
| Fig. 1 (a) Scheme of the surface domain heterojunction (SDH), (b) total density of states (TDOSs) of the rutile TiO2(110) face with and without Cl decoration, and (c) atomic structures and the band diagram of SDH. | ||
Theoretically, SDHs can promote the separation of electron–hole pairs, but it is still difficult to fabricate this special heterojunction in real. Designing a rational structure that exposes an adequate surface to form SDHs is of great significance. To achieve this goal, in this work, we have constructed single-crystal rutile TiO2 nanorods that are assembled into radial microspheres (RT-Cl) with a diameter of ∼2 μm and in situ chemisorbed Cl on the surface through nucleation and the seed-oriented growth approach, which form stable SDHs (Fig. 2a). The single-crystal nanorods assembled into radial microspherical structures with a highly curved surface expose an adequate surface to form SDHs, enhance hole-trapping at the surface and favor efficient contact with water, and this single-crystal structure with scarce crystal-boundary defects in the bulk can inhibit charge recombination, which is good for promoting the photocatalytic hydrogen evolution performance. The formation process of the single-crystal rutile TiO2 nanorod-assembled radial microspheres is illustrated in Fig. 2a. Firstly, the titania oligomer particles were formed through the rapid hydrolysis of TiCl4 in water at a low temperature of 0 °C (Fig. S2, ESI†). The nucleation process occurred first, forming the primary TiO2 nanoparticles. Then, the other titania oligomers originating from Ti(OC4H9)4 (TBOT) were grown oriented on the surface of the initial titania oligomers that acted as seeds and cores at the liquid–liquid phase interface (cyclohexane/water). During the growth process, the large microcrystals were relatively stable, while the smaller ones were reoriented via dissolution and regrown by prolonging the reaction time (Fig. S3, ESI†). Finally, uniform radial rutile TiO2 nanorod-assembled microspheres were formed by the subsequent solvothermal process, leading to further crystal-oriented growth and crystallinity. Chlorine was in situ chemisorbed on the surface of the rutile TiO2 nanorod-assembled radial microspheres. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 2b and d) showed uniform radial microspheres with a diameter of ∼2 μm after the solvothermal process. The enlarged images showed that the microspheres consisted of nanorods with a size of tens of nanometers (Fig. 2c and e). The highly curved surface favors long-lasting hole-trapping, which enables electrons to live longer and subsequently promotes proton reduction.21 The N2 adsorption–desorption isotherms showed typical type IV curves (Fig. S4, ESI†), indicating a mesoporous structure with a pore size of ∼10 nm.15 The nearly constant Brunauer–Emmett–Teller surface area of RT and RT-Cl implied the intact microstructure after Cl decoration (Table S1, ESI†). The high-resolution TEM (HRTEM) image and the selected-area electron diffraction (SAED) pattern (Fig. 2f and g) further revealed that the radially assembled nanorods had single-crystal rutile TiO2 characteristics. The clear lattice fringe of ∼0.325 nm corresponded to the (110) plane of rutile TiO2 (Fig. 2f), indicating that the (110) plane was the dominant exposed facet along the nanorods of the radial microspheres. The nanorods can shorten the charge transfer distance to the surface, which could thus reduce the recombination of charge carriers.22,23 The SAED pattern showed clear diffraction spots with hexagonal symmetry (Fig. 2g), indicating a single-crystal rutile structure. It is well-known that polycrystalline rutile TiO2 with abundant crystal boundaries, which act as recombination centers for the charge carriers, can be easily obtained. However, single-crystal rutile TiO2 can overcome this problem, inhibit charge carrier recombination and promote the photocatalytic activity. Therefore, the resultant single-crystal rutile TiO2 with chemisorbed Cl is favorable for promoting photocatalytic performance.
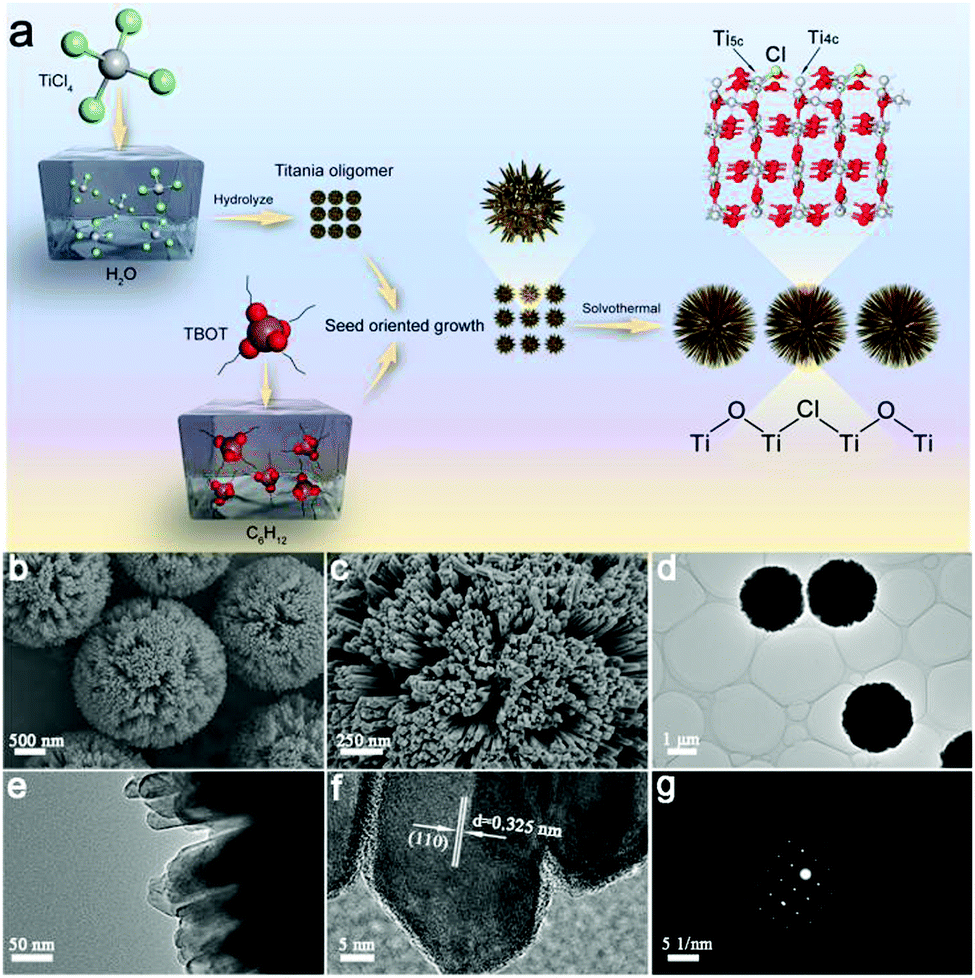 | ||
| Fig. 2 (a) Schematic representation of the formation process, (b) and (c) SEM, (d) and (e) TEM, (f) HRTEM images, and (g) the selected-area electron diffraction (SAED) pattern of the single-crystal rutile TiO2 nanorod-assembled radial microspheres with chemisorbed Cl on the surface. | ||
Typical X-ray diffraction (XRD) patterns of the rutile TiO2 nanorod-assembled radial microspheres before and after calcination showed well-defined diffraction peaks of phase-pure rutile (Fig. 3a), but a stronger diffraction intensity for RT after calcination was confirmed by Raman spectroscopy, which implied that the crystallinity further improved obviously after calcination. The calculated crystal cell volume for RT-Cl was larger than that of RT (Table S1, ESI†), suggesting a slight structural distortion due to chemisorbed Cl. The Raman spectra of RT and RT-Cl are shown in Fig. S5 (ESI†). The peaks at 446 and 610 cm−1 correlated well with the typical rutile TiO2 Raman bands of Eg and A1g. The peak at 239 cm−1 was attributed to the multi-proton scattering process.24 The red-shift of the Raman peaks of RT-Cl also indicated efficient Cl decoration, which is consistent with the XRD analysis. The introduction of Cl was evidenced by X-ray photoelectron spectroscopy (XPS). As shown in the XPS survey spectra (Fig. S6, ESI†), trace Cl species were detected in RT-Cl, but not in RT, illustrating the complete removal of Cl after calcination. To further determine the presence of Cl, high-resolution Cl 2p spectra were obtained (Fig. 3b), which displayed two peaks located at 197.5 and 199.0 eV for RT-Cl corresponding to chemisorbed Cl.19 No Cl peaks for RT could be observed, indicating the thorough removal of Cl through the calcination process. The O 1s and Ti 2p XPS spectra (Fig. S7 and S8, ESI†) indicated the existence of trace amounts of Ti–OH, oxygen vacancy and Ti3+ during the formation process of rutile TiO2. The electron paramagnetic resonance (EPR) spectra (Fig. S9, ESI†) further confirmed the formation of oxygen vacancy defects.25 The valence band XPS is shown in Fig. 3c; the VB maximum (VBM) of RT-Cl was located at 2.08 eV, which was 0.07 eV higher than that of RT (2.01 eV). The result suggested that chemisorbed Cl slightly increased VBM. The UV-vis absorption spectra showed that both samples strongly absorbed UV light, while RT-Cl exhibited a red-shift of the absorption edge compared with that of RT, implying a narrower band gap (Fig. 3d). The band gaps were estimated to be ∼3.03 and 2.93 eV for RT and RT-Cl (inset of Fig. 3d), respectively. The narrowed band gap can be ascribed to the structural variation caused by chemisorbed Cl. According to density functional theory (DFT) calculations (Fig. 1b), the band gap of RT-Cl was a little smaller than that of RT. The VBM and conduction band minimum (CBM) were both slightly decreased. The band positions were also measured by Mott–Schottky (M–T) plots at the frequencies of 1.0, 1.5 and 2.0 kHz (Fig. 3e). Both showed positive slopes, illustrating a typical n-type semiconductor characteristic.26 The flat band positions derived from the intersections were at ∼−0.23 and ∼−0.13 V vs. Ag/AgCl for RT and RT-Cl, respectively. The electron densities of RT-Cl and RT were estimated to be ∼3.6 × 1018 and 1.5 × 1018 cm−3, respectively. The high carrier density in RT-Cl is responsible for the high photocatalytic performance. As for TiO2, the CB potential was similar to that of the flat band potential (<0.2 eV), and the CBM positions of RT and RT-Cl were estimated to be −4.27 and −4.48 eV, respectively. The downshift of CBM for RT-Cl could be mainly ascribed to chemisorbed Cl, which is in good agreement with the DFT calculations. Interestingly, although the CBM of RT-Cl had downshifted obviously, the potential was still negative enough for the reduction of H+/H2.27 The variation in the CBM of RT-Cl did not influence the photocatalytic hydrogen evolution obviously. According to the above UV-vis and M–T analyses, the VBM potentials of RT and RT-Cl were calculated to be −7.30 and −7.41 eV, respectively. The corresponding band gap alignments of RT and RT-Cl are depicted in Fig. 3f. A scanning Kelvin probe (SKP) measured the contact potential difference between the tip and the sample, which was converted into work function under the thermoequilibrium state and showed direct evidence for Fermi-level variations.28 The work functions for RT and RT-Cl were ∼5.29 and 5.33 eV (Fig. 3g), respectively. The relatively large work function of RT-Cl favored easier escape of electrons from the CBM of rutile TiO2 to that of RT-Cl, facilitating photocatalytic hydrogen evolution in Domain I.29 Due to Cl decoration, the structural change affects the behavior of both band gap and the photogenerated charge carrier. The well-established noncontact technique, surface photovoltage spectroscopy (SPS), was employed to reveal the photophysical processes involved in charge-transfer or separation.30 Strong SPS peaks at around 380 nm could be observed for both RT and RT-Cl (Fig. 3h) and were ascribed to the electron transitions from VB to CB (band-to-band transitions, O2p–Ti3d).31 The SPS response intensity of RT-Cl increased remarkably compared with that of RT, indicating highly efficient separation of the photoexcited electron–hole pairs and long excitation lifetimes.32 Compared with RT, the redshift of RT-Cl illustrates its visible-light photoactivity. The photoelectrochemical properties of the RT and RT-Cl photoelectrodes were also measured to investigate the charge transfer efficiency. The chronoamperometry response of RT-Cl showed a photocurrent density of 212 μA cm−2 (Fig. S10, ESI†), which was much higher than that of RT (110 μA cm−2), indicating that charge carrier separation and transfer were enhanced greatly, consistent with the SPS analysis. In addition, both showed a rapid and symmetrical photocurrent response within four on–off cycles, implying high stability of the obtained rutile TiO2 materials. The dramatic decrease in the photoluminescence response of RT-Cl indicated efficient inhibition of charge recombination (Fig. S11, ESI†), which is in good agreement with the SPS results. The electrochemical impedance spectrum (EIS) of RT-Cl showed smaller interface resistance than that of RT (Fig. 3i), indicating faster interface charge transfer and electron diffusion mobility in the former.15 The highly efficient charge transfer and separation in RT-Cl is favorable for photocatalytic hydrogen evolution.
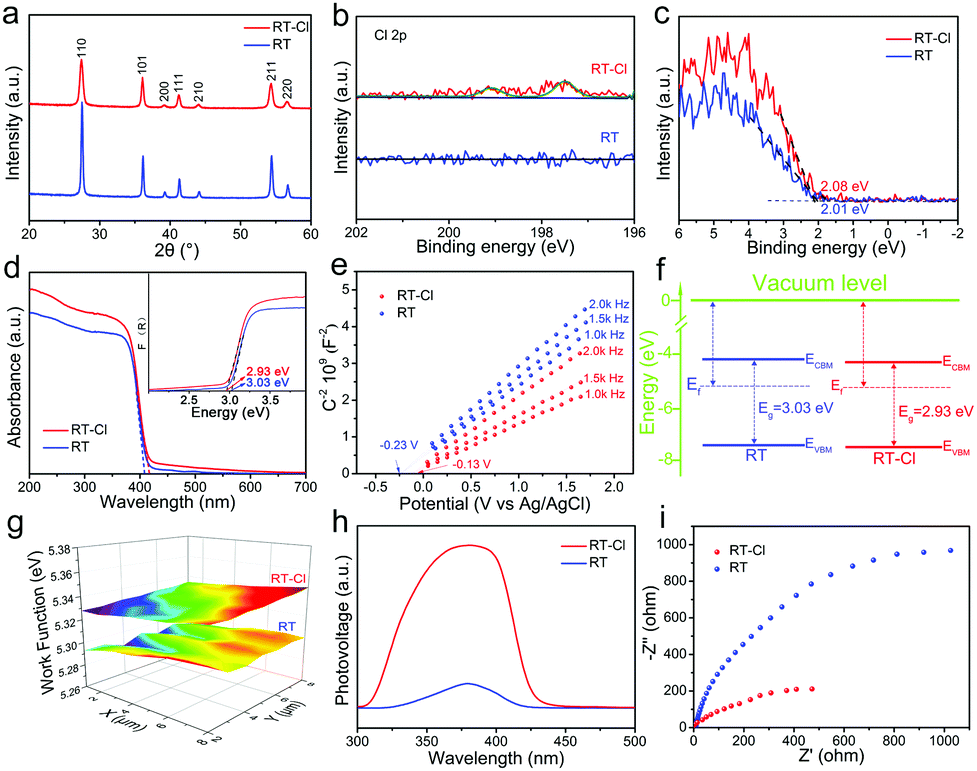 | ||
| Fig. 3 (a) The typical XRD patterns, (b) Cl 2p XPS spectra, (c) XPS VB, (d) UV-vis absorption spectra and (inset) the corresponding band gap energies, (e) Mott–Schottky plots collected at various frequencies versus the saturated Ag/AgCl reference electrode (pH = 7), (f) electronic band structures, (g) SKP maps, (h) SPS, and (i) EIS Nyquist plots of RT-Cl and RT. | ||
Photocatalytic hydrogen evolution was evaluated in a methanol/water mixture with Pt (0.15%) as the cocatalyst under AM 1.5G irradiation based on 100 mg of the photocatalysts (Fig. S12, ESI†). Pt nanoparticles with a diameter of 2–3 nm were dispersed on the surface of RT and RT-Cl uniformly so as to maximise the use of the cocatalyst. The HRTEM image indicated that surface Cl-decoration did not disturb Pt-loading obviously (Fig. S13 and S14, ESI†). A photocatalytic hydrogen rate of 1.59 mmol h−1 was obtained per 100 mg RT-Cl with a power density of 100 mW cm−2 (Fig. 4a), which was about 8-fold higher than that of RT (0.19 mmol h−1) and the commercial catalyst Degussa P25. No obvious decrease in H2 evolution was observed with both RT and RT-Cl materials within five cycles, and the structures of the cycled samples were almost similar to those of the fresh ones (Fig. S15 and S16, ESI†), indicating high stability. The hydrogen evolution rate of RT-Cl at 365 nm was 2.04 mmol h−1, which was much higher than that of 420 nm (0.27 mmol h−1), implying that UV light was still the main contributor to the high photocatalytic activity of RT-Cl under AM 1.5G irradiation, which accords with the UV-vis absorption analysis. The solar-to-hydrogen (STH) conversion efficiency of RT-Cl was around 2.58%, which was about eight times higher than that of RT (∼0.33%), implying the high photon-to-electron efficiency for the former.33 Moreover, RT-Cl achieved a high turnover frequency (TOF) of 2054.3 h−1, which is significantly higher by an enhancement factor of 8 compared with that of RT (247 h−1). As shown in Fig. 4b and Table S2 (ESI†), the apparent quantum efficiency (AQE) of RT-Cl at 365 nm (75.2%) was several times higher than that of RT (9.01%), which coincides with the optical absorption results. A similar trend could be observed at a single wavelength of 420 nm (9.4% for RT-Cl and 1.12% for RT). This photocatalytic hydrogen evolution is much higher compared to those reported in the literature (Table S3, ESI†). These results confirm that Cl decoration and the special single-crystal radial structure indeed improved the photocatalytic performance greatly. Interestingly, the ultra-low loading amount of the uniform tiny Pt cocatalyst (0.15%) on this special radial RT-Cl structure loaded via the liquid nitrogen freeze method could make the best use of Pt to achieve much better photocatalytic hydrogen activity than those reported earlier (Table S4, ESI†). In addition, splitting pure water to produce hydrogen and hydrogen peroxide without sacrificial reagents could also be achieved with RT-Cl (Fig. S17, ESI†), indicating superior photocatalytic performance. With organic contaminants as sacrificial reagents, such as Methylene Blue, Methyl Orange and Congo Red, photocatalytic hydrogen evolution using RT-Cl could be further improved obviously (Fig. S18, ESI†), implying that it is a promising candidate for degrading contaminants in wastewater treatment and producing solar fuel simultaneously. In order to get insights into the charge separation efficiency, experiments on active species trapping were performed to clarify the contribution of the electrons and holes towards photocatalytic hydrogen evolution. EPR experiments were performed to confirm the reactive species. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was employed as the trapping agent for ˙OH in water and O2˙− in methanol.34 Typical signals of ˙OH and O2˙− could be observed for both RT-Cl and RT (Fig. S19, ESI†), which confirmed the existence of photogenerated holes and electrons. Obviously, the intensities of both the RT-Cl peaks were stronger than those of RT, illustrating highly efficient charge separation in the former. Positron annihilation spectrometry was performed to study the structural variations in the solid (Fig. 4c), which provided information about the type and concentration of the structural defects based on the positron lifetime.35 The positron lifetime spectra of both RT and RT-Cl showed three lifetime components, τ1, τ2 and τ3, relative to intensities of I1, I2 and I3, respectively (Table S5, ESI†). The longest lifetime components (τ3) of both samples could be ascribed to positron annihilation in the big voids present in the radial structures.36 The shortest components (τ1) could be ascribed to the bulk defects in the primary rutile TiO2 microspheres.37 The longer one (τ2) should be attributed to positron annihilation trapped at the surface-chemisorbed Cl sites or the trace oxygen vacancy defects that are formed during crystallization. The relative intensity (I) is associated with the concentration of defects.38 Obviously, τ2 was the main contributor to positron annihilation. It was noted that the τ2 of RT-Cl was shorter than that of RT but its I2 was stronger than that of RT, which may be due to the promotion of annihilation by the relatively high concentration of oxygen vacancies and the surface anionic polarization of chemisorbed Cl. The activation of oxygen vacancies in water, the surface anionic polarization of chemisorbed Cl and the efficient charge separation in SDHs all contributed to the high photocatalytic hydrogen evolution. The electron density difference (EDD) diagrams also showed obvious charge differences due to decoration with surface chemisorbed Cl (Fig. 4d). The existence of lone-pairs of electrons in surface-chemisorbed Cl favors the trapping of the photogenerated holes and subsequently prolongs the lifetime of the electron–hole pairs,39 which is beneficial for improving the photocatalytic performance. Interestingly, rutile TiO2 materials decorated with other halogens, such as F, Br and I, also showed improved photocatalytic hydrogen evolution compared with the pristine one (Table S6, ESI†), indicating similar roles of the halogens in promoting the photocatalytic hydrogen evolution performance (Fig. S20, ESI†).
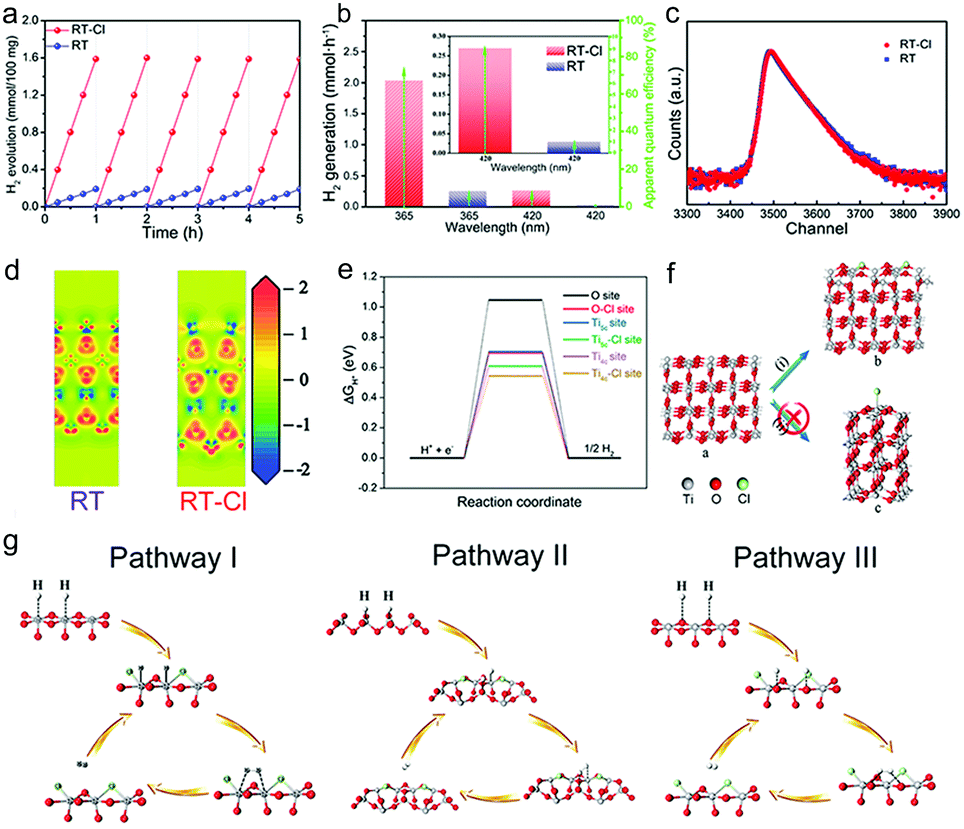 | ||
| Fig. 4 (a) Time-course H2 evolution plots, (b) wavelength-dependent efficiency of H2 evolution and AQE, (c) positron lifetime spectra, and (d) the electron density difference (EDD) diagrams for RT and RT-Cl. (e) Energy profiles for different catalytic sites, (f) types of chlorine chemisorption, and (g) hydrogen evolution pathways of chemisorbed Cl on rutile TiO2 surfaces. | ||
To further reveal the role of Cl, the rutile TiO2 sample was treated hydrothermally using hydrochloric acid to recover chemisorbed Cl. As shown in Fig. S21 (ESI†), chemisorbed Cl could be formed successfully on rutile TiO2 (RT-HCl). Further, the amounts of surface chemisorbed chlorine and the H2 evolution efficiency were also evaluated using different concentrations of HCl (Table S7, ESI†). The photocatalytic hydrogen evolution of RT-HCl (1.23 mmol h−1 per 100 mg catalysts) was much higher than that of RT but a little lower compared with that of RT-Cl (Fig. S22, ESI†). This may be ascribed to the lower concentrations of Cl, surface Ti–OH and surface-active sites in RT-HCl than those in RT-Cl (Fig. S7, ESI†). This also implies that the excellent photocatalytic performance of rutile TiO2 can be recovered by controlling the chemisorbed Cl species. The slightly increased water contact angle on RT-HCl also confirmed the decrease in surface Ti–OH from another point of view (Fig. S23, ESI†), which was further proven by the Fourier transform infrared spectroscopy (FT-IR) results (Fig. S24, ESI†). The consideration of surface Ti–OH is in good agreement with the present calculation results after further DFT calculations (Fig. S25, ESI†). The above factors of RT-HCl resulted in photocatalytic hydrogen evolution yet could not achieve the efficiency of RT-Cl. Importantly, the water contact angle on RT-Cl was obviously smaller than that on RT (Fig. S23, ESI†), implying enhanced surface polarity in RT-Cl due to the formation of chemisorbed Cl on the surface of RT, which favors photocatalytic hydrogen evolution. Notably, the photocatalytic performance could be promoted greatly through this facile Cl decoration strategy, which could be extended to other TiO2 materials (Fig. S26, ESI†). As shown in the SKP maps of anatase TiO2 and anatase TiO2–Cl (Fig. S27, ESI†), the work function of anatase TiO2–Cl (∼0.12 eV) was higher than that of anatase TiO2, and the variation in the Fermi energy indicated the successful formation of SDHs on anatase TiO2. The above results imply that this is a universal approach to improve photocatalytic hydrogen evolution.
To reveal the mechanism of the photocatalytic hydrogen evolution reaction (HER), DFT calculations were performed. Theoretically, the HER pathway can be described by the energy of three states (the initial state of H+ + e−, the intermediate state of adsorbed H*, and the final state of H2).40,41 Generally, an ideal hydrogen evolution catalyst requires the free energy of H* adsorption (|ΔGH*|) to be close to zero, and this value determines the proton/electron-transfer steps, as well as the hydrogen release process.42 For an ideal rutile TiO2(110) surface, one O and two Ti sites (4-coordinated and 5-coordinated sites, Ti4c and Ti5c) are considered respectively, as depicted in Fig. S28 (ESI†). Fig. 4e shows that the Gibbs free energy (ΔGH*) for the adsorption of H species was about 1.04 eV, suggesting that it is difficult for these species to attach to the O sites during the catalytic process, thus leading to poor HER performance. In comparison, the values for the Ti sites were much smaller. This result indicated that these Ti sites on the ideal TiO2 surface can act as active centers, despite their unsatisfactory catalytic activities. To enhance their catalytic activity, modification on the surface is considered to be an effective strategy.43,44 The calculation results suggested that when Cl species are introduced, the electronic structures of the surfaces changed significantly (Fig. 4d and e), which also affected their adsorption and desorption behaviors. According to geometry optimization, Cl tends to be bonded with two neighboring Ti rather than one, forming a Ti–Cl–Ti bridge at the surface (Fig. 4f(i)), rather than attaching to one Ti (Fig. 4f(ii) and Movie S1, ESI†). Because of decoration with Cl, |ΔGH*| of the O sites near the Cl species was reduced obviously (Fig. 4e), leading to surface reconfiguration and making pathway III possible (Fig. 4g). Furthermore, the introduction of Cl also results in the enhancement of the catalytic activities of both Ti4c and Ti5c sites through pathway I and II (Fig. 4g), as both their |ΔGH*| values decrease to a large extent (Fig. 4e). The calculation results confirmed that the increasing number of active centers (O site) and the enhancement of the catalytic activities (Ti4c and Ti5c sites) are together responsible for enhanced photocatalytic hydrogen evolution.
In summary, we have demonstrated that chemisorbed Cl on single-crystal rutile TiO2 nanorod-assembled radial microspheres with an SDH structure promotes the spatial separation of photogenerated electron–hole pairs. The solar-driven photocatalytic hydrogen rate of RT-Cl was up to 1.59 mmol h−1 per 100 mg catalyst, which was about 8-fold higher than that of RT. Both STH and TOF of RT-Cl were much higher than those of RT. This could be ascribed to the formation of SDHs that facilitate charge separation and surface reconfiguration after the introduction of chemisorbed Cl by increasing the active centers (O sites) and enhancing the catalytic activities (Ti4c and Ti5c), which together enhance the photocatalytic hydrogen evolution. In addition, the scarce crystal boundaries and increased surface polarity of the single-crystal rutile TiO2 nanorod-assembled radial microsphere structure of RT-Cl could be other reasons for the exceptional photocatalytic hydrogen evolution. The excellent photocatalytic performance can be easily enhanced via controlling the chemisorbed Cl. These findings suggest that the solar-to-fuel conversion efficiency can be promoted through the rational design of the structure and surface decoration of oxide photocatalysts.
Experimental methods
Chemicals
Titanium tetrachloride (TiCl4) and tetrabutyltitanate (TBOT) were purchased from Aladdin Industrial Inc. Cyclohexane was of analytical grade and purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. Deionized water (DI) was used for all experiments.Preparation of the single-crystal rutile TiO2 nanorod-assembled radial microspheres
Single-crystal rutile TiO2 radial microspheres (RT-Cl) were prepared through nucleation and the seed-oriented growth strategy. Typically, 1 mL TiCl4 was dissolved in deionized water in an ice-bath to obtain a 25% aqueous solution. Then, 5 mL TBOT was dissolved in 30 mL cyclohexane at room temperature, and subsequently, 5 mL aqueous TiCl4 solution was added to the above TBOT/cyclohexane solution dropwise. After stirring for 1 h, the obtained mixtures were transferred into a 100 mL stainless steel autoclave lined with Teflon and maintained at 150 °C for 18 h. The as-synthesized samples were washed with ethanol 3 times and dried at 60 °C for 8 h to obtain the resultant materials (RT-Cl). After calcination at 500 °C for 3 h, rutile TiO2 without Cl, denoted as RT, was obtained.Uniform photodeposition of Pt nanoparticles as cocatalysts
50 mg of the photocatalyst was dispersed in 40 mL of deionized water containing 66.43 μL of an aqueous H2PtCl6 solution (4 g L−1) under magnetic stirring for 30 min. Next, the mixed solution was ultrasonicated for 30 min and stirred for 12 h. Then, the mixed solution was rapidly frozen using liquid nitrogen, followed by irradiation under a 300 W Xe light with a light filter of 420 nm. The icy layer naturally melted after about 10 min of irradiation. The formed solution was centrifuged and separated. The obtained precipitates were further washed with deionized water three times and dried at 60 °C for 8 h. The Pt loading amount was measured by ICP-OES (Optima 8300 (PerkinElmer, USA)) as 0.15 wt%.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2018YFE0201704), the National Natural Science Foundation of China (21631004, 21871078 and 51672073) and the Natural Science Foundation of Heilongjiang Province (JQ2019B001).References
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- W. Li, A. Elzatahry, D. Aldhayan and D. Zhao, Chem. Soc. Rev., 2018, 47, 8203–8237 RSC.
- J. Ran, J. Qu, H. Zhang, T. Wen, H. Wang, S. Chen, L. Song, X. Zhang, L. Jing, R. Heng and S. Qiao, Adv. Energy Mater., 2019, 9, 1803402 CrossRef.
- J. Ran, B. Zhu and S. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS.
- B. Xia, Y. Zhang, B. Shi, J. Ran, K. Davey and S. Qiao, Small Methods, 2020, 4, 2000063 CrossRef.
- Z. Zhang, Q. Wu, G. Johnson, Y. Ye, X. Li, N. Li, M. Cui, J. Lee, C. Liu, S. Zhao, S. Li, A. Orlov, C. B. Murray, X. Zhang, T. B. Gunnoe, D. Su and S. Zhang, J. Am. Chem. Soc., 2019, 141, 16548–16552 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. Waterhouse, L. Zhu, C. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef.
- B. Liu, S. Feng, L. Yang, C. Li, Z. Luo, T. Wang and J. Gong, Energy Environ. Sci., 2020, 13, 221–228 RSC.
- C. Günnemann, C. Haisch, M. Fleisch, J. Schneider, A. V. Emeline and D. W. Bahnemann, ACS Catal., 2019, 9, 1001–1012 CrossRef.
- K. Zhang, L. Wang, J. K. Kim, M. Ma, G. Veerappan, C. Lee, K. Kong, H. Lee and J. H. Park, Energy Environ. Sci., 2016, 9, 499–503 RSC.
- C. Pang, R. Lindsay and G. Thornton, Chem. Rev., 2013, 113, 3887–3948 CrossRef CAS.
- S. He, J. Huang, J. L. Goodsell, A. Angerhofer and W. D. Wei, Angew. Chem., Int. Ed., 2019, 58, 6038–6041 CrossRef CAS.
- Y. Zhang, Z. Xu, G. Li, X. Huang, W. Hao and Y. Bi, Angew. Chem., Int. Ed., 2019, 58, 14229–14233 CrossRef CAS.
- X. Song, D. He, W. Le, Z. Ke, J. Liu, C. Tang, L. Cheng, C. Jiang, Z. Wang and X. Xiao, Angew. Chem., Int. Ed., 2019, 58, 16660–16667 CrossRef CAS.
- W. Zhou, W. Li, J. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- L. Huang, R. Li, R. Chong, G. Liu, J. Han and C. Li, Catal. Sci. Technol., 2014, 4, 2913–2918 RSC.
- A. A. Chilkalwar, P. A. Mangrulkar, A. A. Moinuddin, N. Penumaka and S. S. Rayalu, Sol. Energy, 2018, 174, 1019–1025 CrossRef CAS.
- Y. Luan, L. Jing, Q. Meng, H. Nan, P. Luan, M. Xie and Y. Feng, J. Phys. Chem. C, 2012, 116, 17094–17100 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- K. Shirai, G. Fazio, T. Sugimoto, D. Selli, L. Ferraro, K. Watanabe, M. Haruta, B. Ohtani, H. Kurata, C. Valentin and Y. Matsumoto, J. Am. Chem. Soc., 2018, 140, 1415–1422 CrossRef CAS.
- T. D. Phan, S. Luo, D. Vovchok, J. Llorca, J. Graciani, J. F. Sanz, S. Sallis, W. Xu, J. Bai, F. J. Piper, D. E. Polyansky, E. Fujita, S. D. Senanayake, D. J. Stacchiola and J. Rodriguez, ACS Catal., 2016, 6, 407–417 CrossRef.
- Y. Yu, X. Yang, Y. Zhao, X. Zhang, L. An, M. Huang, G. Chen and R. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8550–8554 CrossRef CAS.
- Y. Zhang, C. X. Harris, P. Wallenmeyer, J. Murowchick and X. Chen, J. Phys. Chem. C, 2013, 117, 24015–24022 CrossRef CAS.
- Y. Li, Y. Peng, L. Hu, J. Zheng, D. Prabhakaran, S. Wu, T. J. Puchtler, M. Li, K. Wong, R. A. Taylo and S. Tsang, Nat. Commun., 2019, 10, 4421 CrossRef.
- B. Sun, W. Zhou, H. Li, L. Ren, P. Qiao, W. Li and H. Fu, Adv. Mater., 2018, 30, 1804282 CrossRef.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS.
- G. Zhou, Y. Shan, Y. Hu, X. Xu, L. Long, J. Zhang, J. Dai, J. Guo, J. Shen, S. Li, L. Liu and X. Wu, Nat. Commun., 2018, 9, 3366 CrossRef.
- W. Hu, W. Zhou, K. Zhang, X. Zhang, L. Wang, B. Jiang, G. Tian, D. Zhao and H. Fu, J. Mater. Chem. A, 2016, 4, 7495–7502 RSC.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509–9549 RSC.
- L. Kronik and Y. Sapira, Surf. Sci. Rep., 1999, 37, 1–206 CrossRef CAS.
- W. Zhou, F. Sun, K. Pan, G. Tian, B. Jiang, Z. Ren, C. Tian and H. Fu, Adv. Funct. Mater., 2011, 21, 1922–1930 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS.
- Z. Li, C. Xiao, S. Fan, Y. Deng, W. Zhang, B. Ye and Y. Xie, J. Am. Chem. Soc., 2015, 137, 6587–6593 CrossRef CAS.
- L. Liang, K. Li, C. Xiao, S. Fan, J. Liu, W. Zhang, W. Xu, W. Tong, J. Liao, Y. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2015, 137, 3102–3108 CrossRef CAS.
- X. Zhang, H. Cui, M. Humayun, Y. Qu, N. Fan, X. Sun and L. Jing, Sci. Rep., 2016, 6, 21430 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef.
- Y. Liu, G. Yu, G. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752–10757 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS.
- Q. Guo, Z. Ma, C. Zhou, Z. Ren and X. Yang, Chem. Rev., 2019, 119, 11020–11041 CrossRef CAS.
- C. Gao, T. Wei, Y. Zhang, X. Song, Y. Huan, H. Liu, M. Zhao, J. Yu and X. Chen, Adv. Mater., 2019, 31, 1806596 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00491j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |