
High compression strength single network hydrogels with pillar[5]arene junction points†
Xiaowen
Xu
 a,
Florica Adriana
Jerca
ab,
Kristof
Van Hecke
c,
Valentin Victor
Jerca
*ab and
Richard
Hoogenboom
*a
a,
Florica Adriana
Jerca
ab,
Kristof
Van Hecke
c,
Valentin Victor
Jerca
*ab and
Richard
Hoogenboom
*a
aSupramolecular Chemistry Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281-S4, 9000 Ghent, Belgium
bCentre of Organic Chemistry “Costin D. Nenitzescu” Romanian Academy, Spl. Independentei 202B, 060023, Bucharest, Romania
cXStruct, Department of Chemistry, Ghent University, Krijgslaan 281-S3, B-9000, Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be; victor_jerca@yahoo.com
First published on 24th October 2019
Abstract
The present study highlights a straightforward and versatile strategy for the synthesis of strong poly(2-isopropenyl-2-oxazoline) hydrogels with tunable properties by using a bifunctional macrocyclic pillar[5]arene host having two carboxylic acid groups as cross-linker. This new strategy provides access to materials with tailored properties from soft and flexible to rigid and strong. The mechanical properties and water uptake of the hydrogels could be effortlessly controlled during the synthesis step through variation of the cross-linker content and after cross-linking by guest–host interactions. The hydrogels displayed strongly enhanced mechanical properties (i.e., compression and tensile modulus, energy dissipation, stress at break and storage modulus) compared to their counterparts cross-linked with linear dicarboxylic acids. The remarkable properties of the pillar[5]arene cross-linked hydrogels were assigned to the transfer of the external stress to the rigid and bulky pillar[5]arene residues that contribute to the overall dimensional stability of the hydrogels and allow energy dissipation. Moreover, we demonstrate the applicability of these materials for water purification. The hydrogels showed high adsorption performance for phenols and dyes such as methylene blue and methyl red and they could be easily regenerated, by washing with an organic solvent for reuse.
New conceptsIn this article, we introduce the concept of cross-linking polymer hydrogels using rigid macrocycles, which gives access to hydrogels with remarkable mechanical properties. Our simple, innovative and robust strategy tackles, on the one hand, the mechanical weakness of the majority of the hydrogels, which drastically restricts their applications and, on the other hand, the generally challenging and laborious synthesis of hydrogels with improved mechanical properties, such as sliding-ring gels, nanocomposites, and double network hydrogels. Hydrogels with superior and tunable mechanical properties as well as water uptake were developed by covalent cross-linking of a well-defined polymer matrix (e.g. poly(2-isopropenyl-2-oxazoline)) with a bulky bifunctional dicarboxylic macrocyclic pillar[5]arene host. The strongly enhanced mechanical properties are proposed to result from the transfer of external stress to the rigid and bulky pillar[5]arene units that contribute to the overall dimensional stability of the hydrogels and allow energy dissipation. The utility of these hydrogels for water purification, which is a challenging problem in our society, has also been demonstrated. The significance of this approach has been proven by in-depth mechanical characterization (e.g., rheology, compression and tensile measurements) and it can open new avenues not only in the synthesis of hydrogels but also in their applications. |
Introduction
Hydrogels represent one of the most investigated classes of materials due to their broad applications in various areas and can be obtained from synthetic and/or naturally occurring polymers through chemical or physical cross-linking processes.1–8 Each cross-linking strategy has specific advantages as well as drawbacks.2,9–11 However, in most of the cases regardless of the synthesis method, the hydrogels have relatively poor tunability and low mechanical properties. Consequently, considerable efforts have been devoted to the development of more complex polymer networks with a hierarchical structure such as sliding-ring gels,12,13 nanocomposites,14,15 and double network hydrogels.16–18 While elegant examples of hydrogel materials with improved mechanical properties based on these strategies have been reported, their synthesis is challenging and laborious and presents some limitations.19–21 For example, sliding-ring hydrogels, require the preparation of pre-polymer chains threaded through the rings of the cyclodextrin molecules and then subsequent cross-linking, while the current methods to produce double networks (DN) hydrogels also require slow sequential preparation steps. The processes are tedious and time-consuming, and the reproducibility could represent an important issue, especially for DN hydrogels. Therefore, in view of practical applications, finding a simple and robust synthetic protocol which would give access to hydrogels with tunable swelling and improved mechanical properties is of utmost importance. A plethora of examples can be found in the literature regarding the use of macrocycles for the synthesis of supramolecular hydrogels (i.e., physical cross-linking), via host–guest complexation.10,22,23 Moreover, dual cross-linked hydrogels formed by covalent and noncovalent bonds exhibiting enhanced mechanical performance were reported.6,24–27 Nevertheless, the macrocycles were incorporated as pendent groups and used to alter the cross-linking density of the hydrogel materials by host–guest complexation, thus serving as non-covalent supramolecular cross-linker. A method that was overlooked and could represent a viable and superior alternative for improvement of mechanical properties is the use of macrocycles possessing functional groups as covalent cross-linkers for the formation of covalent hydrogels with reactive, functional polymers.Recently two studies have shown the advantage of such an approach for host–guest interaction. Sessler et al.28 reported poly(vinyl alcohol) hydrogels cross-linked with a tetracationic macrocycle which could effectively be used for purification of aqueous solutions containing organic or inorganic salts. Zhao et al.29 investigated the drug delivery applications of hydrogels by copolymerization of N-isopropyl acrylamide and acrylic acid in the presence of β-cyclodextrin dimethacrylate cross-linker. However, none of these studies investigated the influence of the host on the mechanical properties of such hydrogels.
It is anticipated that through incorporation of macrocyclic hosts as cross-linkers hydrogels with tailored mechanical properties and water-uptake could be readily prepared by simply varying the cross-linker content, while it is expected that the host will play an active and essential role in the dissipation of the stress at the junction points. Moreover, by judiciously choosing the polymeric precursor and the host, further tuning of their properties could be achieved even at a high cross-linker ratio. While in the previous studies relatively flexible cyclodextrin hosts were used, we hypothesize that rigid macrocycles such as pillar[n]arenes are more suitable as cross-linkers due to their symmetric pillar-shaped architectures, rigid and π-rich cavities.30,31 Furthermore, pillar[n]arenes have a high affinity for a variety of guest and were used in the construction of a wide range of supramolecular systems including sensors,32–34 hybrid materials,35 membranes,36 molecular valves,37 and so on. They are relatively easy to synthesize, and the selective introduction of functional groups in targeted position is less complicated as compared to the most other macrocyclic hosts.38–41
Poly(2-isopropenyl-2-oxazoline) (PiPOx) has recently emerged as a versatile and functional platform for the preparation of thermoresponsive copolymers with tunable lower critical solution temperature (LCST) behavior, hydrogels, molecular brushes and materials for photonics.42–48 Furthermore, PiPOx can be prepared with well-defined characteristics, is highly hydrophilic, biocompatible49 and it can be easily modified by post-polymerization reactions with carboxylic acids. Even though PiPOx hydrogels cross-linked with aliphatic dicarboxylic acid showed tunable water uptake, their mechanical properties were rather weak.43
Herein we report a simple method that overcomes challenges for the design and synthesis of conventional hydrogels with high mechanical strength. Strong and robust PiPOx hydrogels with controllable properties are synthesized through ring-opening addition of the 2-oxazoline pendent groups with a specially designed dicarboxylic pillar[5]arene (PA5) macrocycle. Due to the unique network structure, of the resulting pillar[5]arene cross-linked hydrogels the external stress is hypothesized to be transferred to the PA5 residues, endowing the hydrogels with superior mechanical toughness. Moreover, their properties could be further tuned by guest–host interaction, even at a high cross-linking degree. A potential application for water purification and solvent-mediated regeneration has been identified for these hydrogels and proof of concept data is provided. These findings could open new avenues in the synthesis and applications of hydrogels.
Discussion
The water soluble PiPOx with a number average molar mass of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1 and a dispersity of 1.15 was prepared using our previously reported method.42 The dicarboxylic acid functionalized PA5 was synthesized by cyclooligomerization of 1,4-dimethoxybenzene, diethyl 2,2′-(1,4-phenylenebis(oxy))diacetate and paraformaldehyde using boron trifluoride diethyl etherate as Lewis acid, followed by hydrolysis of the ester. The structure of the obtained PA5 was confirmed by FT-NMR, HRMS, while the ester intermediate was unambiguously assigned based on single crystal X-ray diffraction (for further synthetic and characterization details, see ESI†). The PiPOx-PA5 hydrogel synthesis was performed by adapting a previously developed method from our group, namely heating a solution of PiPOx with PA5 (Fig. 1a).43 Note that in the current study the temperature was lowered to 100 °C to avoid any degradation of PA5, the solvent was N,N′-dimethylformamide (DMF) and the reaction time was 13 h. The stability of the PA5 was tested by H-NMR spectroscopy in DMF-d7. The H-NMR spectrum after heating for 13 h at 100 °C was identical with the initial one (Fig. S9, ESI†) thus demonstrating the thermal stability of the PA5 under the conditions that were used for preparing the hydrogels. The COOH/iPOx molar ratio was varied between 0.01 and 0.2 corresponding to 0.5 and 10 mol% PA5 cross-linker (G0–G6). No shape stable hydrogels could be obtained when 0.5 mol% of PA5 was used. PiPOx hydrogels cross-linked with azelaic (AAz), and 2,2′-(1,4-phenylenebis(oxy))diacetic acid (PhDA), which is the constitutive unit of the PA5, were also synthesized as a control system. When the cross-linker content was 1.5 mol% (i.e., COOH/iPOx ratio was 0.03), no stable hydrogels could be obtained with these linear acids, regardless of the acid used. However, increasing the content to 2.5 mol% resulted in shape stable hydrogels in the case of AAz and PhDA acid.
900 g mol−1 and a dispersity of 1.15 was prepared using our previously reported method.42 The dicarboxylic acid functionalized PA5 was synthesized by cyclooligomerization of 1,4-dimethoxybenzene, diethyl 2,2′-(1,4-phenylenebis(oxy))diacetate and paraformaldehyde using boron trifluoride diethyl etherate as Lewis acid, followed by hydrolysis of the ester. The structure of the obtained PA5 was confirmed by FT-NMR, HRMS, while the ester intermediate was unambiguously assigned based on single crystal X-ray diffraction (for further synthetic and characterization details, see ESI†). The PiPOx-PA5 hydrogel synthesis was performed by adapting a previously developed method from our group, namely heating a solution of PiPOx with PA5 (Fig. 1a).43 Note that in the current study the temperature was lowered to 100 °C to avoid any degradation of PA5, the solvent was N,N′-dimethylformamide (DMF) and the reaction time was 13 h. The stability of the PA5 was tested by H-NMR spectroscopy in DMF-d7. The H-NMR spectrum after heating for 13 h at 100 °C was identical with the initial one (Fig. S9, ESI†) thus demonstrating the thermal stability of the PA5 under the conditions that were used for preparing the hydrogels. The COOH/iPOx molar ratio was varied between 0.01 and 0.2 corresponding to 0.5 and 10 mol% PA5 cross-linker (G0–G6). No shape stable hydrogels could be obtained when 0.5 mol% of PA5 was used. PiPOx hydrogels cross-linked with azelaic (AAz), and 2,2′-(1,4-phenylenebis(oxy))diacetic acid (PhDA), which is the constitutive unit of the PA5, were also synthesized as a control system. When the cross-linker content was 1.5 mol% (i.e., COOH/iPOx ratio was 0.03), no stable hydrogels could be obtained with these linear acids, regardless of the acid used. However, increasing the content to 2.5 mol% resulted in shape stable hydrogels in the case of AAz and PhDA acid.
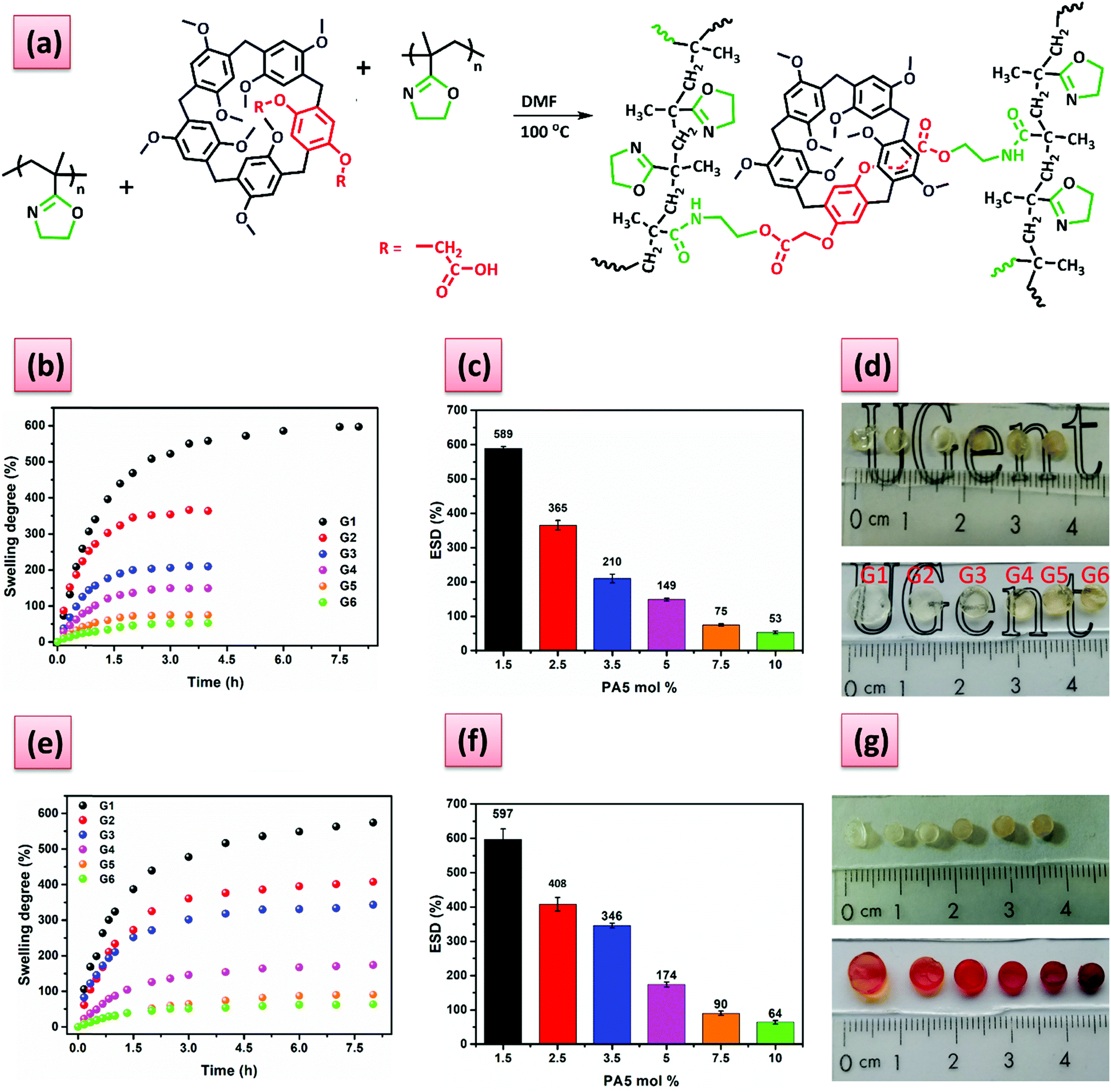 | ||
| Fig. 1 (a) Schematic representation of the reaction path to obtain PiPOx based hydrogels by cross-linking with PA5 dicarboxylic acid (b) swelling degree-time plots, (c) ESD dependence on the PA5 mol% and (d) photographs of the dry (up) and swollen (bottom) G1–G6 gels in de-ionized water at 25 °C, respectively; (e) swelling degree-time plots, (f) ESD dependence on the PA5 mol%, and (g) photographs of the dry (up) and swollen (bottom) G1–G6 gels in 5 mM MV solution at 25 °C, respectively. | ||
The structure of the PiPOx-PA5 dry gels was investigated using FT-IR spectroscopy. The signal corresponding to the carbonyl stretching vibration of the newly generated ester and to the phenyl plane bending from the PA5 are present at 1757 cm−1 and 1500 cm−1, respectively, accounting for the ester-amide cross-linked structure (Fig. S10, ESI†). Gel fraction values of 0.95 or higher (Table S1, ESI†) were determined in all cases, which is indicative of the highly efficient nature of the cross-linking reaction under the used conditions.
Further on we analyzed the influence of the amount of cross-linker (PA5) on the water absorption capacity of the hydrogels. The swelling degree (SD) in de-ionized water decreased as the molar percentage of the cross-linker increased from 1.5 mol% (G1) to 10 mol% (G6) (see Fig. 1b–c), because of the formation of more densely cross-linked network structures, that absorb less water as swelling is limited. The equilibrium swelling degree (ESD) of the hydrogels could be easily tuned between 600% and 50% by simply increasing the amount of PA5 cross-linker. As expected, the cross-linking density (ρc) increased with the amount of PA5. The hydrogels cross-linked with AAz and PhDA were found to have a higher ρc as compared to G2 (Table S1, ESI†), despite that the gel fraction of PhDA is lower than for G2. The time necessary for the hydrogels to reach equilibrium is an important parameter that predetermines their future applications and was found to be dependent on the amount of PA5 (Fig. 1b). G1 reaches equilibrium after 7 h, while the G2–G6 hydrogels require only 2.5 h. However, compared to our previously reported PiPOx hydrogels cross-linked with aliphatic dicarboxylic acids that required 24 h43 to reach ESD the time is much shorter for the G1–G6 hydrogels. This faster swelling may be related to the presence of the bulky PA5 that may facilitate a more open network structure. Next, we investigated the host–guest complexation with methyl viologen diiodide (MV), which is a known guest molecule that can complex strongly with pillar[5]arenes, and its impact on the SD. Swelling the gels in 5 mM MV aqueous solution led to a change in both the color and SD (Fig. 1e–g), due to the formation of the inclusion complexes between the PA5 and the MV groups. All the complexed hydrogels displayed the characteristic orange-red color of the PA5-MV complex and an increase in the SD, except for G1 (Fig. 1e–g). The increase in swelling upon MV complexation may be ascribed to the charge repulsion. Apparently, the lower ρc of G1 allows enough space between the complexed MV so that no significant increase in SD due to charge repulsion is observed.
Rheological investigations were carried out to investigate the mechanical properties of the hydrogels. The fully swollen hydrogels have high mechanical strength ranging from 4.6 to 608 kPa. The dynamic frequency sweep curves showed that G′ remained larger than G′′ and did not significantly change with the fixed 1% strain (Fig. S11a, ESI†), indicating the excellent stability of the hydrogel towards frequency oscillation. G′ and G′′ increased with increasing PA5 content as expected (Fig. S11a, ESI†), because of the higher cross-link density. The value of G′ of the hydrogels increased several orders of magnitude as the cross-linker content increased from 3 to 10 mol%. The dynamic strain sweep curves showed that G′ was always larger than G′′ with the strain from 0.1% to 25% (Fig. S11b, ESI†), indicating that the hydrogels were stable and remained undamaged. The hydrogels showed, however, a significant strain-dependent viscoelastic response. The linear viscoelastic behavior (LVE), was dependent on the amount of PA5 used. For G1 the LVE behavior persisted up to γ ∼ 60%, while for G2–G6 lower LVE behavior was registered, varying from 9% to 0.2% shear strain (Fig. S11b, ESI†). The G2 hydrogel displayed superior mechanical properties as compared to the AAz or PhDA hydrogels (Fig. S12 and S13, ESI†), indicating the importance of the 3D structure of PA5 for the mechanical properties. The registered value of G′ for the G2 hydrogel was 4 times higher than for the other two hydrogels (Fig. S13, ESI†), even though G2 had a lower cross-linking density (Table S1, ESI†). Using PhDA resulted in a minor increase of the G′ as compared to AAz, due to higher rigidity of the PhDA. These findings are in accordance with our design strategy regarding the enhancement of mechanical properties when PA5 is used as a junction point in the hydrogel network. One may assume that the hydrogels cross-linked with PA5 would have higher excluded volume effect, than the ones cross-linked with AAz or PhDA. Consequently, applying a mechanical stress would lead to steric repulsions and hence improved mechanical properties.
Swelling the hydrogels in a solution of MV led to an increase in the mechanical properties (Fig. S14, ESI†), despite that a higher ESD was previously determined. Both G′ and G′′ increased after complexation for all the investigated hydrogels (Fig. S15, ESI†), which proves that the complexation process contributes to the mechanical strength of the hydrogels through the generation of electrostatic repulsive interactions.
Compression tests were carried out to further investigate the mechanical properties of the PiPOx-PA5 hydrogels. The compression modulus (EC) of the gels swollen at equilibrium in de-ionized water gradually increased from 0.13 kPa for G1 to 102.8 kPa for G6 (Table S2 and Fig. S16a, ESI†). Moreover, the EC strongly increased when the PA5 content was higher than 7.5 mol% (Fig. S17, ESI†). This effect can be ascribed to the formation of less hydrophilic hydrogels at high PA5 content, where the mesh size is lower than 1 nm (Table S1, ESI†), thus increasing the overall rigidity of the gel matrix. The stress resistance followed the same trend as EC, that is increasing with the content of PA5 (Fig. S16a, ESI†). Hydrogels with ultrahigh stress resistance (>3 MPa) were obtained when the cross-linker content was higher than 5 mol% (Fig. S16a, ESI†). Consequently, both soft and ductile as well as rigid and brittle hydrogels could be easily obtained by controlling the PA5 cross-linker amount. Further enhancement of the hydrogel's toughness was achieved by swelling them in an aqueous solution containing 5 mM MV. Compared with the original hydrogels the modulus increased by almost a factor of two for G1–G4 after swelling in MV solution; while the G5–G6 hydrogels only displayed a factor of 0.2 increase (Table S2, ESI†).
Further tensile investigations were performed only for G1–G3, because these samples could be nicely obtained as plate-shaped hydrogels, which could be further cut into dumbbell shapes (Fig. S18, ESI†). The plate-shaped hydrogels could be bent and twisted without affecting their mechanical integrity (Fig. S18, ESI†). The tensile modulus (ET) and stress at breaking in tension increased, yet the breaking strain decreased with increasing the PA5 content from 1.5 mol% to 3.5 mol% (Fig. 2 and Table S2, ESI†). An increase in the ET and breaking stress, similar to compression, was observed for all the hydrogels after swelling in MV solution (Fig. 2b–e).
 | ||
| Fig. 2 (a) The photograph of the ongoing tensile test experiment. The tensile stress–strain curves: (b) de-ionized water and (c) 5 mM MV aqueous solution. The breaking stress for the hydrogels swelled in: (d) de-ionized water and (e) 5 mM MV aqueous solution. (f) The photograph of G3 after swelling in 5 mM MV aqueous solution. | ||
Compressive and tensile curves (Fig. 2 and 3) clearly demonstrate the striking enhancement of mechanical strength of the hydrogels cross-linked with PA5. G2 reached a high compression stress of 0.9 MPa at a strain of 75% and remained undamaged, while the breaking stress for the PhDA and the AAz hydrogels was 0.1 MPa (Fig. 3). Moreover, the tensile elastic modulus (ET) increased dramatically from 0.18 kPa for PhDA hydrogel to 1.25 kPa for G2 (Table S2, ESI†). Therefore, we can undoubtedly assign these improvements of strength and modulus to the presence of PA5.
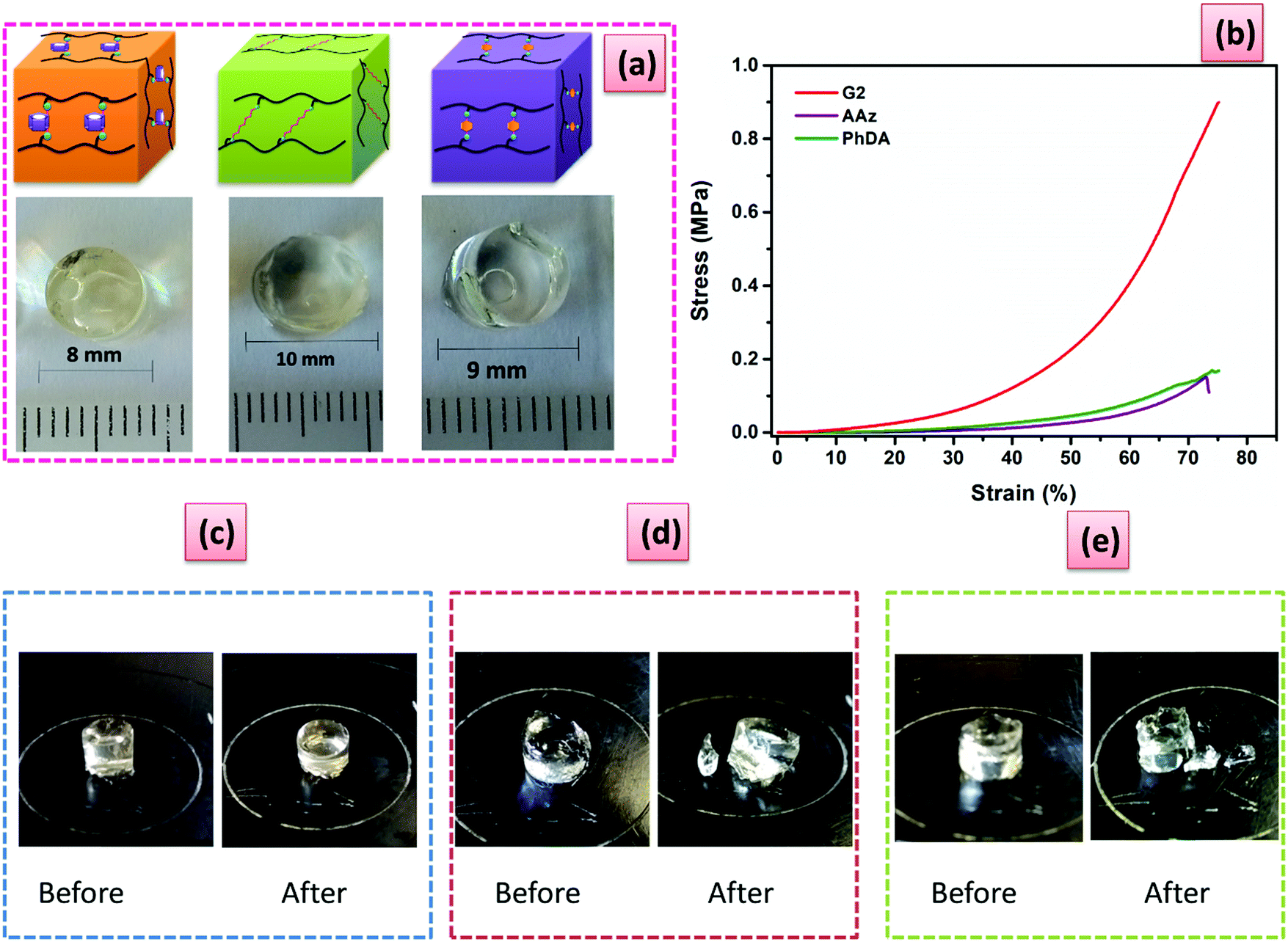 | ||
| Fig. 3 PiPOx hydrogels cross-linked using 2.5 mol% of PA5, AAz, and PhDA, respectively: (a) photographs after swelling in de-ionized water; (b) stress–strain curves under compressive mode. Photographs before and after compression measurements for: (c) G2, (d) PiPOx-AAz and (e) PhDA hydrogels. | ||
Successive loading–unloading tests were also performed to investigate the energy dissipation process of the PiPOx hydrogels (Fig. S19, ESI†). During loading–unloading tests, no resting time was given between the consecutive loading cycle. Therefore, the extension ratio of each cycle is calculated using the initial length of gel specimen from the first loading-unloading cycle. Upon cyclic loading of the G2 hydrogel with varied compression strain without resting interval between cycles, efficient energy dissipation was manifested as pronounced hysteresis loops due to the structural rearrangements (Fig. S19, ESI†) attributed to the breaking of physical associations (i.e., hydrophobic, π–π stacking) of the PA5 units. At 50% strain the dissipated energy was 2506 kJ m−3, (48% of the total input work). In sharp contrast, the AAz and PhDA hydrogels showed much smaller hysteresis loops, and only 139 kJ m−3 (23% of the total input work) and 173 kJ m−3 (25% of the total input work), respectively were dissipated at a strain of 50% (Fig. S19, ESI†). The continuous loading-unloading cycles (Fig. S20, ESI†) at fixed strain (i.e., 25%) demonstrate the strain hardening behavior of G2. The hysteresis loop of the second cycle becomes larger than that of the first one and the dissipated energy increased from 159.2 kJ m−3 in the first cycle to 254.3 kJ m−3 on the fifth cycle (Table S3, ESI†). The difference between loading and unloading paths of the stress–strain diagrams under cyclic deformation cannot be ascribed to the time-dependent effects as the duration of a cyclic test does not exceed 60 s, while no water expelling was noticed at 25% strain deformation. The pronounced strain hardening of G2 may arise from the steric repulsions upon compression together with a distortion of the PA5 units. Moreover, considering the high strength and rigidity of the junction zones (e.g., PA5) which are formed by laterally combined PiPOx polymer chains, a small amount of deformation would cause a considerable restoring force, which increased with the strain.
The hydrogel cross-linked with AAz showed only a minor variation of the dissipated energy during the five repetitive cycles (Table S3, ESI†). Furthermore, the dissipated energy of G2 swollen in MV solution only slightly increased in the second cycle, while continuously decreasing in the next cycles due to the electrostatic repulsive interactions between guest molecules, thus accounting for the proposed mechanism. If the G2 hydrogel is re-swollen for 60 min in de-ionized water, the loading-unloading cycle (Fig. S20a, ESI†) nearly overlaps with the first one, suggesting that the hydrogen bonds can be quickly recovered. Moreover, high recovery efficiency (e.g., 96%) was found for G2 hydrogel after swelling in DW for 1 h.
To demonstrate the versatility of our hydrogels, we tested them in a practical application, namely water purification based on the host–guest complexation ability. The hydrogel containing the highest amount of PA5 was, therefore, chosen. The performance of G6 for organic micropollutant removal was evaluated towards a series of contaminants with adverse effects on human health and the environment. G6 can effectively remove the pollutants with large uptake and uptake efficiency, especially for methylene blue and methyl red (Fig. 4b–d). The role of the PA5 in the adsorption process was demonstrated by performing reference adsorption experiments with the PiPOx-AAz and PiPOx-PhDA hydrogels. A striking difference can be seen between the PiPOx-AAz, and PiPOx-PhDa on the one hand and G6 on the other hand, after swelling for 24 h in a 0.05 mM solution of methylene blue (Fig. S21, ESI†). The G6 hydrogel was intensively blue-colored and completely opaque, while the PiPOx-AAz as well as PiPOx-PhDA showed a minor blue coloration while keeping their transparency (Fig. S21, ESI†). The uptake of the PiPOx-AAz hydrogel was very low, regardless of the micropollutant chemical structure thus proving that the 2-oxazoline groups have minimum interactions with the studied dyes (Table S4, ESI†). The PiPOx-PhDA hydrogel showed similar micropollutants uptake as PiPOx-AAz demonstrating that the water purification properties of the G6 are solely due to the guest complexation ability of PA5.
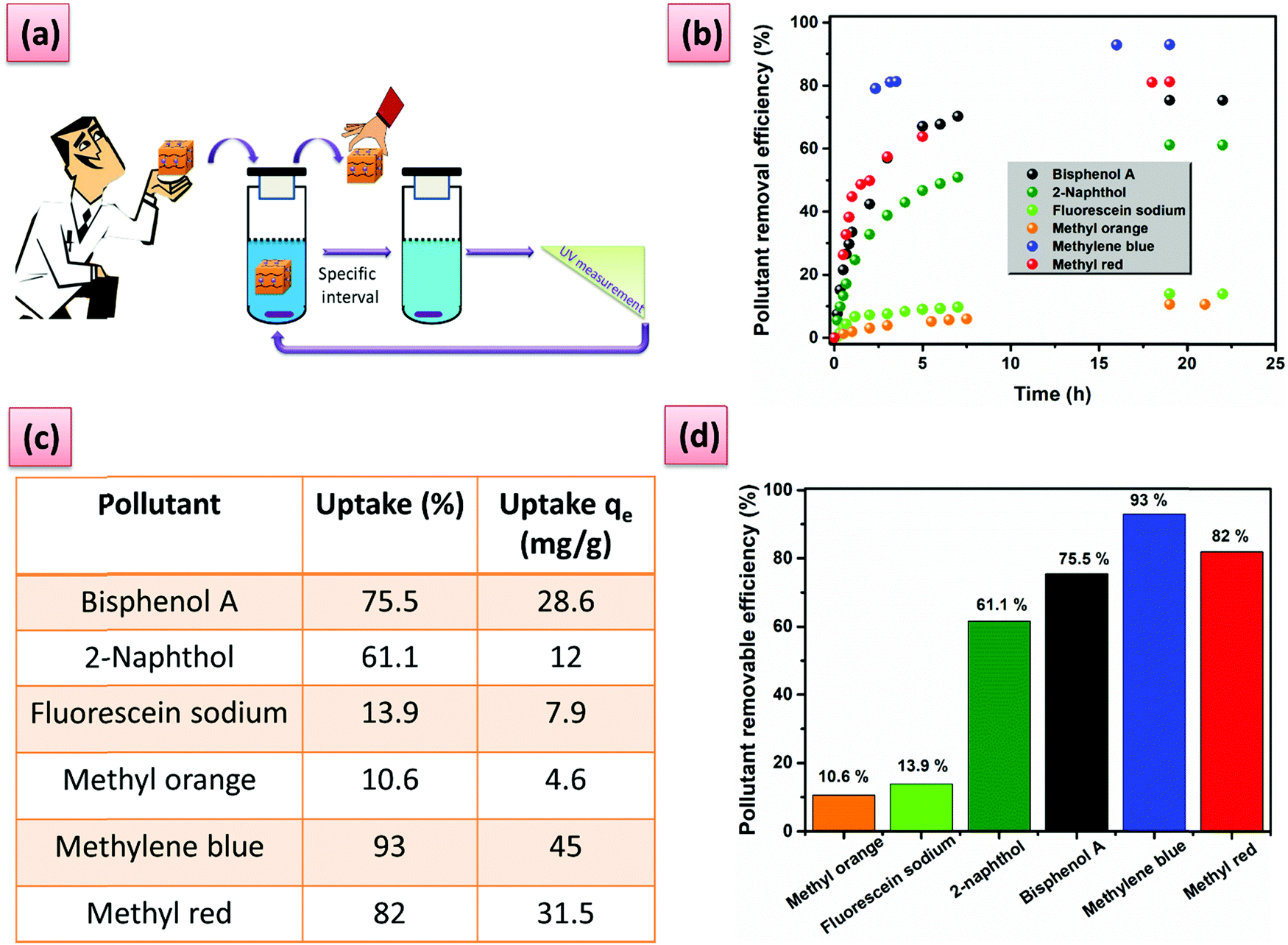 | ||
| Fig. 4 (a) Cartoon representation of time-dependent adsorption process; (b) the time-dependent adsorption process of each organic micropollutant (0.05 mM); (c) the equilibrium uptake percentage and amount of micropollutant adsorbed per gram of G6; (d) the percentage removal efficiency of each organic micropollutant determined after 24 h contact. | ||
Furthermore, the high removal efficiency can be visually observed and monitored for these two dyes (Fig. 5a and b). Phenol derivatives, which are common contaminants can also be removed with an efficiency higher than 60% (Fig. 4d). Moderate adsorption kinetics are registered with the adsorption reaching a maximum in approximately 7 h (Fig. 4b). The hydrogel regeneration and reuse was demonstrated through five repetative cycles in the case of methylene blue by using a washing procedure with N,N′-dimethylformamide without loss in performance (Fig. 5c and d). The process could be easily followed by the naked eye (Fig. 5c).
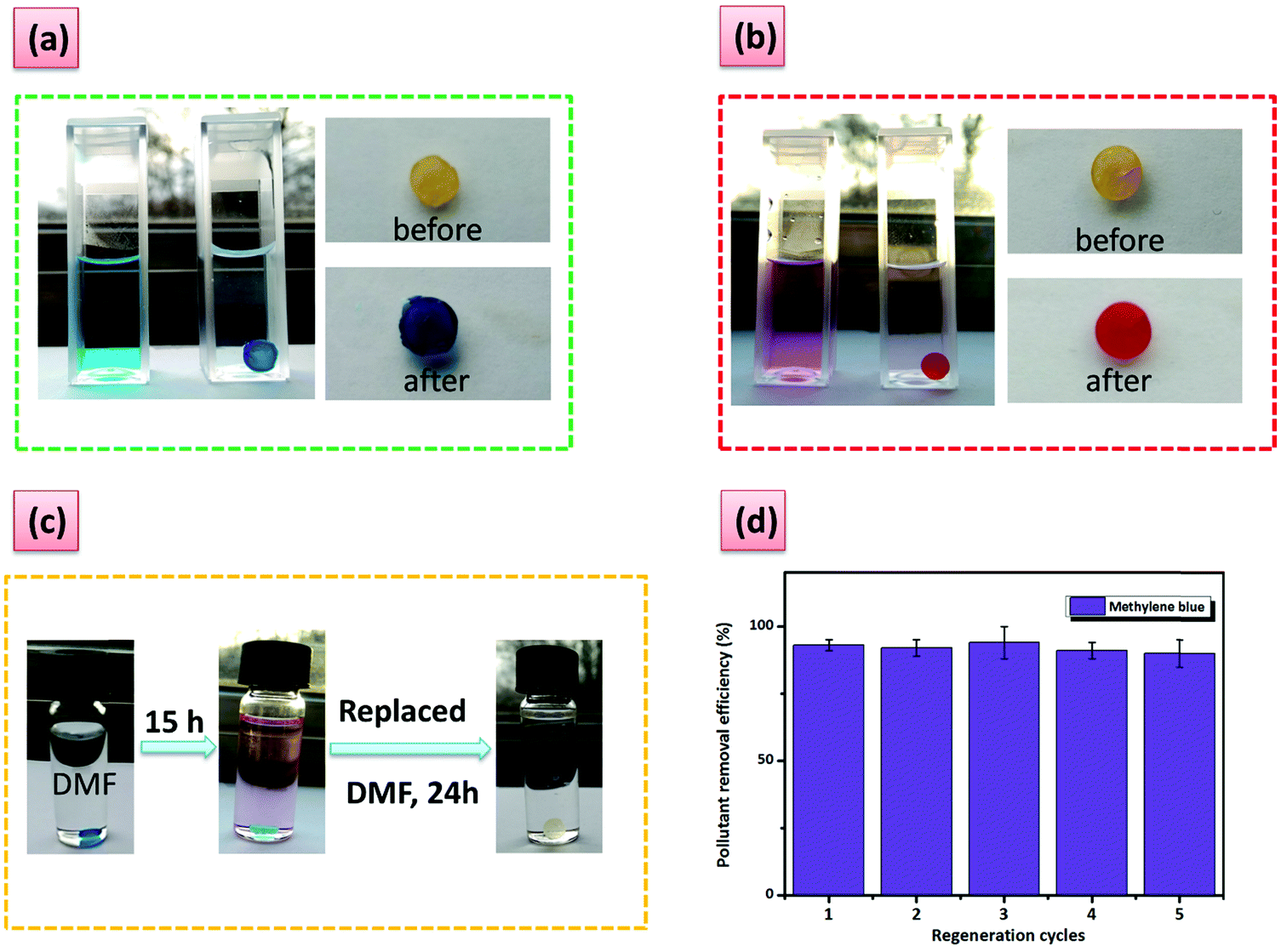 | ||
| Fig. 5 Color changes after 24 h for aqueous solutions containing: (a) methylene blue and (b) methyl red hydrochloride. (c) Color changes for G6 before and after washing. (d) The regeneration cycles of G6 after the adsorption of methylene blue and washing with DMF. | ||
Conclusions
In summary, hydrogels with tunable mechanical properties and swelling behavior were obtained by covalently cross-linking PiPOx with a specifically constructed dicarboxylic acid pillar[5]arene derivative. Superior resistance to external stress was demonstrated for these homogeneous single network hydrogels, induced by the higher rigidity of the PA5 cross-linker as compared to “regular” cross-linkers. Moreover, the properties of the resulting PA5-containing hydrogels could be controlled by supramolecular interactions. Their potential applications as adsorbents for water purification have also been demonstrated. The simplicity, versatility, and robustness of the present method lead us to suggest that such a strategy could have an impact on the synthesis of multi-functional hydrogels with superior and controllable properties. Most importantly this general synthetic strategy could be further applied to other types of polymers such as poly(vinyl alcohol) or poly(2-hydroxyethyl methacrylate), provided that pillar[n]arene derivative possesses a proper functional group giving access to a variety of hydrogels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Xu acknowledges the Chinese Scholarship Council (CSC, File No. 201506780014) and Ghent University (BOF, File No. 01SC1717) for the financial support for his PhD program; R. Hoogenboom thanks FWO Flanders and Ghent Unviersity for continuous financial support. KVH thanks the Hercules Foundation (project AUGE/11/029 “3D-SPACE: 3D Structural Platform Aiming for Chemical Excellence”) and the Special Research Fund (BOF) – UGent (project 01N03217) for funding.References
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS.
- R. Edri, I. Gal, N. Noor, T. Harel, S. Fleischer, N. Adadi, O. Green, D. Shabat, L. Heller, A. Shapira, I. Gat-Viks, D. Peer and T. Dvir, Adv. Mater., 2019, 31, 1803895 CrossRef.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS.
- M. Nakahata, Y. Takashima, A. Hashidzume and A. Harada, Angew. Chem., 2013, 125, 5843–5847 CrossRef.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS.
- C. Li, M. J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman and D. Liu, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS.
- E. Ye, P. L. Chee, A. Prasad, X. Fang, C. Owh, V. J. J. Yeo and X. J. Loh, Mater. Today, 2014, 17, 194–202 CrossRef CAS.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- H. Xing, Z. Li, Z. L. Wu and F. Huang, Macromol. Rapid Commun., 2018, 39, 1700361 CrossRef.
- L. Jiang, C. Liu, K. Mayumi, K. Kato, H. Yokoyama and K. Ito, Chem. Mater., 2018, 30, 5013–5019 CrossRef CAS.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef.
- J. Yang, C.-R. Han, J.-F. Duan, F. Xu and R.-C. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 3199–3207 CrossRef CAS.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441–453 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171–4176 CrossRef CAS PubMed.
- C. Katsuno, A. Konda, K. Urayama, T. Takigawa, M. Kidowaki and K. Ito, Adv. Mater., 2013, 25, 4636–4640 CrossRef CAS.
- A. B. Imran, T. Seki and Y. Takeoka, Polym. J., 2010, 42, 839 CrossRef.
- H. Xin, S. Z. Saricilar, H. R. Brown, P. G. Whitten and G. M. Spinks, Macromolecules, 2013, 46, 6613–6620 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13 CrossRef.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- M. Nakahata, Y. Takashima and A. Harada, Macromol. Rapid Commun., 2016, 37, 86–92 CrossRef CAS.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef.
- J. Liu, C. S. Y. Tan, Z. Yu, Y. Lan, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1604951 CrossRef.
- X. Xu, F. A. Jerca, V. V. Jerca and R. Hoogenboom, Adv. Funct. Mater., 2019, 1904886 CrossRef CAS.
- X. Ji, R.-T. Wu, L. Long, C. Guo, N. M. Khashab, F. Huang and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 2777–2780 CrossRef CAS.
- H. Zhao, J. Gao, R. Liu and S. Zhao, Carbohydr. Res., 2016, 428, 79–86 CrossRef CAS.
- T. Ogoshi, T.-A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS.
- T. Kakuta, T.-A. Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS.
- Y. Yao, M. Xue, J. Chen, M. Zhang and F. Huang, J. Am. Chem. Soc., 2012, 134, 15712–15715 CrossRef CAS.
- L. Shao, J. Sun, B. Hua and F. Huang, Chem. Commun., 2018, 54, 4866–4869 RSC.
- K. Jie, Y. Zhou, Y. Yao, B. Shi and F. Huang, J. Am. Chem. Soc., 2015, 137, 10472–10475 CrossRef CAS.
- N. Song, T. Kakuta, T.-A. Yamagishi, Y.-W. Yang and T. Ogoshi, Chem, 2018, 4, 2029–2053 CAS.
- Q. Li, X. Li, L. Ning, C.-H. Tan, Y. Mu and R. Wang, Small, 2019, 15, 1804678 CrossRef.
- T. Ogoshi, S. Takashima and T.-A. Yamagishi, J. Am. Chem. Soc., 2018, 140, 1544–1548 CrossRef CAS.
- N. L. Strutt, H. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2631–2642 CrossRef CAS.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS.
- H. Ju, F. Zhu, H. Xing, Z. L. Wu and F. Huang, Macromol. Rapid Commun., 2017, 38, 1700232 CrossRef.
- Y. Liu, B. Shi, H. Wang, L. Shangguan, Z. Li, M. Zhang and F. Huang, Macromol. Rapid Commun., 2018, 39, 1800655 CrossRef.
- F. A. Jerca, V. V. Jerca, A. M. Anghelache, D. M. Vuluga and R. Hoogenboom, Polym. Chem., 2018, 9, 3473–3478 RSC.
- F. A. Jerca, A. M. Anghelache, E. Ghibu, S. Cecoltan, I.-C. Stancu, R. Trusca, E. Vasile, M. Teodorescu, D. M. Vuluga, R. Hoogenboom and V. V. Jerca, Chem. Mater., 2018, 30, 7938–7949 CrossRef CAS.
- M. C. Spiridon, F. A. Jerca, V. V. Jerca, D. S. Vasilescu and D. M. Vuluga, Eur. Polym. J., 2013, 49, 452–463 CrossRef CAS.
- N. Zhang, S. Huber, A. Schulz, R. Luxenhofer and R. Jordan, Macromolecules, 2009, 42, 2215–2221 CrossRef CAS.
- N. Zhang, T. Pompe, I. Amin, R. Luxenhofer, C. Werner and R. Jordan, Macromol. Biosci., 2012, 12, 926–936 CrossRef CAS.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 1963–1969 CrossRef CAS.
- A. F. Nicolescu, V. V. Jerca, A.-M. Albu, D. M. Vuluga and C. Draghici, Mol. Cryst. Liq. Cryst., 2008, 486, 38/[1080]–1049/[1091] Search PubMed.
- Z. Kroneková, M. Mikulec, N. Petrenčíková, E. Paulovičová, L. Paulovičová, V. Jančinová, R. Nosál', P. S. Reddy, G. D. Shimoga, D. Chorvát Jr. and J. Kronek, Macromol. Biosci., 2016, 16, 1200–1211 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1910255. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9mh01401b |
| This journal is © The Royal Society of Chemistry 2020 |