
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected reactivity of a PONNOP ‘expanded pincer’ ligand†
Arthur R.
Scheerder
 a,
Martin
Lutz
b and
Daniël L. J.
Broere
*a
a,
Martin
Lutz
b and
Daniël L. J.
Broere
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: d.l.j.broere@uu.nl
bCrystal and Structural Chemistry, Bijvoet Centre for Biomolecular Research Faculty of Science, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
First published on 1st May 2020
Abstract
We report the synthesis, characterization and coordination chemistry of a new naphthyridine-derived phosphinite PONNOP expanded pincer ligand. As envisioned, the dinucleating ligand readily binds two copper(I) centers in close proximity, but undergoes an unexpected rearrangement in the presence of nickel(II) salts to form an interesting PONNP pincer platform.
The use of well-defined bimetallic complexes wherein two metal atoms are bound in close proximity is an emerging area in homogeneous catalysis.1 The cooperative activation of substrate molecules by two metal centers in such bimetallic systems can enable distinct reactivity or improved catalytic performance when compared to monometallic analogues.2 The design and exploration of new dinucleating ligands that enable such metal–metal cooperativity is key to further explore the potential of bimetallic catalysis. A motif well-suited to bind two metals is the 1,8-naphthyridine scaffold.3 Yet, relatively few ligands derived thereof have been reported that are capable of both binding two metal centers in close proximity while also providing accessible adjacent coordination sites on both metals to enable metal–metal cooperativity.4 In the pursuit of such new platforms we recently reported a dinucleating ‘expanded PNNP pincer’ ligand,5 which was inspired by the mononucleating PNP pincer ligands6 (Scheme 1, top). Similar to the PNP ligands the CH2 linkers in the PNNP ligand can be deprotonated concomitant with dearomatization of the naphthyridine core, and this enabled the cooperative activation of H2 to give a tetranuclear copper dihydride cluster.7
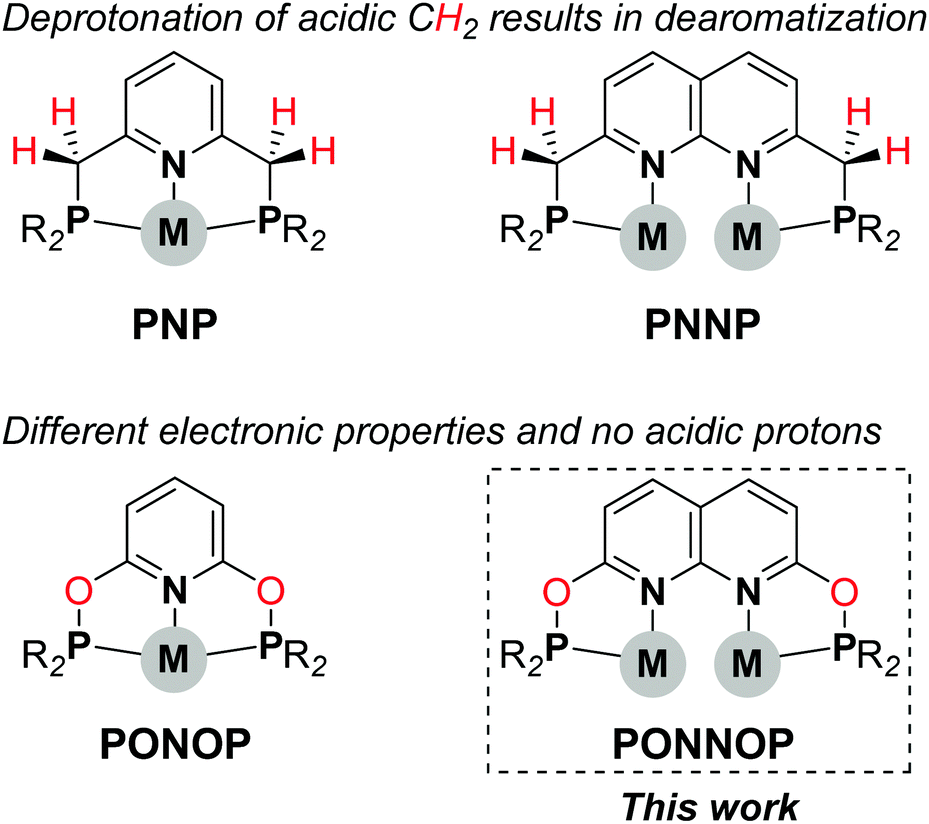 | ||
| Scheme 1 Comparison of the PNP and PONOP pincer ligands with the PNNP and PONNOP expanded pincer ligands. | ||
Exchanging the acidic CH2 linkers in the PNP pincer ligands by O atoms affords the diphosphinite PONOP pincer ligand (Scheme 1, bottom), which was developed independently by the Milstein8 and Goldberg & Brookhart9 groups. The PONOP pincer ligand has been utilized to stabilize various metal complexes and, for example, has enabled the detailed study of dihydrogen complexes of Rh and Ir,10 σ-alkane complexes,11 and more recently photolytic N2 reduction to NH3 using Re.12 Given the distinct differences between the PNP and PONOP pincer ligands we were interested to study a PNNP analogue wherein the reactive methylene linkers are replaced by oxygen atoms. Herein, we describe the synthesis, characterization and coordination chemistry of a new expanded pincer ligand, i-PrPONNOP. Similar to the PNNP analogue, i-PrPONNOP can bind two copper atoms in close proximity. However, we found that this ligand undergoes an unusual rearrangement in the presence of nickel salts. Although this rearrangement provides access to a unique ‘regular’ pincer ligand, it can prohibit binding two metal centers in close proximity, and should therefore be taken into consideration in the design of naphthyridine-derived dinucleating ligands featuring phosphinite donors.13
The phosphinite expanded pincer ligand i-PrPONNOP was prepared in a three-step procedure from commercially available starting materials in an overall yield of 37%. The first two steps comprise the synthesis of 2,7-dihydroxy-1,8-naphthyridine according to a modified literature procedure.14 Subsequent phosphorylation was achieved by a reaction with a slight excess of chlorodiisopropylphosphine in the presence of excess Et3N in THF at 65 °C (Scheme 2) affording i-PrPONNOP as a brown oil in 82% yield and 90% purity after workup. Several attempts to further enhance the purity solely resulted further decrease in purity due to the facile hydrolysis of the P–O bonds in the expanded pincer ligand (see ESI† for more detail). The related 2,6-dihydroxypyridine-derived i-PrPONOP pincer ligand was also reported in 90% purity.8 Fortunately, the unidentified impurities in the expanded pincer ligand are readily removed in subsequent reactions (see below). The 1H, 13C and 31P NMR spectra of i-PrPONNOP in C6D6 at 298 K show the expected number of resonances for a C2v symmetric species. The 31P{1H} NMR spectrum shows a singlet at δ = 148.2 ppm, similar to the observed resonance for the i-PrPONOP pincer (δ = 149.1 ppm).8
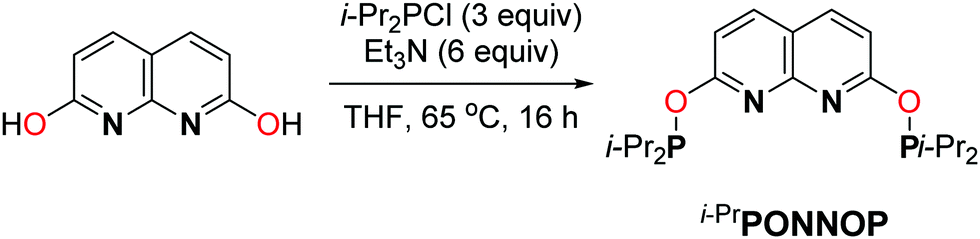 | ||
| Scheme 2 The synthesis of the i-PrPONNOP “expanded pincer” ligand. | ||
Reacting i-PrPONNOP with 2 equiv. of CuCl in THF or CH2Cl2 (Scheme 3) gives the dicopper(I) complex 1, which was isolated as yellow-orange powder in 77% yield. The 1H, 13C and 31P NMR spectra of 1 in CD2Cl2 show that the C2v symmetry is retained upon binding the Cu centers. The 31P NMR spectrum of 1 in CD2Cl2 features a single broad resonance at δ = 123.7 ppm. VT NMR analysis (Fig. S14–S19, ESI†) showed that the broadness is not due to a fluxional process on the NMR time scale. This suggests that it originates from quadrupolar relaxation arising from 63Cu and 65Cu (both I = 3/2) nuclei, which is not uncommon for Cu(I) phosphinite complexes.15 Crystals suitable for single-crystal X-ray diffraction were grown by vapor diffusion of hexane into a THF solution of 1 at ambient temperature. The solid-state structure of 1 (Fig. 1) revealed a nearly flat dinucleating phosphinite ligand bound to a diamond-shaped Cu2Cl2 core. The structure is similar to the previously reported t-BuPNNPCu2Cl27 but features a Cu–Cu distance (2.7224(9) Å) that is ∼0.14 Å longer, and PN bite angles (∠P1–Cu1–N1 = 83.33(14)° and ∠P2–Cu2–N2 = 83.00(14)°) that are smaller by ∼3° (see Tables S3–S5, ESI† for more detail).
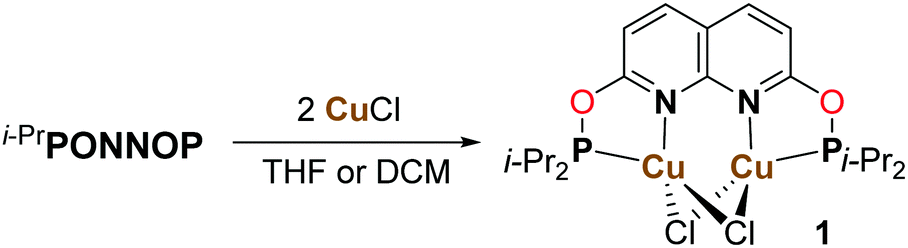 | ||
| Scheme 3 Synthesis of the dicopper(I) complex 1. | ||
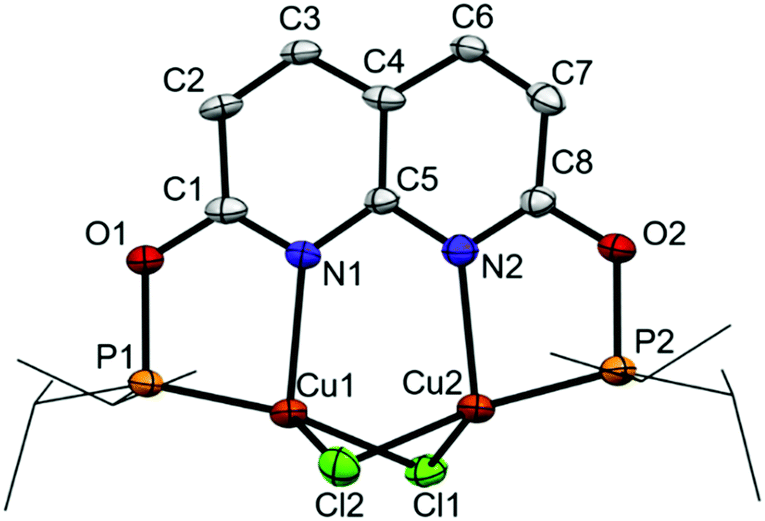 | ||
| Fig. 1 Displacement ellipsoid plot (50% probability) of complex 1 in the crystal. Hydrogen atoms and THF solvent molecule are omitted, and i-Pr groups on P are depicted as wireframe for clarity. | ||
As the P–O bonds in the phosphinite expanded pincer ligand are highly susceptible to hydrolysis, the use of solvents containing trace water typically results in precipitation of 2,7-dihydroxy-1,8-naphthyridine and the formation of a partial hydrolysis product i-PrPONNOH. The latter monophosphinite displays four characteristic aromatic resonances in 1H NMR spectra of reaction mixtures and is also observed as an intermediate in the synthesis of i-PrPONNOP (see ESI† for more detail). Both these undesired naphthyridine containing products were also observed in several reactions as a result of alcoholysis or aminolysis. For example, a reaction in THF of i-PrPONNOP with 2 equiv. NiCl2 containing residual EtOH resulted in the formation of an insoluble 2,7-dihydroxy-1,8-naphthyridine-derived species and trans-NiCl2(P(OEt)(i-Pr)2)2, which was isolated and crystallographically characterized (see ESI† for more detail). Nonetheless, when using rigorously dried solvents these undesired side reactions can be prevented.
Reacting i-PrPONNOP with 2 equiv. NiBr2 in THF results in the formation of an insoluble precipitate and a single species in solution that features four aromatic resonances in the 1H NMR spectrum, indicative of a loss of C2v symmetry. Unlike the partial hydrolysis product i-PrPONNOH, this unsymmetrical species contains two diisopropylphospinite moieties, which appear as two doublets (2JPP = 326 Hz) at δ = 192.0 and 131.5 ppm in the 31P NMR spectrum in CD2Cl2. To our surprise single-crystal X-ray structure determination (Fig. 2) did not reveal a dinuclear complex but showed mononuclear complex 2, which is consistent with the 1H, 13C and 31P NMR spectra as well as the CHN elemental analysis of the obtained solid. Furthermore, complex 2 can also be prepared in 89% yield by a reaction of i-PrPONNOP ligand with 1 equiv. NiBr2 in CH2Cl2 (Scheme 4). The solid state structure (Fig. 2) shows that the expanded pincer ligand underwent a rearrangement to form an interesting naphthyridone-like i-PrPONNP ‘regular’ pincer ligand bound to nickel. The nickel atom exhibits a slightly distorted square pyramidal geometry with a Br atom in the apical position. Nickel(II) pentacoordination is uncommon in pincer chemistry16 and all reported NiX2 (X = Cl or Br) complexes bearing related lutidine-derived PNP,17–19PONOP20 and PNNNP21 pincer ligands display square planar geometries with a non-coordinated halide anion. The nickel bromide bond lengths in 2 differ greatly, and the distance is significantly longer for the bromide in the apical position (Ni1–Br1 = 2.7852(8) Å) than what is observed for the bromide in the basal position (Ni1–Br2 = 2.3200(8) Å). A similar observation was made for a dibromo{bis[2-(diphenylphosphanyl)ethyl]amine} Ni(II) complex, which showed an apical Ni–Br bond of 2.698(7) Å and a basal Ni–Br bond of 2.333(7) Å.22 A rare example of a related pentacoordinated Ni(PNNNP) pincer complex with an anionic phosphinite instead of a basal Br ligand, showed an significantly shorter apical Ni–Br bond of 2.535(4) Å.23 The intraligand bond metrics are distinctly different from those in complex 1 (Table S6, ESI†). The C7–O2 bond length of 1.214(6) Å is significantly shorter than the C1–O1 bond length of 1.346(5) Å. This observation indicates a dearomatization of the six-membered heterocycle containing N2 to form a localized naphthyridone, and this is further underlined by the clear localization of single and double bonds in this ring (Fig. S29, ESI†). We reason that the rearrangement of the i-PrPONNOP ligand to the pincer ligand present in complex 2 is also enabled by the ability to form a naphthyridone structure.
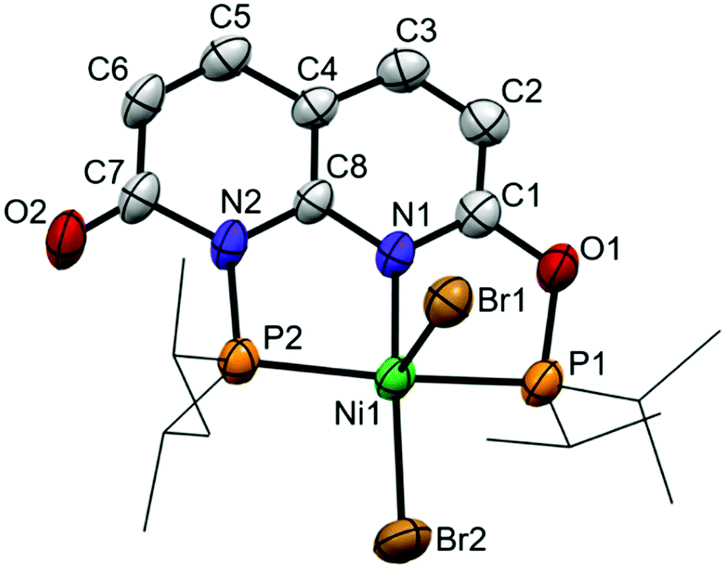 | ||
| Fig. 2 Displacement ellipsoid plot (50% probability) of complex 2 in the crystal. Hydrogen atoms and THF solvent molecules are omitted, and i-Pr groups on P are depicted as wireframe for clarity. | ||
 | ||
| Scheme 4 Synthesis of the mononuclear Ni complex 2. | ||
To gain insight into why the ligand rearrangement is only observed with NiBr2 and not with CuCl we studied the interconversion of the PONNOP and PONNP isomers computationally. Both isomers and the transition state geometries for their conversion were optimized (BP86-D3, def2-TZVP) without a metal, bound to CuCl or bound to NiBr2 (see ESI† for more details and discussion). Unfortunately, the heterogeneous nature of the studied reactions and the high sensitivity of the i-PrPONNOP ligand prohibited experimental validation of the computational methods beyond a good agreement of the experimental and computational geometries of complex 2. Hence, we refrain from putting much value on the absolute energies and choose to only look at the general trends, which provide some insight. The calculated free energy profile for the free ligand (Fig. 3, top) shows that the PONNOP and PONNP isomers are similar in energy and that there is a relatively high barrier for their interconversion. Heating a toluene solution of i-PrPONNOP for multiple hours at 110 °C showed no conversion into a new species based on NMR analysis indicating that either the barrier for the interconversion is too high or that the PONNOP isomer is the thermodynamic product.24 Notably, when the PONNOP ligand is bound to either CuCl or NiBr2 the calculated free energies of the PONNP isomer are significantly more stable than the PONNOP isomer. Similarly, the calculated energy barrier for the conversion of the PONNOP isomer to the PONNP is also significantly lower. Upon analysis of the corresponding transition state geometries (Fig. 3, bottom) it can be seen that the P atom, which migrates from O to N is bound to the transition metal atom. Although this also results in significant bending of the naphthyridine core (Fig. S33, ESI†), we reason that this provides the additional stabilization of the transition state to enable the isomerization towards the more stable PONNP pincer motif. We hypothesize that these effects are most pronounced when the ligand is bound to NiBr2 because the TS and PONNP geometries are closer to the preferred coordination geometry of Ni(II) than of that of Cu(I) (see ESI† for more detail). Although dinuclear complex 1 was obtained in high yield, we reacted i-PrPONNOP with one equiv. of CuCl since the calculated free energy profile indicated facile formation of a PONNP pincer complex of CuCl. Unfortunately, no conclusions can be drawn from this experiment as it resulted in the formation of an inseparable mixture of unidentified unsymmetrical species. The broadness of the resonances due to coordination to Cu also obscured the observation of a potential characteristic trans coupling like that observed in the 31P NMR spectrum of 2. We reason that the facile formation of 2 is likely fast and irreversible under the experimental conditions, and this is in agreement with the calculated free energy profile that features a low barrier and is very exergonic. In contrast, it is conceivable that with Cu the binding of a second CuCl is associated with a lower energy barrier, and therefore kinetically favored over the rearrangement. An alternative explanation is that the rearrangement is reversible in the presence of excess CuCl, and perhaps mediated by binding to a second Cu center. Unfortunately, since various attempts at reactions of i-PrPONNOP with 1 equiv. CuCl or 2 equiv. NiBr2 did not give a monocopper or dinickel species, respectively, we have thus far been unable to verify these hypotheses.
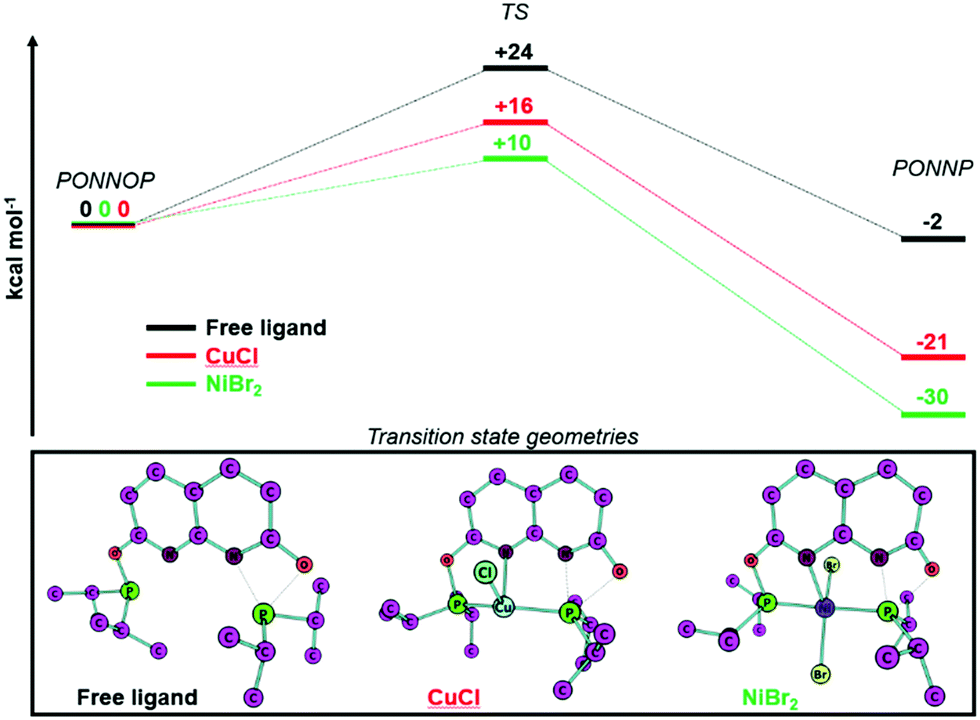 | ||
| Fig. 3 DFT calculated (BP86-D3, def2-TZVP) free energy profiles (ΔG0298K in kcal mol−1) for the rearrangement of PONNOP to PONNP (top) and the transition state geometries (bottom). | ||
In conclusion, we have described the synthesis, characterization and coordination chemistry of a new diphosphinite expanded pincer ligand, i-PrPONNOP. This new dinucleating ligand does not feature the acidic methylene linkers that are present in the related PNNP ligand, and binds two copper centers in close proximity in a similar fashion. In contrast, attempts to bind two nickel centers resulted in a ligand rearrangement concomitant with the formation of a unique naphthyridone PONNP pincer ligand. This work adds a new, readily synthesized dinucleating ligand to the bimetallic catalysis toolkit. Moreover, the results herein demonstrate that naphthyridine-derived ligands featuring phosphinite donors should be used with caution as they are able to undergo a rearrangement under certain conditions to form a ‘regular’ pincer ligand. Although the latter is undesired for bimetallic chemistry, it does provide access to an otherwise inaccessible ligand class for mononuclear chemistry.
This work was supported by Utrecht University (tenure track start-up package to D. L. J. B.), The Netherlands Organization for Scientific Research (START-UP grant 740.018.019 to D. L. J. B.) and the European Union's Horizon 2020 research and innovation program (agreement 840836, MSCA-IF to D. L. J. B., BiMetaCat). Access to supercomputer facilities was sponsored by NWO Exacte en Natuurwetenschappen (Physical Sciences). The X-ray diffractometer was financed by the NWO. Lada Dabranskaya is acknowledged for her artistic input for the TOC graphic. NMR and ORCA data files can be obtained free of charge from: http://doi.org/10.4121/uuid:8fd78884-01b0-4c37-a4ca-fb1cf4af8b61.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Xiong, G. Zhang, X. Sun and R. Zeng, Chin. J. Chem., 2020, 38, 185–201 CrossRef CAS; (b) R. B. Ferreira and L. J. Murray, Acc. Chem. Res., 2019, 52, 447–455 CrossRef CAS PubMed; (c) S. Rej, H. Tsurugi and K. Mashima, Coord. Chem. Rev., 2018, 355, 223–239 CrossRef CAS; (d) I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS; (e) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1717 RSC; (f) N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed; (g) M. Iglesias, E. Sola and L. A. Oro, in Homo and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, 2016, pp. 31–58 Search PubMed.
- Selected recent examples: (a) H.-C. Yu, S. M. Islam and N. P. Mankad, ACS Catal., 2020, 10, 3670–3675 CrossRef CAS; (b) Y.-Y. Zhou and C. Uyeda, Science, 2019, 363, 857–862 CrossRef CAS PubMed; (c) K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS; (d) I. G. Powers, J. M. Andjaba, X. Luo, J. Mei and C. Uyeda, J. Am. Chem. Soc., 2018, 140, 4110–4118 CrossRef CAS PubMed.
- J. K. Bera, N. Sadhukhanb and M. Majumdar, Eur. J. Inorg. Chem., 2009, 4023–4038 CrossRef CAS.
- Selected examples of ligand platforms: (a) M. S. Ziegler, D. S. Levine, K. V. Lakshmi and T. D. Tilley, J. Am. Chem. Soc., 2016, 138, 6484–6491 CrossRef CAS PubMed; (b) T.-T. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Inorg. Chem., 2014, 53, 11770–11777 CrossRef PubMed; (c) E. Binamira-Soriaga, N. L. Keder and W. C. Kaska, Inorg. Chem., 1990, 29, 3167–3171 CrossRef CAS; (d) T.-P. Cheng, B.-S. Liao, Y.-H. Liu, S.-M. Penga and S.-T. Liu, Dalton Trans., 2012, 41, 3468–3473 RSC; (e) C. He, A. M. Barrios, D. Lee, J. Kuzelka, R. M. Davydov and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 12683–12690 CrossRef CAS; (f) A. Nicolay and T. D. Tilley, Chem. – Eur. J., 2018, 24, 10329–10333 CrossRef CAS PubMed.
- E. Kounalis, M. Lutz and D. L. J. Broere, Organometallics, 2020, 39, 585–592 CrossRef CAS.
- (a) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin, 2013, vol. 40 Search PubMed; (b) The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications Top Organometallic Chem, ed. G. van Koten and R. A. Gossage, Springer, 2016, vol. 54 Search PubMed; (c) Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabó and O. Wendt, Wiley, 2014 Search PubMed.
- E. Kounalis, M. Lutz and D. L. J. Broere, Chem. – Eur. J., 2019, 25, 13280–13284 CrossRef CAS PubMed.
- H. Salem, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Organometallics, 2009, 28, 4791–4806 CrossRef CAS.
- W. H. Bernskoetter, S. Kloek Hanson, S. K. Buzak, Z. Davis, P. S. White, R. Swartz, K. I. Goldberg and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603–8613 CrossRef CAS PubMed.
- M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS PubMed.
- (a) M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed; (b) J. Campos, S. Kundu, D. R. Pahls, M. Brookhart, E. Carmona and T. R. Cundari, J. Am. Chem. Soc., 2013, 135, 1217–1220 CrossRef CAS PubMed.
- Q. J. Bruch, G. P. Connor, C.-H. Chen, P. L. Holland, J. M. Mayer, F. Hasanayn and A. J. M. Miller, J. Am. Chem. Soc., 2019, 141, 20198–20208 CrossRef CAS PubMed.
- While this manuscript was in preparation an interesting ligand system for the selective synthesis of heterobimetallic complexes that features such a naphthyridine-derived phosphinite system was reported: S. Deolka, O. Rivada Wheelaghan, S. Aristizábal, R. Fayzullin, S. Pal, K. Nozaki, E. Khaskin and J. Khusnutdinova (2020): Metal–Metal Cooperative Bond Activation by Heterobimetallic Alkyl, Aryl, and Acetylide PtII/CuI Complexes. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.11742687.v1.
- P. S. Lemport, P. N. Ostapchuk, A. A. Bobrikova, P. V. Petrovskii, N. D. Kagramanov, G. V. Bodrin and E. E. Nifant’ev, Mendeleev Commun., 2010, 20, 223–225 CrossRef CAS.
- A. Marker and M. J. Gunter, J. Magn. Reson., 1982, 47, 118–132 CAS.
- D. M. Roddick and D. Zargarian, Inorg. Chim. Acta, 2014, 422, 251–264 CrossRef CAS.
- Z. Yang, D. Liu, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Organometallics, 2015, 34, 1228–1237 CrossRef CAS.
- M. Vogt, O. Rivada-Wheelaghan, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Organometallics, 2013, 32, 300–308 CrossRef CAS.
- S. Lapointe, E. Khaskin, R. R. Fayzullin and J. R. Khusnutdinova, Organometallics, 2019, 38, 1581–1594 CrossRef CAS.
- S. Kundu, W. W. Brennessel and W. D. Jones, Inorg. Chem., 2011, 50, 9443–9453 CrossRef CAS PubMed.
- D. Benito-Garagorri, E. Becker, J. Wiedermann, W. Lackner, M. Pollak, K. Mereiter, J. Kisala and K. Kirchner, Organometallics, 2006, 25, 1900–1913 CrossRef CAS.
- P. L. Orioli and C. A. Ghilardi, J. Chem. Soc. A, 1970, 1511–1516 RSC.
- D. Benito-Garagorri, K. Mereiter and K. Kirchner, Eur. J. Inorg. Chem., 2006, 4374–4379 CrossRef CAS.
- Although the computations suggest that the PONNP isomer is the thermodynamic product (by only 2 kcal mol−1), we consider this subtle difference to be within the uncertainty of our non-benchmarked DFT method.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR and X-ray crystallographic data and synthetic procedures for all compounds. CCDC 1990856–1990858. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc02166k |
| This journal is © The Royal Society of Chemistry 2020 |