DOI:
10.1039/C8SC03186J
(Edge Article)
Chem. Sci., 2019,
10, 359-369
Controlling the speciation and reactivity of carbon-supported gold nanostructures for catalysed acetylene hydrochlorination†
Received
18th July 2018
, Accepted 22nd November 2018
First published on 22nd November 2018
Abstract
Carbon-supported gold catalysts have the potential to replace the toxic mercuric chloride-based system applied industrially for acetylene hydrochlorination, a key technology for the manufacture of polyvinyl chloride. However, the design of an optimal catalyst is essentially hindered by the difficulties in assessing the nature of the active site. Herein, we present a platform of carbon supported gold nanostructures at a fixed metal loading, ranging from single atoms of tunable oxidation state and coordination to metallic nanoparticles, by varying the structure of functionalised carbons and use of thermal activation. While on activated carbon particle aggregation occurs progressively above 473 K, on nitrogen-doped carbon gold single atoms exhibit outstanding stability up to temperatures of 1073 K and under reaction conditions. By combining steady-state experiments, density functional theory, and transient mechanistic studies, we assess the relation between the metal speciation, electronic properties, and catalytic activity. The results indicate that the activity of gold-based catalysts correlates with the population of Au(I)Cl single atoms and the reaction follows a Langmuir–Hinshelwood mechanism. Strong interaction with HCl and thermodynamically favoured acetylene activation were identified as the key features of the Au(I)Cl sites that endow their superior catalytic performance in comparison to N-stabilised Au(III) counterparts and gold nanoparticles. Finally, we show that the carrier (activated carbon versus nitrogen-doped carbon) does not affect the catalytic response, but determines the deactivation mechanism (gold particle aggregation and pore blockage, respectively), which opens up different options for the development of stable, high-performance hydrochlorination catalysts.
Introduction
The growing demand for plastics coupled with the volatility of oil and natural gas prices and concerns over supply security have revived interest in coal as a low-cost and long-term feedstock for the chemical industry.1 In particular, this development gives a new impulse to acetylene hydrochlorination, a key technology for the production of polyvinyl chloride (PVC). Already today, China, the global player in the PVC market, exploits its vast coal reserves to produce more than 70% of this commodity (13 Mton per year) from acetylene.2–4 Unfortunately, the present process still relies on a highly toxic and volatile mercuric chloride catalyst supported on activated carbon (10–15 wt% HgCl2/AC), the use of which will be banned from 2022.5 Among possible alternatives, carbon-supported gold catalysts stand out as the most promising candidates to ultimately replace the mercuric chloride-based system and enable a sustainable expansion of the technology.3 However, wide industrial implementation is still hindered by the low durability of the gold catalysts under reaction conditions (T = 403–453 K, P = 1.5 bar, HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C2H2 = 1.1:1). Specifically, the deactivation has been ascribed to the reduction of the Au(I)/Au(III) species to Au(0), while also sintering of gold clusters into particles and the formation of carbonaceous deposits have been reported.6–9 To guide the rational design of a stable gold catalyst, it is crucial to gain a deeper understanding on the nature of the active site. While to date, various gold species have been identified to be potentially active, including particles,6,10,11 clusters,12,13 and recently also single atoms,14 there remains considerable controversy on their relative activity and the prevailing reaction and deactivation mechanisms. The foremost barriers for a systematic assessment of the influence of gold dispersion and speciation as central catalytic descriptors are: (i) the co-existence of single atoms, clusters and particles in carbon supported gold-based catalysts and (ii) their tendency to sinter under reaction conditions.6–9 The development of suitable synthetic approaches to overcome these issues is restricted by the generally limited understanding on the interaction of single atoms with carbon-based carriers.15 The intrinsic stability of single-atom catalysts is determined by the ability of the host to provide suitable anchoring sites (e.g., N or O) for metal coordination.16 For example, gold single atoms hosted on zeolites or metal oxides exhibit remarkable stability, which can be attributed to the presence of oxygen linkages.17–19 However, under the harsh reaction conditions of acetylene hydrochlorination metal oxide-supported systems (e.g., Al2O3 or TiO2) quickly deactivate due to bulk chlorination or coking. In fact, only amorphous carbon materials (e.g., activated carbon, carbon black) are practically applicable supports for gold-based catalysts in acetylene hydrochlorination.3,7 In this respect, doping of amorphous carbon materials with p-block elements could provide the necessary sites to entrap gold single atoms and improve their thermal stability and durability under reaction conditions.15,20 In particular, nitrogen-doped carbon (NC) has demonstrated unique suitability to entrap atomically dispersed transition metals (e.g., Pd, Pt, Fe, Co).21–23 Interestingly, also gold nanoparticles showed improved catalytic performance on N-doped carbon in comparison to activated carbon in acetylene hydrochlorination.24,25
:C2H2 = 1.1:1). Specifically, the deactivation has been ascribed to the reduction of the Au(I)/Au(III) species to Au(0), while also sintering of gold clusters into particles and the formation of carbonaceous deposits have been reported.6–9 To guide the rational design of a stable gold catalyst, it is crucial to gain a deeper understanding on the nature of the active site. While to date, various gold species have been identified to be potentially active, including particles,6,10,11 clusters,12,13 and recently also single atoms,14 there remains considerable controversy on their relative activity and the prevailing reaction and deactivation mechanisms. The foremost barriers for a systematic assessment of the influence of gold dispersion and speciation as central catalytic descriptors are: (i) the co-existence of single atoms, clusters and particles in carbon supported gold-based catalysts and (ii) their tendency to sinter under reaction conditions.6–9 The development of suitable synthetic approaches to overcome these issues is restricted by the generally limited understanding on the interaction of single atoms with carbon-based carriers.15 The intrinsic stability of single-atom catalysts is determined by the ability of the host to provide suitable anchoring sites (e.g., N or O) for metal coordination.16 For example, gold single atoms hosted on zeolites or metal oxides exhibit remarkable stability, which can be attributed to the presence of oxygen linkages.17–19 However, under the harsh reaction conditions of acetylene hydrochlorination metal oxide-supported systems (e.g., Al2O3 or TiO2) quickly deactivate due to bulk chlorination or coking. In fact, only amorphous carbon materials (e.g., activated carbon, carbon black) are practically applicable supports for gold-based catalysts in acetylene hydrochlorination.3,7 In this respect, doping of amorphous carbon materials with p-block elements could provide the necessary sites to entrap gold single atoms and improve their thermal stability and durability under reaction conditions.15,20 In particular, nitrogen-doped carbon (NC) has demonstrated unique suitability to entrap atomically dispersed transition metals (e.g., Pd, Pt, Fe, Co).21–23 Interestingly, also gold nanoparticles showed improved catalytic performance on N-doped carbon in comparison to activated carbon in acetylene hydrochlorination.24,25
Herein, we show that gold single atoms hosted on N-doped carbon, derived by the controlled pyrolysis of polyaniline, exhibit remarkable stability in a wide temperature range of 413–1073 K and in acetylene hydrochlorination. This contrasts with the progressive aggregation of gold observed on the Au/AC reference systems above 473 K and under reaction conditions. The exclusive presence of gold single atoms on NC was confirmed by aberration-corrected scanning transmission electron microscopy (STEM) and extended X-ray absorption fine structure spectroscopy (EXAFS). Changes in the metal-carrier interaction upon thermal activation further enable a gradual tuning of the gold oxidation state as confirmed by X-ray photoelectron spectroscopy (XPS) and X-ray absorption near edge spectroscopy (XANES), and rationalised by density functional theory (DFT). With a platform of AC- and NC-supported gold catalysts of varying speciation, from single atom to nanoparticle, we systematically assess the relation between the metal speciation, electronic properties, and catalytic activity in acetylene hydrochlorination. Finally, we combine DFT and transient mechanistic studies from temporal analysis of products (TAP) to rationalise the structure-performance relationships of different gold sites.
Results and discussion
Evolution of the gold speciation during thermal activation
Gold particle size.
The impact of nitrogen doping on the stability of gold single atoms on carbon-based hosts was examined with commercial activated carbon (NORIT, ROX 0.8) and polyaniline-derived N-doped carbon (ca. 10 wt% N). Both carriers were loaded with 0.5 wt% gold via the well-established impregnation technique with aqua regia, using a drying temperature of T = 413 K (see Experimental section in the ESI†).26 In both samples (Au/NC-413 and Au/AC-413) gold was fully atomically dispersed, as evident from high-angle annular dark-field (HAADF) STEM (Fig. 1, S2, S3 and S4 of the ESI†) and corroborated by powder X-ray diffraction (XRD) analysis (Fig. S5†). In order to probe the individual ability of the carriers to stabilise gold single atoms, the temperature of thermal treatment was gradually increased in the range of 473–1073 K, yielding two series of catalysts Au/AC-T and Au/NC-T, with T indicating the respective temperature applied during the thermal activation. Isolated gold atoms were still present on both series of catalysts, but with increasing temperature more and more dimers, clusters and nanoparticles were found on AC-based materials. Already from 473 K on gold single atoms start to agglomerate or sinter (e.g., via particle coalescence or Ostwald ripening), which is evident from the development of the characteristic Au(111) and Au(200) diffraction peaks at 2θ = 38.25° and 44.46° for the Au/AC-T series with rising temperature (Fig. S5†). While the particle size distribution on the catalyst calcined at 573 K is still relatively narrow with a mean value of 5.6 nm, severe particle aggregation occurs at 673 K, yielding a sample with a very heterogeneous particle size distribution from single atoms to nanoparticles of >200 nm (Fig. S4†). In stark contrast, no build-up of gold diffraction peaks is observed for the NC-based catalysts over a temperature range of 413–1073 K and indeed, only single atoms were observed by aberration-corrected STEM, demonstrating the high suitability of N-doped carbon to stabilise gold single atoms. Additional evidence of the low propensity to sintering was obtained by monitoring the evolution of the metal dispersion on Au/NC-413 and Au/NC-1073 with time (Videos S1 and S2†). Consistent with the expected spatial and energetic heterogeneity of the metal coordination sites, anomalous diffusion is evident in both samples. Nonetheless, a slightly higher diffusivity is appreciable for NC-413 and is confirmed by analysis of the trajectories of individual gold atoms (Fig. S6†).27
 |
| | Fig. 1 Synthesis of gold single-atom catalysts hosted on activated carbon (AC) and N-doped carbon (NC), accompanied by STEM (top) and aberration-corrected STEM images (bottom) of selected samples of both series. The critical departing temperature of gold single atoms on AC is ca. 473 K, albeit even at 673 K single atoms are still present (Fig. S4†). On NC, gold single atoms are stable up to 1073 K. | |
Metal speciation.
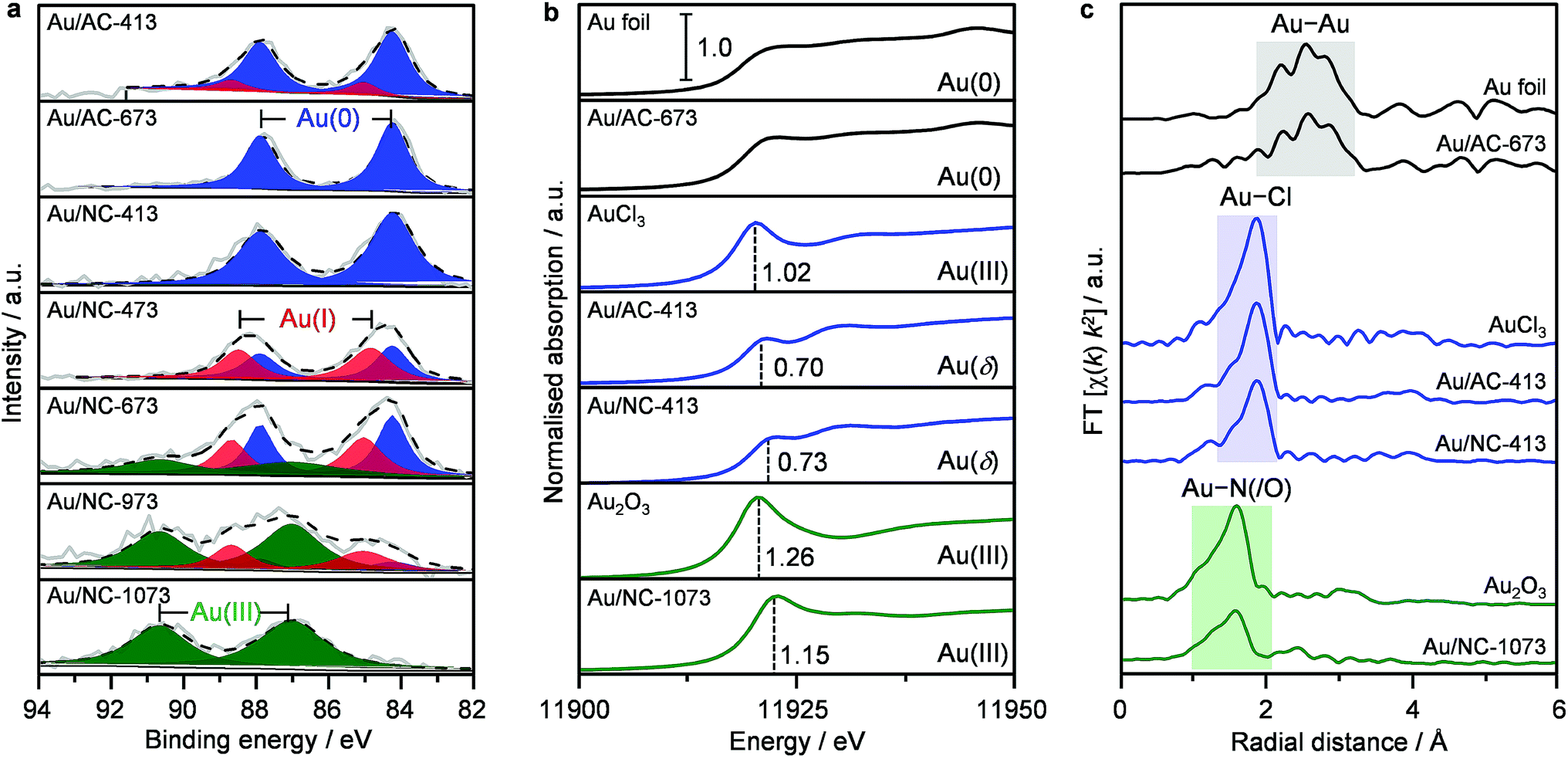
To rationalise the exceptional stability of gold single atoms on NC and to gain a deeper understanding into their chemical nature, all catalysts were analysed by XPS (Fig. 2a, Table S1†). Since the incident X-ray radiation during spectrum acquisition can potentially lead to the photoreduction of gold chloride species, which frequently causes an overestimation of the metallic gold content,3,28,29 we first studied the impact of decisive parameters, including the incident photon energy, irradiation time and beam flux, on AuCl3 as a reference sample (see Experimental section in the ESI†). Indeed, the decay of Au(III) was found to be highly dependent on the applied X-ray dose. Within the first hour of acquisition, it is possible to minimise photoreduction effects by increasing the measuring spot to 1000 × 500 μm, consequently lowering the X-ray dose (Fig. S7†). Accordingly, all XPS spectra were recorded under these optimised conditions. Unexpectedly, the Au 4f XPS raw spectra of all Au/AC catalysts are very similar (Fig. S8†), even though their speciation varies from predominately single atoms to an increasing population of nanoparticles. A detailed fitting of the XPS spectra revealed a significant contribution at a binding energy (BE) of 84.2 eV that is commonly assigned to Au(0). On the contrary, the Au 4f XPS raw spectra of the Au/NC-T series clearly show a steady shift to higher binding energies with increasing catalyst treatment temperature. Only the sample Au/NC-413 shows a very similar Au 4f XPS spectrum to the Au/AC-T series with an exclusive contribution at BE = 84.2 eV. An increase in the treatment temperature to 473 K leads to the build-up of Au(I) (BE = 85.0 eV). Above 973 K Au(III) species, located at 87.0 eV, predominate. Overall, these results evidence the superior ability of the NC carrier to stabilise charged gold species and inhibit their photoreduction. However, the assignment of distinct metal oxidation states for Au/NC-413 and the whole Au/AC-T series must be treated with caution, as the initially performed evaluation of the photoreduction effect on bulk AuCl3 might not be directly extrapolated to single cationic gold species. Since the effect of photoreduction is less prevailing with X-ray absorption spectroscopy (XAS), a selection of key catalysts was analysed to gain more insight into the gold speciation (Fig. S9†).3,14
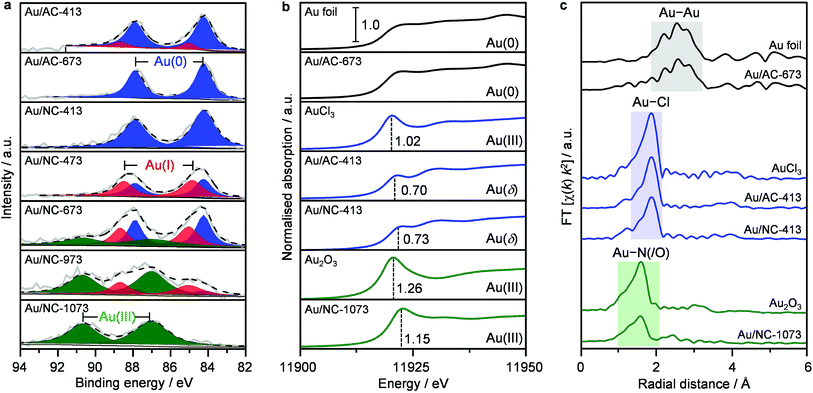 |
| | Fig. 2 Analysis of the gold speciation of selected catalysts based on (a) Au 4f XPS, (b) Au L3 edge XANES, and (c) Fourier transformed EXAFS spectra. Dashed black lines in (a) show the result of fitting the raw data (solid grey lines). Vertical dashed lines in (b) indicate the white line intensity of the distinct cationic gold species. I < δ <III. | |
From the normalised Au L3-edge XANES it is possible to derive the gold oxidation states by comparing the white line intensity and other spectral features with appropriate standards (e.g., Au foil, AuCl3). In accordance to expectations, the white line intensities of the low-temperature samples Au/AC-413 and Au/NC-413 are 0.70 and 0.73, respectively. These values are lower than the reference AuCl3 intensity of 1.02 (Fig. 2b), but close to the reported value of 0.6 for AuCl,14 thus indicating the predominant presence of AuCl. With linear combination fitting, the Au(I):Au(III) ratio was estimated to be 2.6 and 4.0, respectively, for Au/NC-413 and Au/AC-413, which agrees well with previous literature reports.14 The extended X-ray absorption fine structure (EXAFS) analysis of Au/NC-413 and Au/AC-413, indicates 2.0 and 2.5 Cl neighbours, respectively, at distances similar to those in AuCl2 (RAu–Cl = 2.272(5) Å and RAu–Cl = 2.269(3) Å, respectively) while no Au–Au contribution was found (Fig. 2c and Table S3†). In accordance with the XPS results, Au/NC-1073 has a much higher white line intensity of 1.15, suggesting the presence of Au(III) species. Interestingly, EXAFS FT analysis of Au/NC-1073 does not indicate the presence of Au–Cl entities, but rather Au–O or Au–N species. A more specific assignment is not possible with the XAS technique, as oxygen and nitrogen scattering signals cannot be distinguished. Fitting of the first coordination shell revealed 4.0 N/O neighbours around Au at a distance of RAu–N/O = 1.96(5) Å. Importantly to notice, no Au–Au characteristic peaks were detected for the whole Au/NC-T series, highlighting the exclusive presence of gold single atoms on the NC carrier. On the contrary, for Au/AC-673, a significant Au–Au contribution (RAu–Au = 2.99(1) Å) predominates in the EXAFS spectrum, while no white line feature at 11919 eV is observed in XANES, corroborating with the metallic nature of gold in this catalyst according to the microscopic observations. The integrated approach of combining XPS and XAS techniques enables clear distinction of the nature of different gold species on the carbon hosts.
Role of the gold anchoring site
Structural and elemental changes in Au/NC-T during thermal activation.
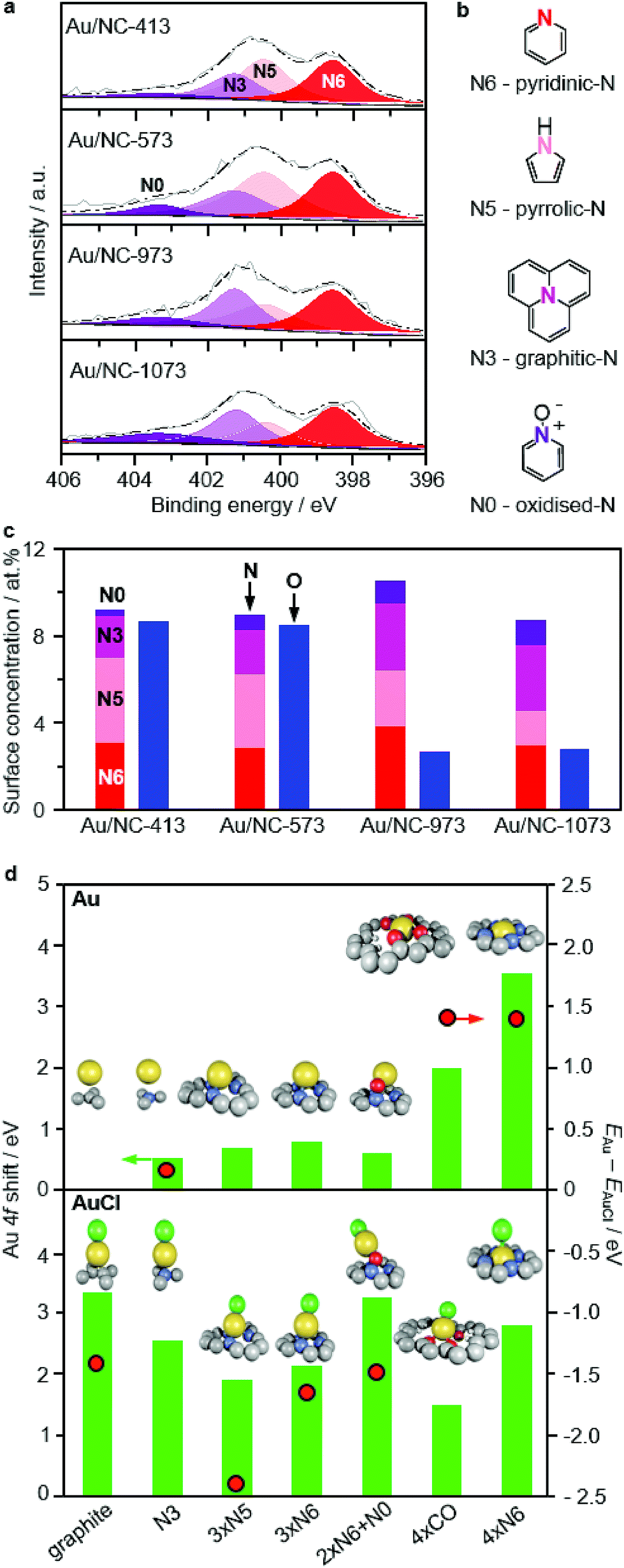
To elucidate the gradual change of gold single atoms on NC from predominant Au(I)Cl to Au(III)N/O species with increasing activation temperature, we analysed the Cl, O, and N surface concentrations by XPS (Tables 1 and S2, Fig. 3a and S10†). The Cl 2p core level spectra revealed a major contribution of C–Cl (BE ≈ 200 eV) rather than Au–Cl (BE ≈ 198 eV),30 likely due to the low gold content, in the low-temperature catalysts with significantly over-stoichiometric molar Cl:Au ratios of ca. 37. This ratio first decreases to 25 at 673 K, and further drops to <2 at above 973 K accompanied with the disappearance of the C–Cl peaks for the two high-temperature catalysts (Fig. S10†). A very similar trend is found for the oxygen content of the Au/NC-T series, as for T = 413–573, the total content is ca. 9 at% and for T = 973–1073 it drops below 3 at% (Fig. 3c). On the contrary, the N content (ca. 10 at%) is remarkably constant over the whole investigated temperature range. Overall, these insights from XPS substantiate the above conclusions from EXAFS and XANES and further help to rationalise the evolution of gold species with rising catalyst treatment temperatures. Since nitrogen functionalities have clearly superior thermal stability to oxygen functionalities, we can speculate that the gold species in Au/NC-1073 rather comprises nitrogen than oxygen binding sites. On the basis of the N 1s fitting results it is further possible to foreshadow a leading role in stabilising gold single atoms to pyridinic-N, based on their superb stability and high surface concentration in the Au/NC-1073 sample.
Table 1 Characterisation data of selected Au catalysts
| Sample |
Au speciationa |
Aub at% |
Nb at% |
Ob at% |
Clb at% |
V
total
cm3 g−1 |
V
micro
cm3 g−1 |
S
BET
m2 g−1 |
|
SA: single atom, NP: nanoparticle.
XPS.
Volume of Ar at p/p0 = 0.98.
t-Plot method.
BET method.
|
| NC |
— |
0.00 |
9.3 |
6.4 |
0.05 |
0.42 |
0.21 |
517 |
| AC |
— |
0.00 |
0.3 |
4.4 |
0.00 |
0.84 |
0.69 |
1795 |
| Au/NC-413 |
SA |
0.05 |
9.2 |
8.7 |
1.8 |
0.32 |
0.16 |
376 |
| Au/NC-473 |
SA |
0.04 |
10.4 |
8.6 |
1.5 |
0.20 |
0.14 |
356 |
| Au/NC-673 |
SA |
0.02 |
9.8 |
7.6 |
0.8 |
0.33 |
0.20 |
551 |
| Au/NC-973 |
SA |
0.05 |
10.6 |
2.7 |
0.1 |
0.17 |
0.16 |
389 |
| Au/NC-1073 |
SA |
0.02 |
8.7 |
2.8 |
0.02 |
0.39 |
0.22 |
496 |
| Au/AC-413 |
SA |
0.04 |
0.2 |
9.5 |
1.1 |
0.69 |
0.56 |
1278 |
| Au/AC-473 |
SA |
0.08 |
0.5 |
7.8 |
0.7 |
0.60 |
0.54 |
1150 |
| Au/AC-673 |
SA + NP |
0.04 |
0.3 |
7.7 |
0.6 |
0.84 |
0.67 |
1773 |
| Au/NC-473-12 h |
SA |
0.03 |
9.1 |
8.1 |
1.3 |
0.07 |
0.02 |
90 |
| Au/AC-473-12 h |
SA + NP |
0.04 |
0.6 |
6.6 |
0.9 |
0.58 |
0.50 |
1100 |
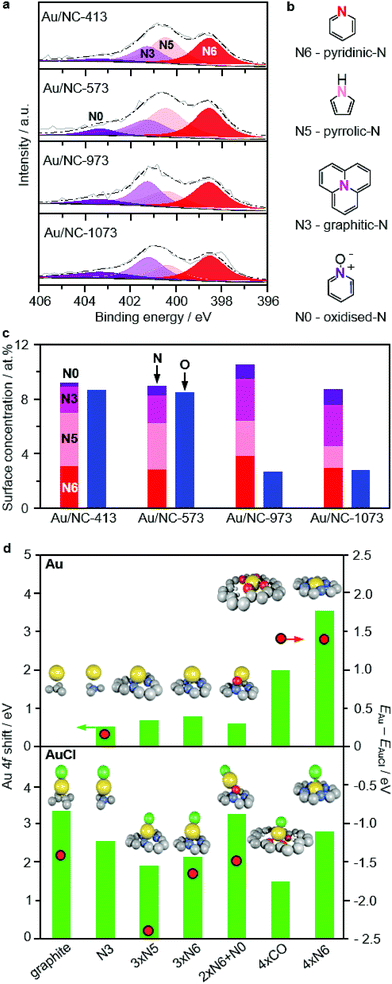 |
| | Fig. 3 (a) N 1s XPS spectra of selected Au/NC samples, (c) oxygen and nitrogen surface concentration, and (b) the structures of all observed nitrogen species. (d) Comparative overview of Au and AuCl species on selected defects with corresponding XPS shifts and the difference in formation energies ΔE as the relative stability descriptor. | |
The remarkable ability of the NC carrier to stabilise gold atoms was rationalised by DFT. A set of 21 graphite defects (Fig. S11†) was used to represent the AC and NC support materials. The models were built in the basal plane according to stoichiometry: regular, N-containing: N-pyridinic (N6), N-pyrrolic (N5); O-containing: epoxide, keto, hydroxyl; plus combinations of those and the corresponding step models. These structures were evaluated for their ability to bind Au and AuCl. The differences in their formation energies, calculated as ΔE = EAu − EAuCl, (Fig. 3d; Table S4†) indicate that in general AuCl species are thermodynamically favoured and thus more stable than their Au counterparts. In accordance with the XAS analysis, simulation of Au 4f XPS shift for these AuCl species suggests they are significantly oxidised Au(δ) (I < δ < III), but are likely observed as Au(0) by XPS due to photoreduction effects. Similar results are obtained for the AuCl2 and AuCl3 species (Table S5†). Exceptions to the general trend occur only when the cavity has a suitable size and geometry to provide a square planar coordination pattern (four pyridinic-4xN6 or four keto environments 4xCO) for the gold atom (Fig. S12†). On the basis of the results presented above, AuCl species are dominantly present in the low-temperature Au/NC-T catalysts and are responsible for the XPS signal. With increasing catalyst treatment temperature, the Cl content is lowered and the mobility of the gold atoms increases (Fig. S13†). This leads to a larger population of configurations observed as more oxidised gold species since these are deeper wells in the potential energy surface of the doped carbons (Table S4†). A further increase in temperature enhances the diffusion of gold species as they are able to escape local diffusion minima and enter the 4xN6 cavities, adopting a four-fold N-coordinated planar geometry where they are trapped. With an Au 4f XPS shift of 3.67 eV, this structure represents the global diffusion minimum (adsorption E = −3.61 eV with respect to gas-phase Au) and is observed in Au/NC-1073 by XPS and XANES as Au(III) that is more resistant to X-ray reduction (Fig. S7†).
Catalytic performance in acetylene hydrochlorination
Catalytic descriptors.
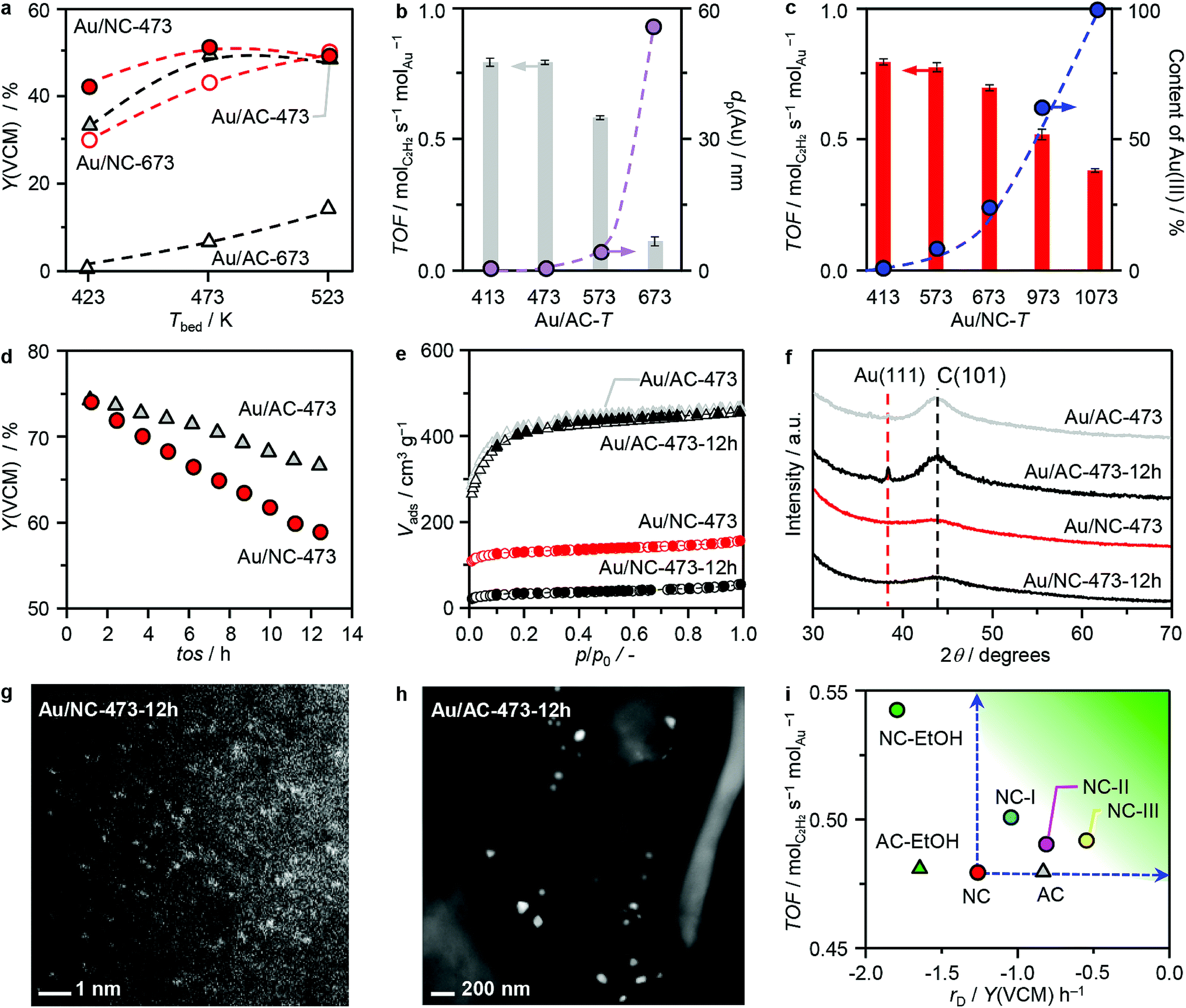
Acetylene hydrochlorination over the two series of Au/AC-T and Au/NC-T catalysts was studied at bed temperatures (Tbed) of 423–523 K with a gas hourly space velocity based on acetylene (GHSV(C2H2)) of 1500 h−1. Initially, a temperature ramp experiment was performed over the catalysts of both series prepared at 473 K and 673 K to identify the optimal reaction temperature (Fig. 4a). To exclude the influence of catalyst deactivation, each point was obtained in an independent experiment. The activity of the surveyed catalysts follows the order of Au/NC-473 ≈ Au/AC-473 > Au/NC673 ≫ Au/AC-673, and for the two best-performing catalysts was the highest at Tbed = 473 K, which was selected for all the following studies. In accordance to expectations, the catalytic activity of the Au/AC-series clearly decreases with the increasing activation temperature (Fig. 4b). A dependency of the catalytic activity on the gold dispersion is apparent for the whole Au/AC series. In particular, catalysts exhibiting nanoparticles display activity losses with increasing mean particle size, which can in turn be related to a decreasing population of gold single atoms, demonstrating their central role in acetylene hydrochlorination. In literature, gold nanoparticles and clusters are frequently reported as the active site, but the possible coexistence of gold single atoms was not assessed and conclusions were exclusively based on XPS10,31 or computational results.12,13 Interestingly, the metal dispersion does not suffice as the only catalytic descriptor for gold catalysts in acetylene hydrochlorination, as is evident from the performance of the Au/NC-T series (Fig. 4c). Despite the common single-atom nature of all samples in this series, the catalytic activity clearly drops with increasing temperature of thermal activation, though slower compared to the Au/AC series, suggesting that also the metal oxidation state and/or the gold anchoring sites are determining the overall catalytic performance. With an increasing content of Au(III), as derived from the XPS fitting results, the catalytic activity decreases, indicating that Au(I) is more active than Au(III). By comparing the TOF of Au/NC-473 (only Au(I)) and Au/NC-1073 (only Au(III)), it can be further concluded that Au(I) is ca. 2-fold more active than Au(III) (Fig. 4c). The origins of the distinct behaviour of the two single-atom catalysts will be further analysed by DFT (vide infra).
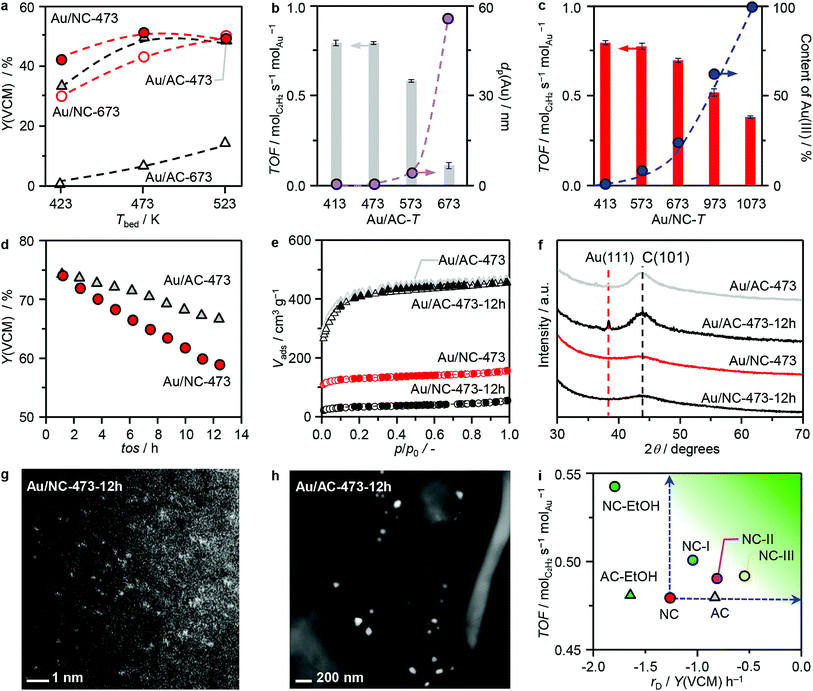 |
| | Fig. 4 (a–d) Catalytic performance of AC- and NC-supported gold catalysts in acetylene hydrochlorination, expressed as the yield of vinyl chloride monomer Y(VCM) or the turnover frequency (TOF) at (a) different reaction temperatures and as a function of (b) the mean particle size of gold (dp) for Au/AC and (c) the percentage of Au(III) for Au/NC, as well as (d) during a short-term stability test. (e) Argon sorption isotherms, (f) XRD, and (g and h) HAADF-STEM images of the catalysts after 12 h time on stream (tos). (i) Comparison of TOFs and deactivation rate, rD, of the catalysts developed in this work with Au/AC-473 and Au/NC-473 as reference systems (Fig. S15a†). Reaction conditions: Tbed = 473 K, Wcat = 0.1 g (a–c) and 0.25 g (d and i), HCl:C2H2:Ar = 44:40:16, FT = 15 cm3 min−1, P = 1 bar. | |
Catalyst stability and deactivation.
To evaluate the stability of the gold single atoms on both carriers under practically relevant reaction conditions of acetylene hydrochlorination, the two best-performing catalysts of both series, Au/AC-473 and Au/NC-473, were studied in a 12 h test at 473 K with a GHSV(C2H2) of 610 h−1 (Fig. 4d). Clearly, the performance of both catalysts drops over the course of the test, with a conversion loss of 11% for Au/AC-473 and 22% for Au/NC-473. To evaluate on possible reasons for the observed deactivation we applied an integrated approach combining XRD, STEM, XPS, XAS, elemental analysis, and sorption measurements. Remarkably, XRD and STEM analysis confirmed the exclusive presence of gold single atoms in the used Au/NC catalyst, whereas on AC also nanoparticles were found, which likely accounts for the observed decrease in activity (Fig. 4f–h). These results further confirm the high suitability of N-doped carbon to stabilise Au single atoms, even under the harsh reaction conditions. The XPS and XAS spectra of the used catalysts (Fig. S14†) did not reveal any major changes to the fresh catalysts, which indicates that the largest fraction of gold sites was not altered. Hence, it can be concluded that the deactivation of Au/NC-473 does not stem from alterations of the active sites. Instead, significant differences were found for the porous properties and elemental composition between the fresh and used catalysts (Fig. 4e, Tables 1 and S6†). While in the case of Au/AC neither the porous properties nor the elemental composition changed markedly, a significant decrease in surface area and pore volume (376 to 90 m2 g−1 and 0.32 to 0.07 cm3 g−1, respectively), accompanied by an increase in the carbon content (from 68.1 to 72.2 wt%) was found for Au/NC. These findings suggest pore blockage, possibly as a consequence of coking, as the major reason for catalyst deactivation for Au/NC. Overall, these results demonstrate that while the AC carrier lacks the suitable anchoring sites to stabilise gold single atoms, it still exhibits superior stability under hydrochlorination conditions. On the other hand, the NC carrier provides suitable anchoring sites that can effectively entrap gold single atoms under reaction conditions, but suffers from severe pore blockage.
Challenges and opportunities for Au/NC catalysts.
Since the deactivation of Au/NC likely originates from the structural changes in the support under reaction conditions, it is important to identify and alter the specific carrier properties that cause the coking and/or pore blockage, without compromising the ability to anchor the gold single atoms. Based on the remarkable stability of activated carbon under hydrochlorination conditions, two factors were envisioned to determine the overall stability of the carrier under reaction conditions: (i) the total nitrogen content, and (ii) the porous properties, including pore size and surface area. The impact of these two potential stability descriptors was studied with the catalysts Au/NC-I, featuring a relatively low nitrogen content of 4.4 wt%, and Au/NC-II, exhibiting a ca. 2.5 fold increased surface area and pore volume in comparison to the pristine Au/NC-473 catalyst (Table S6†). The overall nitrogen content in NC-I was adapted by increasing the calcination temperature applied in the synthesis of the N-doped carbon material.32 Changes in the porous properties for NC-II were achieved via steam activation of the pristine NC carrier. The time-on-stream performance of both catalysts was evaluated under the same reaction conditions applied for Au/NC-473 and Au/AC-473 (Fig. 3i and S15a†). Both Au/NC-I and Au/NC-II showed slightly higher initial activity and improved stability in comparison to the benchmark Au/NC-473 system, rivalling the performance of Au/AC-473. Importantly to notice, the significant changes in the porous properties and the N content did not affect the ability of the support to stabilise Au single atoms, as indicated by the absence of the characteristic Au(111) diffraction peaks in the XRD patterns of the catalysts before and after use in the reaction (Fig. S15b†). The increase in the carbon content combined with a decrease in the surface area and pore volume suggest that also Au/NC-I and Au/NC-II deactivate due to coking and/or pore blockage, though both effects are less prevailing in comparison to the benchmark Au/NC-473 catalyst (Table S6†). A further improvement of the catalyst stability was achieved by combining the synthesis strategies applied for NC-I and NC-II, yielding a sample with a N content of 4.3 wt% and comparable porous properties to Au/AC-473. In line with the improved catalytic performance, changes in the porous properties and elemental composition were less pronounced in this catalyst in comparison to all other samples in the Au/NC series. These results demonstrate that the pore structure and the nitrogen content are reliable stability descriptors for the N-doped carbon support, guiding the rational design of a stable hydrochlorination catalyst.
Towards the wide implementation of an economically viable and green technology based on gold catalysts, another challenge to face is the elimination of highly corrosive and hazardous aqua regia from the catalyst synthesis protocols. Several studies have demonstrated the central role of this reagent in oxidising and dispersing the active gold species on the carbon support, being the key for high catalytic activity.4,26,33 In fact, no alternative solvents could yield atomic dispersion of gold on carbon supports, without nanoparticle formation. As rationalised in the previous section, the stabilisation effect of gold single atoms on NC originates from the nitrogen dopant and consequently there should be no need to use a strong oxidising acid. In this respect, we applied ethanol, as an environmental benign solvent, in the impregnation step and obtained another catalyst of fully atomically dispersed gold species on N-doped carbon (Au/NC–EtOH) as evident from HAADF-STEM and XRD analysis (Fig. S15b and c†). On the contrary, following the same approach with activated carbon yielded the sample Au/AC–EtOH, which showed Au(111) diffraction peaks in XRD, indicating a contribution of gold nanoparticles. Remarkably, in terms of initial catalytic activity Au/NC–EtOH outperformed all other catalysts developed in this study, demonstrating the potential to replace aqua regia in the synthesis protocol and underlining the central role of the nitrogen dopant rather than the impregnation solvent to disperse and stabilise gold single atoms. However, despite the high initial activity, Au/NC–EtOH deactivates rather quickly, comparable to Au/AC–EtOH. While the latter suffers from severe particle aggregation, as evident from a clear increase in the gold diffraction peaks observed with XRD, Au/NC–EtOH mainly deactivates due to pore blockage and/or coking (Table S6†), solidifying the importance of the carrier properties and the need for careful engineering of the support.
Mechanistic studies
Computational and kinetic studies enable the detailed understanding of catalytic action, guiding the design towards stable high-performance single atom catalysts.34,35 To date, acetylene hydrochlorination is widely believed to follow an Eley–Rideal mechanism, in which HCl reacts from the gas phase with the initially formed C2H2–Au(III) complex.36–38 Based on DFT calculations it was shown that C2H2 is the preferred ligand for the Au(III) site and it was further concluded that the addition of HCl to the C2H2–metal complex is the rate-determining step.39,40 Recently, an alternative mechanism has been proposed that proceeds via the oxidative addition of HCl to Au(I)Cl, followed by the addition of C2H2 to the formed Au(III) complex and finally the reductive elimination of C2H3Cl.14 Except for the dynamic transformation between Au(I) and Au(III) species, observed by in situ EXAFS,14 there is limited experimental evidence to support either of the two proposed mechanisms. To gain further experimental insights into the mechanism of acetylene hydrochlorination over Au single-atom catalysts, we conducted kinetic studies on Au/AC-473 and Au/NC-473 (Fig. S16†). In good agreement with the similar initial catalytic activity, the kinetic parameters were found to be quite comparable as well, which suggests that the two catalysts promote the same reaction mechanism. Specifically, apparent activation energies of 28.0 and 30.4 kJ mol−1 were derived for Au/NC-473 and Au/AC-473, respectively. The partial reaction orders of C2H2 and HCl were determined to be 1.05 and 0.72 for Au/NC-473 and 1.02 and 0.76 for Au/AC-473, which indicates a dependence of the reaction rate on the surface coverage of both reactants. In this respect, a detailed analysis of the interactions of HCl, C2H2, and C2H3Cl with distinct gold sites could provide further insights.
Temporal analysis of products.
The temporal analysis of products (TAP), a powerful technique to quantitatively compare the adsorption and desorption properties of reactive gases on a catalyst surface,41,42 was performed on a range of selected samples (Fig. S17†). The experiments were conducted in high vacuum to exclude external mass transfer limitations and minimise collisions between gas-phase molecules, ensuring that their transient response is a measure of gas–solid interactions. To derive mechanistic and kinetic insights into acetylene hydrochlorination over various gold sites, two types of experiment were conducted: (i) single pulsing of individual reactants/product and (ii) simultaneous pulsing of both reactants (see Experimental section in the ESI†). In the latter case no reaction product was observed, which likely originates from the significantly shorter contact time in the pulse experiment and a lower partial pressures of the reactants in comparison to the steady-state catalytic tests. Nevertheless, the TAP experiments enable to derive information about the mechanism and kinetics of the interaction of HCl, C2H2, and C2H3Cl with the catalysts studied. A characteristic crossing of the dimensionless response of all gases with the standard diffusion curve (dimensionless response of inert gases) was observed for the majority of samples. This is a fingerprint of reversible adsorption43 irrespective of the presence or speciation of gold. Only HCl adsorbed irreversibly on the bare supports (AC and NC) and Au/NC-1073, as no response signal of HCl was detected at the reactor outlet. The strong interaction of the NC-samples with HCl likely originates from favourable N–H interaction, as rationalised by DFT (Fig. S18†). To enable a more detailed and quantitative analysis of the influence of the gold speciation on the adsorption/desorption properties, experimental responses of HCl, C2H2 or C2H3Cl were kinetically evaluated (see ESI†). A simple reversible adsorption model (eqn (2)†) was applied to describe the experimental data (Fig. S19†). The adsorption (kads,eff) and desorption constants (kdes) and their ratio K derived for all gases are given in Table 2. In general, the interactions between the reactive gases and the catalyst surface decrease in the order of HCl ≫ C2H3Cl > C2H2, as reflected by their respective K values. Interestingly, this parameter for C2H2 and C2H3Cl is relatively insensitive towards the type of catalyst, but varies for HCl depending on the gold speciation, with single-atom catalysts (Au/AC-473, Au/NC-473, and Au/NC-1073) showing significantly stronger HCl interaction in comparison to the nanoparticle analogues (Au/AC-673 and Au/AC-473-12 h). Based on this finding the decrease in catalytic activity of the gold nanoparticle-containing catalysts could originate from the weakened HCl adsorption ability due to a shrinking population of gold single atoms. In the case of Au/NC-473-12 h both HCl and C2H2 adsorption are weakened, which likely originates from the significant decrease in surface area in this catalyst. Interestingly, the effect is much more pronounced for C2H2, likely due to the generally weaker interaction with the support in comparison to HCl. In fact, over Au/NC-473-12 h hardly any C2H2 adsorption could be detected, which might explain the gradual deactivation evidenced for Au/NC-473. These results suggest that the adsorption of both reactants on the catalyst surface is essential for optimal catalytic performance, as is valid for reactions proceeding through a Langmuir–Hinshelwood mechanism.
Table 2 Kinetic parameters of HCl, C2H2, and C2H3Cl adsorption and desorption over bare supports and selected gold catalysts
| Catalyst |
HCl |
C2H2 |
C2H3Cl |
|
k
ads,eff, s−1 |
k
des, s−1 |
K
, — |
k
ads,eff, s−1 |
k
des, s−1 |
K
, — |
k
ads,eff, s−1 |
k
des s−1 |
K
, — |
|
Complete consumption of HCl due to irreversible adsorption.
C2H2 simply diffused through the reactor.
The ratio of kads,eff:kdes.
|
| NC |
—a |
—a |
—a |
1.2 × 103 |
1.4 × 102 |
8.6 |
1.2 × 103 |
14 |
90.6 |
| Au/NC-473 |
8 × 103 |
2 × 10−3 |
4 × 106 |
9.3 × 102 |
1.3 × 102 |
7.2 |
1.2 × 103 |
17 |
70.9 |
| Au/NC-1073 |
—a |
—a |
—a |
4.7 × 102 |
87 |
5.4 |
1.2 × 103 |
14 |
84.6 |
| Au/NC-473-12 h |
5 × 103 |
2 × 10−2 |
3 × 105 |
—b |
—b |
—b |
6.7 × 102 |
19 |
34.9 |
| AC |
—a |
—a |
—a |
5.5 × 103 |
4.5 × 102 |
12.3 |
8.1 × 104 |
13 |
6.2 × 103 |
| Au/AC-473 |
3 × 103 |
1 × 10−6 |
3 × 109 |
5.0 × 102 |
1.6 × 102 |
3.1 |
7.1 × 106 |
1.2 × 105 |
58.0 |
| Au/AC-673 |
5 × 102 |
4 × 102 |
1.3 |
4.1 × 102 |
1.2 × 102 |
3.4 |
6.7 × 106 |
7.7 × 104 |
87.0 |
| Au/AC-473-12 h |
1 × 102 |
0.5 |
2 × 102 |
1.8 × 102 |
1.0 × 102 |
1.8 |
2.3 × 103 |
44 |
53.0 |
Density functional theory.
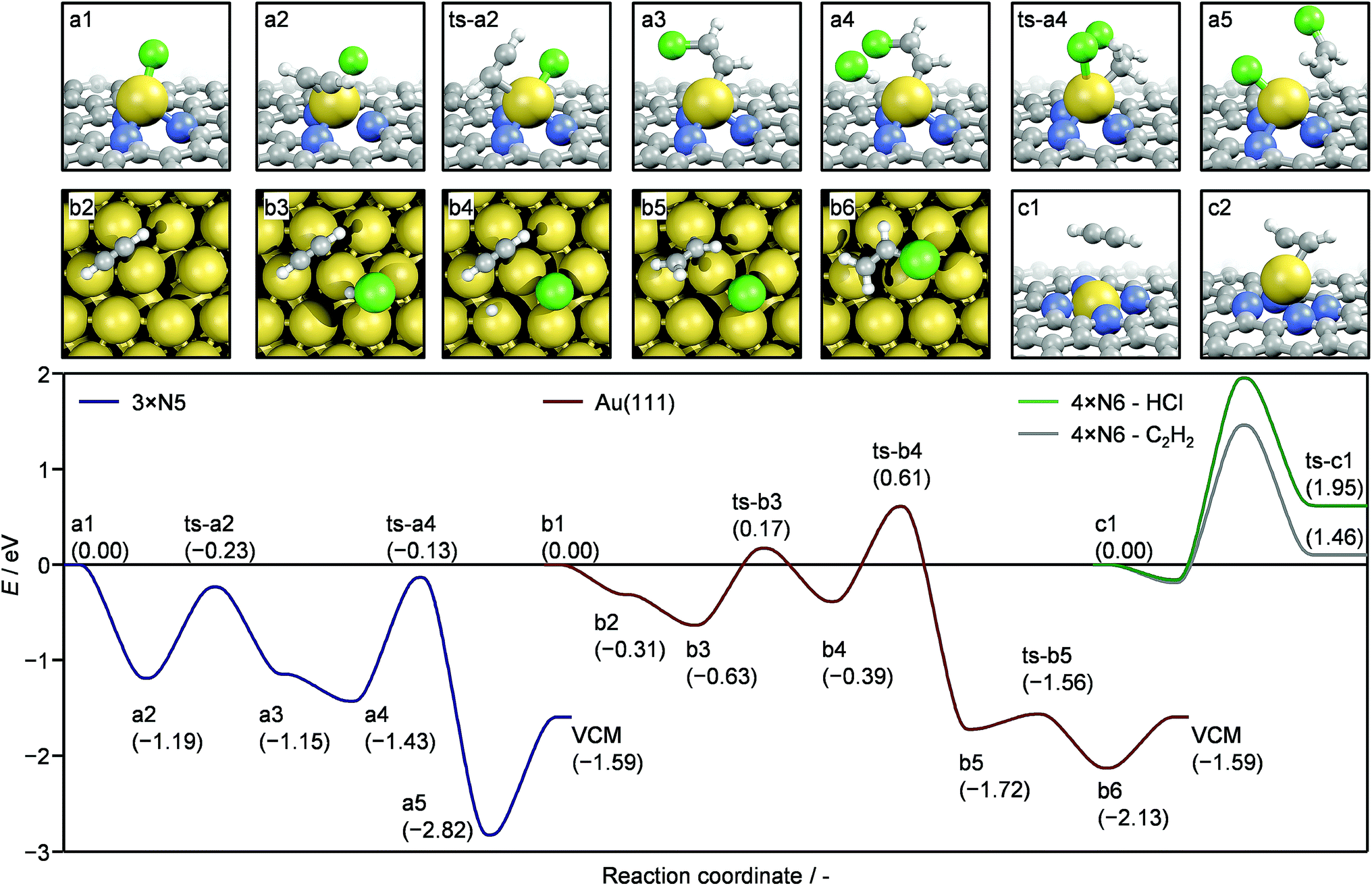
Based on the detailed experimental and computational analysis of the various gold species present in the platform of catalysts, the mechanism of acetylene hydrochlorination was simulated (Fig. S20†). All investigated structures are presented in Fig. S11† and can be retrieved from the ioChem-BD database.44,45 For simplicity, only three selected structures, serving as model systems, are presented here (Fig. 5): (i) a defect containing three pyrrolic N (3xN5) was chosen as the carrier for Au(I)Cl single atoms; (ii) the 4-fold N6 cavity (4xN6) for Au(III), and (iii) Au(111) surface to represent a gold nanoparticle. As suggested by TAP, the reaction path over Au(I)Cl and the Au(111) surface can follow a Langmuir–Hinshelwood scheme with both reactants co-adsorbed on the active gold species (Table S7†). Initially C2H2 is adsorbed and the reaction occurs on the Au(I)Cl sites. Although the interaction of acetylene with gold depends on the nature of the metal coordination environment and the presence of Cl ligands, acetylene adsorption on the 3xN5-AuCl single atom ensemble a2 is significantly more exothermic (−1.19 eV) compared to the adsorption on Au(111) b2 (−0.31 eV). In its adsorbed state C2H2 is activated, (159° C–C–H angle), which leads to the transfer of Cl from AuCl to the adsorbed C2H2, creating an intermediate a3. In the next step HCl is adsorbed (a4). The H transfer occurs as the Au–Cl bond is broken and a new Au–Cl bond is formed, yielding the adsorbed vinyl chloride molecule (a5). This is the preferred product (−2.82 eV) as the formation of 1,2-dichloroethane is thermodynamically less favoured (−2.13 eV). Once the product desorbs, the NC–Au–Cl site is recovered (a1). The described mechanism for Au(I)Cl single atom species relies on the availability of gold with unsaturated coordination spheres, hence stabilised AuClx (x > 1) species would negatively impact the activity due to a lowered acetylene adsorption ability.46 On the other hand, the reaction is hindered by the activation of HCl as this reaction is endothermic by more than 1.3 eV. While the presented mechanism infers that the reaction should be of first order with respect to both reactants, the strong interaction of HCl with empty cavities of the NC support (vide supra) reduces the apparent HCl reaction order, since the NC carrier acts as a HCl reservoir. Consequently, the question arises whether HCl adsorbed on such cavities could participate in the reaction cycle. HCl adsorption was found to proceed either via a non-dissociative or a dissociative mechanism, depending on the type of empty site (Fig. S21a†). In the latter case, a new N–H bond is formed and the Cl ion can either be adsorbed on the cavity or become mobile on the surface, if sufficient thermal energy is provided. Molecular HCl can also exit the cavity in an endothermic process. Once mobile, the surface HCl can participate in the catalytic cycle like the gaseous counterpart as described in Fig. 5. The source of HCl (coming from the gas phase or adsorbed on the surface) only affects steps a4–a6 of the reaction cycle. Specifically, the reaction profile is shifted upwards energetically by a value equal to the HCl adsorption energy (Fig. S21b†). The reaction on the gold surfaces is schematically represented by structures b1–b6 (on Au(111)). Both Au(111) and Au(211) show relatively low C2H2 adsorption energies, −0.31 and −0.29 eV, respectively. In combination with the relatively poor surface to volume ratio when compared to single atoms, the weak acetylene adsorption renders gold nanoparticles comparatively inferior catalysts. The poor activity of Au(III)-rich systems comes from the high stability of the 4xN6–Au ensemble. In order to complete a catalytic cycle, the adsorption of C2H2/HCl (c1) must be followed by its activation (c2). These processes are hindered both thermodynamically and kinetically.
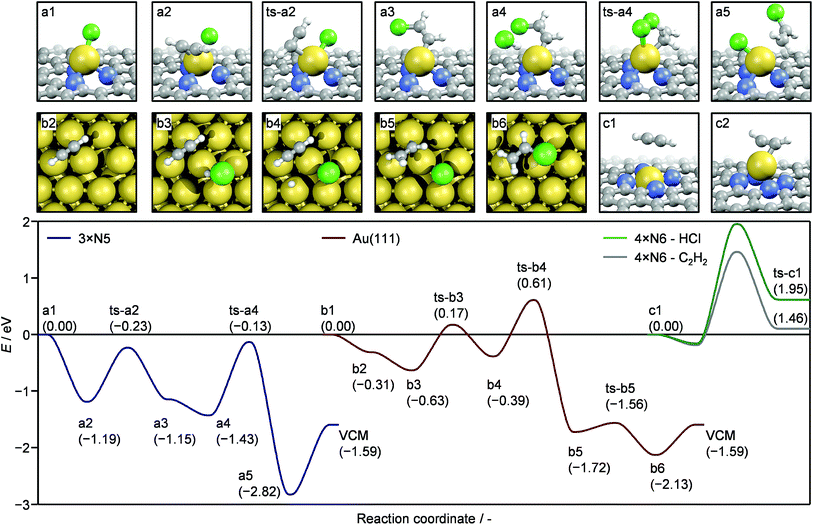 |
| | Fig. 5 The complete reaction cycle of acetylene hydrochlorination over the AuCl@pyrrolic defect (3xN5) (a1–a5, blue line), Au@4-fold pyridinic defect (4xN6) (c1, c2, gray and green lines), and Au (111) (b1–b5, red line). The geometries for intermediates and transition states (ts) are shown in the top row for the 3xN5 defect; intermediates are shown in the bottom row on Au (111) and for the 4xN6 defect. | |
Conclusions
In this study we presented a strategy to gradually tune the electronic properties and the stability of individual gold atoms by varying the structure of functionalised carbons and use of thermal activation. Owing to the presence of suitable metal anchoring sites in polyaniline-derived N-doped carbon, gold remains atomically dispersed up to 1073 K, but experiences a gradual increase in the metal oxidation state with rising activation temperature, as is evident from aberration-corrected STEM, XANES, and EXAFS analysis. In the absence of nitrogen sites, progressive agglomeration of gold atoms occurs already above 473 K. This distinct evolution of gold atoms as a function of the carbon carrier enabled the development of a unique platform of gold nanostructures suitable to assess the relation between the metal speciation, electronic properties, and catalytic performance in acetylene hydrochlorination. On the basis of steady-state catalytic tests, computational and transient mechanistic studies, we showed that the activity of gold-based catalysts correlates with the population of Au(I)Cl single atoms and the reaction follows a Langmuir–Hinshelwood mechanism. Accordingly, strong interaction with HCl and the thermodynamically favourable activation of acetylene are the key features of the Au(I)Cl sites that endow their superior catalytic performance. In line with the substantially lower catalytic activity, gold nanoparticles and N-coordinated Au(III) single atom ensembles were found to exhibit higher energy barriers for the activation of both reactants. Remarkably, the choice of carbon host does not affect the catalytic response, but determines the deactivation mechanism (pore blockage for Au/NC and gold particle aggregation for Au/AC), suggesting different strategies for catalyst optimisation. Going beyond acetylene hydrochlorination, the herein developed approach of controlling the reactivity of individual atoms provides exiting perspectives for the design of metal species for targeted applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by ETH Reserach Grant ETH-40 17, the Swiss National Science Foundation (project no. 200021-169679), and Spanish MINECO (CTQ2015-68770-R). E. F. thanks MINECO La Caixa Severo Ochoa for a predoctoral grant through Severo Ochoa Excellence Accreditation 2014 2018 (SEV 2013 0319). BSC-RES for providing generous computational resources. V. A. K and E. V. K acknowledge the State of Mecklenburg-Vorpommern for financial support. S. M. C. acknowledges support from the Henslow Research Fellowship and Girton College, Cambridge. P. A. M. acknowledges the EPSRC for funding under grant number EP/R008779/1. The Scientific Centre for Optical and Electron Microscopy (ScopeM) at ETH Zurich is acknowledged for the use of their facilities.
References
- I. T. Trotus, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761 CrossRef CAS PubMed.
- R. Lin, A. P. Amrute and J. Pérez-Ramírez, Chem. Rev., 2017, 117, 4182 CrossRef CAS PubMed.
- G. Malta, S. J. Freakley, S. A. Kondrat and G. J. Hutchings, Chem. Commun., 2017, 53, 11733 RSC.
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137, 14548 CrossRef CAS PubMed.
- United Nations Environment Programme, Minamata Convention on Mercury, www.mercuryconvention.org/, accessed July 2018.
- M. Conte, C. J. Davies, D. J. Morgan, T. E. Davies, A. F. Carley, P. Johnston and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 128 RSC.
- B. Nkosi, J. Catal., 1991, 128, 366 CrossRef CAS.
- B. Dai, Q. Wang, F. Yu and M. Zhu, Sci. Rep., 2015, 5, 10553 CrossRef PubMed.
- K. C. O'Connell, J. R. Monnier and J. R. Regalbuto, Appl. Catal., B, 2018, 225, 264 CrossRef.
- X. H. Tian, G. T. Hong, B. B. Jiang, F. P. Lu, Z. W. Liao, J. D. Wang and Y. R. Yang, RSC Adv., 2015, 5, 46366 RSC.
- J. Oliver-Meseguer, A. Doménech-Carbó, M. Boronat, A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2017, 56, 6435 CrossRef CAS PubMed.
- F. Zhao, Y. Wang and L. Kang, Can. J. Chem., 2016, 94, 842 CrossRef CAS.
- Y. Wang, M. Zhu, L. Kang and B. Dai, RSC Adv., 2014, 4, 38466 RSC.
- G. Malta, S. A. Kondrat, S. J. Freakley, C. J. Davies, L. Lu, S. Dawson, A. Thetford, E. K. Gibson, D. J. Morgan, W. Jones, P. P. Wells, P. Johnston, C. R. Catlow, C. J. Kiely and G. J. Hutchings, Science, 2017, 355, 1399 CrossRef CAS PubMed.
- H. Li, H.-X. Zhang, X.-L. Yan, B.-S. Xu and J.-J. Guo, New Carbon Mater., 2018, 33, 1 CrossRef.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65 CrossRef CAS.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498 CrossRef CAS PubMed.
- M. Yang and M. Flytzani-Stephanopoulos, Catal. Today, 2017, 298, 216 CrossRef CAS.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913 CrossRef CAS.
- D. Cazorla-Amorós, Frontiers in Materials, 2014, 1, 1 Search PubMed.
- Y. J. Chen, S. F. Ji, Y. G. Wang, J. C. Dong, W. X. Chen, Z. Li, R. A. Shen, L. R. Zheng, Z. B. Zhuang, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2017, 56, 6937 CrossRef CAS PubMed.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu, W.-C. Cheong, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2017, 139, 17269 CrossRef CAS PubMed.
- D. A. Bulushev, M. Zacharska, E. V. Shlyakhova, A. L. Chuvilin, Y. N. Guo, S. Beloshapkin, A. V. Okotrub and L. G. Bulusheva, ACS Catal., 2016, 6, 681 CrossRef CAS.
- J. Zhao, J. Xu, J. Xu, T. Zhang, X. Di, J. Ni and X. Li, Chem. Eng. J., 2015, 262, 1152 CrossRef CAS.
- X. Li, M. Zhu and B. Dai, Appl. Catal., B, 2013, 142, 234 CrossRef.
- M. Conte, C. J. Davies, D. J. Morgan, T. E. Davies, D. J. Elias, A. F. Carley, P. Johnston and G. J. Hutchings, J. Catal., 2013, 297, 128 CrossRef CAS.
- T. Furnival, R. K. Leary, E. C. Tyo, S. Vajda, Q. M. Ramasse, J. M. Thomas, P. D. Bristowe and P. A. Midgley, Chem. Phys. Lett., 2017, 683, 370 CrossRef CAS.
- Y. Y. Fong, B. R. Visser, J. R. Gascooke, B. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine and H. H. Harris, Langmuir, 2011, 27, 8099 CrossRef CAS PubMed.
- F. Karadas, G. Ertas, E. Ozkaraoglu and S. Suzer, Langmuir, 2005, 21, 437 CrossRef CAS PubMed.
- X. Liu, M. Conte, D. Elias, L. Lu, D. J. Morgan, S. J. Freakley, P. Johnston, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 5144 RSC.
- G. Hong, X. Tian, B. Jiang, Z. Liao, J. Wang, Y. Yang and J. Zheng, RSC Adv., 2016, 6, 3806 RSC.
- R. Lin, S. K. Kaiser, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2018, 8, 1114 CrossRef CAS.
-
P. T. Bishop, N. A. Carthey and P. Johnston, U.S. Pat. 9,409, vol. 161, 2016.
- T. Z. H. Gani and H. J. Kulik, ACS Catal., 2018, 8, 975 CrossRef CAS.
- J. C. Liu, Y. G. Wang and J. Li, J. Am. Chem. Soc., 2017, 139, 6190 CrossRef CAS PubMed.
- S. Wang, B. Shen and Q. Song, Catal. Lett., 2009, 134, 102 CrossRef.
- J. Zhong, Y. Xu and Z. Liu, Green Chem., 2018, 20, 2412 RSC.
- M. Conte, A. Carley, C. Heirene, D. Willock, P. Johnston, A. Herzing, C. Kiely and G. Hutchings, J. Catal., 2007, 250, 231 CrossRef CAS.
- J. Ma, S. Wang and B. Shen, React. Kinet., Mech. Catal., 2013, 110, 177 CrossRef CAS.
- T. V. Krasnyakova, I. V. Zhikharev, R. S. Mitchenko, V. I. Burkhovetski, A. M. Korduban, T. V. Kryshchuk and S. A. Mitchenko, J. Catal., 2012, 288, 33 CrossRef CAS.
- J. Pérez-Ramírez and E. Kondratenko, Catal. Today, 2007, 121, 160 CrossRef.
- K. Morgan, N. Maguire, R. Fushimi, J. T. Gleaves, A. Goguet, M. P. Harold, E. V. Kondratenko, U. Menon, Y. Schuurman and G. S. Yablonsky, Catal. Sci. Technol., 2017, 7, 2416 RSC.
- J. T. Gleaves, G. S. Yablonskii, P. Phanawadee and Y. Schuurman, Appl. Catal., A, 1997, 160, 55 CrossRef CAS.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95 CrossRef PubMed.
-
E. Fako, ioChem-BD Collection, DOI:10.19061/iochem-bd-1-74.
- M. García-Mota, N. Cabello, F. Maseras, A. M. Echavarren, J. Pérez-Ramírez and N. López, ChemPhysChem, 2008, 9, 1624 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, additional computational details, TAP experiments, microscopy images and videos, and catalytic data. See DOI: 10.1039/c8sc03186j |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sharon
Mitchell
a,
Sharon
Mitchell