
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA lithium-ion-active aerolysin nanopore for effectively trapping long single-stranded DNA†
Zheng-Li
Hu
,
Meng-Yin
Li
,
Shao-Chuang
Liu
,
Yi-Lun
Ying
 * and
Yi-Tao
Long
* and
Yi-Tao
Long
Key Laboratory for Advanced Materials, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: yilunying@ecust.edu.cn; Tel: +86-21-64252339
First published on 13th November 2018
Abstract
Wild-type aerolysin (AeL) nanopores allow direct single nucleotide discrimination of very short oligonucleotides (≤10 nt) without labelling, which shows great potential for DNA sensing. To achieve real applications, one major obstacle of AeL is its poor capture ability of long single-stranded DNA (ssDNA, >10 nt). Here, we have proposed a novel and robust strategy for the electrostatic focusing of long ssDNA into a lithium-chloride (LiCl)-active AeL. By using this method, for the first time we have demonstrated AeL detection of ssDNA longer than 100 nt. Due to screening more negative charges, LiCl improves AeL capture ability of long ssDNA (i.e. 60 nt) by 2.63- to 10.23-fold compared to KCl. Further calculations and molecular dynamics simulations revealed that strong binding between Li+ and the negatively charged residue neutralized the AeL, leading to a reduction in the energy barrier for ssDNA capture. These findings facilitate the future high-throughput applications of AeL in genetic and epigenetic diagnostics.
Introduction
Nanopore techniques have been developed into powerful platforms for single molecule sensing in a rapid and low-cost way. Aerolysin (AeL), a rivet-shaped protein nanopore with an inner diameter of ∼1.0 nm, was initially used to investigate the structural variation of α-helical peptides,1 then applied to discriminate polymer sizes, study protein folding/unfolding, describe enzyme kinetics and monitor chemical bond making/breaking.2–6 In particular, AeL demonstrated its superior resolution in discrimination of very short oligonucleotides (≤10 nt).7–9 The high sensitivity and long reads are the two important parameters for DNA sensing and sequencing. Compared with other biological nanopores such as α-hemolysin (α-HL) and Mycobacterium smegmatis porin A (MspA),10–13 AeL has high sensitivity in direct discrimination of single nucleotides without chemical labelling, the incorporation of molecular motors or recognition probes.9,14 Previous work showed that AeL was able to recognise subtle differences between methylated cytosine and cytosine under serum conditions.14 Therefore, AeL shows great potential for direct DNA sensing and sequencing in a simple and cheap manner. However, all these achievements were based on very short oligonucleotides (≤10 nt). Few real applications have been achieved via AeL because it is difficult for long single-stranded DNA (ssDNA, >10 nt) to enter into the AeL. For instance, AeL analysis of a 50 nt ssDNA shows an event frequency as low as ∼0.8 s−1 at 100 mV at a concentration of 2.0 μM.15 Most recently, our study demonstrated the discrimination of A, T, C and G in a 14 nt oligonucleotide but showed a low AeL capture rate of the ssDNA.16 This challenge largely restricts nanopore techniques from achieving direct sensing of long stranded DNA. To improve the capture ability of AeL for long stranded DNA, several strategies have been proposed. By increasing the applied voltage to a value as high as 70 mV, a 60 nt ssDNA showed an increased event frequency to 0.4 s−1.17 In our previous work, by modulating the G-quadruplex DNA into an extended form using magnesium ions (Mg2+), the subsequent capture of a 30 nt structured DNA was observed at an event frequency of 0.6 s−1.18 By raising the temperature from 20 to 60 °C, the event frequency of a 50 nt ssDNA increased from 0.8 to 2.2 s−1.15 By protonating AeL in an asymmetric pH environment, the translocation of a 16 nt ssDNA was improved to 5.6 s−1.19 However, these methods increase the difficulty of the experiment and reduce the stability of the sensing system. The high voltage destabilizes the lipid bilayer membrane, and the high temperature causes variation in the salt concentration due to the non-negligible evaporation of water and might irreversibly denature the protein nanopores. The asymmetric pH environment induces spontaneous current blocks for AeL at applied voltages higher than 100 mV.19 Thus, a novel and robust approach is urgently needed to improve AeL’s ability to capture long stranded DNA.According to previous studies,19–24 various possible effects could modulate DNA translocation dynamics, such as the geometry of the nanopore, the pH of the solution, the salt type and the electrolyte concentration gradient. These effects mainly regulate the energy barrier for the entry of DNA, in particular they reduce the interaction between DNA and the entrance of the nanopores. As for aerolysin nanopores, they emerge as negatively-charged protein nanopores (−52e) with numerous aspartic (Asp) and glutamic (Glu) residues around the cis entrances, such as Asp216, Asp207, Glu307 and Glu415.25 The strong electrostatic repulsion between the negatively charged nucleotides and the amino acids at the entrance contributes to a large energy barrier for DNA entering into the AeL. Therefore, a potential solution is to reduce the negative charges at the cis entrance of the AeL. The local charge distribution of a biological nanopore could be changed using site-directed mutagenesis and chemical modification.26–28 However, these methods are always complicated and time-consuming. Previous studies have demonstrated that lithium ions (Li+) exhibit a specific preference over other alkali metal ions due to their high charge density and small size.23,29 As Li+ shows a stronger binding affinity and a longer binding time with charged residues (i.e. Asp) compared with other alkali metal ions,30 it could be used to screen the negative charges of a biological nanopore. Herein, we adopted lithium chloride (LiCl) to improve AeL’s ability to capture long ssDNA. It has been reported for the first time that AeL detection of a 102 nt DNA (ssDNA_102) could be achieved in LiCl. Compared with KCl, LiCl enhanced the capture ability of AeL for ssDNA_102 by 3.89 to 9.07 fold.
Results and discussion
First, we demonstrated that the presented LiCl sensing system shows high stability in a wide voltage range (Fig. S1†). Fig. 1 illustrates a scheme of the nanopore experiment, depicting an individual ssDNA molecule captured in an AeL nanopore in M+Cl− (M+ = Li+ or K+). For both electrolytes, we added the ssDNA in the cis side of the AeL, where the applied voltage is termed as grounded. Here, we adopted the event frequency as a surrogate of capture ability, which is defined as the number of blockades per second and evaluated through the inverse fitting of interevent time. We performed the assays of ssDNA_102 and obtained its real-time current traces in LiCl and KCl. The duration times of ssDNA_102 decrease as the applied voltage ranges from 100 to 160 mV, indicating the successful translocations of ssDNA_102 in M+Cl− (M+ = Li+ or K+) (Fig. 1). Although the signals exhibit similar full blockades, the event frequencies of ssDNA_102 increase apparently when the electrolyte is changed from KCl to LiCl. The event frequency of ssDNA_102 in LiCl at 100 mV is 2.46 ± 0.11 s−1, producing a 9.07-fold increase compared to that in KCl (0.27 ± 0.04 s−1) (Fig. 1 and S2†). When the applied voltage is increased to 160 mV, the event frequency of ssDNA_102 further increases from 2.26 ± 0.23 s−1 in KCl to 8.79 ± 0.54 s−1 in LiCl (Fig. 1 and S2†). | ||
| Fig. 1 Illustration of the detection of a 102 nt ssDNA (ssDNA_102) using an AeL nanopore in M+Cl− (M+ = Li+ or K+). Top: the electrostatic attraction in LiCl and the relative electrostatic repulsion in KCl of a single ssDNA_102 molecule into AeL. The illustration is not to scale. Middle: raw current traces of ssDNA_102. More frequent blockade events were observed clearly in LiCl compared with KCl from the real-time current traces. Bottom: voltage dependence of ssDNA_102 translocation. Data in this work were obtained in 1 M M+Cl− (M+ = Li+ or K+), 1 mM EDTA, 10 mM Tris buffer, and pH 8.0 with the presence of 2.0 μM ssDNA at the cis side of the AeL nanopore. | ||
To figure out the enhancement mechanism in LiCl, we studied the behavior of polydeoxyadenines (dAn) with different lengths in M+Cl− (M+ = Li+ or K+). In LiCl, dA20 and dA40 exhibit a better linear frequency-voltage dependence, suggesting a more dominant diffusion-controlled capture process (Fig. 2a–c).19,31,32 However, dA60 shows an exponential frequency–voltage relationship, indicating a barrier-limited capture process (Fig. 2c). These results unravel the length-dependent capture of dAn in LiCl: diffusion is predominantly responsible for short stranded dAn and an energy barrier is primarily accountable for long stranded dAn. As expected, similar results were obtained for dAn in KCl (Fig. 2a–c).7 According to previous studies,33,34 the energy barrier for DNA capture mainly originates from DNA entropy and DNA-pore electrostatic interactions at the pore entrance. As the length of dAn increases, entropy likely contributes to a portion of the energy barrier for the capture of long stranded dAn. Therefore, the event frequencies reduce from 8.75 ± 0.06 to 5.81 ± 0.31 s−1 with dAn elongating from 20 to 60 nt at 160 mV in LiCl due to an increased energy barrier. Interestingly, LiCl induces higher event frequencies for dAn (n = 20, 40, 60) than KCl at the same applied voltage and the same DNA concentration. LiCl caused a 2.63- to 10.23-fold enhanced capture of dAn (n = 20, 40, 60) compared to that obtained in KCl. The possible explanation could be a lower energy barrier at the cis entrance of the AeL for dAn capture in LiCl than in KCl. As Li+ screens more negative charges than K+, we deduced that the lower energy barrier in LiCl results from weaker DNA-AeL electrostatic repulsions at the cis entrance of the pore.
 | ||
| Fig. 2 AeL detection of dAn with different lengths in M+Cl− (M+ = Li+ or K+). Voltage-dependent (a–c) event frequencies and (d–e) blockade durations of dAn (n = 20, 40, 60) in LiCl (red squares) and KCl (blue circles). The values of event frequencies and blockade durations were evaluated from transformed single-exponential functions (Fig. S3–S5†). | ||
To demonstrate this speculation, we evaluated the electrolyte-dependent difference in energy barriers for dA60, which shows the barrier-limited capture both in LiCl and KCl. The event frequencies of dA60 shown in Fig. 2c are well-fitted with eqn (1):
| f = f0eV/V0 | (1) |
 | (2) |
To better understand the capture enhancement mechanism, we performed molecular dynamics (MD) simulations in M+Cl− (M+ = Li+ or K+). The cations are defined as being bound within a cutoff distance of 3.1 Å from the heavy atoms of the pore.23Fig. 3a illustrates the distributions of the bound Li+, viewed from the cis side of the AeL (K+–AeL binding is shown in Fig. S6†). The binding number of Li+/K+ was averaged over a 10 ns MD simulation, fitting into a Gaussian distribution (Fig. 3b). The average binding numbers are 194 for Li+ and 87 for K+ (Fig. 3b). Although AeL exhibits an identical net charge in the two different electrolytes, the effective charges change due to the cation–AeL electrostatic interactions (Fig. 3a and S6†). The different Li+/K+–AeL binding induced variation of the electrostatic potential distributions at the cis entrance of the pore. Since the Li+/K+ ions created a local concentration of positive ions in the pore cap, the pore vicinity is effectively polarized and the electrostatic potential increases.31,35 In KCl, AeL presents a highly negative electrostatic potential in a confined region at the cis entrance of the pore (Fig. 3c). However, a highly positive electrostatic potential occupies most areas of the pore cap in LiCl (Fig. 3c). As ssDNA is negatively charged, the high positive electrostatic potential facilitates the capture of ssDNA from the cis entrance of AeL in LiCl, in good accordance with our previous discussion.
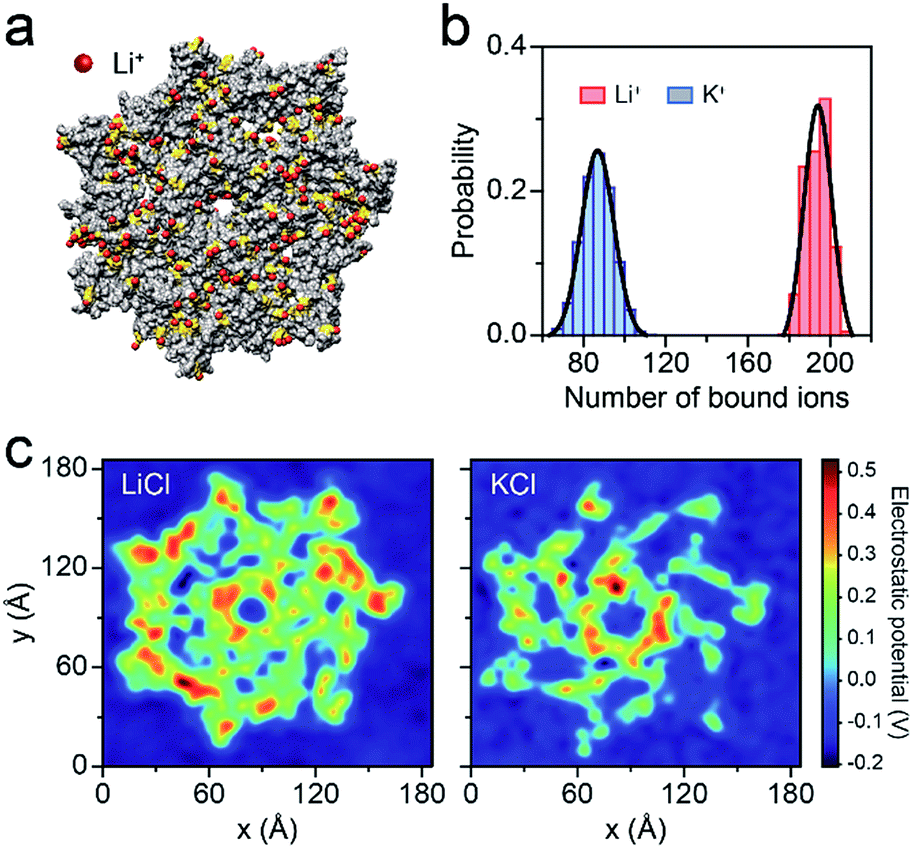 | ||
| Fig. 3 Molecular dynamics (MD) simulations of AeL in M+Cl− (M+ = Li+ or K+). (a) Li+ bound to the AeL in MD simulations. Li+ are represented in red, the negatively-charged amino acids are shown in yellow and the AeL is represented in gray. The illustration is not to scale. (b) The number of M+ (M+ = Li+, K+) bound to the AeL. The ions are regarded as being bound within 3.1 Å from the heavy atoms of the AeL. The binding numbers of Li+/K+ are fitted into a Gaussian function. (c) Average electrostatic potential around the cis entrance of the AeL. | ||
Furthermore, we employed LiCl for different polynucleotides with the same length. The raw current traces of dA20, dT20 and dC20 show more frequent blockades acquired in LiCl than KCl (Fig. 4a). The event frequencies are 8.75 ± 0.06 s−1 for dA20, 9.19 ± 0.68 s−1 for dT20, and 6.91 ± 0.11 s−1 for dC20 at 160 mV in LiCl, providing a 2.39-to-3.15-fold increase compared to those in KCl, respectively (Fig. 4a). The difference in event frequencies might result from the structural variations of dA20, dT20 and dC20 induced by the cation–ssDNA interactions. The duration times are 5.65 ± 0.38 ms for dA20, 0.42 ± 0.01 ms for dT20, and 0.23 ± 0.01 ms for dC20 at 160 mV in LiCl (Fig. 4b). The duration differences in LiCl between dA20 and dT20, dA20 and dC20, and dT20 and dC20 are 5.23 ± 0.39, 5.42 ± 0.39 and 0.19 ± 0.02 ms, respectively. In KCl, the differences between dA20 and dT20, dA20 and dC20, and dT20 and dC20 are 2.34 ± 0.24, 2.48 ± 0.24 and 0.14 ± 0.02 ms, respectively (Fig. 4b). Obviously, these values in KCl are smaller than those gained in LiCl, implying that LiCl provides a higher discrimination ability in the duration time for AeL with polynucleotides. Moreover, we found that LiCl leads to a 1.65-fold increase in event frequency for dA40 at 160 mV using an α-HL (Fig. S8†), which is smaller than the 2.69-fold increase obtained by the AeL. It is reasonable as more negative charges were screened at the cis entrance of the AeL due to the different charge distributions of the two pores.11,25
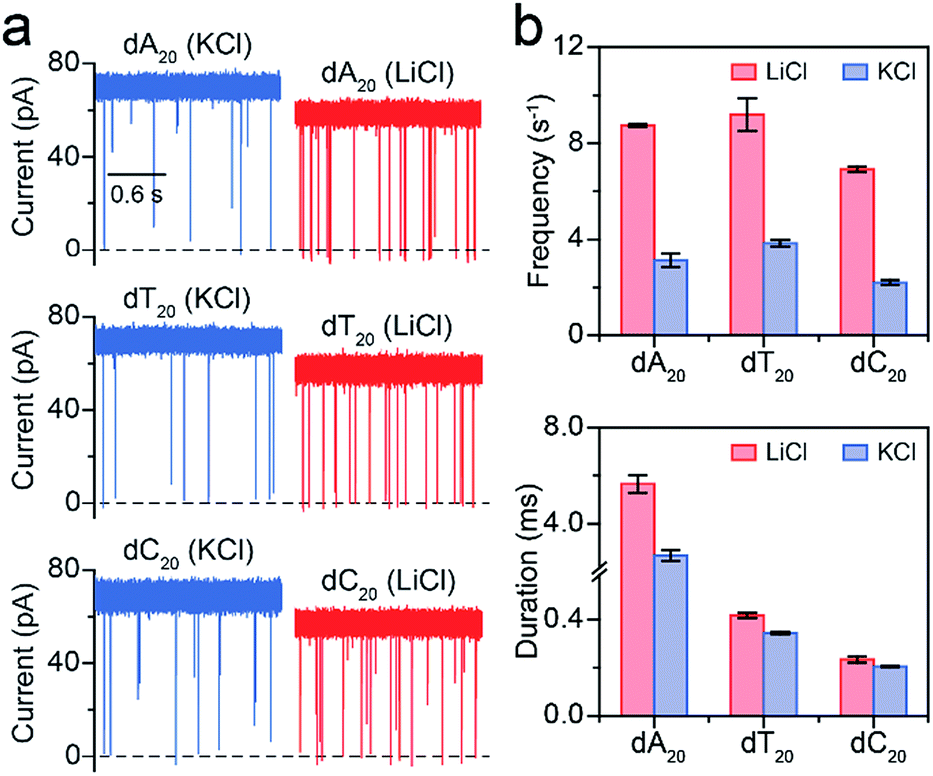 | ||
| Fig. 4 Analysis of different polynucleotides with the same length in M+Cl− (M+ = Li+ or K+). (a) Raw current traces of dA20, dT20 and dC20. (b) The event frequencies and durations of dA20, dT20 and dC20. Event frequencies reveal obvious enhancements for these ssDNA in LiCl. The values of event frequencies and durations were extracted from transformed single-exponential functions (Fig. S7†). Data were obtained at +160 mV in 1 M M+Cl− (M+ = Li+ or K+), 1 mM EDTA, 10 mM Tris buffer, and pH 8.0 with the presence of 2.0 μM ssDNA at the cis side of the AeL nanopore. | ||
Conclusion
In conclusion, we have presented a novel and robust method based on LiCl to enhance AeL capture of long ssDNA. By introducing LiCl, the AeL achieved the detection of a ssDNA longer than 100 nt for the first time. LiCl increased the ssDNA capture ability by 2.63 to 10.23 folds (i.e. 60 nt) compared with KCl at the same potential and DNA concentration. The calculations demonstrated that LiCl leads to an experimental reduction in activation energy of 4.48 kBT for dA60 in LiCl. Further MD simulations revealed that the strong Li+–AeL binding induces a great screen of negative charges on the pore surface, resulting in electrostatic focusing of long ssDNA into the AeL. Our previous work has shown that the event frequency of a 30 nt ssDNA is relatively low (∼0.6 s−1) in 1 M MgCl2, and the effect of divalent cations might be less favorable than monovalent cations for DNA capture. This general strategy is simple and practical without exposing the analyte and nanopore to extreme conditions (i.e. high temperatures and low pH). This work would facilitate various real applications of AeL in life science, such as genetic and epigenetic detection, enzyme activity evaluation and signaling pathway description. These findings also provide new insight into the construction of highly-sensitive AeL arrays for drug separation and screening, and direct DNA sensing and sequencing at high throughput and without labelling.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the “Chen Guang” Project from the Shanghai Municipal Education Commission and Shanghai Education Development Foundation (17CG27), National Natural Science Foundation of China (21421004 and 61871183) and the Fundamental Research Funds for the Central Universities (222201718001, 222201717003, 222201714012).Notes and references
- R. Stefureac, Y. T. Long, H. B. Kraatz, P. Howard and J. S. Lee, Biochemistry, 2006, 45, 9172–9179 CrossRef CAS PubMed.
- F. Piguet, H. Ouldali, M. Pastoriza-gallego, P. Manivet, J. Pelta and A. Oukhaled, Nat. Commun., 2018, 9, 996 CrossRef PubMed.
- G. Baaken, I. Halimeh, L. Bacri, J. Pelta, A. Oukhaled and J. C. Behrends, ACS Nano, 2015, 9, 6443–6449 CrossRef CAS PubMed.
- C. Merstorf, B. Cressiot, M. Pastoriza-gallego, A. Oukhaled, J.-M. Betton, L. Auvray and J. Pelta, ACS Chem. Biol., 2012, 7, 652–658 CrossRef CAS PubMed.
- B. Zhou, Y. Wang, C. Cao, D. Li and Y. Long, Sci. China: Chem., 2018, 61, 1–4 Search PubMed.
- Y. Wang, L. Q. Gu and K. Tian, Nanoscale, 2018, 10, 13857–13866 RSC.
- C. Cao, Y. L. Ying, Z. L. Hu, D. F. Liao, H. Tian and Y. T. Long, Nat. Nanotechnol., 2016, 11, 713–718 CrossRef CAS PubMed.
- C. Cao, J. Yu, M. Li, Y. Wang, H. Tian and Y. Long, Small, 2017, 13, 1702011 CrossRef PubMed.
- Y. Ying, Z. Li, Z. Hu, J. Zhang, F. Meng, C. Cao, Y. T. Long and H. Tian, Chem, 2018, 4, 1893–1901 CAS.
- J. Clarke, H. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1866 CrossRef CAS PubMed.
- M. Faller, M. Niederweis and G. E. Schulz, Science, 2004, 303, 1189–1193 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS PubMed.
- J. Yu, C. Cao and Y. Long, Anal. Chem., 2017, 89, 11685–11689 CrossRef CAS PubMed.
- L. Payet, M. Martinho, C. Merstorf, M. Pastoriza-gallego, J. Pelta, V. Viasnoff, L. Auvray, M. Muthukumar and J. Mathé, Biophys. J., 2015, 109, 1600–1607 CrossRef CAS PubMed.
- C. Cao, M. Li, N. Cirauqui, Y. Wang, M. D. Peraro, H. Tian and Y. Long, Nat. Commun., 2018, 9, 2823 CrossRef PubMed.
- M. Pastoriza-Gallego, M.-F. Breton, F. Discala, L. Auvray, J.-M. Betton and J. Pelta, ACS Nano, 2014, 8, 11350–11360 CrossRef CAS PubMed.
- D. Liao, C. Cao, Y. Ying and Y. T. Long, Small, 2018, 14, 1704520 CrossRef PubMed.
- Y. Wang, K. Tian, X. Du, R. Shi and L. Gu, Anal. Chem., 2017, 89, 13039–13043 CrossRef CAS PubMed.
- N. A. W. Bell, K. Chen, S. Ghosal, M. Ricci and U. F. Keyser, Nat. Commun., 2017, 8, 380 CrossRef PubMed.
- N. A. W. Bell, M. Muthukumar and U. F. Keyser, Phys. Rev. E, 2016, 93, 022401 CrossRef PubMed.
- Y. Zhang, G. Wu, W. Si, J. Ma, Z. Yuan, X. Xie, L. Liu, J. Sha, D. Li and Y. Chen, Nanoscale, 2017, 9, 930–939 RSC.
- S. W. Kowalczyk, D. B. Wells, A. Aksimentiev and C. Dekker, Nano Lett., 2012, 12, 1038–1044 CrossRef CAS PubMed.
- D. Xi, Z. Li, L. Liu, S. Ai and S. Zhang, Anal. Chem., 2018, 90, 1029–1034 CrossRef CAS PubMed.
- I. Iacovache, S. De Carlo, N. Cirauqui, M. Dal Peraro, F. G. van der Goot and B. Zuber, Nat. Commun., 2016, 7, 12062 CrossRef CAS PubMed.
- G. Maglia, M. R. Restrepo, E. Mikhailova and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19720–19725 CrossRef CAS PubMed.
- Y. Wang, C. Cao, Y. Ying, S. Li, M. Wang, J. Huang and Y. Long, ACS Sens., 2018, 3, 779–783 CrossRef CAS PubMed.
- Y. Wang, M. Li, H. Qiu, C. Cao, M. Wang, X. Wu, J. Huang, Y. Ying and Y. T. Long, Anal. Chem., 2018, 90, 7790–7794 CrossRef CAS PubMed.
- M. Boukhet, F. Piguet, H. Ouldali, M. Pastoriza-gallego, J. Pelta and A. Oukhaled, Nanoscale, 2016, 8, 18352–18359 RSC.
- S. Bhattacharya, J. Muzard, L. Payet, J. Mathé, U. Bockelmann, A. Aksimentiev and V. Viasnoff, J. Phys. Chem. C, 2011, 115, 4255–4264 CrossRef CAS PubMed.
- M. Wanunu, W. Morrison, Y. Rabin, A. Y. Grosberg and A. Meller, Nat. Nanotechnol., 2010, 5, 160–165 CrossRef CAS PubMed.
- Y. Wang, K. Tian, L. L. Hunter, B. Ritzo and L. Gu, Nanoscale, 2014, 6, 11372–11379 RSC.
- S. E. Henrickson, M. Misakian, B. Robertson and J. J. Kasianowicz, Phys. Rev. Lett., 2000, 85, 3057–3060 CrossRef CAS PubMed.
- M. Pastoriza-gallego, L. Rabah, G. Gibrat, B. Thiebot, F. G. van der Goot, L. Auvray, J.-M. Betton and J. Pelta, J. Am. Chem. Soc., 2011, 133, 2923–2931 CrossRef CAS PubMed.
- M. Muthukumar, J. Chem. Phys., 2010, 132, 195101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Protocol for aerolysin nanopore experiment, molecular dynamics simulations, histograms of the event frequencies and durations, and supporting figures. See DOI: 10.1039/c8sc03927e |
| This journal is © The Royal Society of Chemistry 2019 |