
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble nucleophilic addition to iminomalonate, leading to the synthesis of quaternary α-amino diesters and desymmetrization of the products†
Makoto Shimizu *ab,
Miki Mushikab,
Isao Mizotab and
Yusong Zhua
*ab,
Miki Mushikab,
Isao Mizotab and
Yusong Zhua
aSchool of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, Jiangsu Province, China
bDepartment of Chemistry for Materials, Graduate School of Engineering, Mie University, Tsu, Mie 514-8507, Japan. E-mail: mshimizu@chem.mie-u.ac.jp
First published on 29th July 2019
Abstract
Alkylation of iminomalonate with Grignard reagents followed by oxidation and allylation gave symmetrical quaternary α-amino diesters in good yields. Subsequent desymmetrization of a diol derivative from these products was conducted via asymmetric carbamylation catalyzed by Cu-Bnbox to give chiral quaternary aminodiol mono-carbamates.
1 Introduction
Quaternary α-amino acid or ester moieties are very important structural units in many biologically active compounds, since the incorporation of rigid amino acid surrogates concerns very useful information on the bioactive conformation and provides intriguing physiological effects.1,2 Quaternary α-amino acid moieties are also found as fundamental skeletons in medicines and agrochemicals such as Ecteinascidin 743, Salinosporamide A, and Neooxazolomycin (Fig. 1).3–5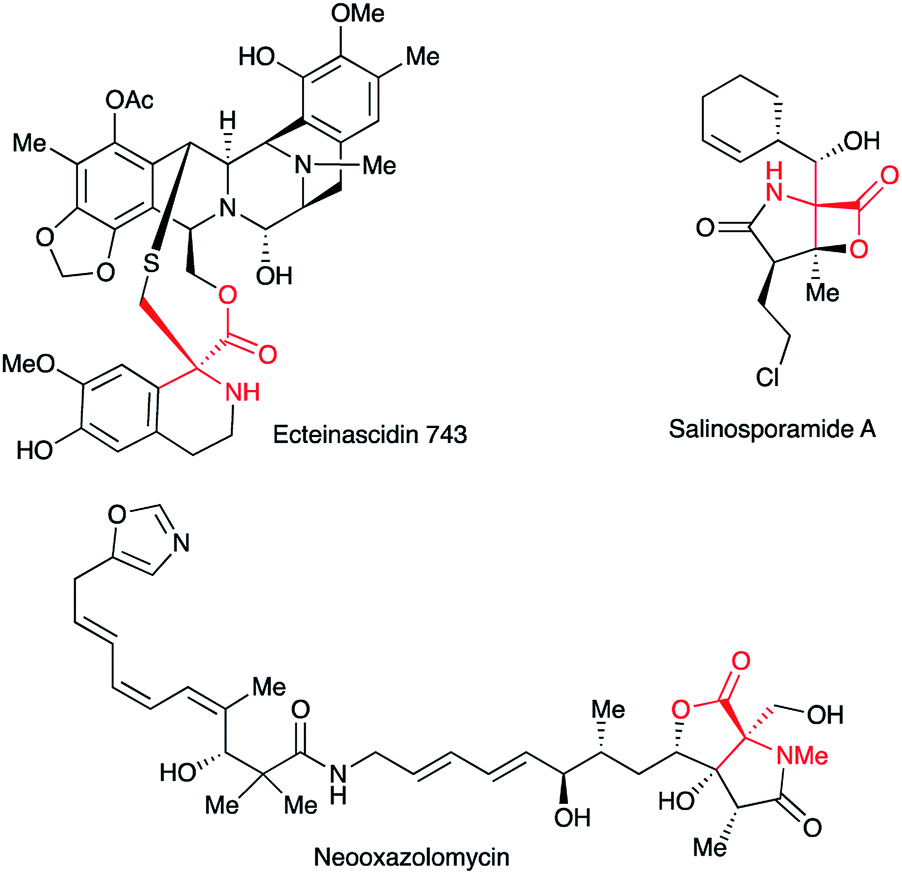 | ||
| Fig. 1 Bioactive compounds possessing a quaternary α-amino acid structure. | ||
However, it is not trivial to construct quaternary α-amino acid frameworks in a stereoselective manner.6 For the synthesis of α-amino acids and their derivatives α-imino esters are useful substrates, since α-imino esters have a unique reactivity: either N- or C-alkylation is possible. Although an umpolung N-alkylation reaction of α-imino ester is difficult due to the electronegativity of the imino group, it can lead to a flexible introduction of substituents into the nitrogen atom of the amino acid frameworks.7,8
We have already reported that umpolung N-alkylation reactions of α-iminoesters 1 followed by C-electrophilic addition or oxidation followed by C-nucleophilic addition gives intriguing N,C-double addition products in good yields with high diastereoselectivities.9 In our previous report, we found that a combined use of diethylaluminum chloride and ethylaluminium dichloride promoted most efficiently the N-alkylation of α-aryl α-iminoester 2, and the subsequent oxidation and allylation were conducted with benzoyl peroxide and allyltributylstannane, respectively (eqn (1), Scheme 2).9b During investigations into amination ability of iminomalonate 1 to organometallics, we found that not only organoaluminum compounds but also Grignard reagents were readily aminated to give 2-aminomalonates in good to high yields (eqn (1), Scheme 1).9a However, the resulting halomagnesium enolate that was an N-addition intermediate was not used for further C–C bond formations. Since the previous study shows that the oxidation of an intermediary α-aminoester enolate readily gives an iminium species that is attacked by a nucleophile, the present intermediary halomagnesium enolate is expected to have a similar reactivity.
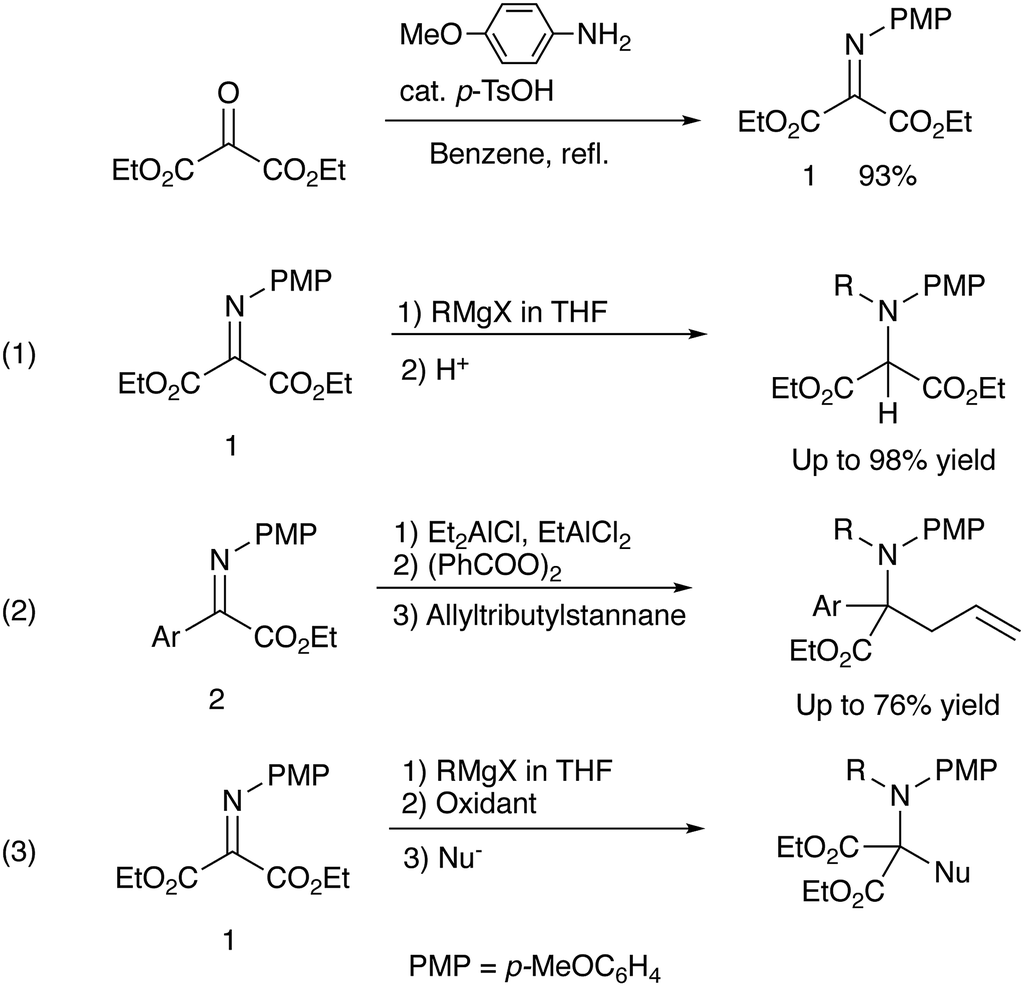 | ||
| Scheme 1 Previous9a,b and present works. | ||
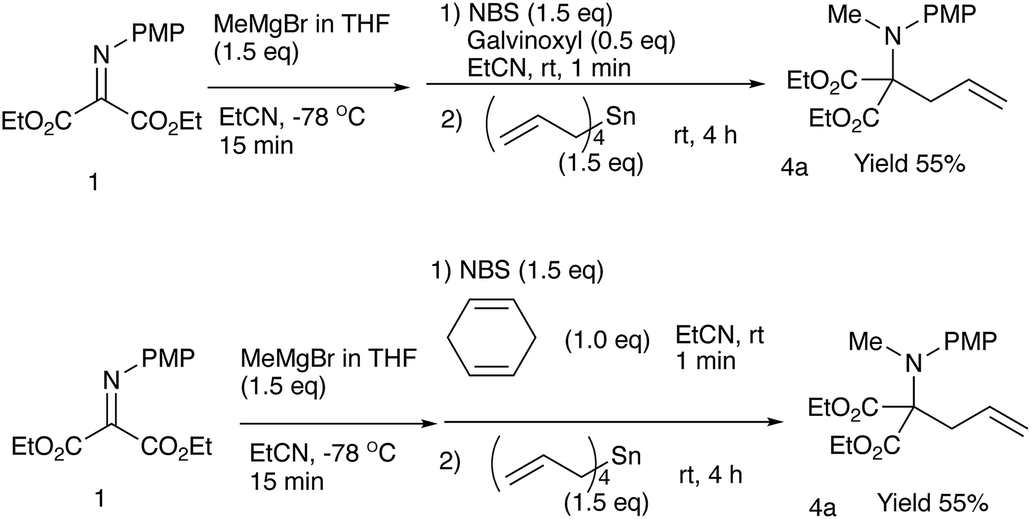 | ||
| Scheme 2 Reactions in the presence of a radical scavenger or a quencher. | ||
We have now found that oxidation of the halomagnesium enolate derived from N-alkylation of the iminomalonate 1 with an appropriate oxidation reagent followed by C-nucleophilic addition leads to the formation of N,C-double addition products, quaternary α-amino diesters in good yields (eqn (3), Scheme 1).
2 Results and discussion
2.1 N-Methylation/C-cyanation reactions
Our previous study leads to the N-ethylation/oxidation/C-cyanation reaction, which involves the formation of an intermediary iminium salt, responsible for the second nucleophilic addition. In the present examination, we carried out the double addition reaction as in the case with the previous one.9b Initially, methylation with methylmagnesium bromide, oxidation, and cyanation with TMSCN were examined regarding the oxidation reagent, and Table 1 summarizes the results.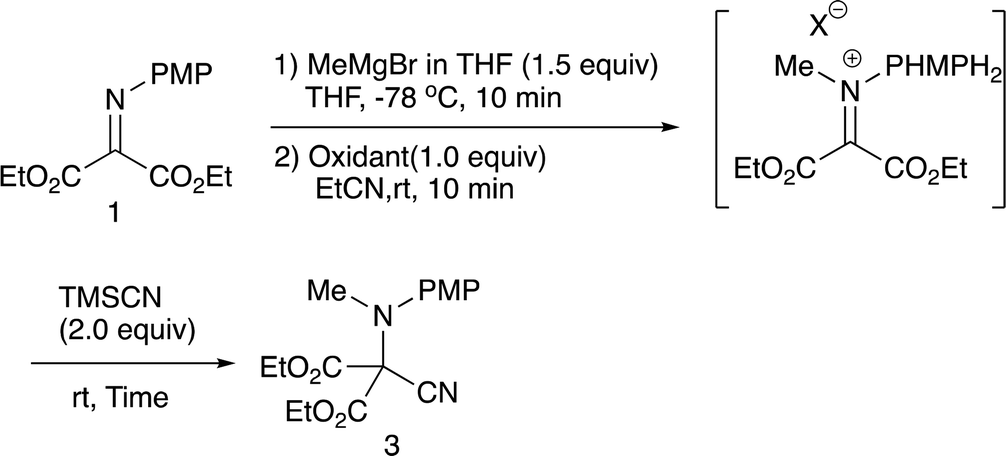
As can be seen from Table 1, benzoyl peroxide (BPO) that was effective in the previous cyanation of α-iminoesters did not work,9b whereas with N-chlorosuccinimide (NCS) or N-bromosuccinimide (NBS) the desired double addition product 3 was obtained, albeit in low yields. The low yield is presumably due to the instability of the intermediary iminium salt, and therefore, the oxidation in the presence of TMSCN was next examined. Table 2 summarizes the results.
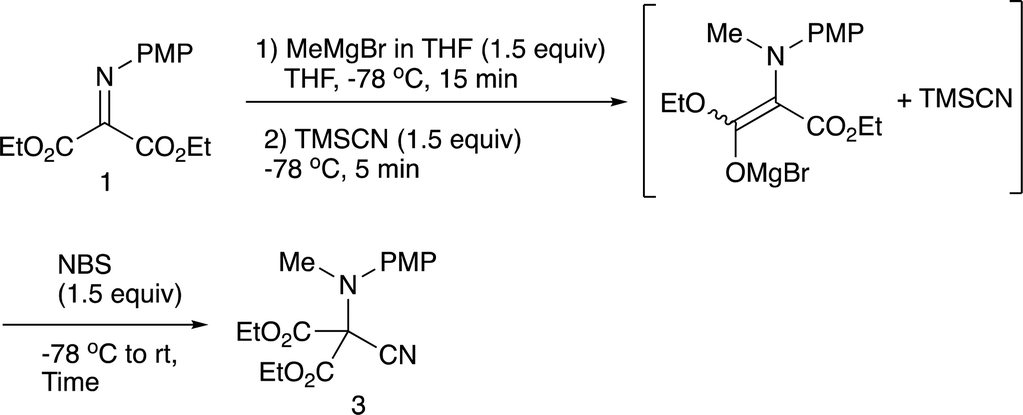
As expected the reaction proceeded relatively well and the desired product 3 was formed in 56% yield after 10 min (entry 1). Further optimization was carried out with respect to the reaction time; a prolonged reaction time of 4 h increased the product yield up to 64% (entry 2). We next examined the allylation reaction that would involve a stronger C–C bond formation, and Table 3 summarizes the results.
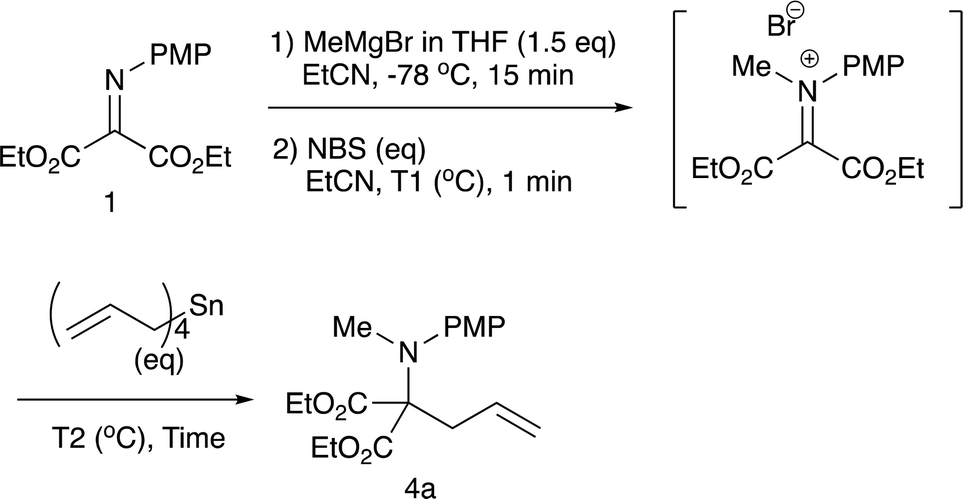
2.2 N-Methylation/C-allylation reactions
Although we initially examined the use of allyltrimethylsilane, allylmagnesium chloride, and ketene silyl acetals, these nucleophiles gave the addition products in only low yields (less than 20%). Among the nucleophiles examined, teteraallylstannane effected the desired double addition reaction most efficiently. Treatment of the iminomalonate 1 with methylmagnesium bromide (1.5 equiv.) at −78 °C followed by oxidation with NBS (1.5 equiv.) and allylation with tetrallylstannane (1.0 equiv.) gave the desired double addition product 4a in 47% yield (entry 1). Further examination into the reactions led to the optimum conditions. Use of NBS (1.5 equiv.) and tetrallylstannane (1.5 equiv.) at room temperature afforded the desired product 4a in 74% yield (entry 3). Under the optimized conditions, various Grignard reagents underwent double addition reactions to give good to high yields of products 4, and Table 4 summarizes the results.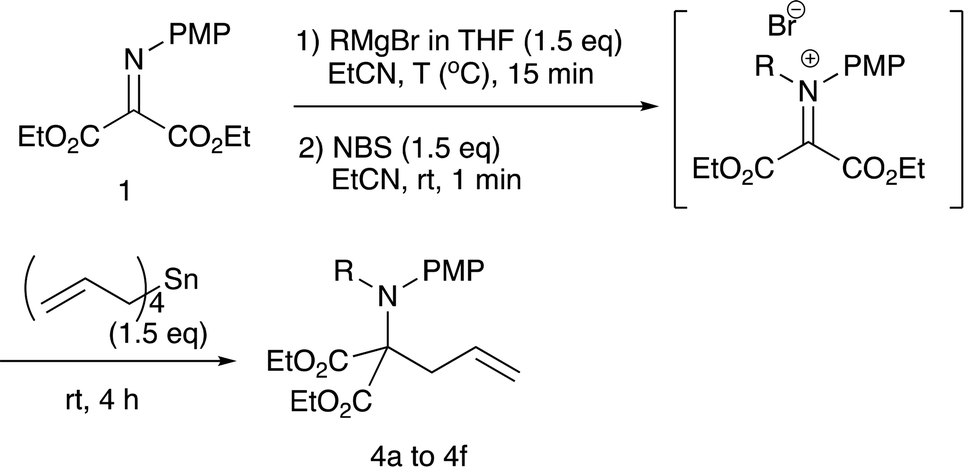
2.3 N-Alkylation/C-allylation reactions
As shown in Table 4, methyl, ethyl, npropyl, and nbutyl Grignard reagents underwent N-addition reaction rapidly and cleanly, and the subsequent oxidation and allylation also proceeded well to give N-alkylation C-allylation products 4a–d in good yields (entries 1 to 4), whereas sterically bulky iso-propyl and tert-butyl counterparts effected the first addition reaction sluggishly to give the desired products 4e–f in moderate to low yields (entries 5 and 6). The present double addition reaction has a limitation; aromatic Grignard reagents did not effect the overall tandem addition, although the first umpolung addition to the nitrogen actually gave the single addition product in 59% yield (entry 7).9a To examine the reaction pathways, these control experiments were carried out (Scheme 2).2.4 Control experiments and a proposed reaction mechanism
The reaction was carried out in the presence of a radical scavenger or a quencher. The yields of the desired product 4a did not noticeably decrease. These results indicate that no radical pathway would be involved in the present reaction. On the basis of these results and our previous investigations,9b the following pathways are proposed (Scheme 3).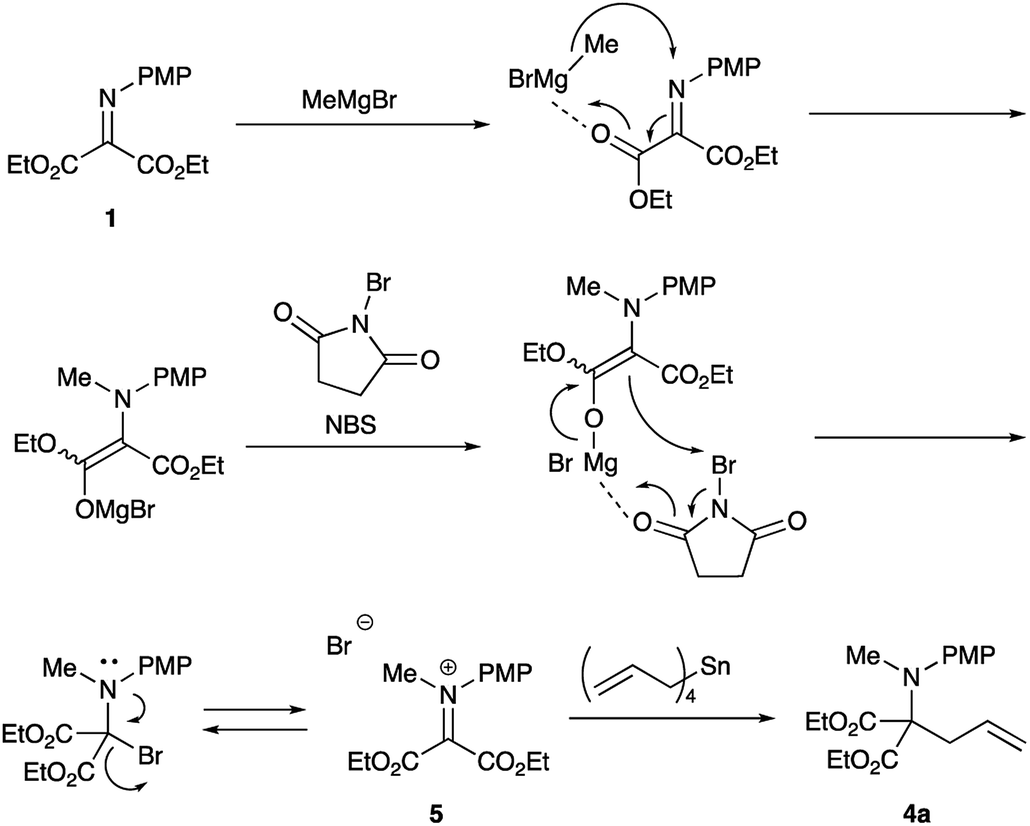 | ||
| Scheme 3 Proposed reaction mechanism. | ||
First, Grignard reagent attacks at the nitrogen atom to form the halomagnesium enolate, which is oxidized with NBS to form the iminium salt 5. This iminium salt 5 is attacked by tetraallylstannane to give the desired double addition product 4a.
2.5 Desymmetrization of the products
For further use of products 4, we carried out desymmetrization reactions of the aminodiesters 4. The initial examination was carried out using biocatalysts such as lipases for the asymmetric hydrolysis of the ester moiety.10 However, none of the satisfactory results was obtained. We next examined chemical transformations via asymmetric acylation of the diol 6a derived from the diester 4a catalyzed by Cu-Bnbox system, which finally worked well to give chiral products 7 with good enantio-purities.11 The reduction of the diester 4a was carried out using LiAlH4 to give the diol 6a in 81% yield (Scheme 4). Table 5 summarizes the results of the subsequent desymmetrization.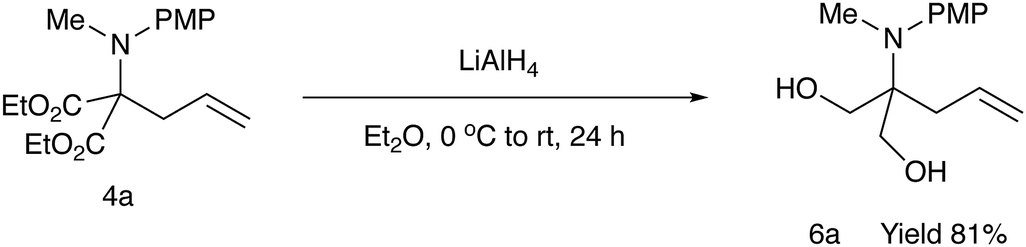 | ||
| Scheme 4 Reduction of the diester 4a. | ||
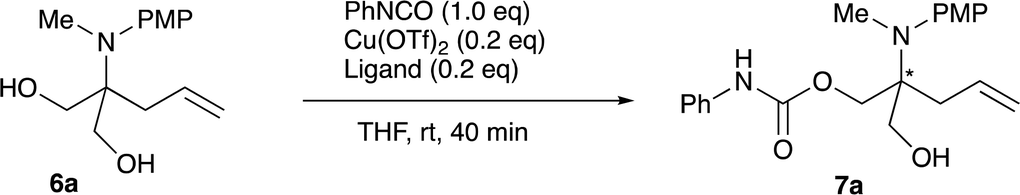
The desymmetrization reaction was carried out with aryl isocyanate in the presence of a catalytic amount of copper(II) triflate and a ligand in THF. Among the ligands examined, the use of Bnbox recorded the best result of 64% ee (entry 1), while tridentate ligands, pybox derivatives, did not work well in the present system (entries 5 and 6). An increase in the amount of both the ligand and Cu(II) to 50 mol% gave a slight increase in the enantiopurity of 66% (entry 2). Further increase in the ees would be possible by changing ArNCO derivatives, and the results will be reported elsewhere.
3 Conclusions
A double nucleophilic addition to iminomalonate was developed using the tandem N-alkylation/oxidation/allylation reaction of diethyl 2-[N-(p-methoxyphenyl)imino]malonate with Grignard reagent/NBS/tetraallylstannane in good to high yields. We also found that the desymmetrization of a diol derivative of the above products with ArNCO/Cu(II)-Bnbox gave a mono-carbamate in good enantiomeric excess.4 Experimental
4.1 General aspects
1H NMR and 13C NMR spectra were recorded with a JEOL ECX-400P, or a JEOL A-500 spectrometer using tetramethylsilane as an internal standard. Mass spectra were recorded on a JEOL MS-700D spectrometer. High performance liquid chromatography was carried out using these apparatuses; HITACHI Pump L-2130, Column Oven L-2300, and UV Detector L-2400. Propionitrile (EtCN) was distilled from phosphorus pentaoxide and then from calcium hydride, and stored over Molecular Sieves 4A. Tetrahydrofuran (THF) and diethyl ether (Et2O) were purified with a Glass Contour Organic Solvent Purification System of Nikko Hansen & Co., Ltd. Benzene was dried over calcium chloride, distilled, and stored over Molecular Sieves 4A. Purification of products was performed by column chromatography on silica gel (Kanto Silica Gel 60N) and/or preparative TLC on silica gel (Merck Kiesel Gel GF254 or Wako Gel B-5F).4.2 Synthesis of diethyl 2-[N-(4-methoxyphenyl)imino]-malonate (1)
This compound 1 was prepared according to the published procedure.9a,12–144.3 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-methylamino]-2-cyanomalonate (3)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) − C3H5O2)+ 320.1372, found 320.1363.
− C3H5O2)+ 320.1372, found 320.1363.4.4 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylmalonate (4a)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added MeMgBr (0.20 mL, 0.23 mmol, 0.99 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (40.0 mg, 0.23 mmol) and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4a (35.4 mg, 74%) as a colorless oil. Rf = 0.5 (ethyl acetate/nhexane = 1/4); 1H NMR (400 MHz, CDCl3) δ: 1.29 (t, J = 7.1 Hz, 6H), 2.60 (d, J = 6.9, 2H), 2.95 (s, 3H), 3.77 (s, 3H), 4.26 (q, J = 7.0), 4.93–5.01 (m, 2H), 5.79–5.89 (m, 1H), 6.77–6.81 (m, 2H), 7.08–7.12 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 14.2, 40.1, 41.9, 55.3, 61.2, 74.6, 113.9, 118.0, 128.2, 133.0, 141.8, 156.9, 169.8; HRMS (EI) calcd for C18H25NO5 (M− C3H5O2)+ 355.1733, found 355.1723.
4.5 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-ethylamino]-2-allylmalonate (4b)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added EtMgBr (0.25 mL, 0.23 mmol, 0.91 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (26.7 mg, 0.15 mmol) and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4b (35.6 mg, 68%) as a colorless oil. Rf = 0.5 (ethyl acetate/nhexane = 1/6, developed twice); 1H NMR (400 MHz, CDCl3) δ: 0.90 (t, J = 7.1 Hz, 3H), 1.30 (t, J = 7.1 Hz, 6H), 2.44–2.46 (m, 2H), 3.17 (q, J = 6.9 Hz, 2H), 3.78 (s, 3H), 4.25 (q, J = 7.2 Hz, 4H), 4.91–4.99 (m, 2H), 5.78–5.89 (m, 1H), 6.78–6.82 (m, 2H), 7.08–7.12 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 14.2, 14.9, 40.4, 48.4, 55.3, 61.1, 75.6, 113.8, 117.7, 131.2, 133.4, 138.4, 157.8, 170.3; HRMS (EI) calcd for C19H27NO5 (M− C3H5O2)+ 349.1889, found 349.1892.
4.6 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-propylamino]-2-allylmalonate (4c)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added nPrMgBr (0.24 mL, 0.23 mmol, 0.93 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (26.7 mg, 0.15 mmol) and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4c (43.3 mg, 79%) as a yellow oil. Rf = 0.5 (ethyl acetate/nhexane = 1/4); 1H NMR (400 MHz, CDCl3) δ: 0.82 (t, J = 18.3 Hz, 3H), 1.28 (m, 8H), 2.44 (d, J = 6.9 Hz, 2H), 3.08 (t, J = 7.4 Hz, 2H), 3.78 (s, 2H), 4.25 (q, J = 6.9 Hz, 4H), 4.92–4.98 (m, 2H), 5.77–5.88 (m, 1H), 6.78–6.82 (m, 2H), 7.09–7.13 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 11.2, 14.2, 22.3, 40.5, 55.3, 55.7, 61.0, 75.6, 113.7, 117.7, 130.9, 133.4, 138.7, 157.7, 170.2; HRMS (EI) calcd for C20H29NO5 (M− C3H5O2)+ 363.2046, found 363.2038.
4.7 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-butylamino]-2-allylmalonate (4d)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added nBuMgBr (0.20 mL, 0.23 mmol, 1.1 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (26.7 mg, 0.15 mmol) and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4d (40.7 mg, 72%) as a yellow oil. Rf = 0.5 (ethyl acetate/nhexane = 1/4); 1H NMR (400 MHz, CDCl3) δ: 0.81 (t, J = 6.8 Hz, 3H), 1.24–1.32 (m, 10H), 2.44 (d, J = 6.9 Hz, 2H), 3.10 (t, J = 6.8 Hz, 2H), 3.79 (s, 3H), 4.25 (q, J = 7.1 Hz, 4H), 4.92–4.98 (m, 2H), 5.78–5.88 (m, 1H), 6.79–6.82 (m, 2H), 7.08–7.12 (m, 2H): 13C NMR (100 MHz, CDCl3) δ: 14.1, 14.2, 20.0, 31.5, 40.5, 53.8, 55.3, 61.0, 75.6, 113.7, 117.8, 130.9, 133.3, 138.7, 157.7, 170.2; HRMS (EI) calcd for C21H31NO5 (M− C3H5O2)+ 377.2202, found 377.2201.
4.8 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-(2-propyl)amino]-2-allylmalonate (4e)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −90 °C. To it was added iPrMgBr (0.27 mL, 0.23 mmol, 0.85 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (26.7 mg, 0.15 mmol) and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4e (22.1 mg, 40%) as a yellow oil. Rf = 0.5 (ethyl acetate/nhexane = 1/4); 1H NMR (400 MHz, CDCl3) δ: 0.97 (d, J = 6.6 Hz, 6H), 1.31 (t, J = 7.1 Hz, 6H), 2.50 (d, J = 7.1 Hz, 2H), 3.60–3.67 (m, 1H), 3.79 (s, 3H), 4.19–4.32 (m, 4H), 4.76–4.92 (m, 2H), 5.61–5.71 (m.1H), 6.77–6.81 (m, 2H), 7.06–7.10 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 11.5, 14.1, 22.6, 40.0, 51.5, 55.2, 61.14, 74.65, 113.0, 117.7, 133.1, 133.9, 157.5, 171.57; HRMS (EI) Calculated for C20H29NO5 (M− C3H5O2)+ 363.2046, found 363.2043.
4.9 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylmalonate (4a) in the presence of galvioxiyl
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added MeMgBr (0.20 mL, 0.23 mmol, 0.99 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (40.0 mg, 0.23 mmol), galvioxyl (7.2 mg, 0.23 mmol), and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4a (26.3 mg, 55%) as a colorless oil.4.10 Synthesis of diethyl 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylmalonate (4a) in the presence of 1,4-cyclohexadiene
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed diethyl 2-[N-(p-methoxyphenyl)imino]malonate (41.9 mg, 0.15 mmol) in THF (1.0 mL) at −78 °C. To it was added MeMgBr (0.20 mL, 0.23 mmol, 0.99 M THF). After the mixture was stirred for 15 min at room temperature, to it were added NBS (40.0 mg, 0.23 mmol), 1,4-cyclohexadiene (80.13 mg, 0.075 mmol), and EtCN (1.0 mL), and the reaction mixture was stirred for 1 min at room temperature. Then, tetraallylstannane (0.054 mL, 0.23 mmol) was added and the mixture was stirred at room temperature for 4 h. The reaction was quenched with sat aq NaHCO3 (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/4) to give the title compound 4a (26.3 mg, 55%) as a colorless oil.4.11 Synthesis of 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylpropan-1,3-diol (6a)
In a 50 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, an argon balloon, and a rubber septum was placed LiAlH4 (0.33 g, 8.7 mmol). To it was added a solution of diethyl 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylmalonate (1.0 g, 3.1 mmol) in Et2O (30 mL) at 0 °C, and the whole mixture was stirred at room temperature for 16 h. The reaction was quenched with sat aq Na2SO4, and the whole mixture was filtered through a pad of Celite. The filtrate was concentrated in vacuo to give a crude oil, which was purified by silica gel column chromatography (ethyl acetate/nhexane = 1/2) to give the title compound 6a (0.64 g, 81%) as a colorless oil. Rf = 0.1 (ethyl acetate/nhexane = 1/2); 1H NMR (400 MHz, CDCl3) δ: 2.25 (d, J = 7.3 Hz, 2H), 2.55 (s, 2H), 2.82 (s, 3H), 3.63 (q, J = 11.5, 8.4 Hz, 4H), 3.80 (s, 3H), 5.05–5.12 (m, 2H), 5.73–5.83 (m, 1H), 6.82–6.86 (m, 2H), 7.21–7.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 34.7, 37.3, 55.4, 63.8, 64.7, 113.8, 118.3, 129.0, 133.8, 141.9, 157.1; HRMS (EI) calcd for C14H21NO3 (M− C3H5O2)+ 251.1521, found 251.1524.
4.12 Desymmetrization of 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylpropan-1,3-diol
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon were placed Cu(OTf)2 (10.9 mg, 0.03 mmol) and Bnbox (10.9 mg, 0.03 mmol). To it were added a solution of 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allylpropan-1,3-diol (37.7 mg, 0.15 mmol) in THF (1.5 mL) and PhNCO(16.0 μL, 0.15 mmol). After the mixture was stirred for 40 min at room temperature, it was quenched with H2O (20.0 mL), and the whole mixture was extracted with ethyl acetate (10 mL × 2). The combined extracts were washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel TLC (ethyl acetate/nhexane = 1/2) to give 2-[N-(4-methoxyphenyl)-N-methylamino]-2-allyl-3-hydroxypropylphenylcarbamate 7a (33.4 mg, 60%, 64% ee) as a colorless oil. Rf = 0.3 (ethyl acetate/nhexane = 1/2); 1H NMR (400 MHz, CDCl3) δ: 2.25–2.35 (m, 2H), 2.83 (s, 3H), 2.92 (s, 1H), 3.54 (s, 2H), 3.75 (s, 3H), 4.12 (d, J = 11.7, 1H), 4.29 (d, J = 11.7, 1H), 5.10–5.14 (m, 2H), 5.78–5.88 (m, 1H), 6.57 (s, 1H), 6.80 (d, J = 8.8, 2H), 7.06–7.10 (m, 1H), 7.17 (d, J = 8.8, 2H), 7.29–7.35 (m, 4H); 13C NMR (100 MHz, CDCl3) δ: 35.4, 37.5, 55.3, 62.0, 63.0, 65.2, 113.6, 118.7, 123.7, 129.1, 129.2, 133.4, 137.5, 141.8, 157.1; HRMS (EI) calcd for C21H26N2O4 (M− C3H5O2)+ 370.1893, found 370.1905; HPLC (Daicel Chiralpak AD, flow rate = 1.0 mL min−1, nhexane/iPrOH = 30/1, detection at 254 nm, set temperature 35 °C).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) and on Innovative Areas “Organic Synthesis Based on Reaction Integration. Development of New Methods and Creation of New Substances” from JSPS and MEXT.Notes and references
- For reviews of α,α-disubstituted amino acids, see: (a) C. Cativiela and M. D. Diaz-de-Villegas, Tetrahedron: Asymmetry, 1998, 9, 3517–3599 CrossRef CAS; (b) C. Cativiela and M. D. Diaz-de-Villegas, Tetrahedron: Asymmetry, 2000, 11, 645–732 CrossRef CAS; (c) C. Spino, Angew. Chem., Int. Ed., 2004, 43, 1764–1766 CrossRef CAS PubMed; (d) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127–5143 CrossRef CAS; (e) H. Vogt and S. Brase, Org. Biomol. Chem., 2007, 5, 406–430 RSC; (f) M. I. Calaza and C. Cativiela, Eur. J. Chem., 2008, 3427–3448 CAS; (g) C. Cativiela and M. Ordóñez, Tetrahedron: Asymmetry, 2009, 20, 1–63 CrossRef CAS PubMed.
- For synthesis of α,α-disubstituted amino acids, see the followings. Alkylation of enolate: (a) Y. N. Belokon, R. G. Davies and M. North, Tetrahedron Lett., 2000, 41, 7245–7248 CrossRef CAS; (b) Y. N. Belokon, D. Bhave, D. D'Addario, E. Groaz, V. M. North and A. Pertrosyan, Tetrahedron Lett., 2003, 44, 2045–2048 CrossRef CAS; Alkylation of cyclic compounds: (c) R. M. Williams and M.-N. Im, J. Am. Chem. Soc., 1991, 113, 9276–9286 CrossRef CAS; (d) D. B. Berkowitz, J. M. McFadden, E. Chisowa and C. L. Semerad, J. Am. Chem. Soc., 2000, 122, 11031–11032 CrossRef CAS PubMed; Asymmetric Strecker reaction: (e) S. Masumoto, H. Usuda, M. Suzuki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 5634–5635 CrossRef CAS PubMed; (f) J. Wang, X. Hu, J. Jiang, S. Gou, X. Huang and X. Feng, Angew. Chem., Int. Ed., 2007, 46, 8468–8470 CrossRef CAS PubMed; Addition to imino ester: (g) M. Mitani, Y. Tanaka, A. Sawada, A. Misu and Y. Matsumoto, Eur. J. Org. Chem., 2008, 1383–1391 CrossRef CAS; Radical reaction: (h) H. Miyabe, R. Asada and Y. Takemoto, Tetrahedron, 2005, 61, 385–393 CrossRef CAS; Memory of chirality: (i) T. Kawabata, T. Wirth, K. Yahiro, H. Suzuki and K. Fuji, J. Am. Chem. Soc., 1994, 116, 10809–10810 CrossRef CAS; (j) K. Fuji and T. Kawabata, Chem.–Eur. J., 1998, 4, 373–376 CrossRef CAS; (k) T. Kawabata, J. Chen, H. Suzuki, Y. Nagae, T. Kinoshita, S. Chancharunee and K. Fuji, Org. Lett., 2000, 2, 3883–3885 CrossRef CAS PubMed; (l) T. Kawabata, S. Kawakami and K. Fuji, Tetrahedron Lett., 2002, 43, 1465–1467 CrossRef CAS; Mitsunobu reaction: (m) J. E. Green, D. M. Bender, S. Jackson, M. J. O'Donnell and J. R. McCarthy, Org. Lett., 2009, 11, 807–810 CrossRef CAS PubMed; Sigmatropic rearrangement: (n) J. C. Anderson and S. Skerrat, J. Chem. Soc., Perkin Trans. 1, 2002, 2871–2876 RSC; Beckmannn rearrangement: (o) R. P. Frutos and D. M. Spero, Tetrahedron Lett., 1998, 39, 2475–2478 CrossRef CAS; Asymmetric hydrolysis: (p) V. Iosub, A. R. Haberl, J. Leung, M. Tang, K. Vembaiyan, M. Parvez and T. G. Back, J. Org. Chem., 2010, 75, 1612–1619 CrossRef CAS PubMed.
- E. J. Corey, D. Y. Gin and R. S. Kania, J. Am. Chem. Soc., 1996, 118, 9202–9203 CrossRef CAS.
- A. Endo and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 8298–8299 CrossRef CAS PubMed.
- A. S. Kende, K. Kawamura and R. J. DeVita, J. Am. Chem. Soc., 1990, 112, 4070–4072 CrossRef CAS.
- (a) K. Maruoka, E. Tayama and T. Ooi, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 5824–5829 CrossRef CAS PubMed; (b) C. Toniolo, F. Formaggio, B. Kaptein and Q. B. Broxterman, Synlett, 2006, 1295–1310 CrossRef CAS; (c) K. Tomohara, T. Yoshimura, R. Hyakutake, P. Yang and T. Kawabata, J. Am. Chem. Soc., 2013, 135, 13294–13297 CrossRef CAS PubMed; (d) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS; (e) A. E. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1–7 CrossRef CAS PubMed.
- For C-alkylation to α-imino esters, see: (a) J.-C. Fiaud and H. B. Kagan, Tetrahedron Lett., 1970, 21, 1813–1816 CrossRef; (b) L. M. Harwood, K. J. Vines and M. G. B. Drew, Synlett, 1996, 1051–1053 CAS; (c) D. Ferraris, B. Young, T. Dudding and T. Lectka, J. Am. Chem. Soc., 1998, 120, 4548–4549 CrossRef CAS; (d) A. Córdova, W. Notz, G. Zhong, J. M. Betancort and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 1842–1843 CrossRef PubMed; (e) K. P. Chiev, S. Roland and P. Mangeney, Tetrahedron: Asymmetry, 2002, 13, 2205–2209 CrossRef CAS; (f) P. Wipf and C. R. J. Stephenson, Org. Lett., 2003, 5, 2449–2452 CrossRef CAS PubMed; (g) M. Mitani, Y. Tanaka, A. Sawada, A. Misu and Y. Matsumoto, Eur. J. Org. Chem., 2008, 1383–1391 CrossRef CAS; (h) P. Fu, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 5530–5541 CrossRef CAS PubMed; (i) S. Fustero, N. Mateu, L. Albert and J. L. Aceña, J. Org. Chem., 2009, 74, 4429–4432 CrossRef CAS PubMed; (j) K. Yeung, F. Talbot, G. P. Howell, A. P. Pulis and D. J. Procter, ACS Catal., 2019, 9, 1655–1661 CrossRef CAS.
- For N-alkylation to α-imino esters, see: (a) J.-C. Fiaud and H. B. Kagan, Tetrahedron Lett., 1971, 12, 1019–1022 CrossRef; (b) Y. Yamamoto and W. Ito, Tetrahedron, 1988, 44, 5415–5423 CrossRef CAS; (c) K. Uneyama, F. Yan, S. Hirama and T. Katagiri, Tetrahedron Lett., 1996, 37, 2045–2048 CrossRef CAS; (d) S. E. Yoo and Y. D. Gong, Heterocycles, 1997, 45, 1251–1255 CrossRef CAS; (e) M. P. Bertrand, L. Feray, R. Nouguier and P. Perfetti, Synlett, 1999, 1148–1150 CrossRef CAS; (f) M. Mae, H. Amii and K. Uneyama, Tetrahedron Lett., 2000, 41, 7893–7896 CrossRef CAS; (g) K. P. Chiev, S. Roland and P. Mangeney, Tetrahedron: Asymmetry, 2002, 13, 2205–2209 CrossRef CAS; (h) J. S. Dickstein, M. W. Fennie, A. L. Norman, B. J. Paulose and M. C. Kozlowski, J. Am. Chem. Soc., 2008, 130, 15794–15795 CrossRef CAS PubMed; (i) J. S. Dickstein and M. C. Kozlowski, Chem. Soc. Rev., 2008, 37, 1166–1173 RSC; (j) J. M. Curto, J. S. Dickstein, S. Berritt and M. C. Kozlowski, Org. Lett., 2014, 16, 1948–1951 CrossRef CAS PubMed.
- (a) Y. Niwa, K. Takayama and M. Shimizu, Bull. Chem. Soc. Jpn., 2002, 75, 1819–1825 CrossRef CAS; (b) Y. Niwa and M. Shimizu, J. Am. Chem. Soc., 2003, 125, 3720–3721 CrossRef CAS PubMed; (c) M. Shimizu, H. Itou and M. Miura, J. Am. Chem. Soc., 2005, 127, 3296–3297 CrossRef CAS PubMed; (d) I. Mizota and M. Shimizu, Chem. Rec., 2016, 16, 688–702 CrossRef CAS PubMed; (e) I. Mizota, Y. Tadano, Y. Nakamura, T. Haramiishi, M. Hotta and M. Shimizu, Org. Lett., 2019, 21, 2663–2667 CrossRef CAS PubMed; (f) M. Shimizu, H. Imazato, I. Mizota and Y. Zhu, RSC Adv., 2019, 9, 17341–17346 RSC.
- (a) T. Miyazawa, Amino Acids, 1999, 16, 191–213 CrossRef CAS PubMed; (b) P. D. Maria, B. Kossmann, N. Potgrave, S. Buchholz, H. Trauthwein, O. May and H. Gröger, Synlett, 2005, 1746–1748 Search PubMed; (c) A. Ghanem, Tetrahedron, 2007, 63, 1721–1754 CrossRef CAS; (d) A. C. L. de Melo Carvalho, T. de Sousa Fonseca, M. C. de Mattos, M. C. de Oliveira, T. L. G. de Lemos, F. Molinari, D. Romano and I. Serra, Int. J. Mol. Sci., 2015, 16, 29682–29716 CrossRef PubMed.
- (a) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed; (b) K. Matsumoto, M. Mitsuda, N. Ushijima, Y. Demizu, O. Onomura and Y. Matsumura, Tetrahedron Lett., 2006, 47, 8453–8456 CrossRef CAS; (c) O. Onomura, M. Mitsuda, M. T. T. Nguyen and Y. Demizu, Tetrahedron Lett., 2007, 48, 9080–9084 CrossRef CAS; (d) A. R. Silva, L. Carneiro, A. P. Carvalho and J. Pires, Catal. Sci. Technol., 2013, 3, 2415–2424 RSC.
- B. K. Banik, M. S. Manhas, E. W. Robb and A. K. Bose, Heterocycles, 1997, 44, 405–415 CrossRef CAS.
- K. Oka and S. Hara, Tetrahedron Lett., 1977, 18, 2939–2942 CrossRef.
- A. K. Bose, M. Tsai and J. C. Kapur, Tetrahedron Lett., 1974, 15, 3547–3548 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04889h |
| This journal is © The Royal Society of Chemistry 2019 |