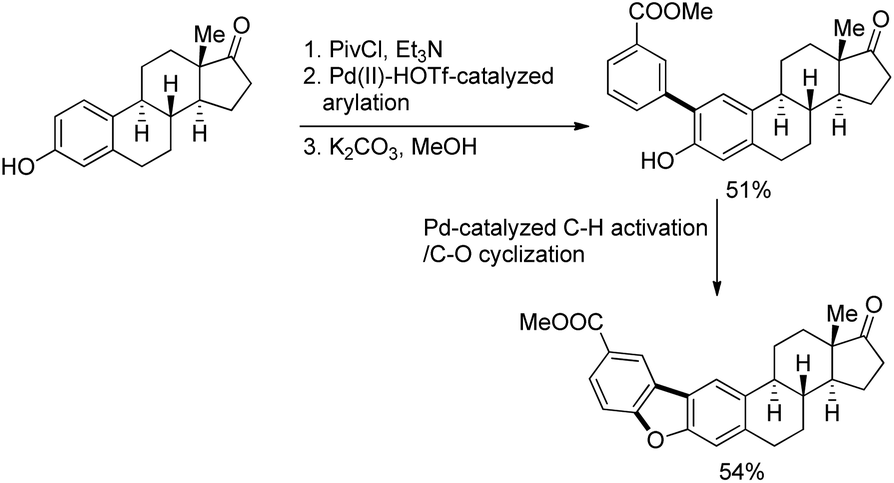
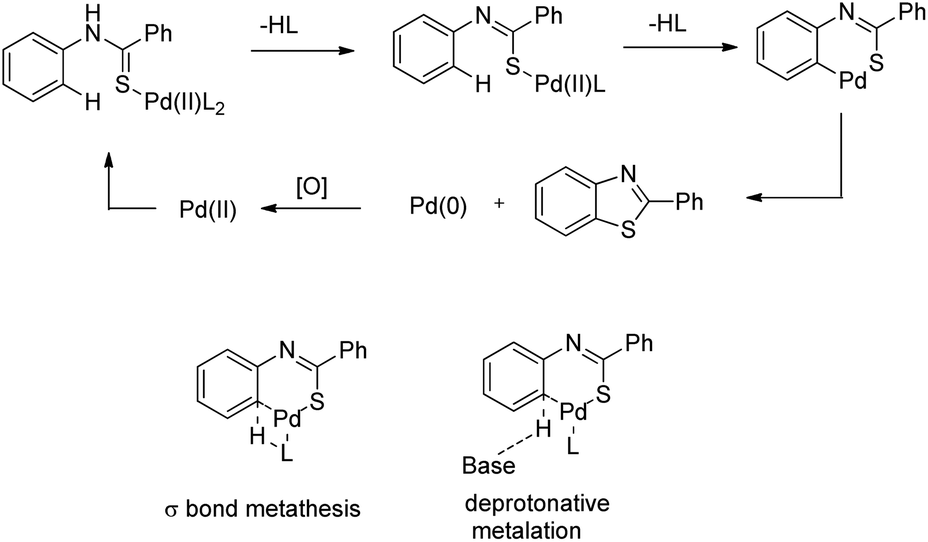
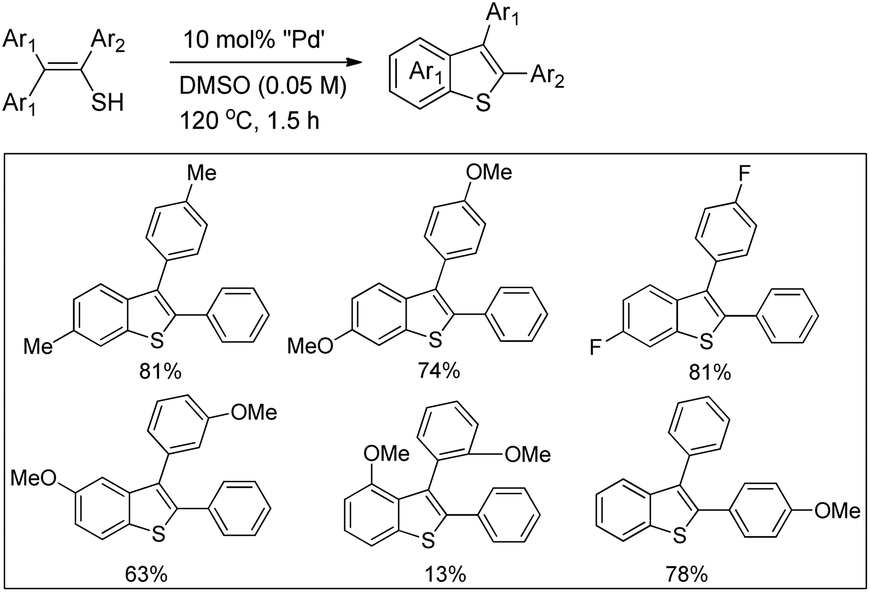
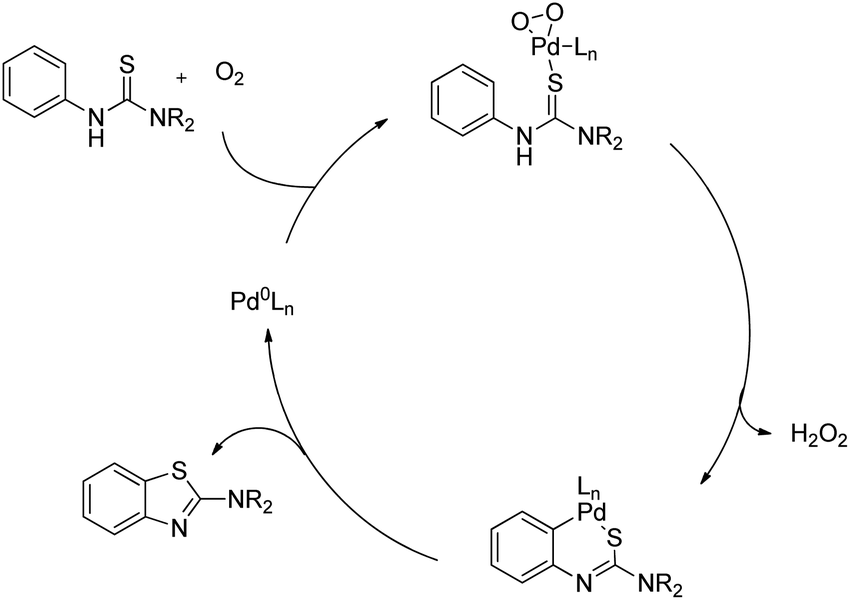
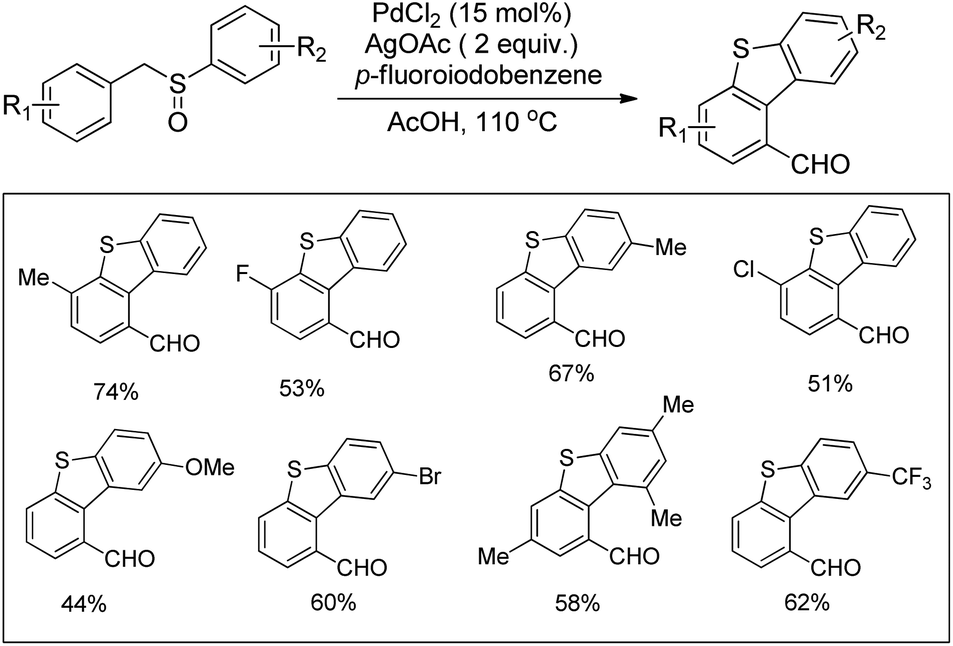
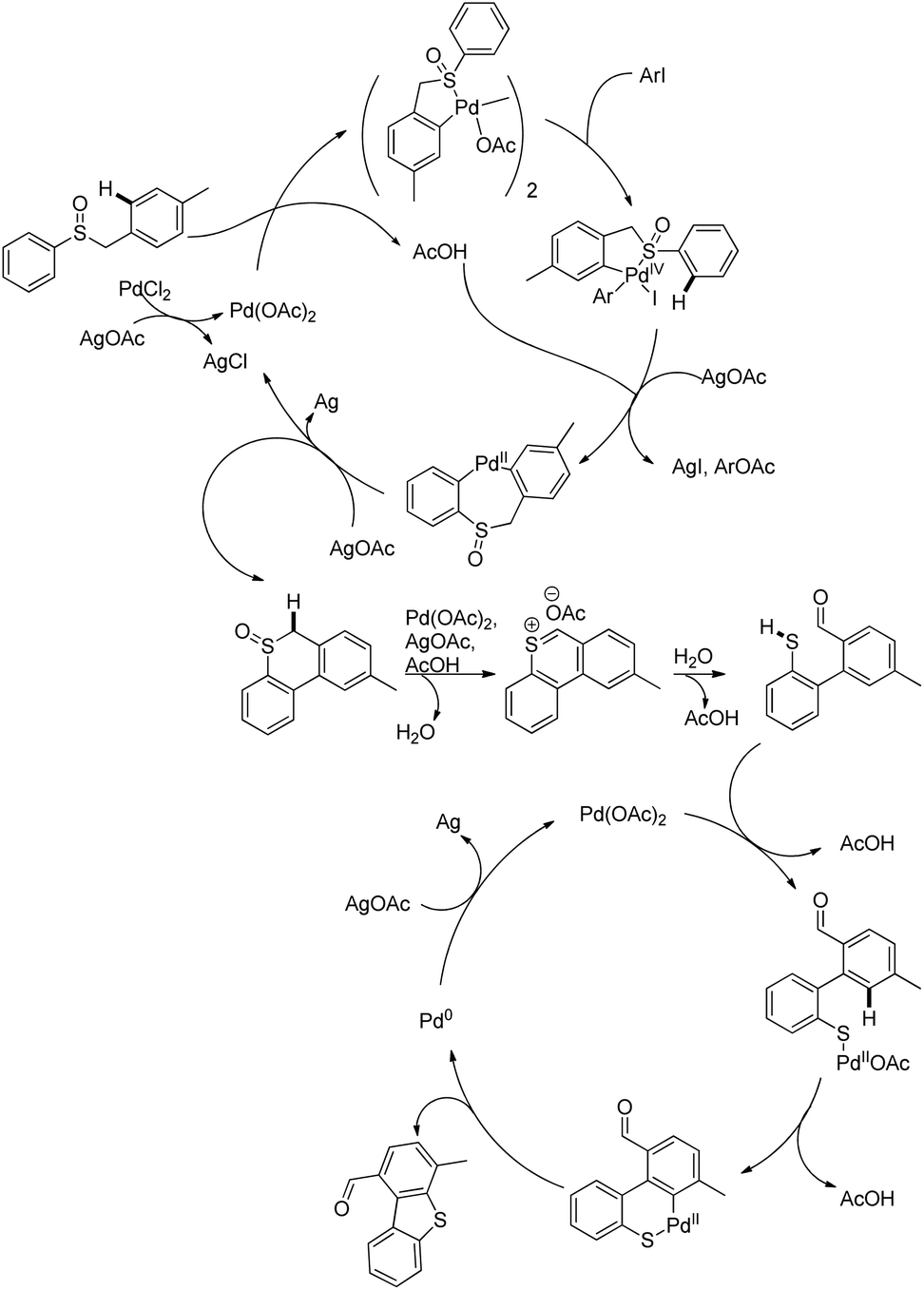
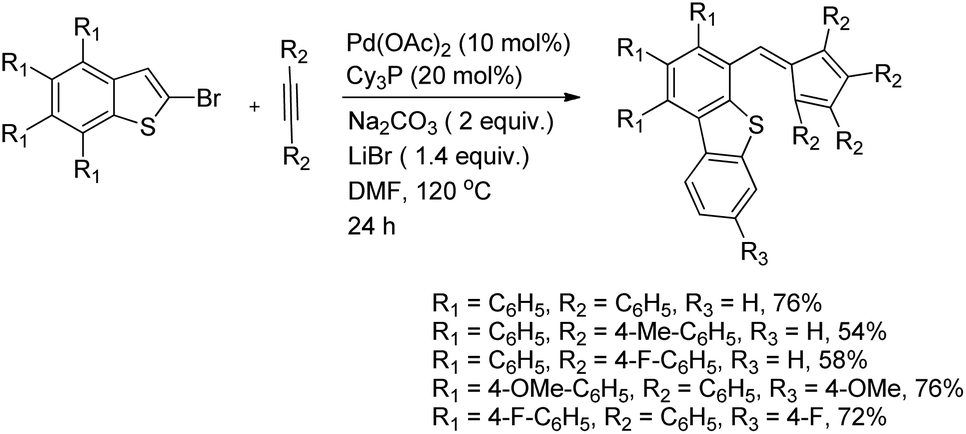
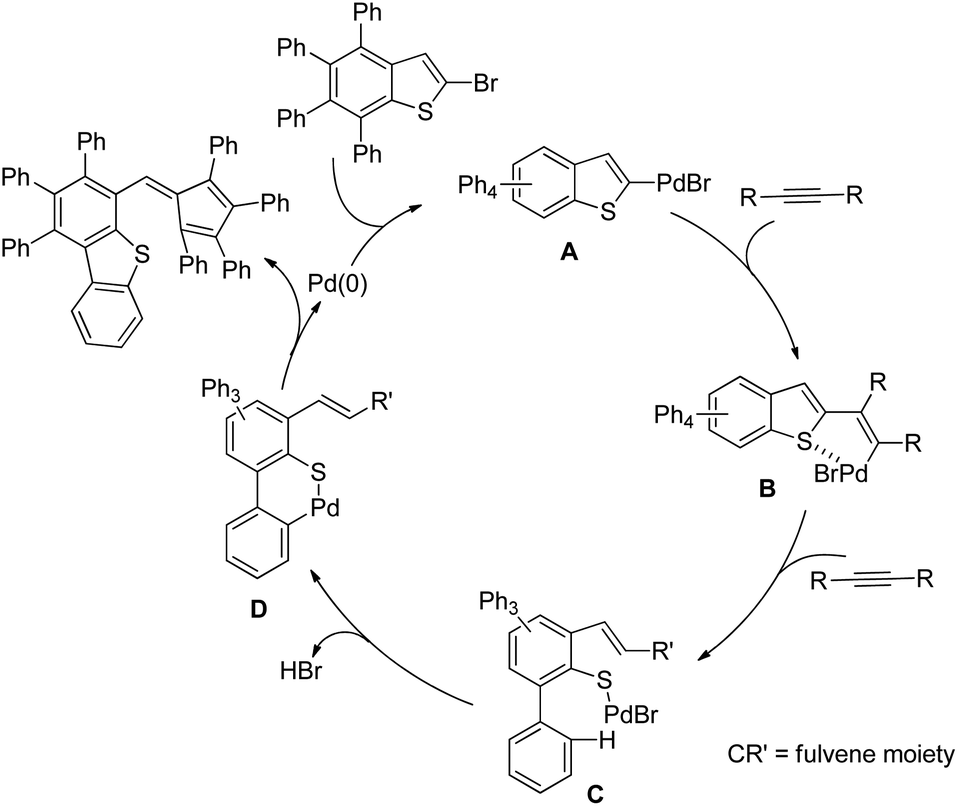
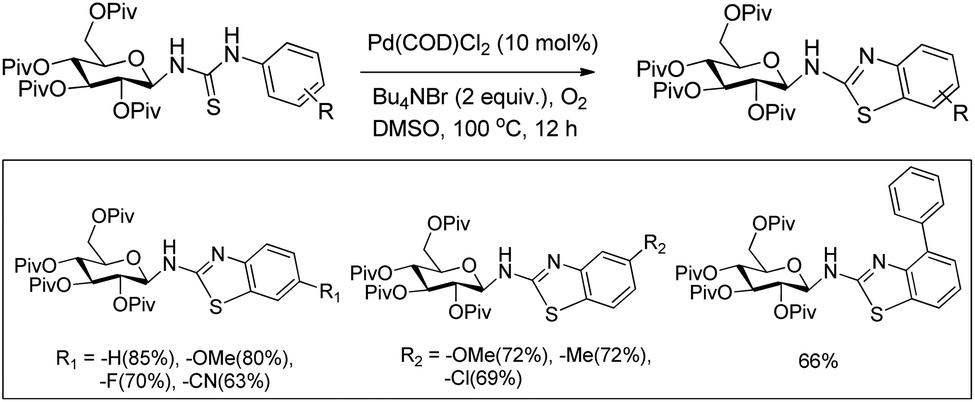
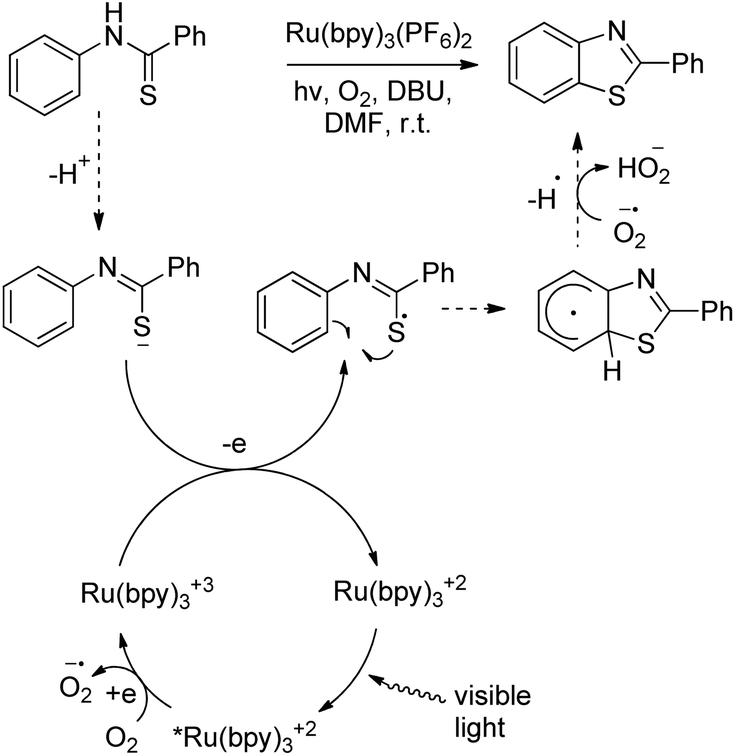
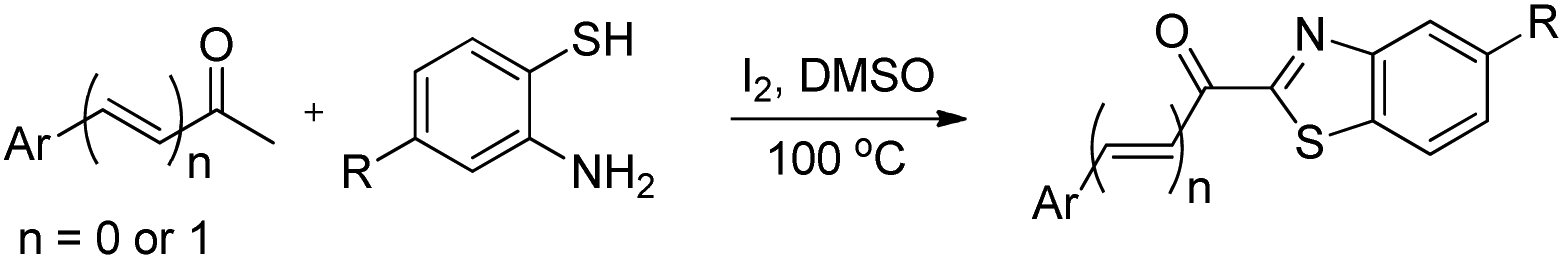
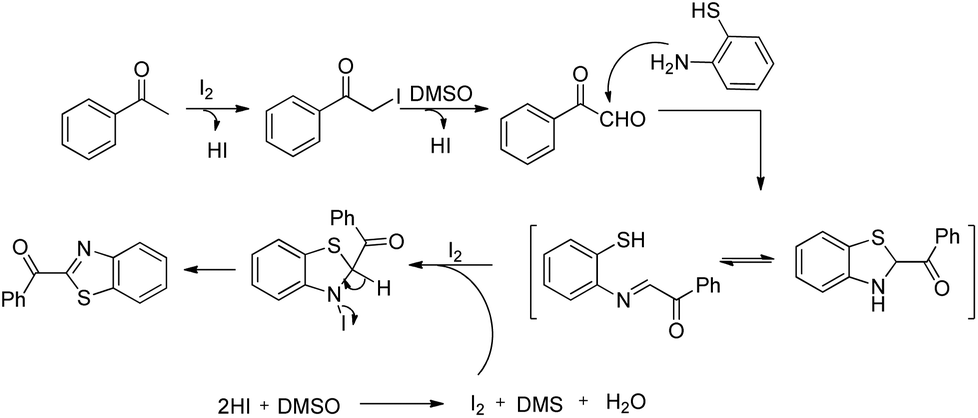
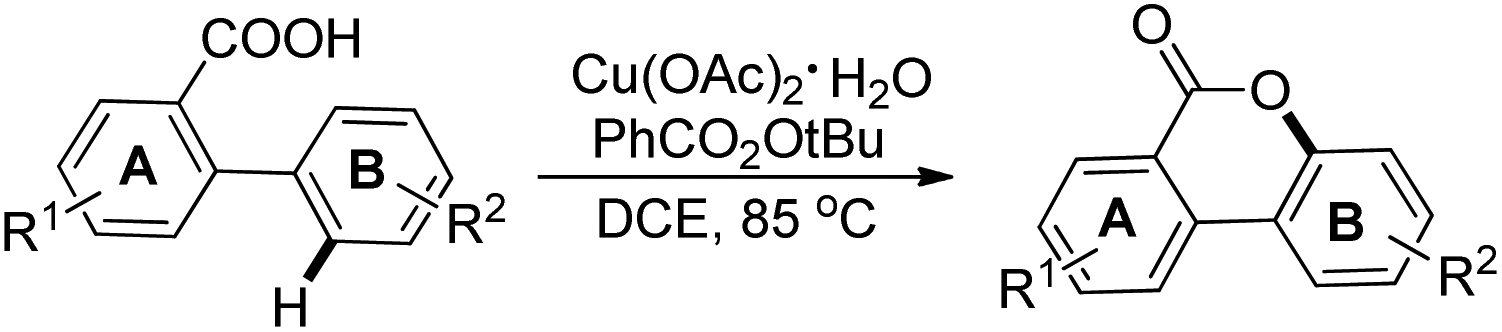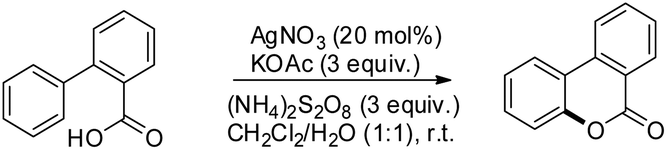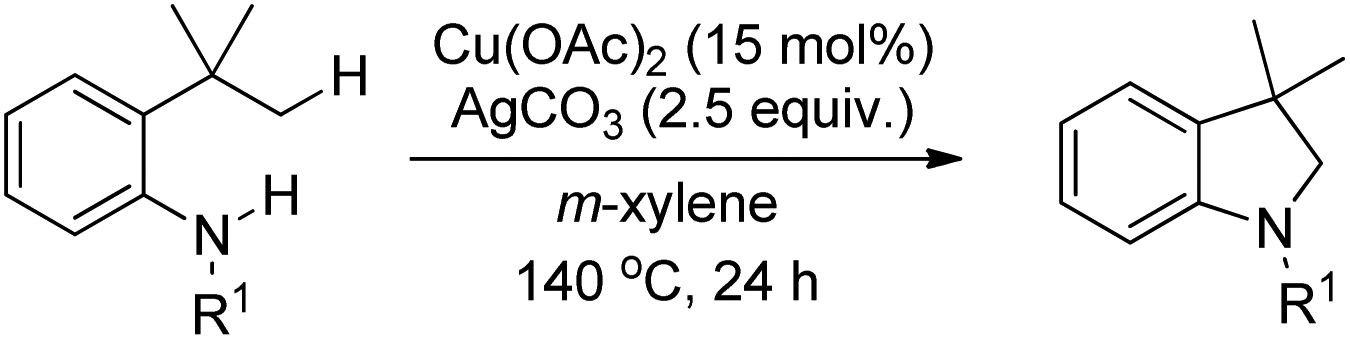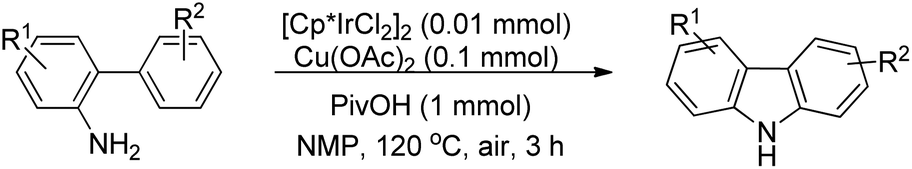DOI:
10.1039/C9QO00863B
(Review Article)
Org. Chem. Front., 2019,
6, 3445-3489
Recent advances in intramolecular C–O/C–N/C–S bond formation via C–H functionalization
Received
7th July 2019
, Accepted 30th August 2019
First published on 10th September 2019
Abstract
This review focuses on the intramolecular cyclization of molecules through C–O, C–N and C–S bonds forming C–H functionalizations with an emphasis on the literature after 2000. Intramolecular C–H functionalization reactions attract much interest because of the ease of reaction due to the proximity of reacting centers and favorable entropy over intermolecular reactions. Less by-product formation, high atom economy, ease of purification, and avoidance of prefunctionalization of substrates are the additional advantages. Hence, a number of research articles have been published in this field, but no review is available so far. This review includes the synthesis of various heterocycles involving C–O, C–N and C–S bond creation. This is the first review on the current topic which will motivate the readers to work in this area further.
 Paran J. Borpatra | Paran J. Borpatra was born in Lakhimpur district of Assam, India. He completed his MSc in chemistry from Cotton College under Gauhati University. He then joined the group of Dr Pranjal K. Baruah for his PhD studies. He is actively pursuing his research in the area of C–H functionalization and development of novel methodologies. |
 Bhaskar Deka | Bhaskar Deka was born in Mangaldoi, Assam, India. He received his MS degree from Gauhati University. He is currently pursuing his PhD under Dr Pranjal K. Baruah. His research interests include the synthesis of indole based compounds and C–H activation. |
 Mohit L. Deb | Dr Mohit L. Deb was born in Tripura, India. He obtained his PhD degree (2008) under the supervision of Dr P. J. Bhuyan at RRL-Jorhat, Assam. He then joined the group of Dr K. R. Prasad (2009) at the Indian Institute of Science as a research associate and worked on asymmetric catalysis. He worked in Jubilant Chemsys Ltd, India as a research scientist. As a FCT postdoctoral fellow at UNL-Lisbon he worked on C–H activation reactions with Professor Christopher D. Maycock. In 2014, he joined Gauhati University as a guest faculty and was awarded the DST-Young Scientist position. His current research area is C–H functionalization and he has >40 publications in reputed international journals. |
 Pranjal K. Baruah | Dr Pranjal K. Baruah did his PhD under Professor G. J. Sanjayan at NCL, Pune, working in the area of molecular recognition. He did his first postdoctoral studies with Professor Harold Kohn (UNC at Chapel Hill, USA) working in medicinal chemistry. He then worked with Professor Martin D. Smith at the University of Oxford, UK as a Marie Curie fellow. There he worked towards the design and synthesis of foldamers and the application of these molecules in asymmetric catalysis. Currently he is an Associate Professor at Gauhati University. His research interests include development of novel methodologies for heterocyclic molecules, catalysis, peptidomimetics and foldamers. |
1. Introduction
Intramolecular carbon–heteroatom (C–X, where X = O, N, and S) bond formation gives heterocycles which are very important in organic chemistry due to their biological activity (Fig. 1).1 They are also used as important synthetic intermediates and in materials science.2 Taxol having an oxetane ring is an important drug used for the treatment of cancer.3a Several other anticancer drugs are reported which have an oxygen-based heterocycle in the core structure.3b Furan based organic compounds are used for the construction of polymeric materials having wide applications in electronics.3c–g Flavonoids are an important class of molecules obtained from plants having a benzopyran ring.3h Heterocyclic compounds having nitrogen or sulphur atoms are of tremendous importance. Pyrimidines, indoles, etc. constitute an important class of natural and non-natural products; many of them possess various useful biological activities and have found medicinal applications.4 In many biological macromolecules such as transfer ribonucleic acids (tRNAs) and enzymes, C–S bond containing compounds are indispensable which control their biological activity. Benzothiophenes are one of the most prominent pharmacophores of many biologically active molecules.5 Inter/intra-molecular C–X bond formation is an efficient technique for the synthesis of these heterocycles. There are several conventional methods (such as Paal–Knorr reaction, Feist–Benary reaction, Diels–Alder reaction, Hofmann–Loffler–Freytag reaction, sulpha-Michael addition reaction, etc.) available for the formation of these compounds. However, intramolecular C–X bond formation through C–H functionalization is an attractive route due to its various advantages. For example, (a) C–H functionalization has become one of the fundamental methods in organic synthesis and has a huge impact on medicinal chemistry, synthetic organic chemistry, and materials science.6 The strategy avoids the prefunctionalization of starting materials which makes the synthetic routes straightforward and more efficient. It is a promising approach towards the minimization of by-product formation and the reduction of the number of steps of organic synthesis.7 (b) Moreover, when the strategy is applied intramolecularly, less reaction time is required because of the ease of reaction due to the proximity of reacting centers and favorable entropy over intermolecular reactions. (c) Intramolecular reactions are usually more atom economical. (d) Purification becomes easier due to the clean reaction. (e) It can be performed under mild conditions. Therefore, the strategy is an efficient and green technique for the synthesis of heterocycles. The salient features of this review are: (i) synthesis of oxygen, nitrogen and sulfur heterocycles through intramolecular C–H functionalization, (ii) discussion of the substrate scope of the reactions and their mechanisms, and (iii) description of up-to-date reactions with no examples taken from the literature earlier than the year 2000. The article is divided into three sections, viz. intramolecular C–O, C–N and C–S bond formation.
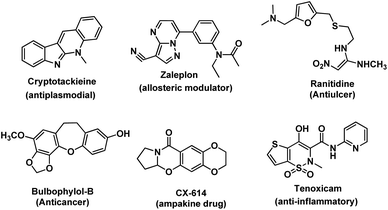 |
| | Fig. 1 Bioactive natural and synthetic heterocycles. | |
In contrast, much research on intermolecular C–X bond formation via C–H functionalization is also conducted across the world and review articles are also available for that.8 However, no review is so far available exclusively on intramolecular C–X bond formation via C–H functionalization although many research articles are published in this area and therefore, we here emphasized only on intramolecular reactions.
2. Intramolecular C–O bond formation
C–O bond formation has much importance in medicinal and industrial fields. Due to the increase of sustainable and environmental considerations, the formation of C–O bonds through the activation of C–H bonds has developed rapidly.9 However, this research area is less explored than C–N bond formation as oxygen has lower nucleophilicity than nitrogen. In this section, we discuss the formation of the C–O bond generated only through intramolecular C–H functionalization. We divide this section of the article according to the nature of the product.
2.1. Synthesis of benzofuran/pyran derivatives
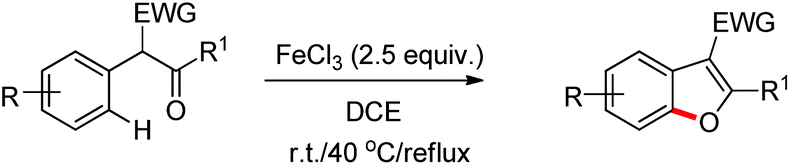
Benzofuran rings are very common structural units in various natural products and pharmaceuticals, including those used to treat traumatic central nervous system (CNS) injury, arteriosclerosis, and hepatopathy.10 Hence, effective methods for the construction of benzofuran derivatives have been actively pursued during the past two decades.11 The synthesis of functionalized benzo[b]furans by FeCl3 mediated intramolecular oxidative C–O bond formation of α-aryl ketones was reported by Zhao and co-workers (Scheme 1).12
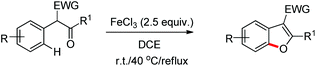 |
| | Scheme 1 Synthesis of functionalized benzofurans. | |
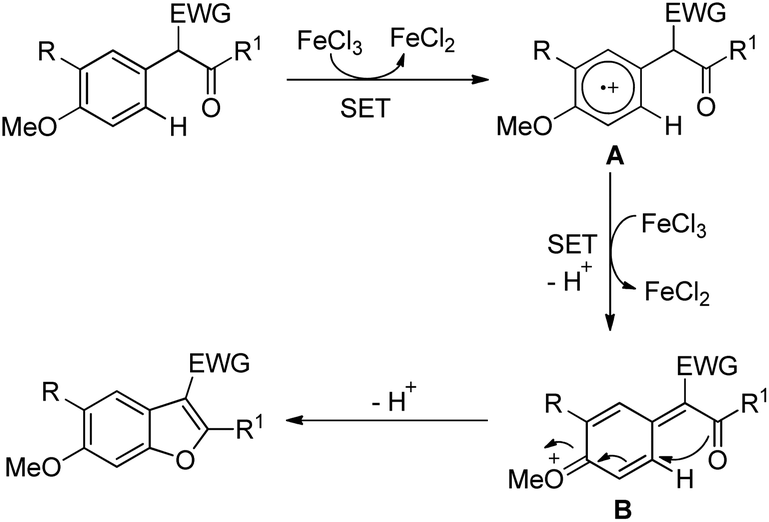
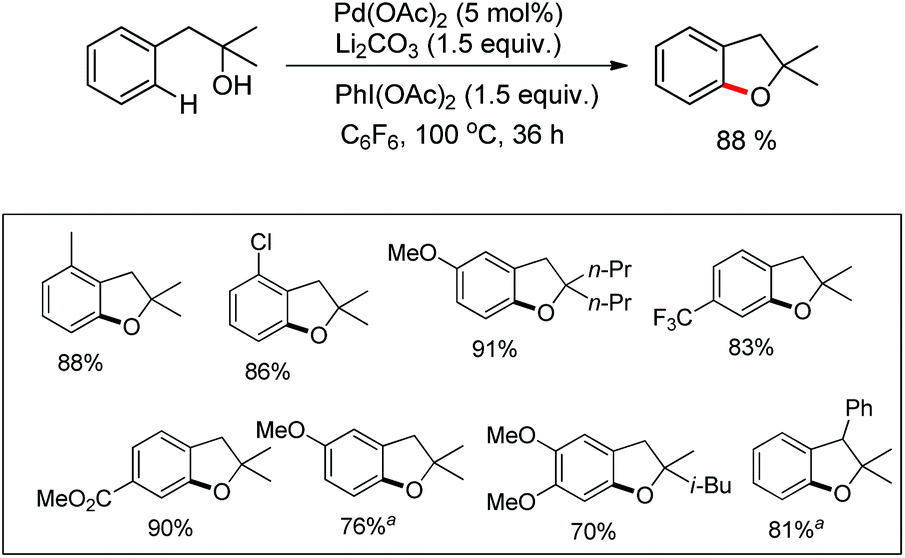
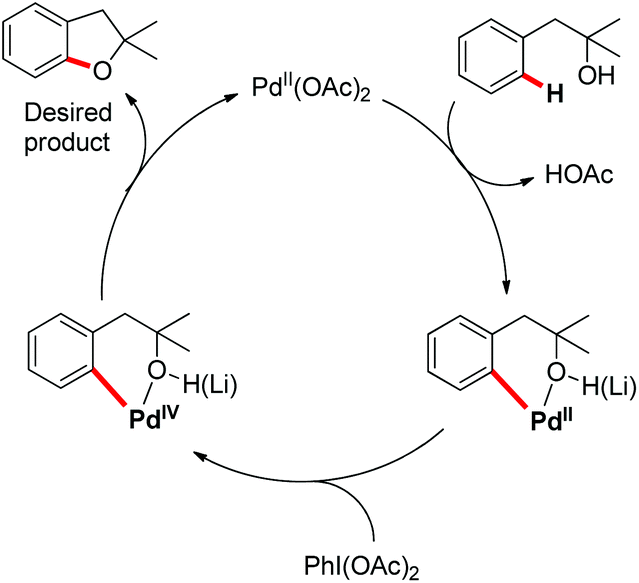
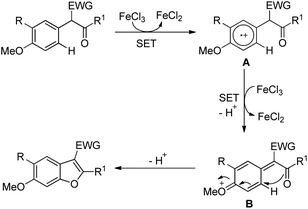
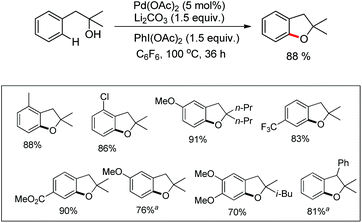
The presence of an alkoxy group in the benzene ring and electron withdrawing group (EWG) α to the keto group is essential for a successful reaction. EWGs such as –CN, ethoxycarbonyl and alkyl acyl groups gave the products in a reasonable yield. The reaction proceeds through one electron abstraction by a Fe(III) catalyst from the electron rich substrate generating the radical cation A (Scheme 2). Furthermore, one more SET process generates the oxonium ion B which subsequently gives the benzofuran product after conjugated cycloaddition and rearomatization. In 2010, Yu et al. developed a hydroxyl group directed intramolecular C–O cyclization catalyzed by Pd(II) to construct dihydrobenzofurans (Scheme 3).13 To optimize the reaction, several oxidants were screened, and PhI(OAc)2 was found to be an effective oxidant. Other higher valent iodonium salts, such as PhI(TFA)2 and PhI(OPiv)2, gave lower yields (13% and 50% respectively). Many bases were also checked and it was found that Li2CO3 was the most effective. In the absence of a base the reaction gave only 45% yield, accompanied by considerable decomposition of the starting material.
 |
| | Scheme 2 Mechanistic pathway for the benzofuran synthesis. | |
 |
| | Scheme 3 Pd(II) catalyzed C–O cyclization. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Na2HPO4 was used instead of Li2CO3. Na2HPO4 was used instead of Li2CO3. | |
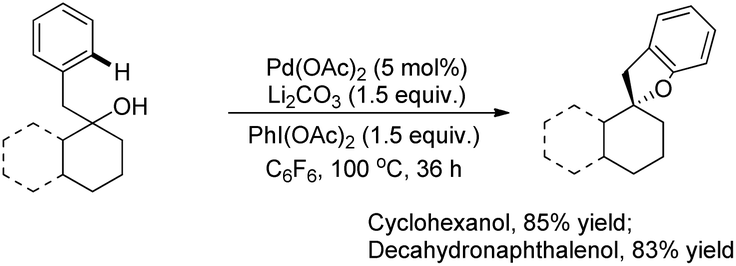
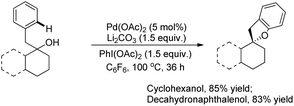
A variety of tertiary alcohols were screened and dihydrobenzofurans were obtained in good to excellent yields. The presence of –Cl and –Br in the products was very useful for further synthetic elaborations. When the α-ester group was present, the yield decreased to 50%. Cyclization of secondary alcohols gave a much lower yield of the desired product because of competitive oxidation of the alcohol to the ketone. This strategy was also applied to synthesize spirocyclic dihydrobenzofurans as these cores are present in many natural products and drug molecules. In both the examples the corresponding spirocyclic products were obtained in good yields (Scheme 4).
 |
| | Scheme 4 Construction of a spirocyclic core via Pd(II)-catalyzed C–O cyclization. | |
A probable mechanism was proposed which involved Pd(II)-catalyzed C–H cleavage followed by oxidation of Pd(II) to a higher oxidation state from which C–O reductive elimination took place (Scheme 5). It was observed that the reaction occurred to some extent in the absence of the base. Since the formation of the [Pd(II)-OR] species as the C–H activation precursor was unfavorable under neutral conditions, it was suggested that the hydroxyl moiety coordinates with Pd(II) as a neutral σ donor.
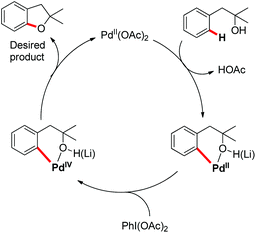 |
| | Scheme 5 Tentative mechanism for the benzofuran synthesis. | |
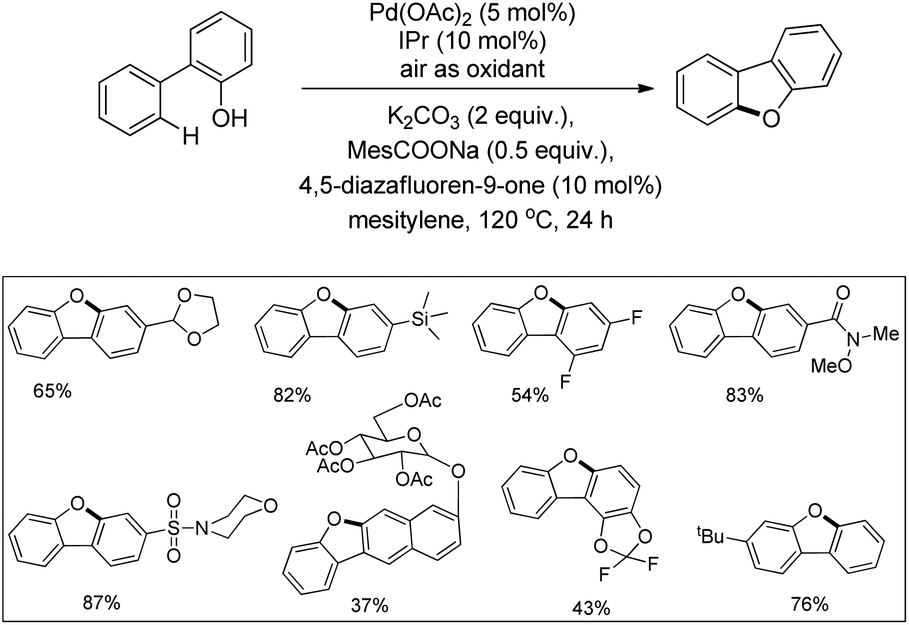
In the next year, Liu and co-workers reported a practical phenol-directed C–H activation/C–O cyclization reaction as an efficient route to dibenzofurans (Scheme 6).14 They tested a variety of weak oxidants for the reaction because a strong oxidant (e.g., PhI(OAc)2) was found to decompose the phenol. However, none of these oxidants were productive and it was observed that air acts an effective oxidant in the transformation. It was found that the addition of sodium pivalate (PivONa) significantly improved the reaction. Then a more bulky RCOONa additive was tested and it was found that sodium 2,4,6-trimethylbenzoate (MesCOONa) provided a higher yield. After examining several bases, K2CO3 was noted as the best for the reaction.
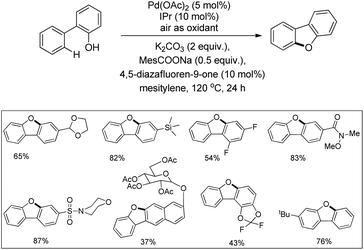 |
| | Scheme 6 Phenol-directed cyclization through a Pd(II)/Pd(0) cycle. | |
They came to the conclusion through a few experiments and believed that the phenol group coordinated with Pd(II) as a neutral σ donor. From the isotope labeling experiment they confirmed that C–O reductive elimination was the turnover-limiting step of the catalytic cycle. Based on the reaction, a synthesis of dibenzofurans directly from phenols was developed in which 54% of the desired product was obtained (Scheme 7).
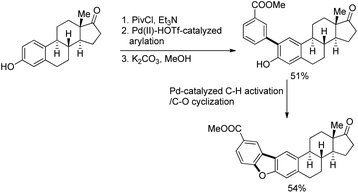 |
| | Scheme 7 Synthesis of dibenzofurans directly from phenols. | |
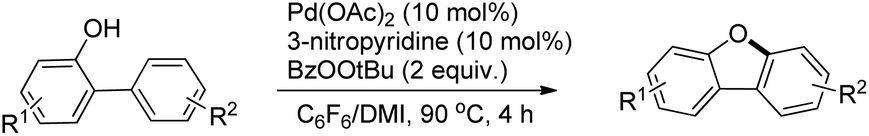
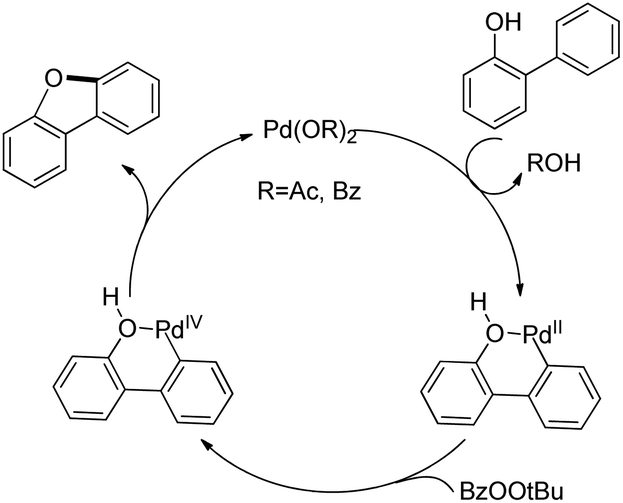
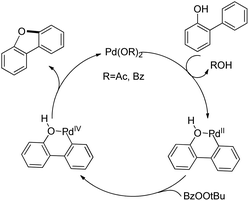
The synthesis of dibenzofurans from 2-arylphenols was reported by Yoshikai et al. using Pd(OAc)2 as a catalyst, 3-nitropyridine as a ligand and tert-butyl peroxybenzoate as an oxidant (Scheme 8).15
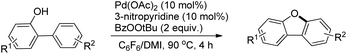 |
| | Scheme 8 Synthesis of dibenzofurans from 2-arylphenols. | |
The kinetic isotope experiments suggested that the C–H bond cleavage was the rate limiting step for the reaction which was in contrast to Liu's work where the reductive C–O bond formation was the rate determining step. The mechanism involves a C–H bond cleavage by a Pd(II) catalyst (Scheme 9). The intermediate then undergoes oxidation to the Pd(IV) state and subsequent reductive C–O bond formation gives the dibenzofuran product.
 |
| | Scheme 9 Tentative mechanism for the dibenzofuran synthesis. | |
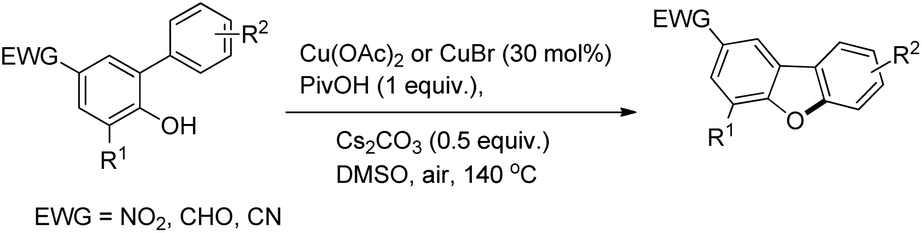
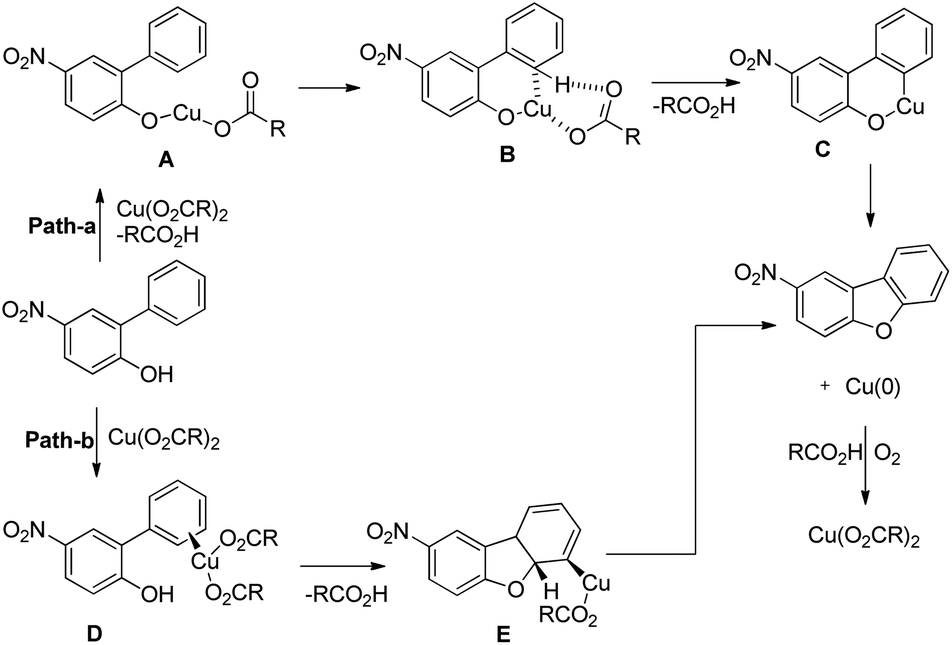
Zhu et al. reported the synthesis of dibenzofurans from substituted o-aryl phenols using copper catalyzed C–H activation and air as an oxidant (Scheme 10).16
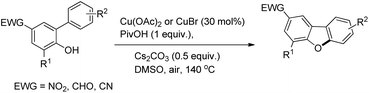 |
| | Scheme 10 Synthesis of dibenzofurans by using a Cu catalyst. | |
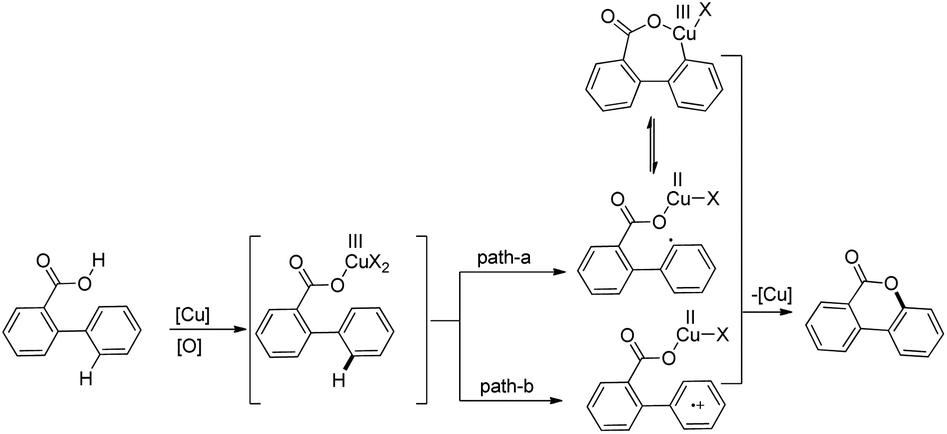
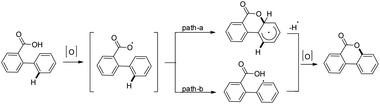
The products were obtained in 44–72% yield. The presence of a strong electron withdrawing group at the para position of phenols was essential for the reaction to be fruitful. In the mechanism, path-a involves coordination of a copper catalyst to the phenolic –OH group to generate intermediate A which undergoes concerted metalation–deprotonation (CMD) to form intermediate B (Scheme 11). Reductive elimination of B gives the dibenzofuran product. The reaction through path-b proceeds through a π-complex. The cyclic product E is obtained by a nucleophilic attack of the –OH group on the activated π-complex. The product dibenzofuran is then obtained by cis hydride elimination.
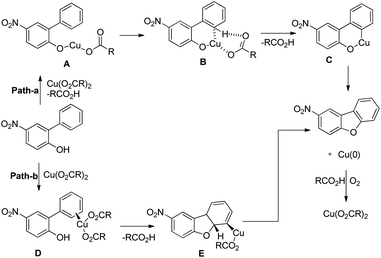 |
| | Scheme 11 Tentative two pathway mechanism for the reaction. | |
2.2. Synthesis of lactone derivatives
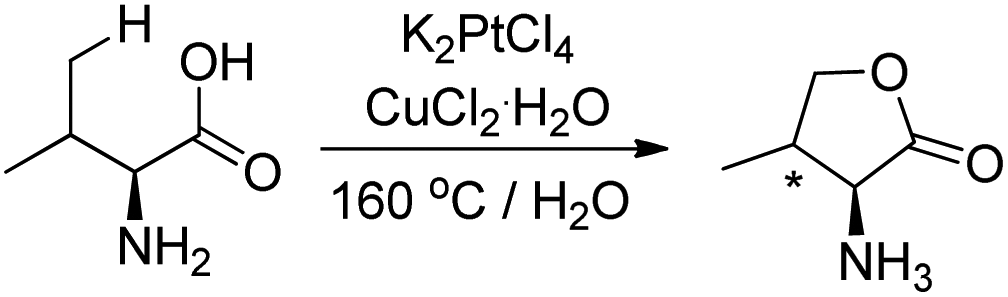
Sames et al. in 2001 reported that when the amino acid L-valine was treated with an aqueous solution of K2PtCl4 and CuCl2 as an oxidant, 67% of the lactone was obtained (Scheme 12).17 The syn/anti ratio for the product is 3:1 suggesting regio- and stereoselectivities of the C–H functionalization reaction.
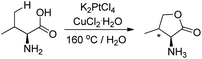 |
| | Scheme 12 Synthesis of lactones by C–H functionalization of an amino acid. | |
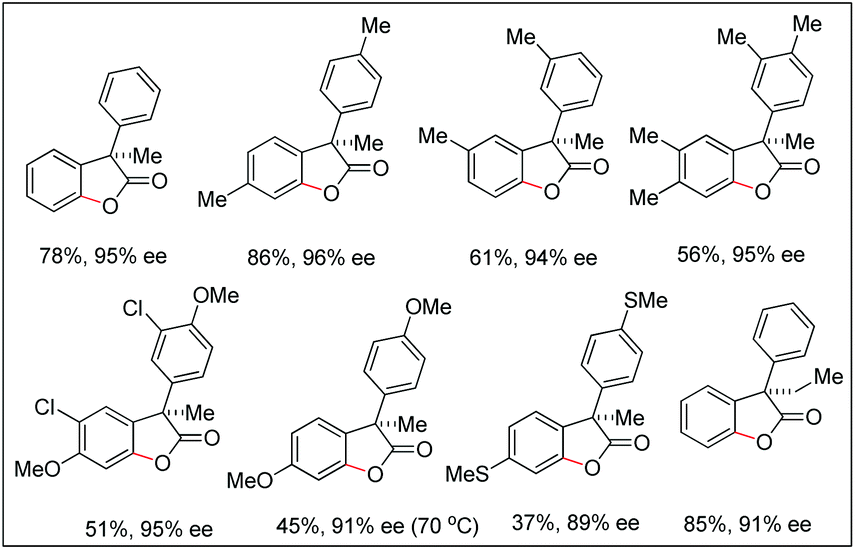
The synthesis of benzofuranones from phenylacetic acids using palladium catalyzed C–H activation followed by intramolecular C–O bond formation was reported by Wang and co-workers (Scheme 13).18 An electron donating group (EDG) and an electron withdrawing group (EWG) on the phenyl ring of the substrate increased and decreased the yield, respectively. Of the various N-protected amino acids used as ligands, Boc-Val-OH gave the best result.
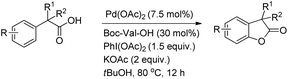 |
| | Scheme 13 Synthesis of benzofuranones. | |
They further extended this strategy to synthesize chiral benzofuranones (89–96% ee) in moderate to good yields starting with diphenylacetic acids (Scheme 14).
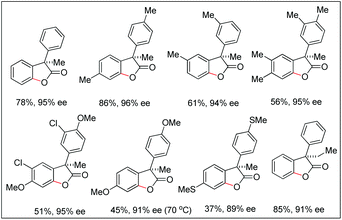 |
| | Scheme 14 Synthesis of chiral benzofuranones. | |
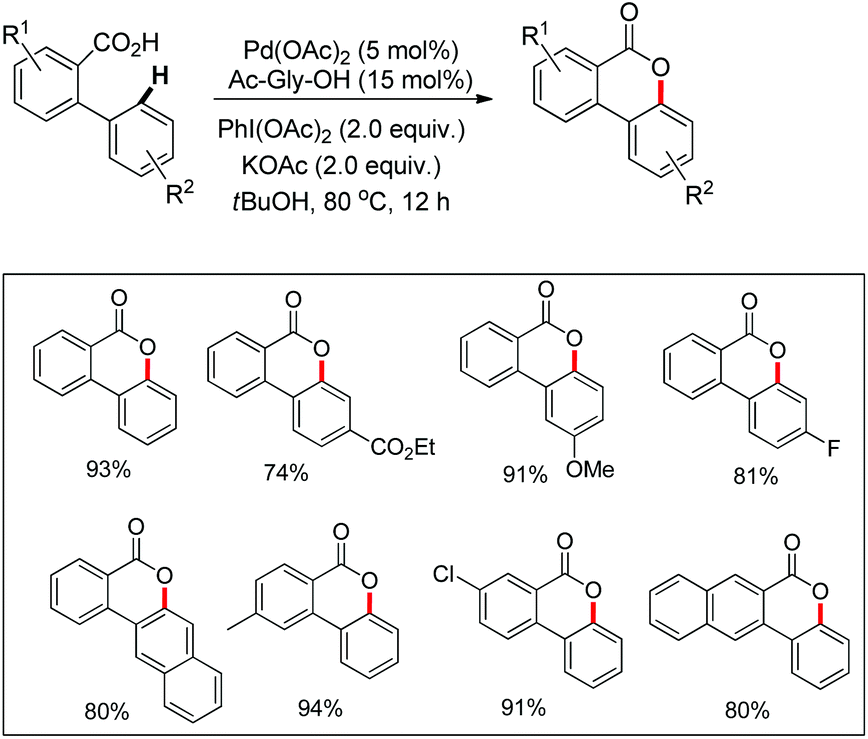
In the same year, Wang et al. reported another palladium catalyzed lactonization via carboxyl-directed C–H activation/C–O cyclization to synthesize biaryl lactones (Scheme 15).19 Biaryl lactones and many of their derivatives are the core structural units found in many natural products and pharmaceuticals and also widely employed as intermediates in the total synthesis of natural products.20
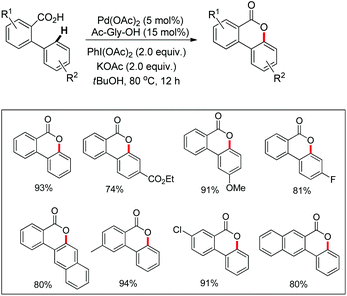 |
| | Scheme 15 Synthesis of biaryl lactones. | |
The synthetic utility of this reaction was established by carrying out a concise total synthesis of the natural product cannabinol from commercially available starting materials (Scheme 16). A plausible mechanism was also suggested for the reaction in which the Pd(II)/Pd(IV) catalytic cycle is involved.
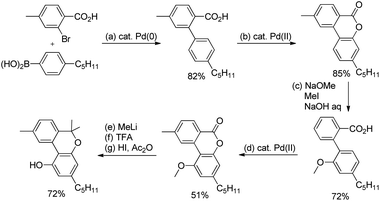 |
| | Scheme 16 Total synthesis of cannabinol. | |
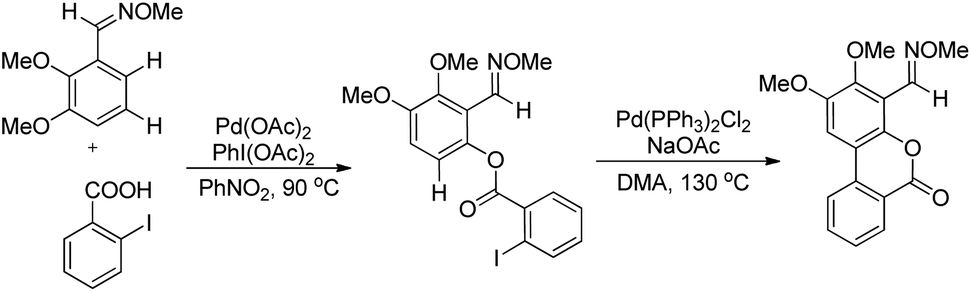
Shi et al. developed Pd-catalyzed double activations of adjacent (sp2)-C–H bonds for the synthesis of 6H-benzo[c]chromen-6-one derivatives.21O-Methyl aryloximes when reacted with iodo benzoic acid undergo ortho benzoyloxylation in the presence of the Pd(OAc)2 catalyst. This product then undergoes a second C–H activation in the presence of the Pd(PPh3)2Cl2 catalyst leading to the formation of the 6H-benzo[c]chromen-6-one product (Scheme 17).
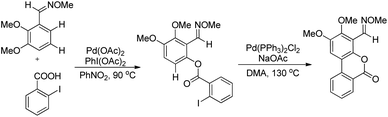 |
| | Scheme 17 Synthesis of 6H-benzo[c]chromen-6-one derivatives. | |
3,4-Benzocoumarins were synthesized by a copper catalyzed intramolecular lactonization reaction of 2-arylbenzoic acids using tert-butyl peroxybenzoate (TBPB) as an oxidant (Scheme 18).22 It was noted that substitution at the meta-position of the B ring (with –F, –Cl, –OMe, and –tBu functional groups) led to cyclization at the less hindered site. However, m-Me substitution of the B ring gave the cyclized product at the more hindered site. EWGs in the B ring lowered the yield and in a few cases the presence of –CHO and –NO2 groups in the substrate inhibited the formation of products.
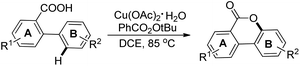 |
| | Scheme 18 Synthesis of benzocoumarins. | |
The reaction is believed to proceed through the initial formation of an active CuIII intermediate which can undergo further reaction either through path-a (oxidative ring closure) or path-b (SET process) giving the cyclized product (Scheme 19).
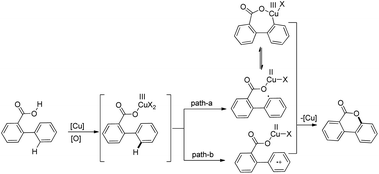 |
| | Scheme 19 Mechanism for the synthesis of benzocoumarins. | |
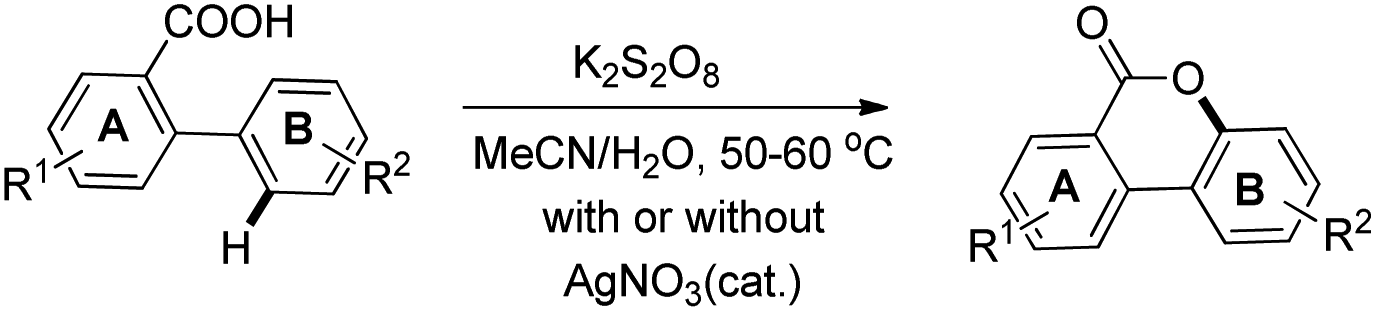
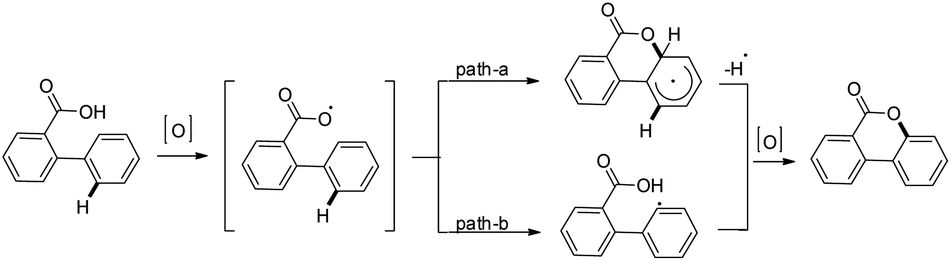
In order to overcome the difficulty of the yield not improving in electron deficient 2-arylbenzoic acids, another method was developed using a K2S2O8-mediated oxygenation reaction in aqueous CH3CN with/without using the AgNO3 catalyst (Scheme 20). Using this method, good yields of cyclized products containing a wide range of EWG and EDG groups in the A or B ring of 2-phenylbenzoic acid were obtained. The product could be synthesized in the gram scale using this method.
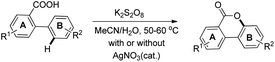 |
| | Scheme 20 Persulfate method of synthesizing benzocoumarins. | |
The K2S2O8 mediated method is likely to proceed via a free radical on the oxygen atom of the carboxyl group which subsequently gives the cyclized product (Scheme 21).
 |
| | Scheme 21 Mechanism for the synthesis of benzocoumarins by the persulfate method. | |
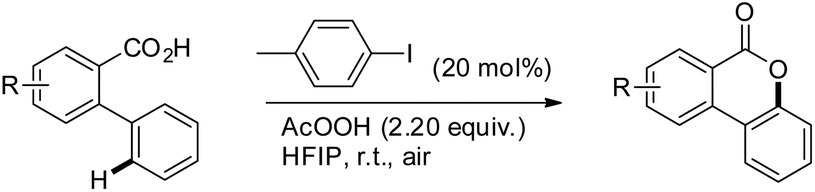
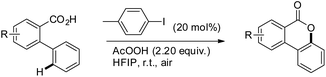
Martin et al. reported a metal-free approach towards the synthesis of benzolactones from 2-arylbenzoic acids using aryl iodides in a catalytic amount in the presence of AcOOH as an oxidant and 1,1,1,3,3,3-hexafluoro-2-propanol as a solvent (Scheme 22).23 This method has the advantage over the Cu catalyzed method reported by Gevorgyan et al.22 since only one regioisomer is obtained in many cases and the reaction is not moisture sensitive. For meta-substituted 2-phenylbenzoic acids the regioselectivity of C–O bond formation could be switched by using a suitable aryl iodide catalyst. The selectivity is dependent on both the aryl iodide used and the concentration of AcOOH. The reaction is catalyzed by in situ generated ArI(OAc)2. The AcOOH used in the reaction has a dual function as an oxidant and an additive.
 |
| | Scheme 22 Synthesis of benzolactones. | |
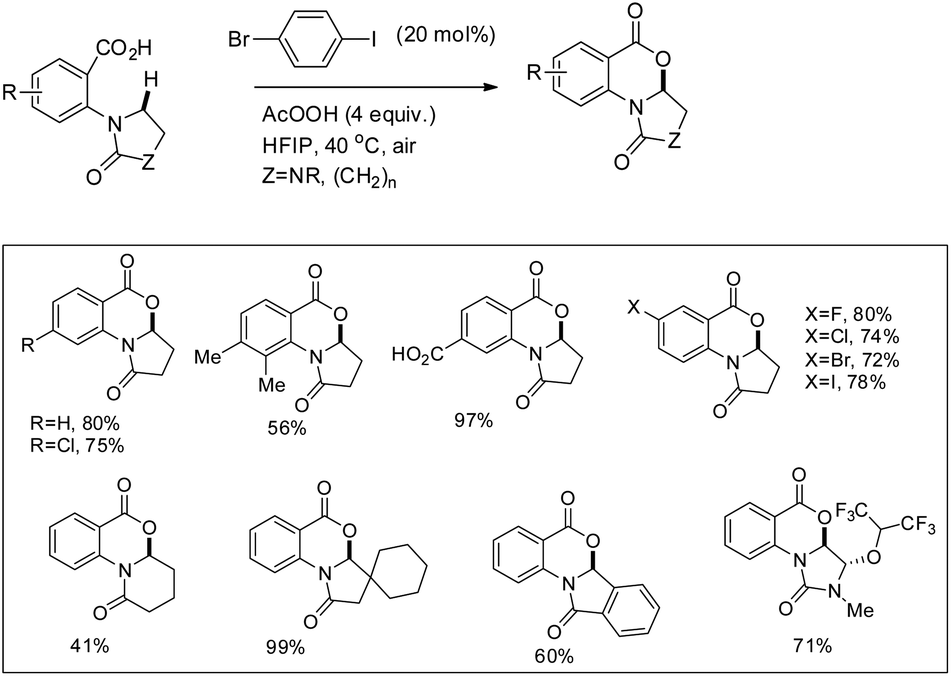
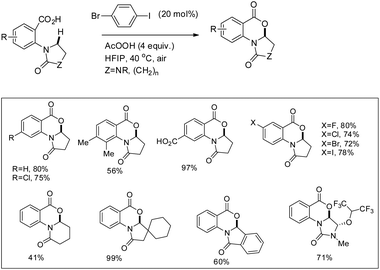
They further achieved C(sp3)–H functionalization α to the tertiary nitrogen atom directed by a carboxylic acid group using another aryl iodide catalyst (Scheme 23). Substrates with various substituents including carboxylic acids and halides gave a good yield.
 |
| | Scheme 23 Lactonisation α to the tertiary nitrogen atom. | |
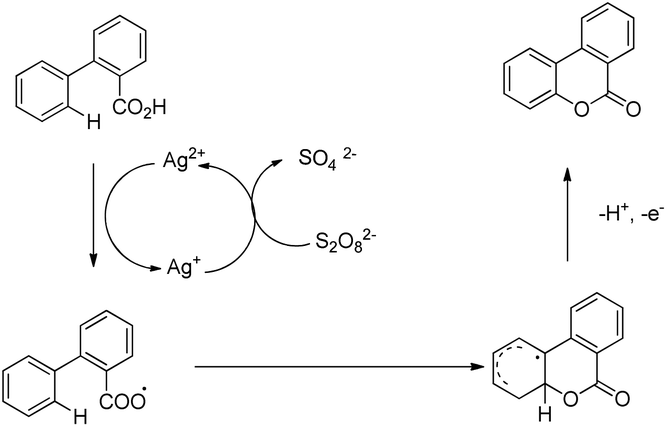
Xu and co-workers reported AgNO3 catalyzed C–O bond formation of 2-aryl carboxylic acids using (NH4)2S2O8 as an oxidant and KOAc as an additive in the CH2Cl2/H2O solvent at room temperature (Scheme 24).24 This study not only provides a convenient, easy-to-handle protocol to the lactone scaffolds but also further authenticates the value of radical-based C–H functionalization for synthetic applications. The reaction has a broad substrate scope and various synthetically useful yet sensitive functional groups are well tolerated. Ag(I) first gets oxidized to Ag(II) which reacts with 2-aryl carboxylic acids generating a carboxyl radical. This radical undergoes cyclization, proton loss and one electron oxidation to give the product (Scheme 25).
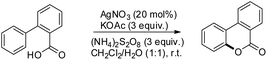 |
| | Scheme 24 Synthesis of benzolactones. | |
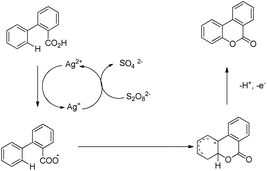 |
| | Scheme 25 Mechanism for the synthesis of benzolactones. | |
A visible light mediated photoredox catalyst [Acr-Mes]ClO4 was used to synthesize benzocoumarins from 2-arylbenzoic acids in the presence of air as a terminal oxidant (Scheme 26).25 For meta-substituted benzoic acids good regioselectivity was observed ranging from 90:10 to 96:4.
 |
| | Scheme 26 Visible light mediated benzocoumarin synthesis. | |
The reaction proceeds through a free radical mechanism. The benzoyloxy radical is generated by the activated photocatalyst through the SET process. This radical undergoes cyclization to form another radical intermediate which after hydrogen atom abstraction (HAT) or SET/deprotonation gives the final product (Scheme 27).
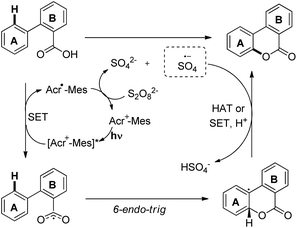 |
| | Scheme 27 Mechanism for the photocatalytic benzocoumarin synthesis. | |
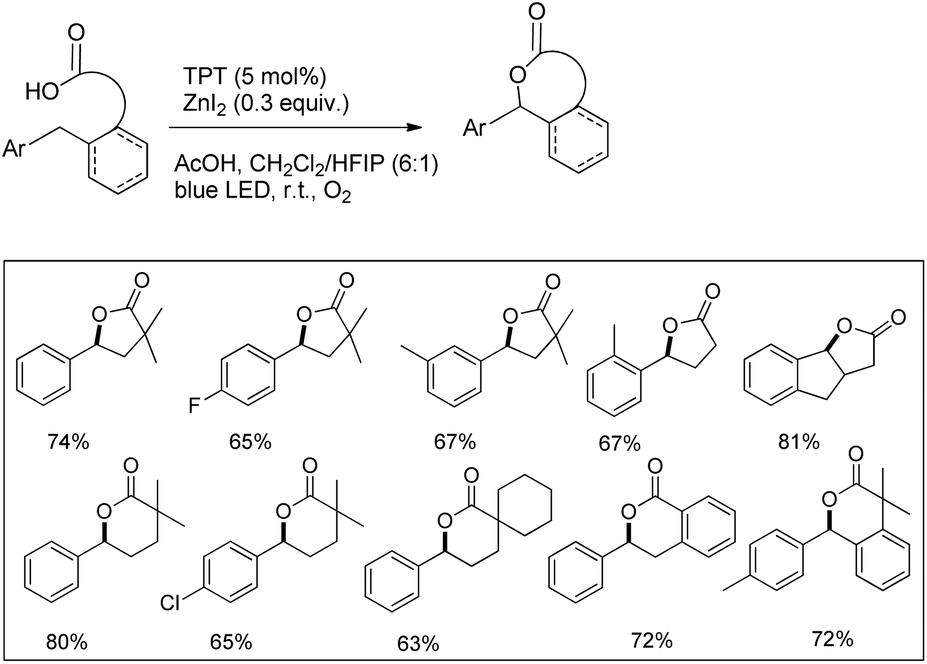
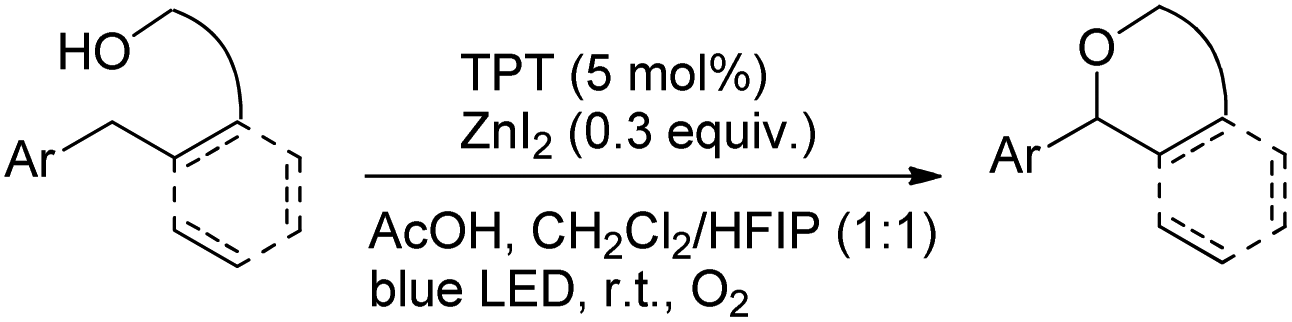
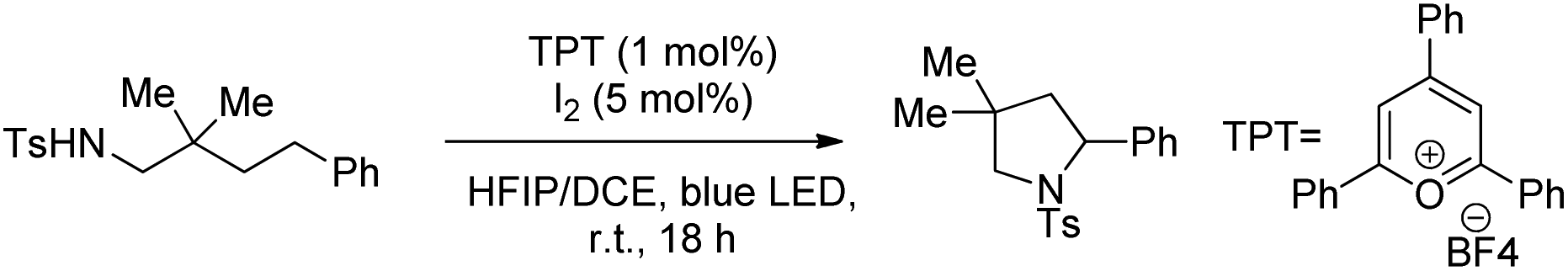
Another visible light catalyzed intramolecular C–O bond formation reaction giving access to γ-/δ-lactones and cyclic ethers was reported by Hong et al. using 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) as a photocatalyst (Schemes 28 and 29).26 The C–O cyclization reaction was promoted by ZnI2 because of the coordination of Zn(II) with the hydroxyl group. The reaction gave the best yield when a mixture of DCM and hexafluoroisopropanol (HFIP) was used as a solvent in the presence of acetic acid.
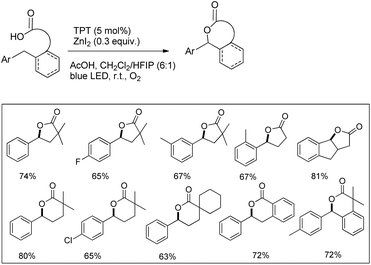 |
| | Scheme 28 Visible light catalyzed synthesis of γ- and δ-lactones. | |
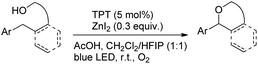 |
| | Scheme 29 Visible light catalyzed synthesis of ethers. | |
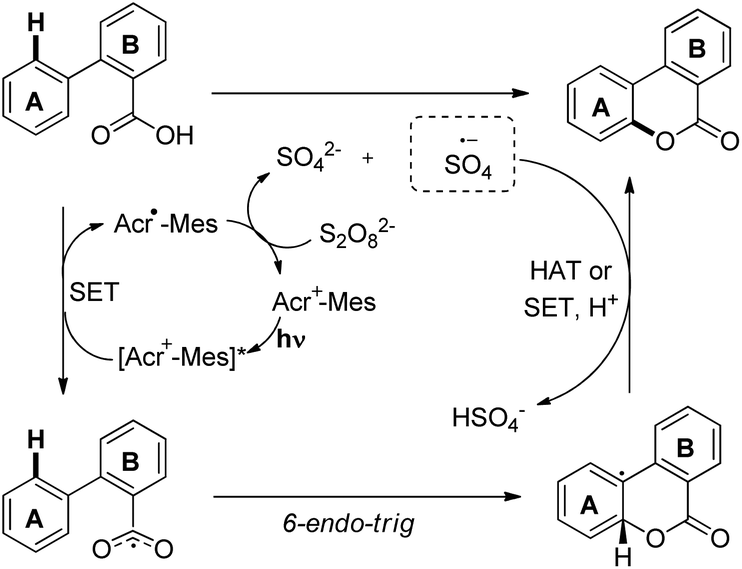
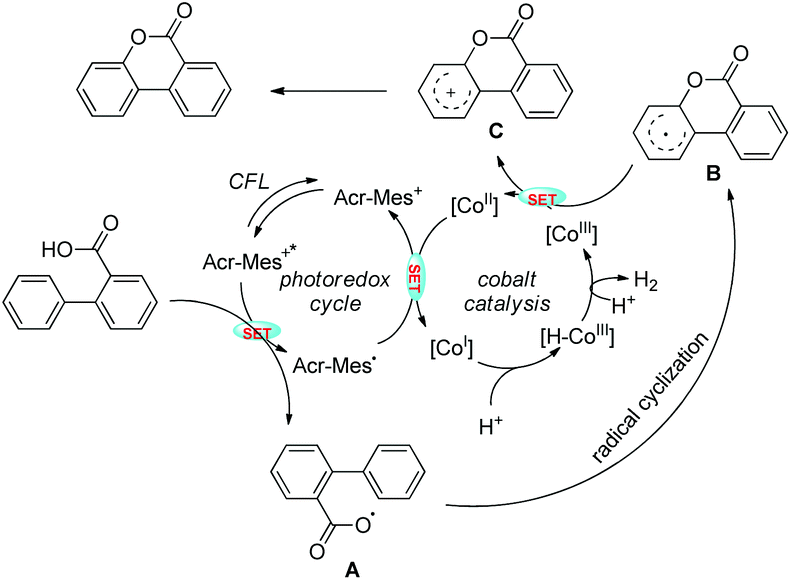
Zhu et al. reported intramolecular C–O bond formation by a C(sp2)–H functionalization reaction for the synthesis of lactones using a combination of photoredox and cobalt catalysts (Scheme 30).27
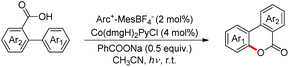 |
| | Scheme 30 Co and photoredox co-catalyzed C–O bond formation. | |
No external oxidant is required for this reaction and the reaction has a broad substrate scope. The photoredox catalyst Arc+-MesBF4− is excited by CFL light and helps in the formation of the aryloxy radical A (Scheme 31). This radical undergoes 6-endo-trig cyclization giving the intermediate B. This radical is then oxidized by CoIII to give the intermediate C which after losing one proton gives the lactone product.
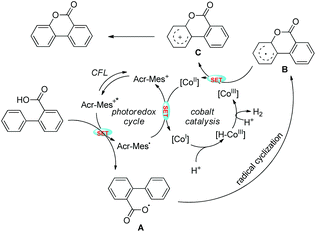 |
| | Scheme 31 Tentative mechanism for the lactone synthesis. | |
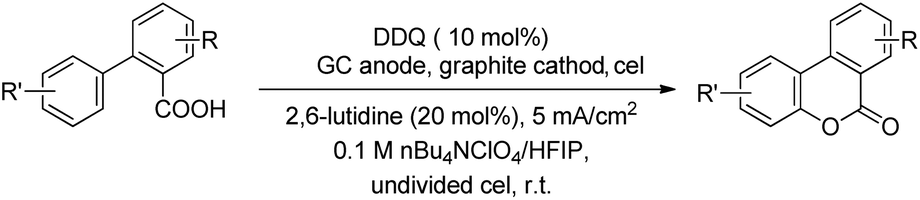
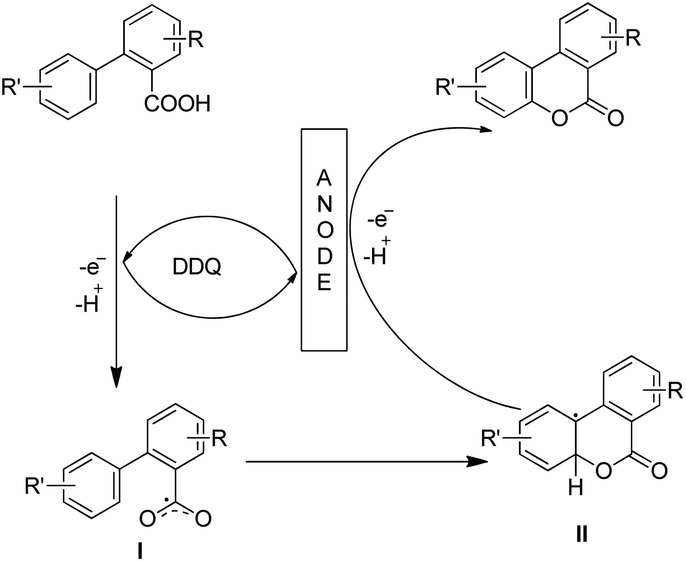
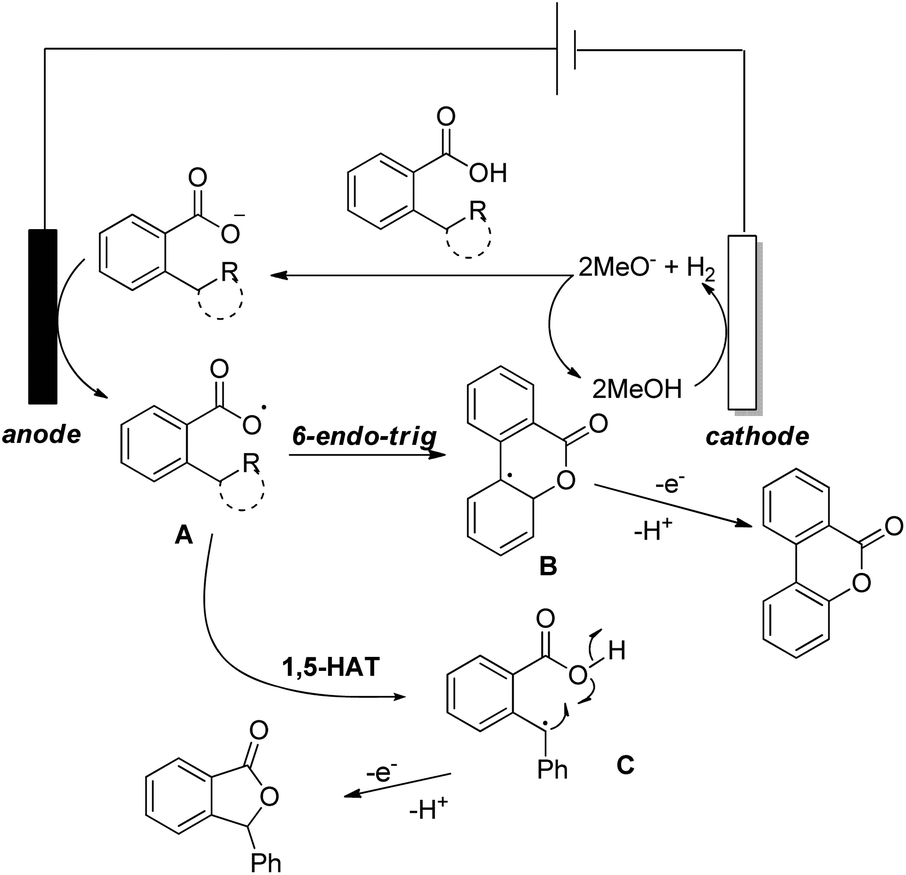
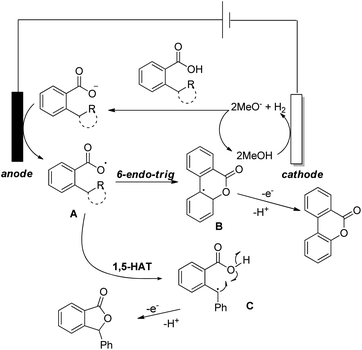
Luo et al. recently used an electrochemical method using a glassy carbon electrode and DDQ as a redox mediator, 2,6-lutidine as an additive and nBu4NClO4/HFIP as an electrolyte solution for the synthesis of 6H-benzo[c]chromen-6-ones by lactonization of biphenyl-2-carboxylic acids (Scheme 32).28
 |
| | Scheme 32 Electrochemical method of lactone formation. | |
The combination of 2,6-lutidine and DDQ is essential for an efficient reaction. The controlled experiments suggest that the reaction proceeds through a radical process. However the kinetic isotope experiments suggest that the C–H bond cleavage is not the rate limiting step. 2,6-Lutidine abstracts a proton from the biphenyl acid and in the presence of DDQ a carboxylic radical I is formed. This radical then undergoes C–O bond formation generating aryl radical intermediate II which finally gives the product (Scheme 33).
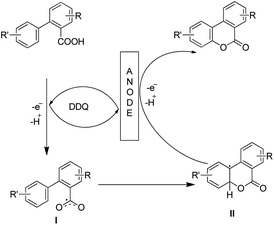 |
| | Scheme 33 Mechanism for electrochemical lactonization. | |
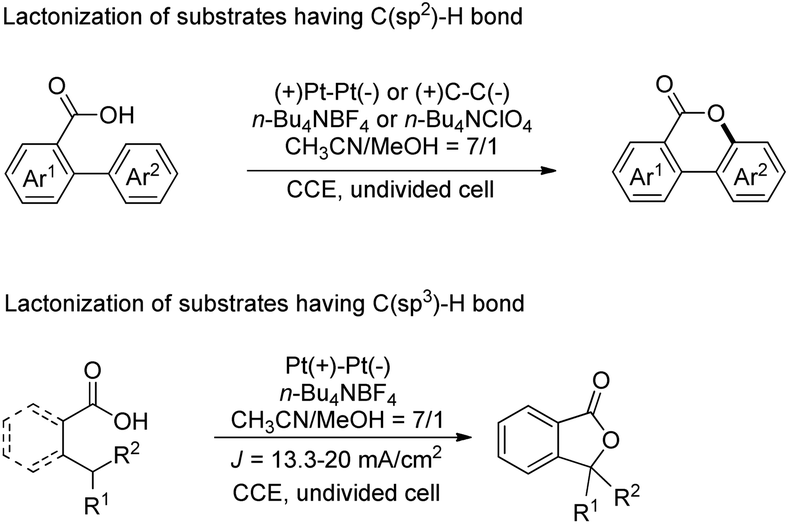
Another electrochemical dehydrogenative method for lactone ring formation was reported by Zhang and co-workers (Scheme 34).29
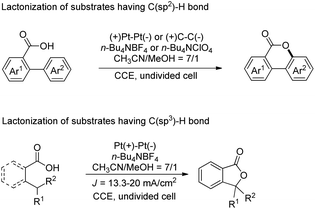 |
| | Scheme 34 Electrochemical dehydrogenative lactonization of substrates having C(sp2)–H and C(sp3)–H bonds. | |
The reaction is regioselective in nature and does not require any external oxidant. It could be applied for diaryl acrylic acids giving coumarin derivatives. The reaction has a broad substrate scope and could be scaled up to the 40 gram scale. The mechanism involves generation of a carboxylate ion which then undergoes electrooxidation generating the carboxylate radical A (Scheme 35). This radical can undergo two different pathways depending on the structure of the substrate. In the case of C(sp2)–H bond lactonization, 6-endo-trig cyclization gives intermediate B whereas for the C(sp3)–H bond substrate the 1,5-HAT process gives intermediate C. Both intermediates B and C after losing one electron and one proton give the respective products.
 |
| | Scheme 35 Tentative mechanism for electrochemical lactonization. | |
2.3. 7-Membered ring synthesis via C–O bond formation
The development of an efficient strategy for 7-membered ring formation is always in demand due to the difficulty in the formation of such ring systems and biological activity of such compounds.30
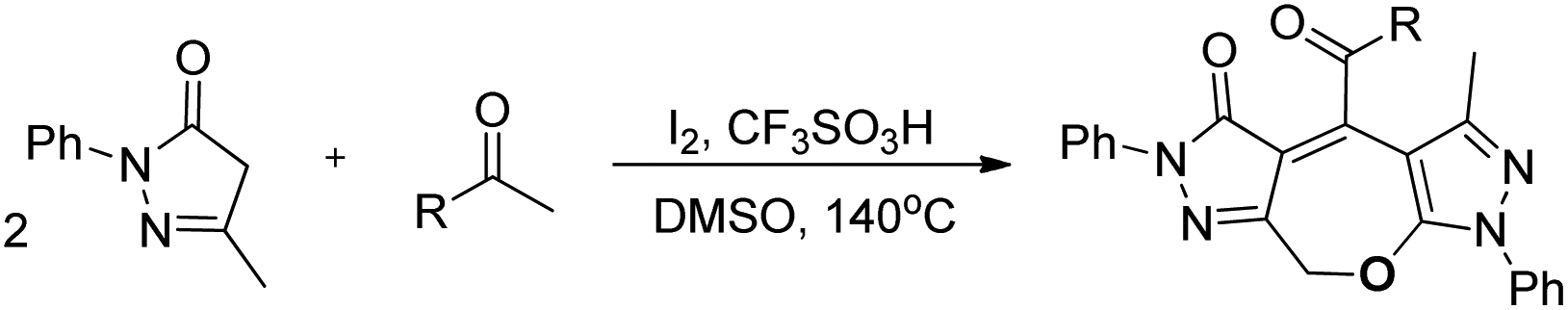
Wu and co-workers disclosed a metal-free cascade approach using acetophenone and 3-methyl-5-pyrazolones for the synthesis of seven-membered 2,3-dihydrooxepines (Scheme 36).31 They used iodine as a catalyst in the presence of 1.5 equivalents of trifluoromethanesulfonic acid and DMSO as a solvent under heating conditions.
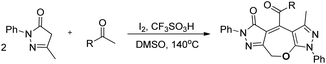 |
| | Scheme 36 I2-Catalysed synthesis of 2,3-dihydrooxepines. | |
2,3-Dihydrooxepines are obtained in moderate to good yields when acetophenone was replaced with different substrates such as phenylacetylene, styrene, 2-bromo-1-phenylethanone, 2-hydroxy-1-phenyl-ethanone, and 1-phenylethanol. The 2,3-dihydrooxepines thus obtained could further lead to more complex heterocycles when reacted with hydrazine hydrate (Scheme 37).
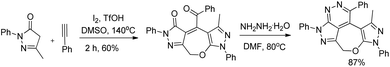 |
| | Scheme 37 Reaction of 2,3-dihydrooxepines with hydrazine hydrate. | |
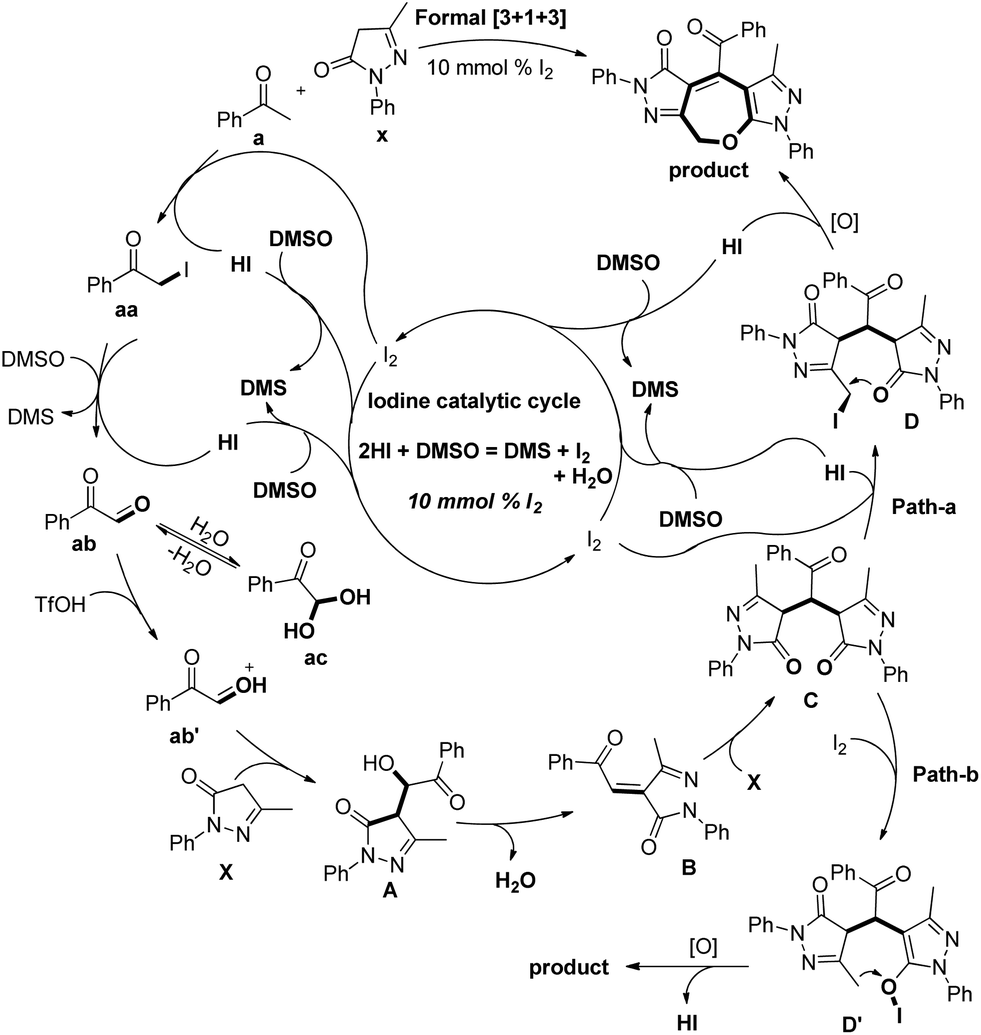
Based on the control experiments the following mechanism was proposed. The iodination of acetophenone and further Kornblum oxidation gave the 2-oxo-2-phenylacetaldehyde ab intermediate. This in the presence of triflic acid reacts with edaravone X to give intermediate A. Elimination of H2O from intermediate A and reaction with another molecule of edaravone X give intermediate C which can undergo iodination through path-a and path-b to furnish intermediate D or D′. The final product could be obtained from them by intramolecular C–O bond formation (Scheme 38).
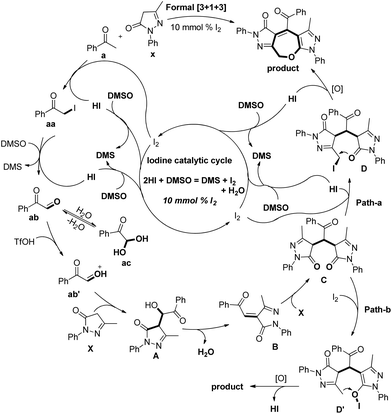 |
| | Scheme 38 Tentative mechanism for the synthesis of 2,3-dihydrooxepines. | |
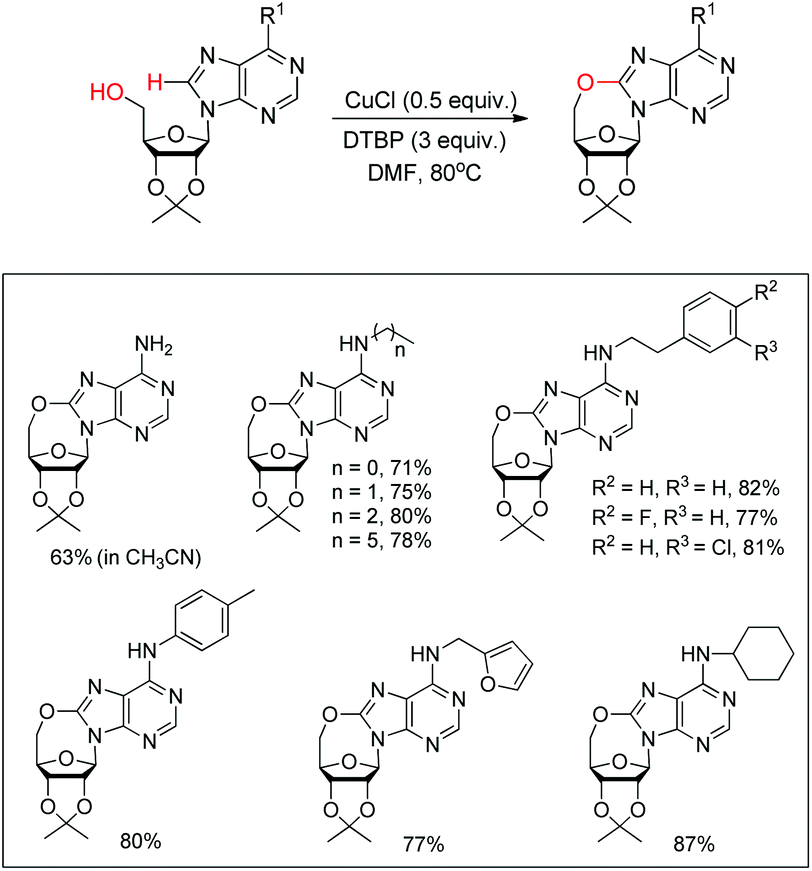
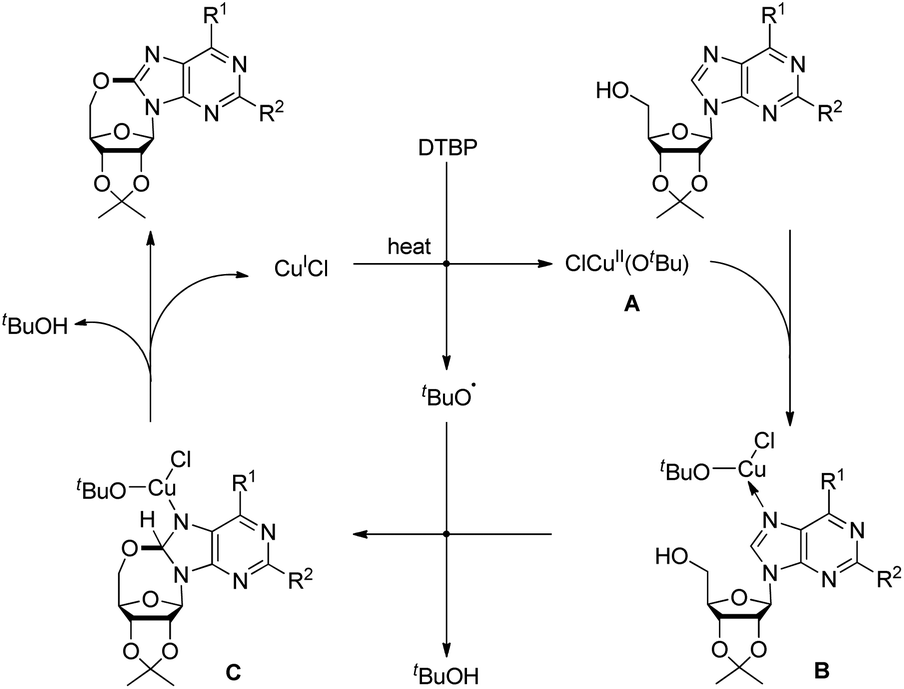
Du et al. developed an intramolecular dehydrogenative alkoxylation of purine nucleosides using CuCl as a catalyst and di-tert-butyl peroxide (DTBP) as an oxidant in DMF as a solvent at 80 °C to form 7-membered ring compounds in good yields (Scheme 39).32 After screening the ratio of DTBP and CuCl, it was found that the best yield was obtained by using 0.5 equiv. of CuCl and 3 equiv. of DTBP. When the substrate has aliphatic amine substitution in the C-6 position the yield was relatively lower than that of cyclic amines. However, when R1 was Ph, Et or H, the reaction was unsuccessful. The reaction proceeds through a free radical mechanism (Scheme 40). Initially the tert-butoxy radical and active Cu(II) catalyst are formed through the reaction of CuCl and DTBP. The Cu(II) catalyst then coordinates with the purine nucleoside which undergoes intramolecular nucleophilic addition of 5′-OH to C-8 eliminating a molecule of tBuOH. The intermediate C undergoes reductive elimination to give the product and regenerates the catalyst CuCl.
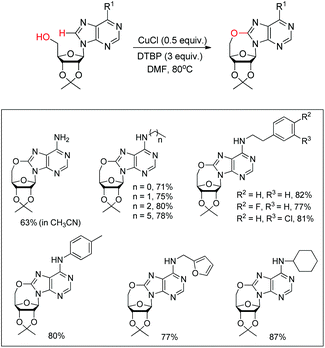 |
| | Scheme 39 Intramolecular alkoxylation of purine nucleosides. | |
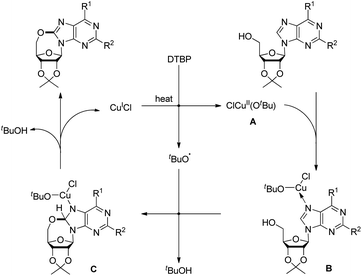 |
| | Scheme 40 Mechanism for the alkoxylation of purine nucleosides. | |
2.4. Synthesis of 1,3-oxazines
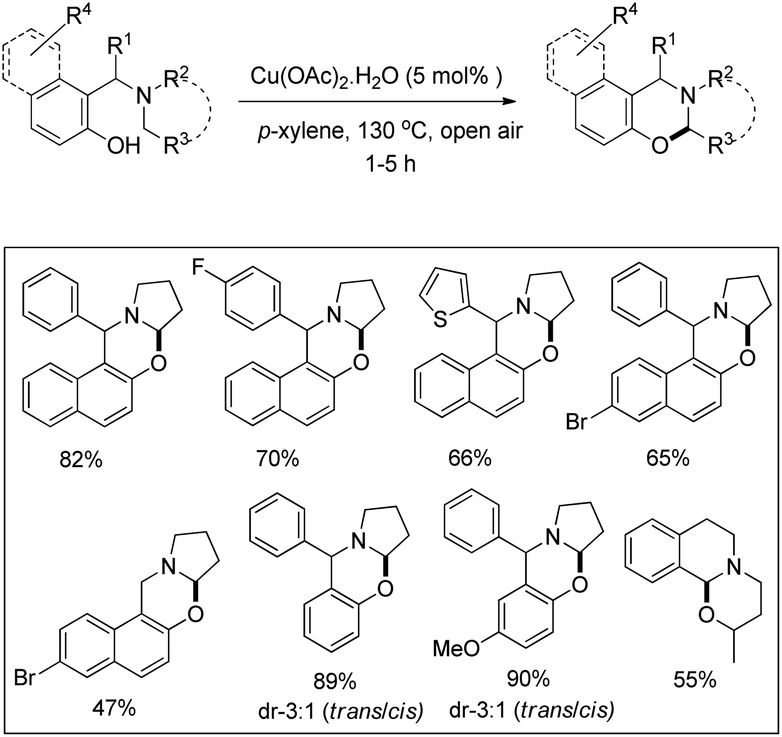
1,3-Oxazines are important heterocyclic compounds which have several biological activities and can be used as intermediates in the synthesis of bioactive natural products.33 Deb et al. reported an elegant synthesis of naphtho- and benzo-2,3-dihydro-1,3-oxazines through the α-C–H functionalization of tertiary amines (Betti base) by using a copper catalyst and air as an oxidant (Scheme 41).34
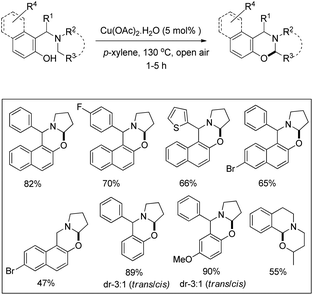 |
| | Scheme 41 Synthesis of 1,3-oxazines using a copper catalyst. | |
The reaction works for substrates having various amine, naphthol and phenolic moieties. The primary C–H bond α to the nitrogen atom was activated regioselectively compared to the secondary one, whereas the benzylic C–H bond reacted readily compared to the non-benzylic primary/secondary C–H bond. A single diastereomer of the desired naphtho-1,3-oxazines was obtained and it was a trans isomer confirmed by single crystal X-ray analysis.
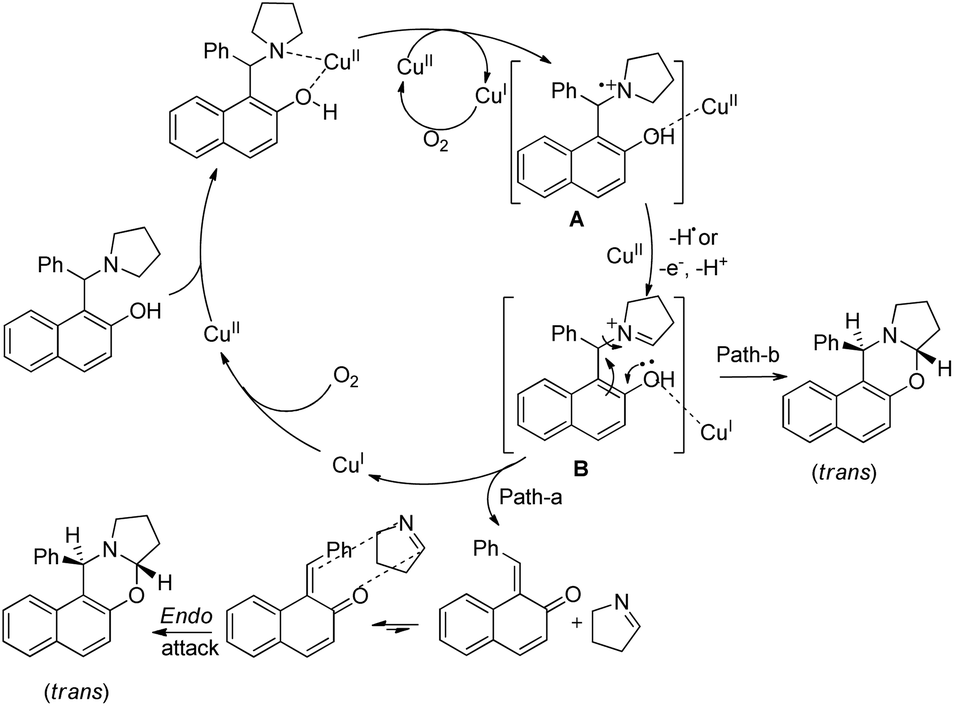
Single electron transfer (SET) from the N-atom of the substrate occurs to generate the radical cation A which subsequently forms the iminium ion B (Scheme 42). The product could be obtained by C–O bond formation through a direct attack of the –OH group on the iminium ion (path-b). Path-a involves the formation of o-quinone methide and cycloaddition of the imine to form the 1,3-oxazine product.
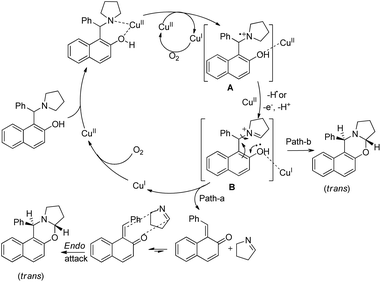 |
| | Scheme 42 Tentative mechanism for the Cu(II) catalyzed oxazine synthesis. | |
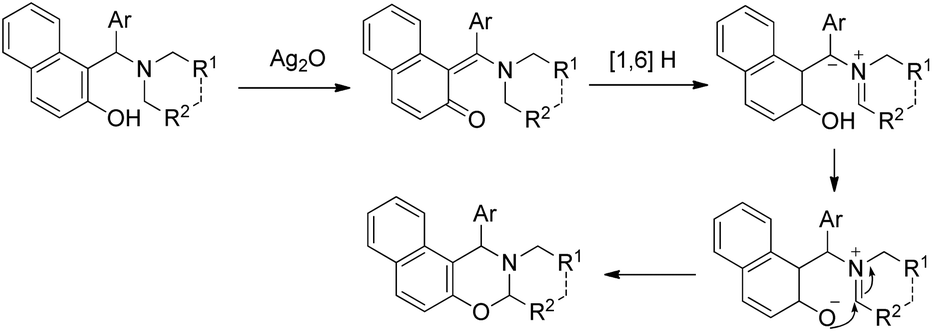
After this report many other groups have reported the synthesis of 1,3-oxazines from Betti bases via C–H functionalization. Jana et al. used Ag2O promoted α-C–H functionalization of tertiary amines to synthesize naphtho-2,3-dihydro-1,3-oxazines in m/p-xylene at 140 °C.35 The products are obtained in moderate to good yields using different amine moieties. In the presence of Ag2O the Betti base forms an o-quinone methide intermediate followed by [1,6]-H transfer generating the zwitterionic intermediate. This then finally undergoes cyclization giving the final product (Scheme 43). Shahrisa et al. reported the synthesis of 1,3-oxazines from alkylamino naphthols using CuI as a catalyst and Cs2CO3 as a base.36 The substrate scope of the reaction is limited and the mechanism is similar to the one proposed by Deb et al.
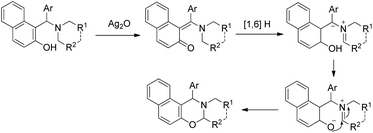 |
| | Scheme 43 Plausible mechanism for the Ag-catalyzed cyclization. | |
Karade et al. disclosed a metal-free approach for the synthesis of 1,3-naphthoxazines from Betti bases via CDC using the PhI(OAc)2 reagent (DIB) in DCM as a solvent.37 The main advantage of this method is that the reaction could be carried out at room temperature. However, the reaction requires more than a stoichiometric amount of catalyst loading (2.2 equiv.).
They proposed a mechanism in which DIB reacts with the Betti base and generates the cyclic iodine intermediate (B). Reductive elimination of PhI and attack of the nucleophilic oxygen on the iminium carbon give the 1,3-oxazine product (Scheme 44).
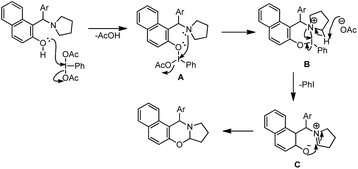 |
| | Scheme 44 Tentative mechanism for the DIB catalyzed 1,3-oxazine formation. | |
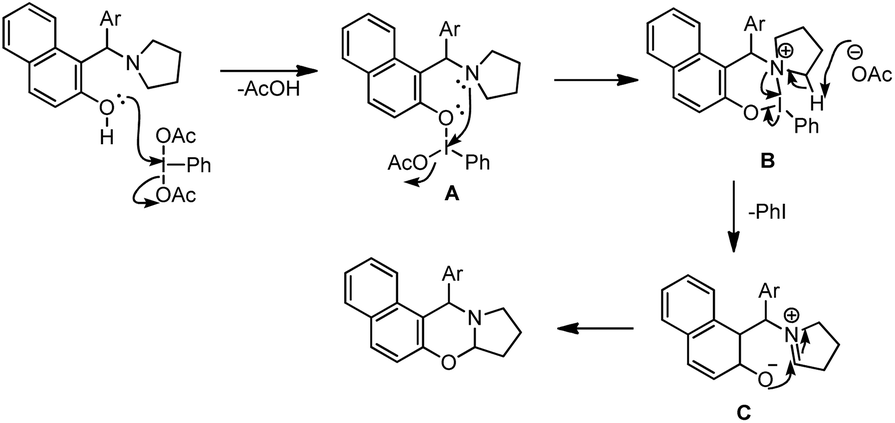
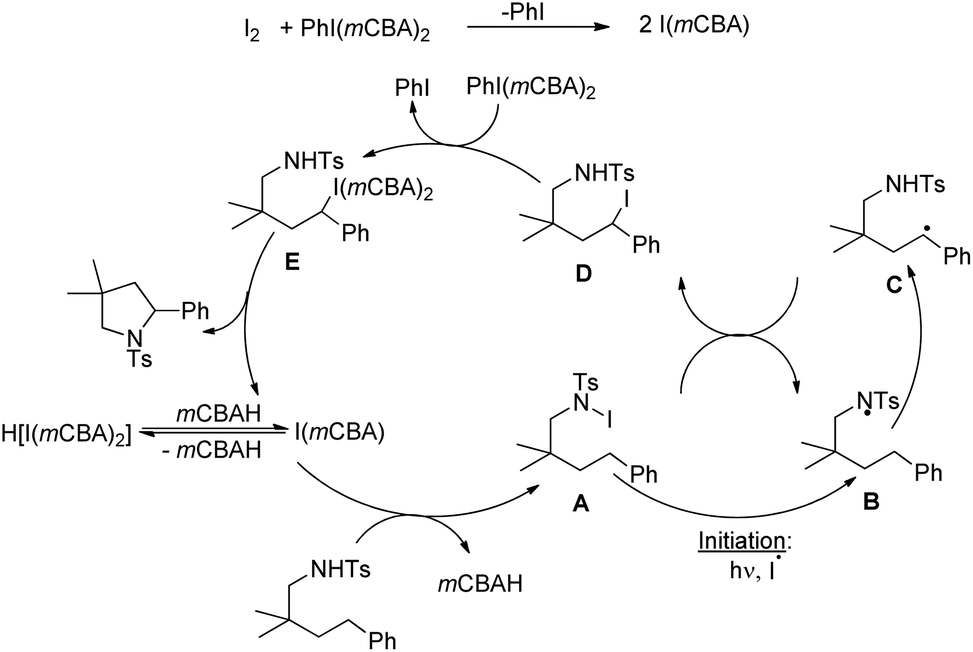
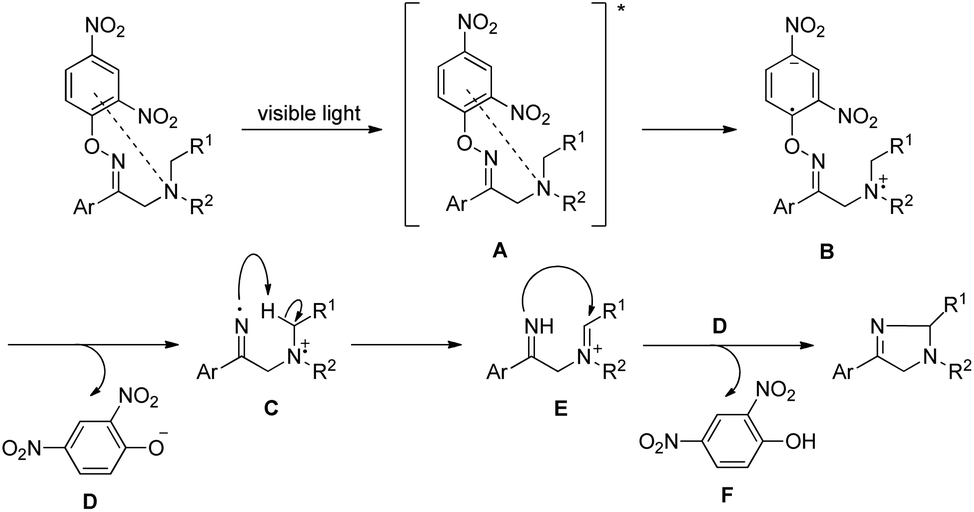
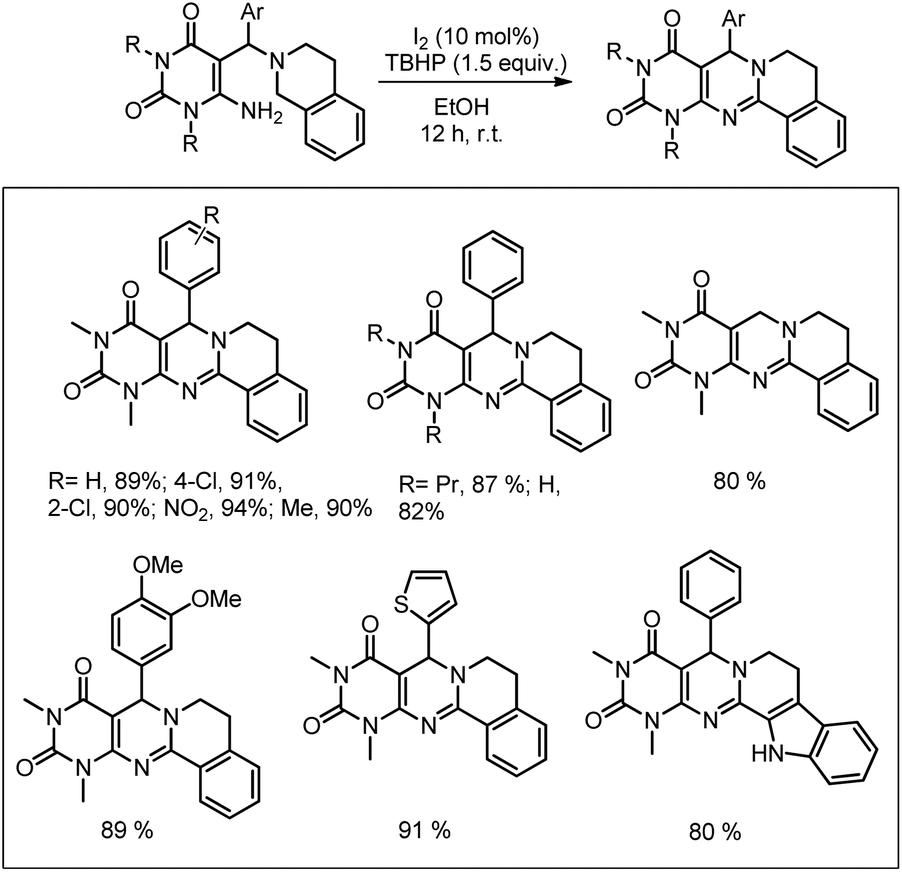
Our group was interested in developing a metal-free approach for the synthesis of 1,3-oxazines. We used I2 as a catalyst and TBHP as an oxidant in DMF at 130 °C for the synthesis of 1,3-oxazines from different Betti bases of phenols and naphthols having cyclic and acyclic tert-amines as starting materials.38 In comparison with the above methods this reaction took a much shorter time for completion (15–30 minutes). We have proposed a non-radical mechanism involving “I+” as the active catalytic species generated from I2 and TBHP. The I+ was then attacked by the nitrogen atom of the substrate generating the intermediate A which then eliminates one molecule of HI to give the iminium ion B. This then undergoes tautomerism to form the intermediate C. Direct attack of the –OH group on the iminium ion (path-a) or generation of an o-quinone methide intermediate and subsequent cycloaddition (path-b) gives the final product (Scheme 45).
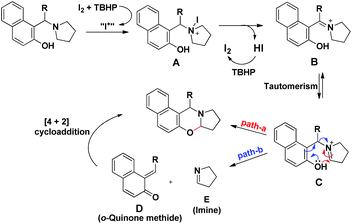 |
| | Scheme 45 Dual pathway mechanism for the reaction. | |
We further developed an environmentally benign and mild method for the synthesis of 1,3-oxazines from Betti bases using I2 as a catalyst, H2O2 as an oxidant and ethanol as a solvent at room temperature.39 The mechanistic study reveals that hypoiodous acid (HOI) generated from I2 and H2O2 is the active catalytic species.
[2-(Pyrrolidin-1-yl)phenyl]methanols in the presence of Co(II) and Cu(II) catalysts and DL-tyrosine as a ligand in the presence of oxygen also gave 1,3-oxazines (Scheme 46).40
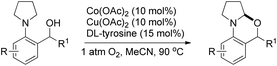 |
| | Scheme 46 Co/Cu catalyzed 1,3-oxazine synthesis. | |
When the cyclic amine moiety is changed to piperidine and morpholine the reaction failed to give the product. A hydrogen atom transfer (HAT) mechanism is proposed for the reaction (Scheme 47). HAT would generate a radical A which in turn generates an iminium ion B by single electron oxidation through path-I and finally a nucleophilic attack by the –OH group on the iminium carbon gives the product. In path-II, a SET would generate a radical cation C followed by HAT and gives intermediate B which is then cyclized to the product.
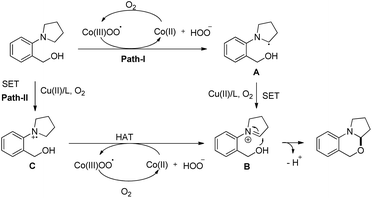 |
| | Scheme 47 Tentative mechanism for Co/Cu catalyzed 1,3-oxazine synthesis. | |
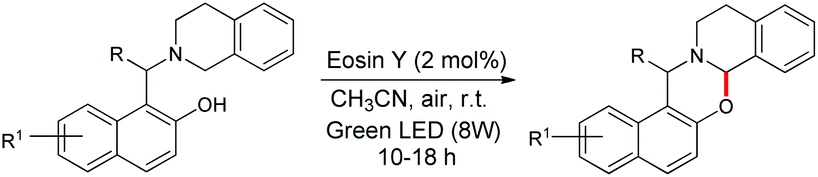
Recently, visible light driven reactions have received considerable attention. The main advantages of using photoredox reactions are that they can be performed under mild conditions and they do not require radical initiators or stoichiometric chemical oxidants or reductants. In 2017 our group reported a visible light promoted metal-free synthesis of 1,3-oxazines having a tetrahydroisoquinoline moiety using eosin Y as a photoredox catalyst (Scheme 48).41
 |
| | Scheme 48 Visible light driven synthesis of 1,3-oxazines. | |
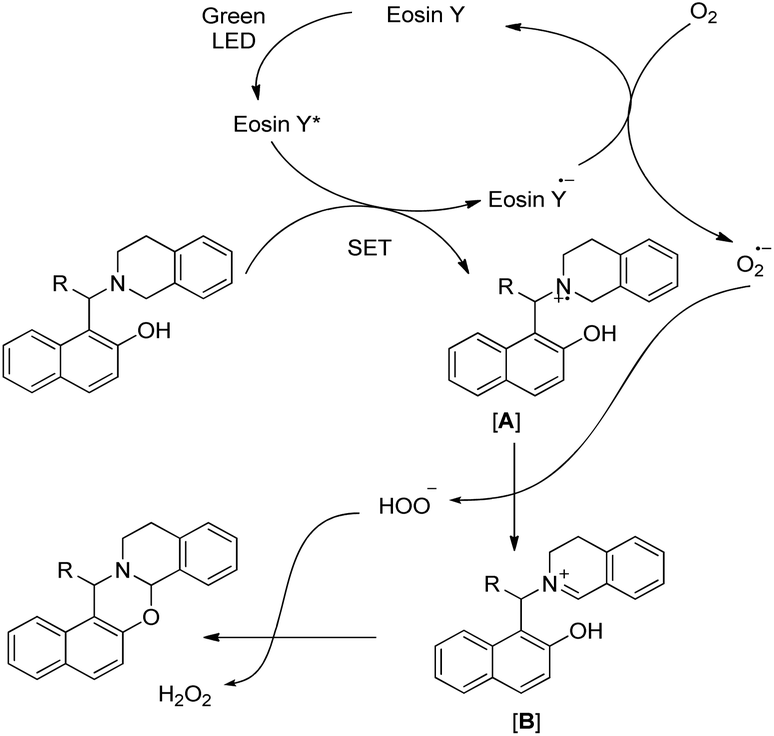
The visible light excites the eosin Y dye which accepts one electron from the substrate generating the radical cation intermediate [A] (Scheme 49). An oxygen radical anion (O2˙−) is generated by accepting one electron from the eosin Y radical anion (eosin Y˙−) regenerating the ground state eosin Y, which later removes a hydrogen atom from [A] to generate iminium ion [B]. Finally the attack of the –OH group on the iminium carbon gives the cyclized 1,3-oxazine.
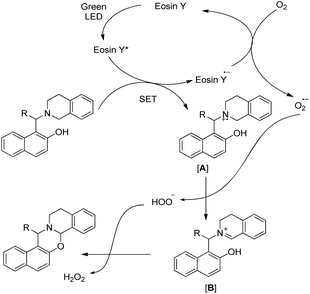 |
| | Scheme 49 Mechanism for the eosin Y catalyzed cyclization. | |
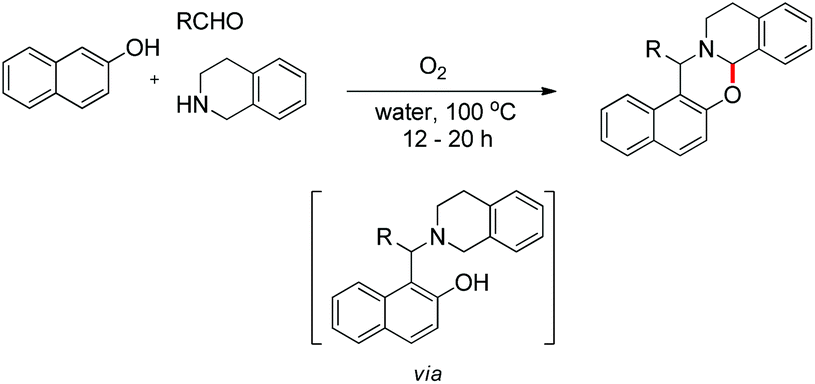
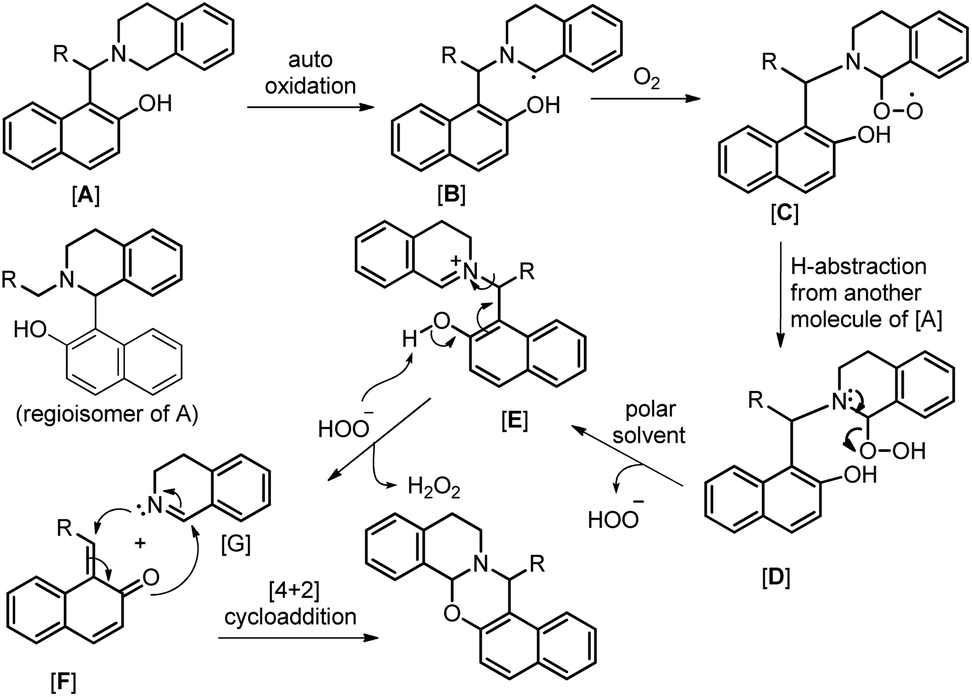
In the same year, we have reported a catalyst-free synthesis of 1,3-oxazines using a multi-component cascade approach in water using O2 as the sole oxidant (Scheme 50).42 Although the reaction is multi-component, it proceeds through an intermediate (Betti base) which we isolated and, therefore, comes under the scope of the current topic.
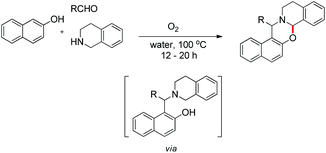 |
| | Scheme 50 O2 mediated 1,3-oxazine synthesis. | |
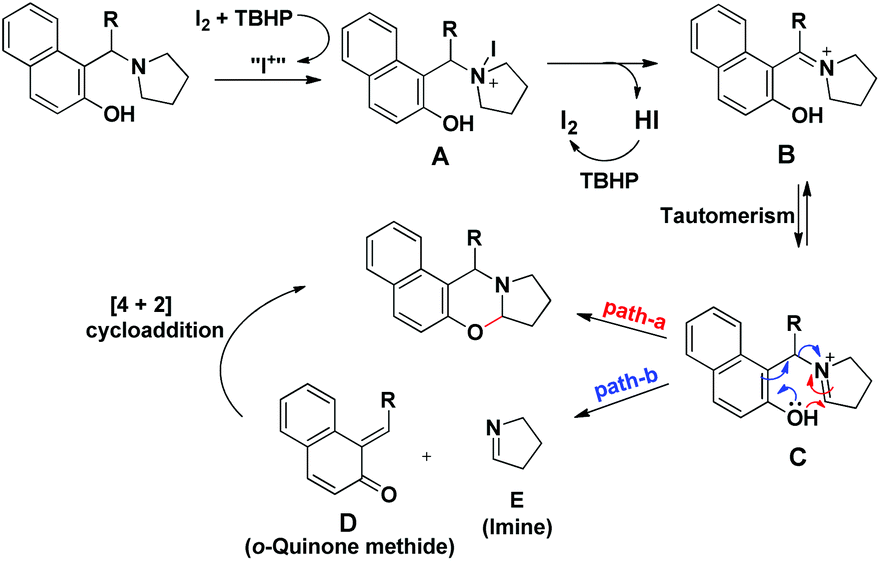
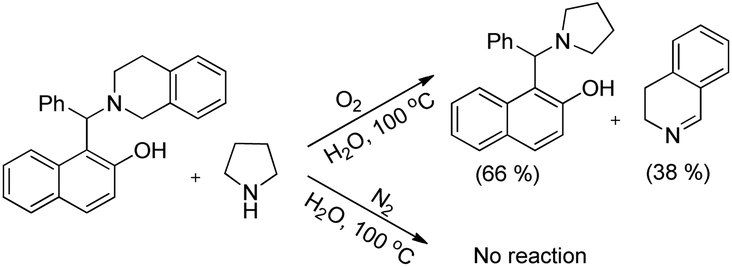
As in the earlier report where it was claimed that 1-(aminoalkyl)-2-naphthol can give 1,3-oxazine via two pathways,34 we carried out a reaction by taking an intermediate compound (Betti base) with pyrrolidines under optimal conditions and observed that the intermediate compound was completely transformed into another 1-(aminoalkyl)-2-naphthol along with the formation of an imine (Scheme 51). The formation of the corresponding oxazine was not detected. On the other hand, no reaction was observed under a nitrogen atmosphere. These two reactions indicate that the cyclization reaction proceeds completely through the formation of quinone methide [F] (Scheme 52). First, two regioisomers of the Betti base are formed from the 3-component reaction of 2-naphthol, THIQ and aldehyde. Only the major isomer [A] undergoes oxidation in the presence of O2 to the free radical [B] which finally gives the iminium ion [E]. Hydrogen abstraction by the HOO− ion generates the o-quinone methide intermediate [F] and the cyclic imine [G] which adds through [4 + 2] cycloaddition giving the final product (Scheme 52).
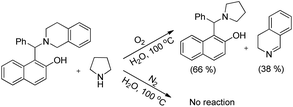 |
| | Scheme 51 Controlled experiment. | |
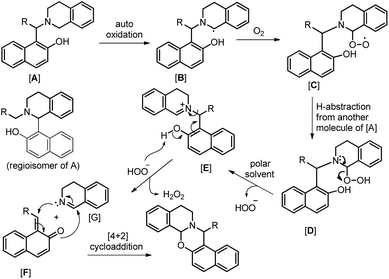 |
| | Scheme 52 Tentative mechanism for the O2 mediated cyclization. | |
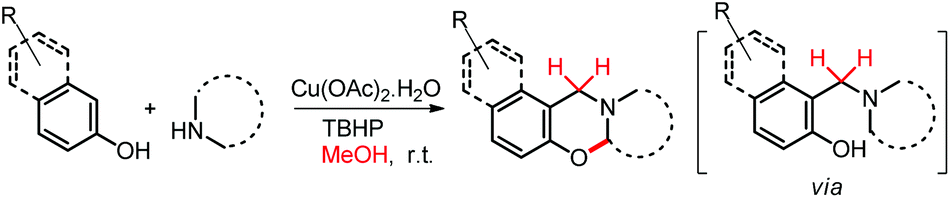
We have also reported a multi-component approach for the synthesis of 1,3-oxazines by using a copper catalyst at room temperature (Scheme 53). Methanol was used as a solvent in the reaction which also acts as the source of the methylene carbon of the oxazine product via formaldehyde.43 The reaction proceeds through the formation of the intermediate 1-(aminoalkyl)-2-naphthol.
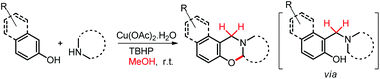 |
| | Scheme 53 Synthesis of 1,3-oxazines using methanol as the C-1 source. | |
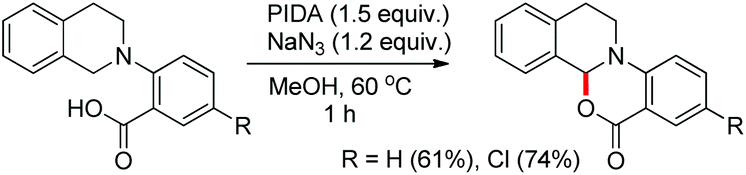
2.5. Synthesis of oxazinones
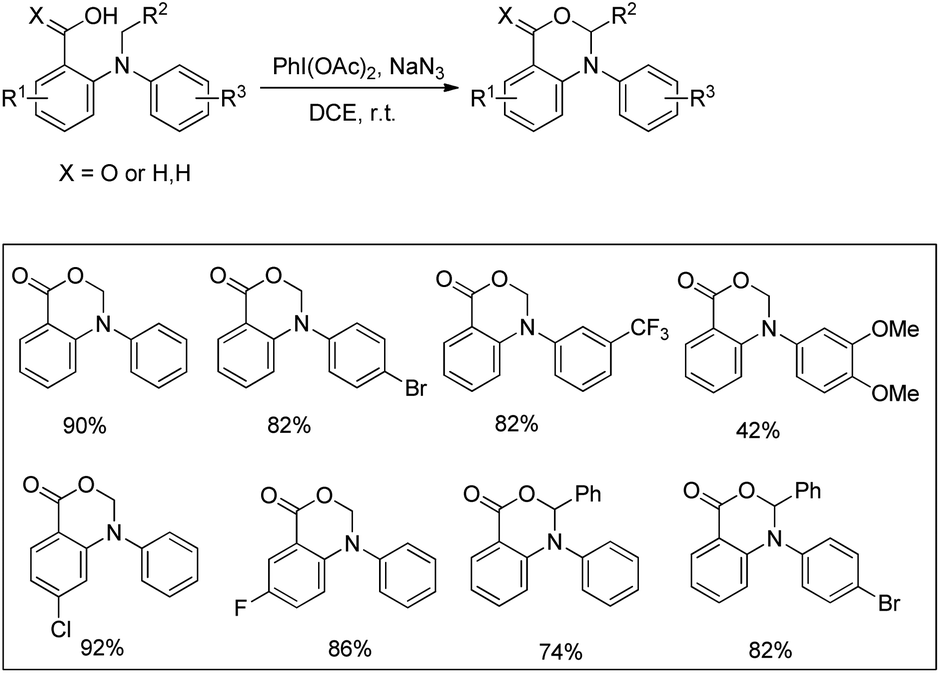
Du and Zhao et al. reported an intramolecular C–O bond formation reaction via PhI(OAc)2/NaN3-mediated C–H functionalization α to the tertiary amine moiety at room temperature (Scheme 54).44 Variation of R1, R2 and R3 groups in N-alkyl-N-aryl anthranilic acid substrates afforded the product in moderate to excellent yields. Both the aryl groups were necessary for the reaction. When the carbonyl group of the substrate was replaced with a methylene group, 1,3-oxazines were obtained although in lower yields than oxazinones.
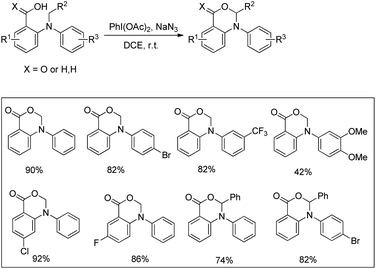 |
| | Scheme 54 PIDA catalyzed synthesis of oxazinones. | |
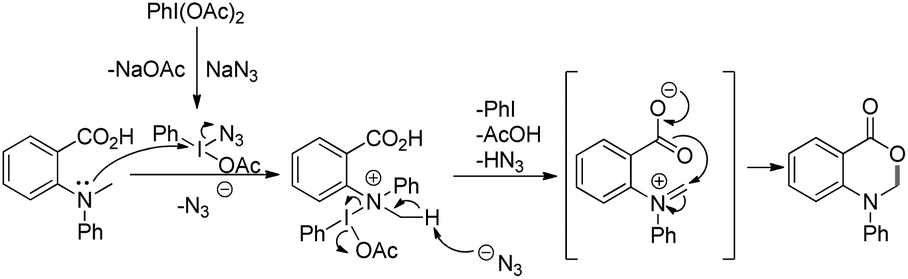
The PhI(OAc)2 first reacts with NaN3 to generate a more powerful iodine(III) species (Scheme 55). Then the nucleophilic attack of the nitrogen of tertiary amines on iodine generates the ammonium intermediate which gets converted into the iminium intermediate and finally cyclizes to the product. The HN3 generated in the reaction decomposes to H2 and N2.
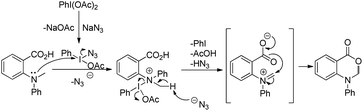 |
| | Scheme 55 Mechanism for the PIDA catalyzed oxazinone synthesis. | |
A couple of years later the same author reported another synthesis of oxazinones using tetrahydroisoquinoline-N-benzoic acid derivatives, in which a moderate yield was obtained (Scheme 56).45 Similar oxazinones were also synthesized by Che et al. induced by visible light using a Pd(II)–porphyrin complex as a catalyst.46
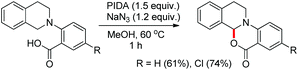 |
| | Scheme 56 Synthesis of oxazinones involving THIQ. | |
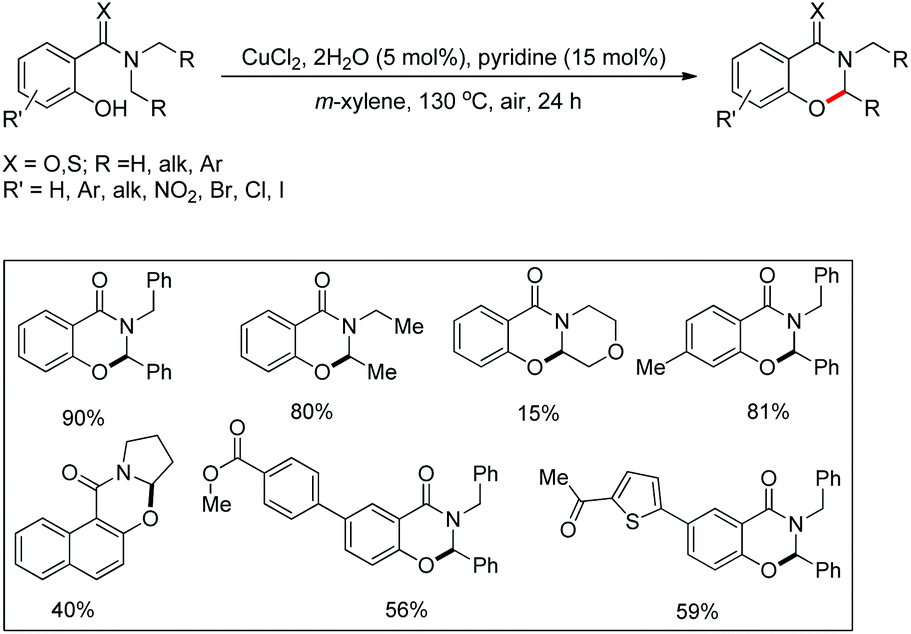
Maiti and co-workers disclosed a copper catalyzed CDC approach for the synthesis of dihydrooxazinone derivatives from symmetrical/unsymmetrical amide substrates (Scheme 57).47 Electron withdrawing groups in the benzene ring reduced the product yield which was due to the deactivation of the nucleophilic –OH group which lowered the efficiency of cyclization. For unsymmetrical amides the site selectivity was found towards the benzylic side of the amide moiety. The cyclization preferentially occurred at 2° C–H bonds compared to 1° C–H bonds.
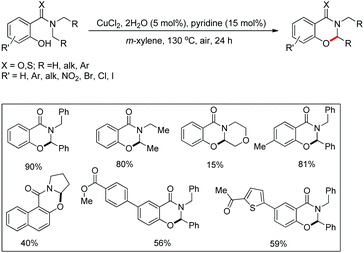 |
| | Scheme 57 Copper catalyzed oxazinone synthesis. | |
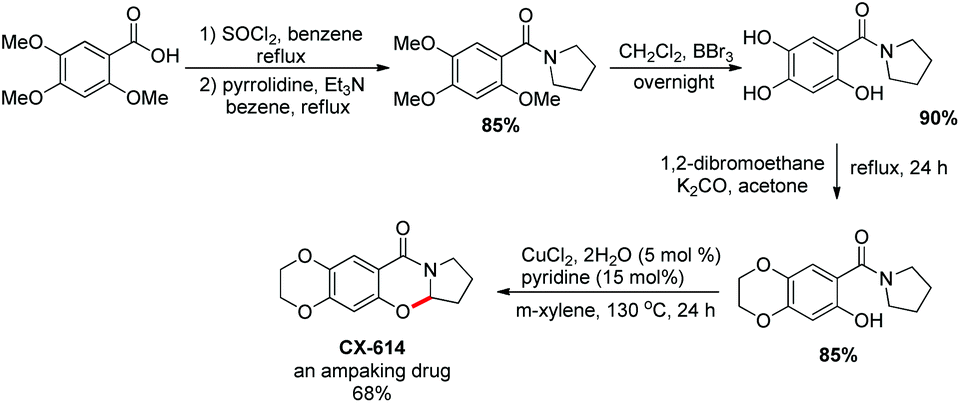
They utilized the methodology to synthesize the drug CX-614 because of its activity on the AMPA receptor. A four step synthesis was designed for this and it provided 44% overall yield of CX-614 (Scheme 58).
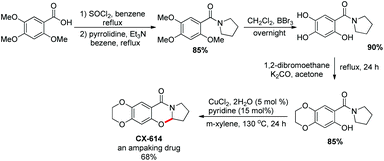 |
| | Scheme 58 Synthesis of CX-614. | |
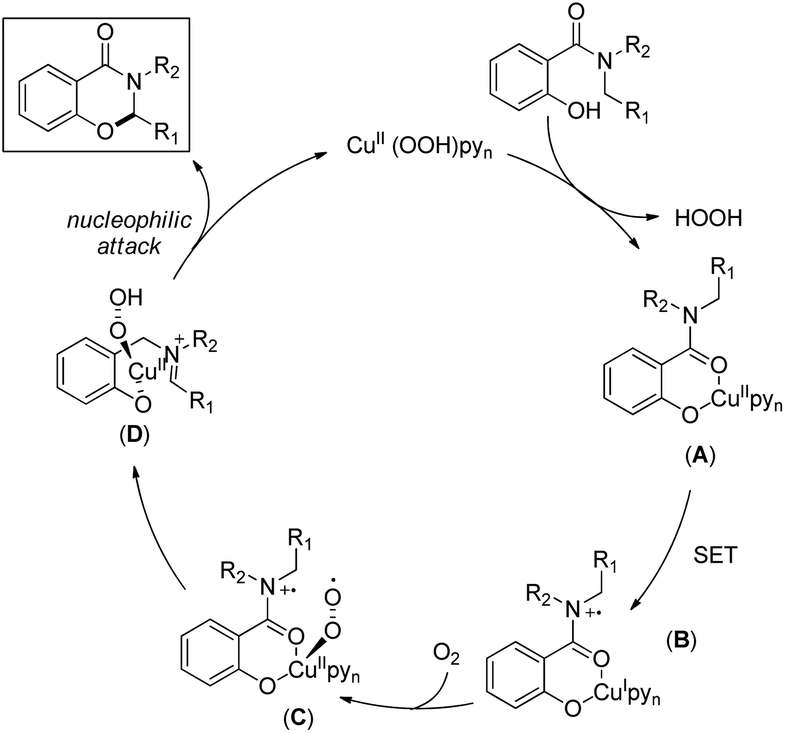
The reaction mechanism involves single electron oxidation of the intermediate A to B (Scheme 59). In the presence of O2, B forms the Cu(II)-superoxo intermediate C which after H-abstraction gives the Cu(II)-hydroperoxo intermediate D. Finally a nucleophilic attack of the oxygen atom of phenolate gives the cyclic product. A small amount of the keto product is also found to be formed from the Cu(II)-hydroperoxo species.
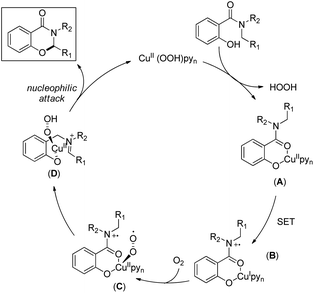 |
| | Scheme 59 Mechanism for the Cu catalyzed oxazinone synthesis. | |
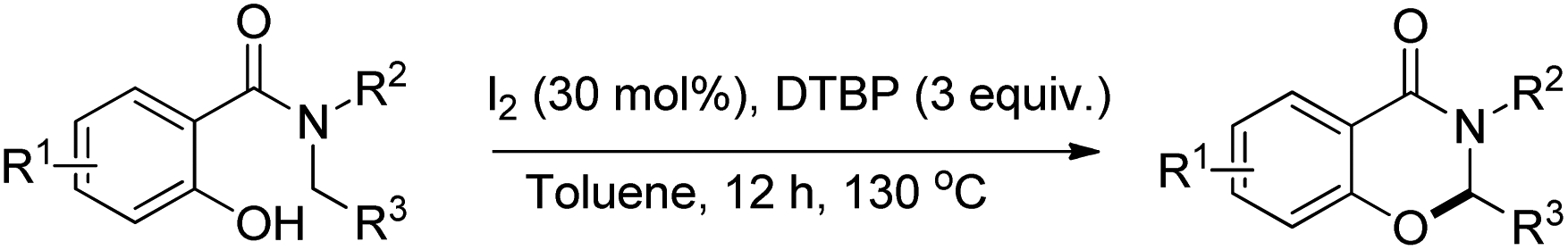
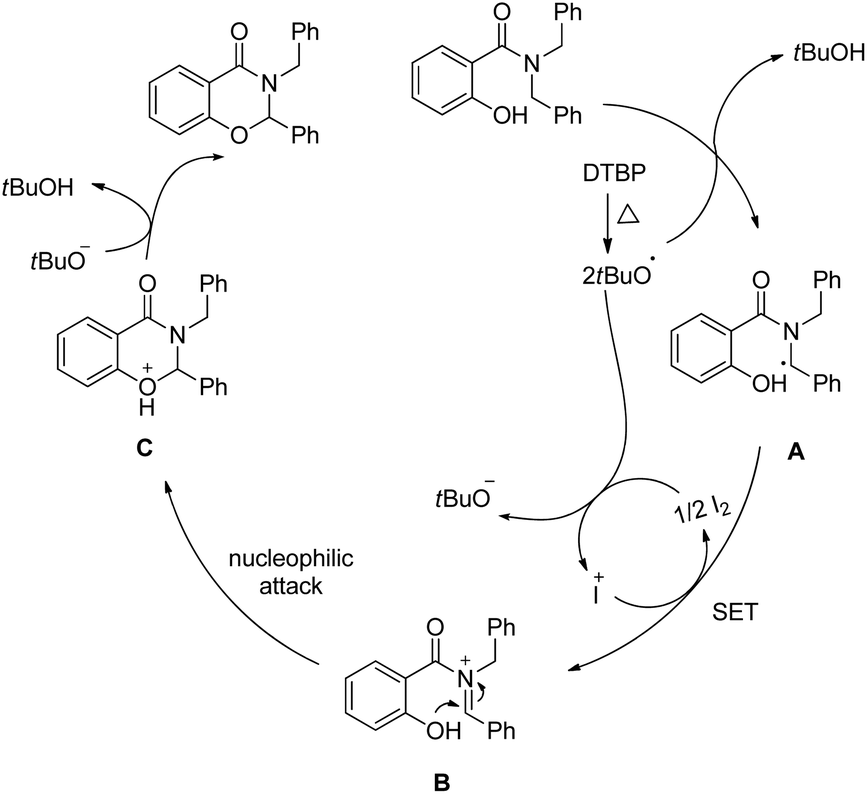
Wang and co-workers reported a similar synthesis of benzoxazinones by an I2-DTBP promoted intramolecular CDC reaction through activation of the C–H bond α to the N atom (Scheme 60).48
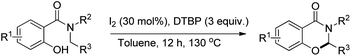 |
| | Scheme 60 I2 catalyzed benzoxazinone synthesis. | |
The reaction follows a free radical pathway. The tBuO˙ radical abstracts one hydrogen atom from the carbon α to the amide nitrogen and subsequently gives the intermediate B through single electron transfer (Scheme 61). Nucleophilic attack of the –OH group on the iminium carbon gives the cyclic intermediate which after elimination of one proton furnishes the final product.
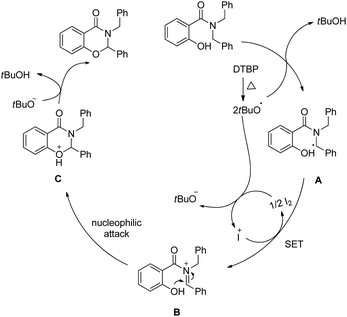 |
| | Scheme 61 Mechanism proposed for the reaction. | |
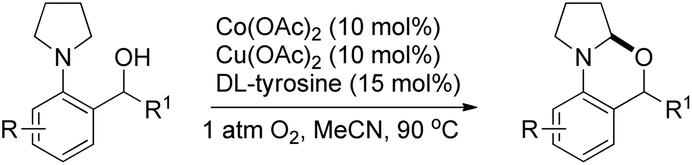
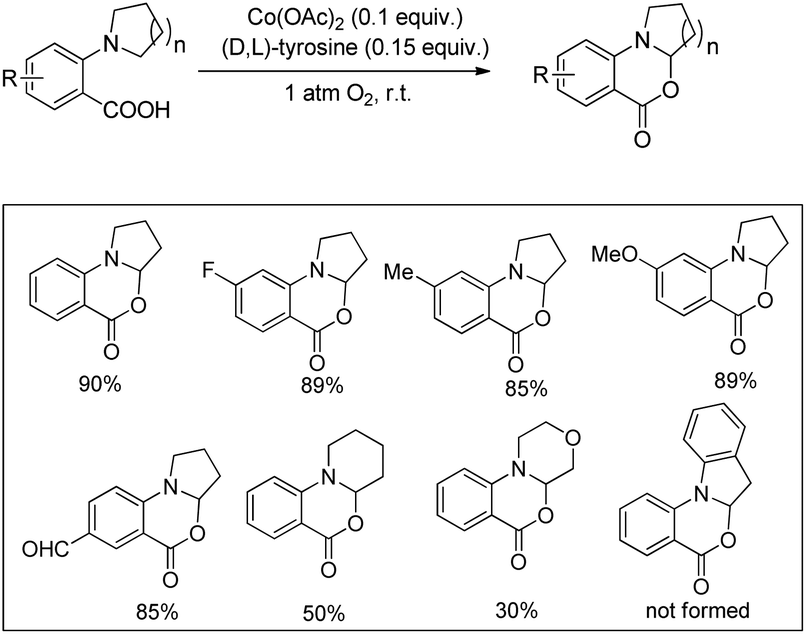
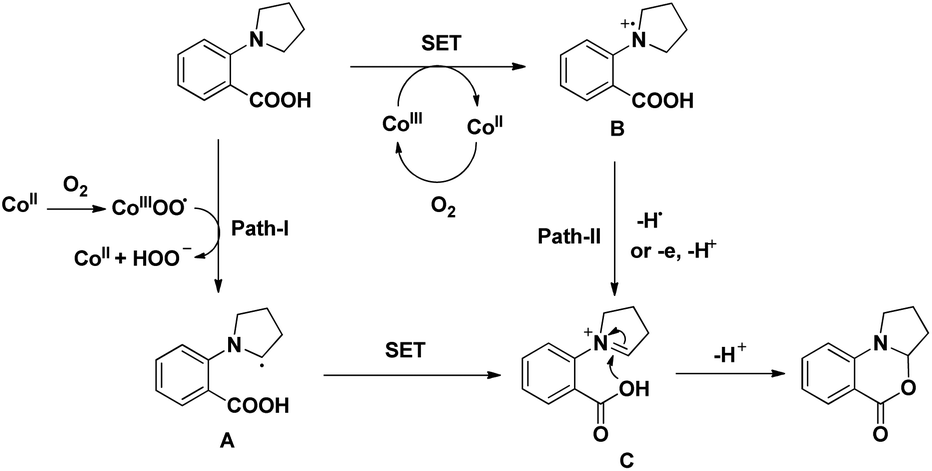
A cobalt(II) catalyzed synthesis of dihydro-benzoxazinones was reported by Shang and Liu through intramolecular C–O bond formation using the CDC approach (Scheme 62).49 The reaction gave a good yield of the products when 2-(pyrrolidin-1-yl)benzoic acids were treated with 0.1 equiv. of Co(OAc)2, 0.15 equiv. of tyrosine in CH3CN as a solvent and O2 as a terminal oxidant. When 2-(piperidin-1-yl)benzoic acid and 2-morpholinobenzoic acid were used as a substrate, lower yields of the products were obtained.
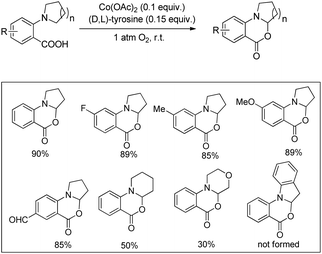 |
| | Scheme 62 Cobalt(II) catalyzed oxazinone synthesis. | |
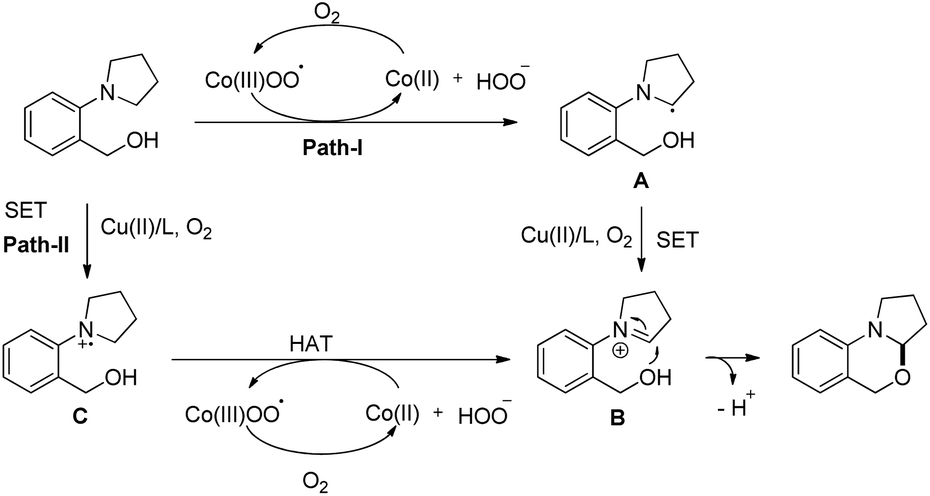
The reaction mechanism involves the formation of iminium intermediate C which could be formed via two pathways (Scheme 63). Path-I generates the radical intermediate through the cobalt(III) peroxo radical which after SET forms the intermediate C. A radical cation B is formed in path-II through SET which subsequently forms the intermediate C. Nucleophilic attack of the –OH group on the inimium ion gives the cyclized product. The role of tyrosine is believed to be stabilizing the Con+ ion by acting as a ligand.
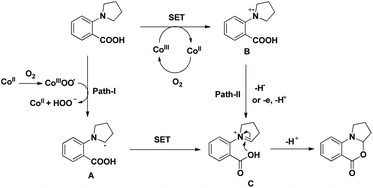 |
| | Scheme 63 Proposed mechanism for the cyclization. | |
2.6. C2-Functionalization of indoles
The C2-functionalization of indoles through C–H activation is an important area of research these days due to the biological activity of such compounds. Sandtorv has extensively reviewed the progress made in the area of C–H functionalization of indoles using transition metal catalysts.50 The C-2 position of indoles is most activated towards metallation and hence many methods to introduce different groups in this position are reported. However, most of the methods reported the formation of C–C, C–N, and C–B bonds but C–O bond formation is rare.
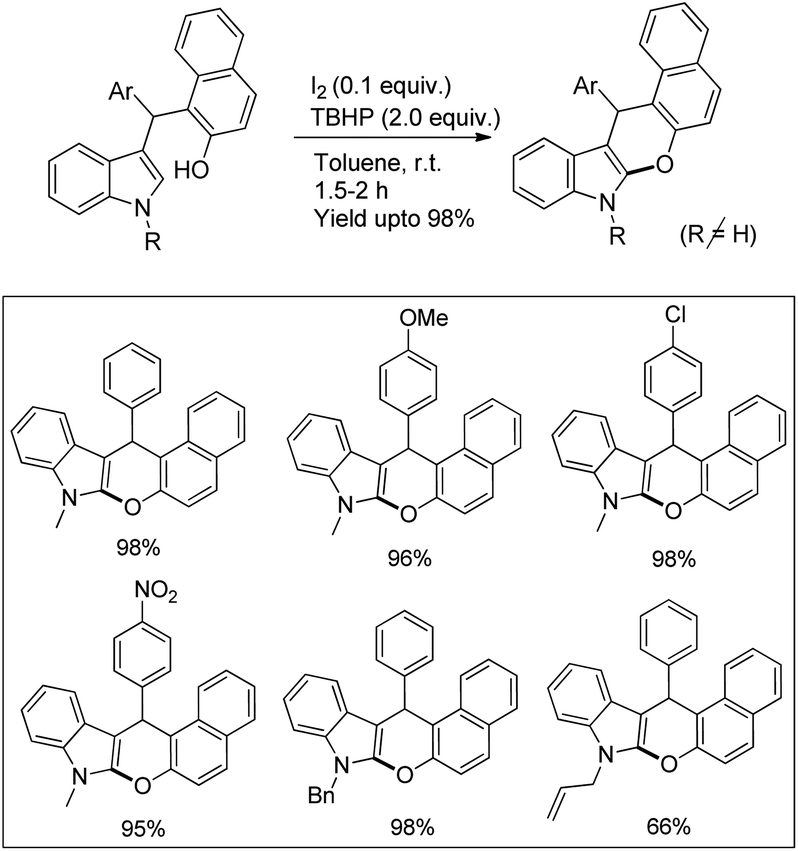
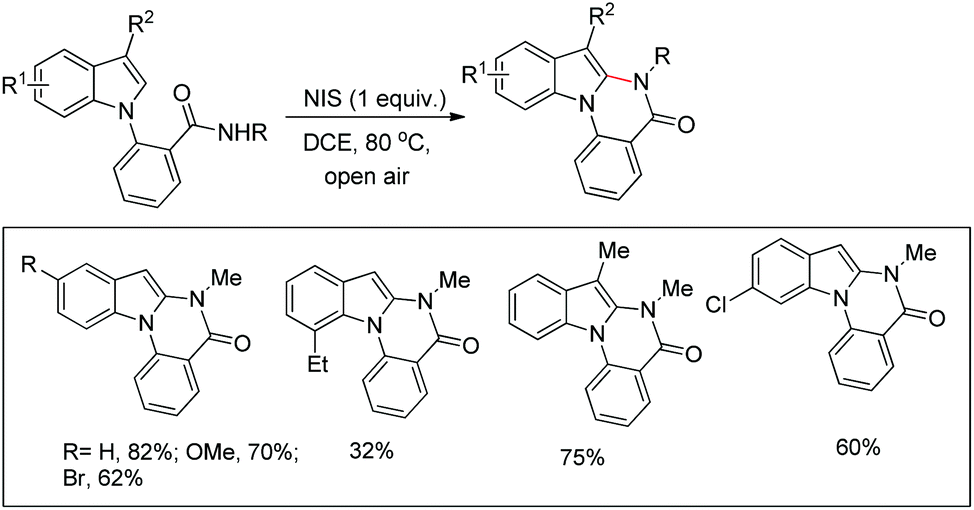
Our group reported an intramolecular C-2 functionalization of indoles using a metal-free approach. 3-(α,α-Diarylmethyl)indoles underwent cyclization in the presence of I2 as a catalyst and TBHP as an oxidant to furnish chromeno[2,3-b]indole derivatives (Scheme 64).51 The reaction did not require any inert atmosphere or dry solvents. We have tried a one pot reaction also but the yields of the products were lower in this case. The intramolecular cyclization was unsuccessful when phenolic substrates were used. Moreover, when NH-indoles were used no cyclization reaction took place. The substrates of the reaction were synthesized from the reaction of 1-(α-aminoalkyl)-2-naphthols (Betti bases) with N-substituted indoles under acidic conditions.
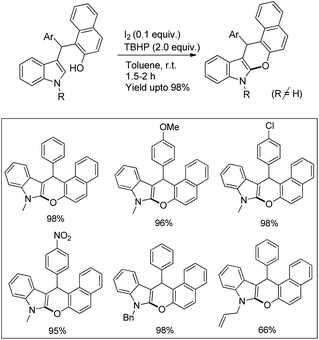 |
| | Scheme 64 Synthesis of chromeno[2,3-b]indoles. | |
By using radical scavengers in the reaction it was confirmed that the reaction was not a free radical mediated process. When the reaction was performed by using an equivalent amount of iodine in the absence of TBHP, it was noticed that the reaction did not occur. Therefore, it was assumed that the purpose of TBHP is not only to re-oxidize the eliminated HI to I2 but also to generate the electrophilic iodine species (“I+”) from the reaction of iodine and TBHP. To gain more insight into the mechanism, a reaction was performed by using a stoichiometric amount of both iodine and NaHCO3 in the absence of TBHP. The reaction gave a good yield but took a longer time compared to the I2/TBHP method. This result suggested that in situ generation of “I+” was not essential for the reaction. Iodination of the substrate may have taken place either by direct attack on molecular I2 (path-a) or on “I+” (path-b) (Scheme 65). Therefore, it was strongly believed that the expulsion of HI from the reaction is necessary for a successful reaction either by means of neutralization by using a base or re-oxidation to I2 by using an oxidant.
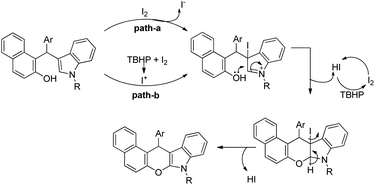 |
| | Scheme 65 Mechanism proposed for the synthesis of chromeno[2,3-b]indoles. | |
2.7. Miscellaneous C–O bond formation strategies
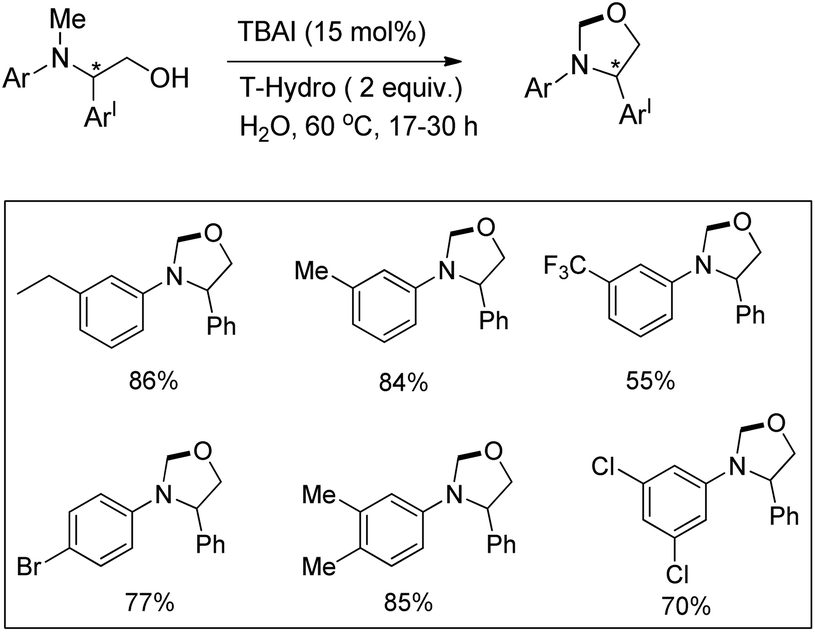
Punniyamurthy et al. reported a metal-free regioselective C–H functionalization reaction for the synthesis of oxazolidines in water using tetrabutylammonium iodide as a catalyst and TBHP in water (T-Hydro) as an oxidant (Scheme 66).52
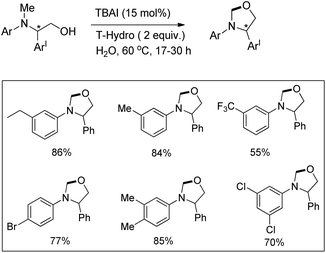 |
| | Scheme 66 Synthesis of oxazolidines. | |
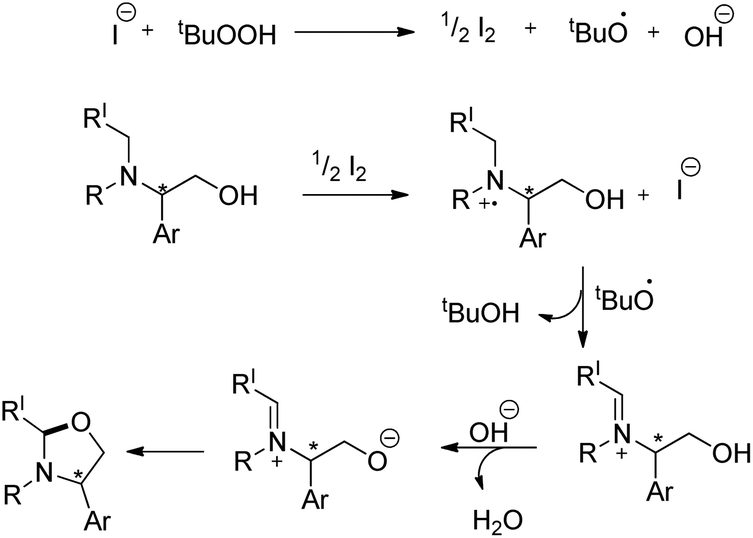
The reaction could be further extended to synthesize imidazolidines using the corresponding diamine substrates. The reaction allows the conversion of optically active substrates into respective optically pure oxazolidines and imidazolidines in good yields. The reaction follows a free radical pathway (Scheme 67). TBHP generates iodine from TBAI which undergoes reduction through single electron transfer from the N-atom of the substrate generating the radical cation. Subsequently, the iminium ion is formed and the nucleophilic attack of the O− anion on the iminium carbon finally gives the cyclized product.
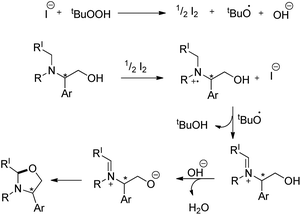 |
| | Scheme 67 Mechanism for the oxazolidine formation. | |
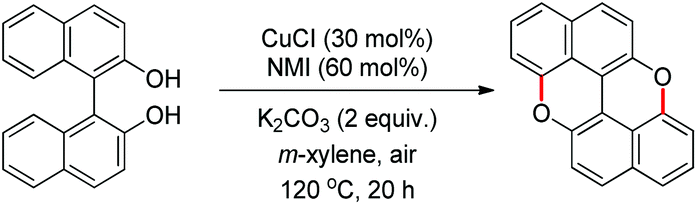
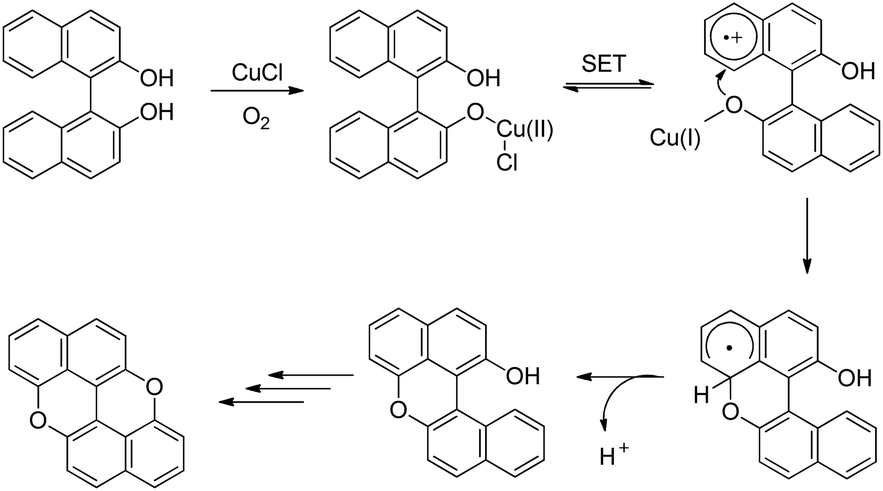
The synthesis of peri-xanthenoxanthene (PXX) was reported by Shimada et al. through copper catalyzed intramolecular C–O cyclization of 1,1′-bi-2-naphthol in the presence of air (Scheme 68).53 The use of N-methylimidazole (NMI) as a ligand gave the best result.
 |
| | Scheme 68 Synthesis of PXX. | |
peri-Xanthenoxanthene (97%) could be synthesized in one pot using 2-naphthol as a starting material under the same reaction conditions but at a temperature of 140 °C. The mechanism involves oxidation of the Cu(I) catalyst and formation of the Cu(II) intermediate (Scheme 69). This intermediate then undergoes single electron transfer and finally intramolecular C–O bond formation and deprotonation give the desired product. The role of K2CO3 is to deprotonate the –OH group of bi-naphthol and accelerate the ligand exchange of the copper catalyst.
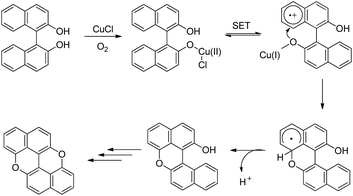 |
| | Scheme 69 Tentative mechanism for the synthesis of PXX. | |
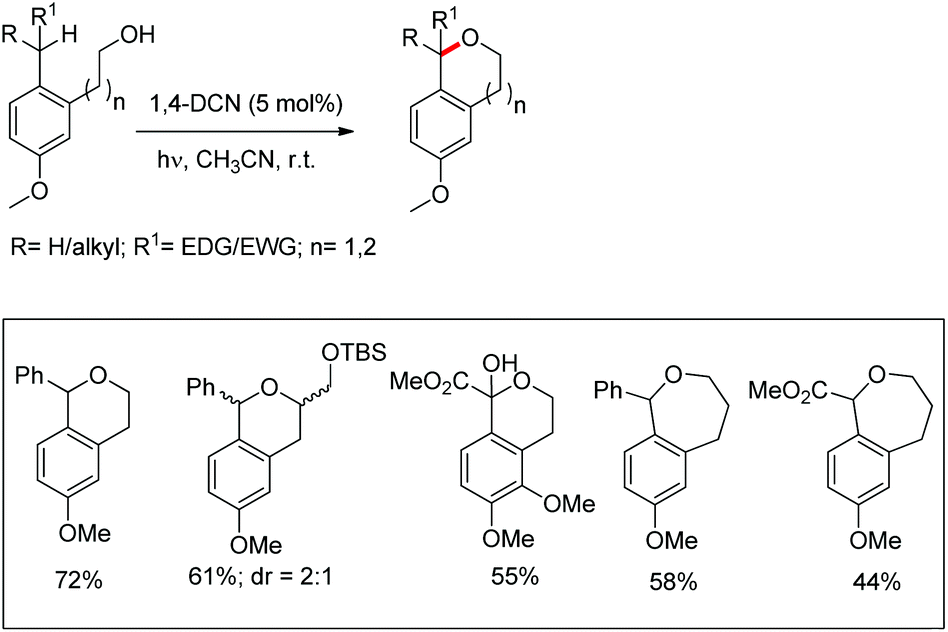
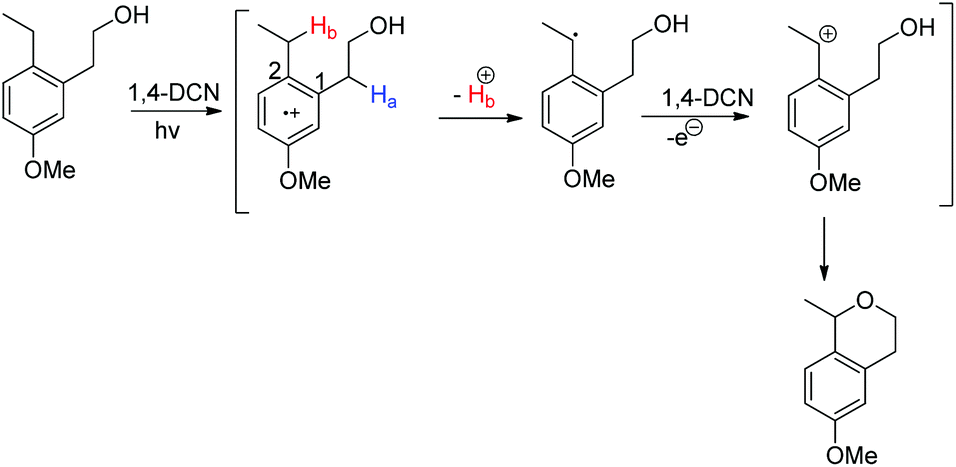
Pandey et al. used 1,4-dicyanonaphthalene (1,4-DCN) as a photoredox catalyst for regioselective intramolecular C–O bond formation by benzylic C–H functionalization of alkylarene substrates (Scheme 70).54 The reaction gave a moderate yield of the cyclic ethers (6- and 7-membered) when the corresponding alkylarene and DCN were photolyzed in acetonitrile at room temperature.
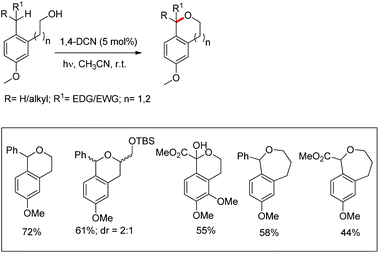 |
| | Scheme 70 Photocatalytic synthesis of cyclic ethers. | |
The mechanism involves the initial formation of the radical cation and deprotonation of the Hb atom to form the benzylic radical (theoretical calculation showed that C2 has more positive charge than C1 facilitating the deprotonation) (Scheme 71). This benzylic radical undergoes one electron transfer to 1,4-DCN and forms the benzyl cation. Then intramolecular nucleophilic substitution of the –OH group gives the cyclized product.
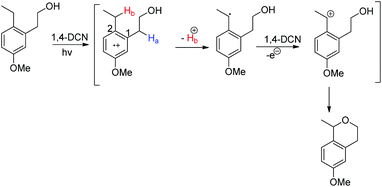 |
| | Scheme 71 Mechanism for the ether ring synthesis. | |
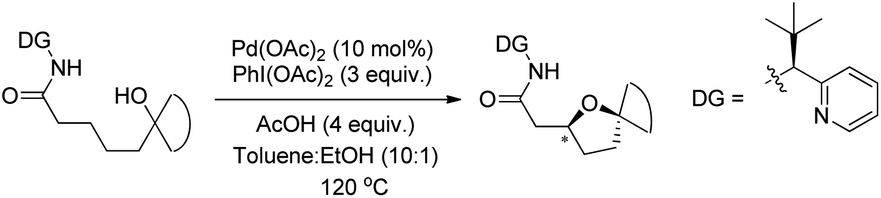
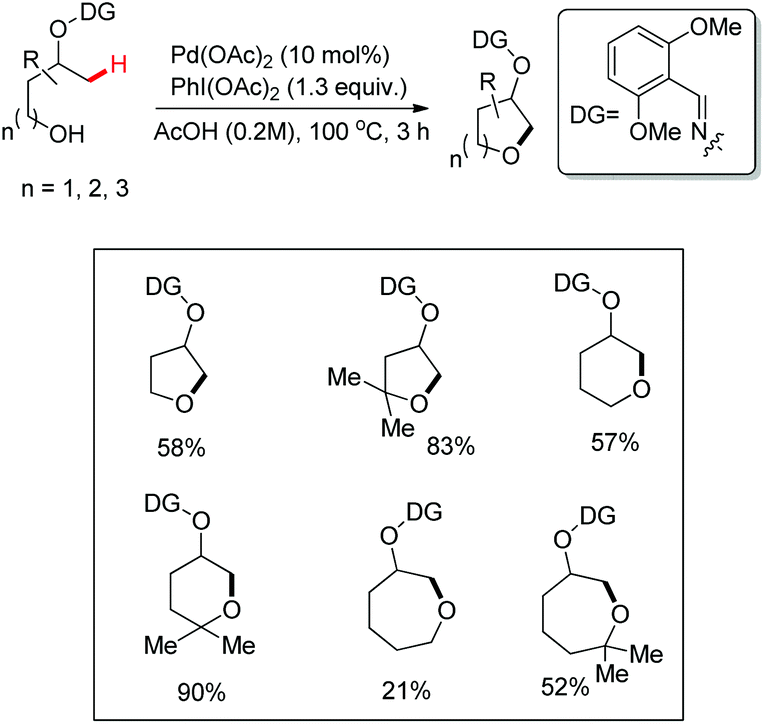
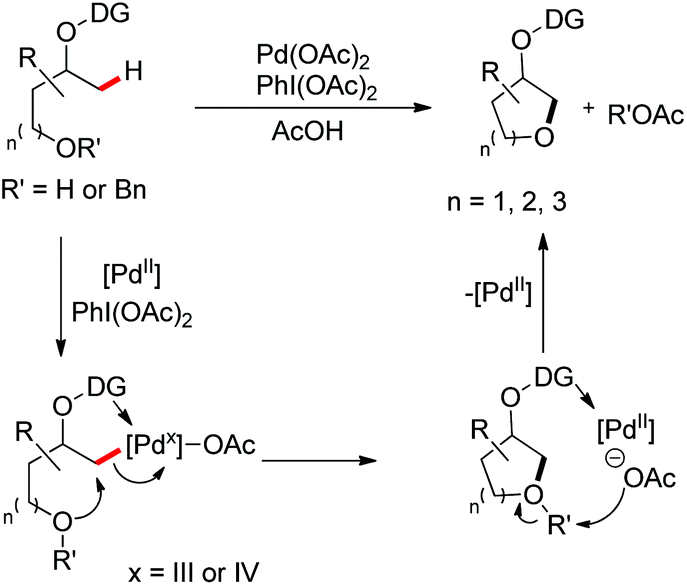
A chiral bidentate directing group (DG) mediated stereoselective C–O bond formation reaction catalyzed by Pd(II) was reported by Hong and co-workers (Scheme 72).55 The reaction required PhI(OAc)2 as an oxidant and acetic acid as an additive. A binary solvent system of toluene and ethanol gave the best yield. Many directing groups were screened and the presence of a bulky t-butyl group on the DG improved the diastereoselectivity.
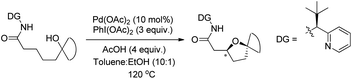 |
| | Scheme 72 Directing group mediated cyclization. | |
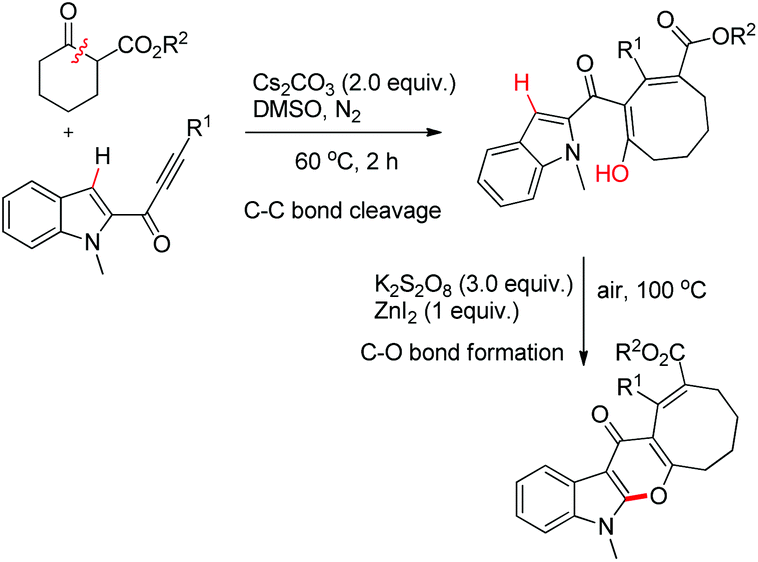
Li et al. reported the synthesis of pyranoindolones using one pot sequential C–C bond cleavage and C–O bond formation reactions (Scheme 73).56 The reaction of an alkynone with a β-ketoester in the presence of Cs2CO3 proceeds through C–C σ-bond insertion to give the ring expanded product. Furthermore, when it was treated with ZnI2 in the presence of K2S2O8 as an oxidant the corresponding pyranoindolone was obtained via C–H/O–H coupling.
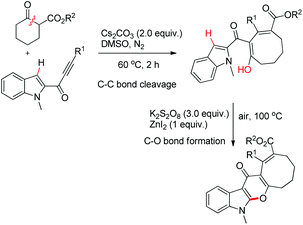 |
| | Scheme 73 Synthesis of pyranoindolones. | |
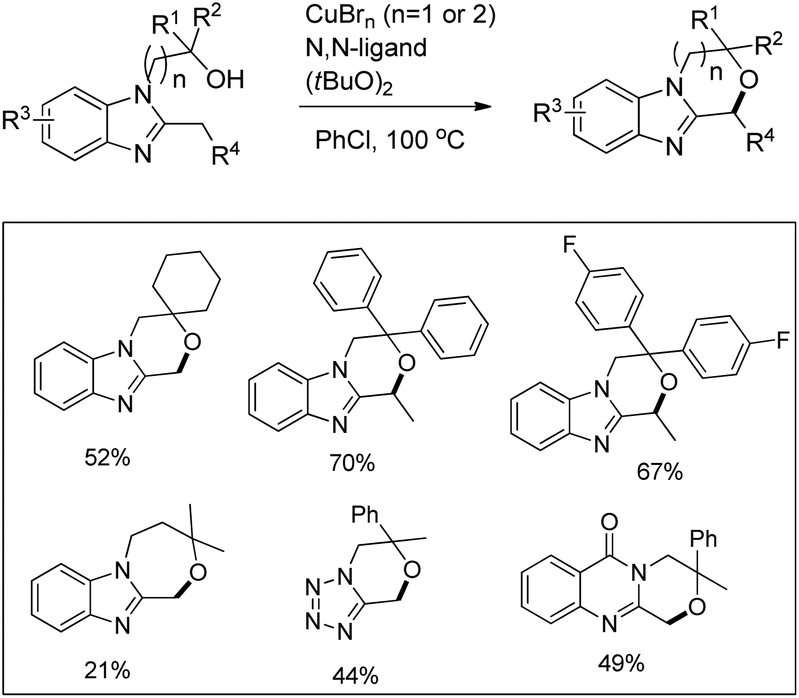
Kanai and co-workers reported a copper catalyzed intramolecular C–H alkoxylation of heterocyclic compounds at the benzylic position in moderate to good yields (Scheme 74).57
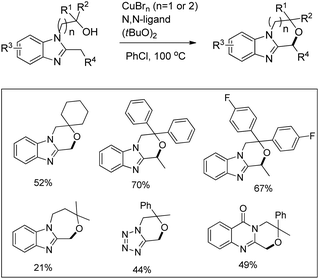 |
| | Scheme 74 Copper catalyzed C–O bond formation. | |
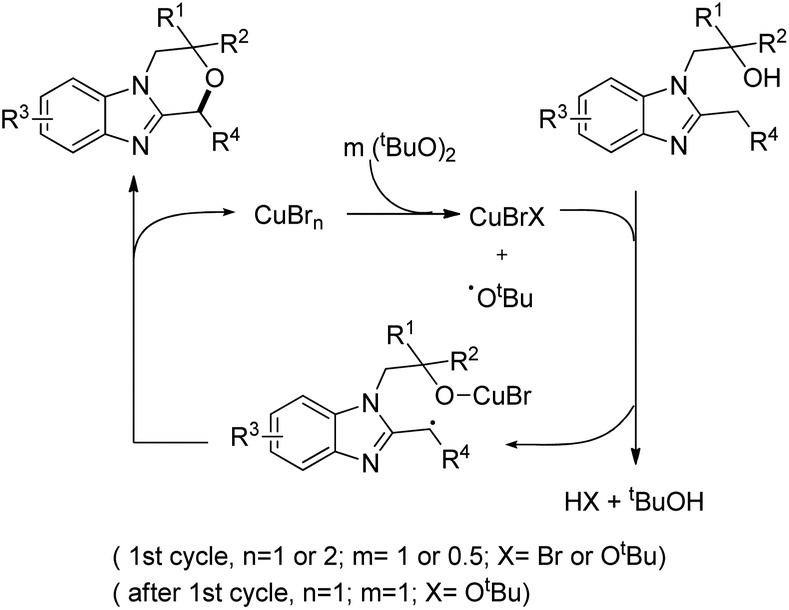
The substrate was treated with a copper catalyst, a ligand (5,6-dimethylphenanthroline or 4,7-dimethoxyphenanthroline) and (tBuO)2 as an oxidant. Heterocyclic substrates other than benzimidazoles also gave a moderate yield of the product. Seven membered ring cyclization was also achieved in a low yield (21%). The authors proposed a mechanism where the t-BuO˙ radical and CuBrX (X = Br or tBu) are formed first. Elimination of HX and tBuOH resulted in the formation of an alkoxycopper intermediate with a benzylic radical which undergoes intramolecular C–O bond formation to give the final product (Scheme 75).
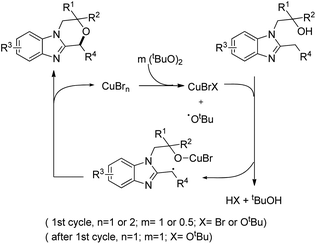 |
| | Scheme 75 Tentative mechanism for the C–H alkoxylation. | |
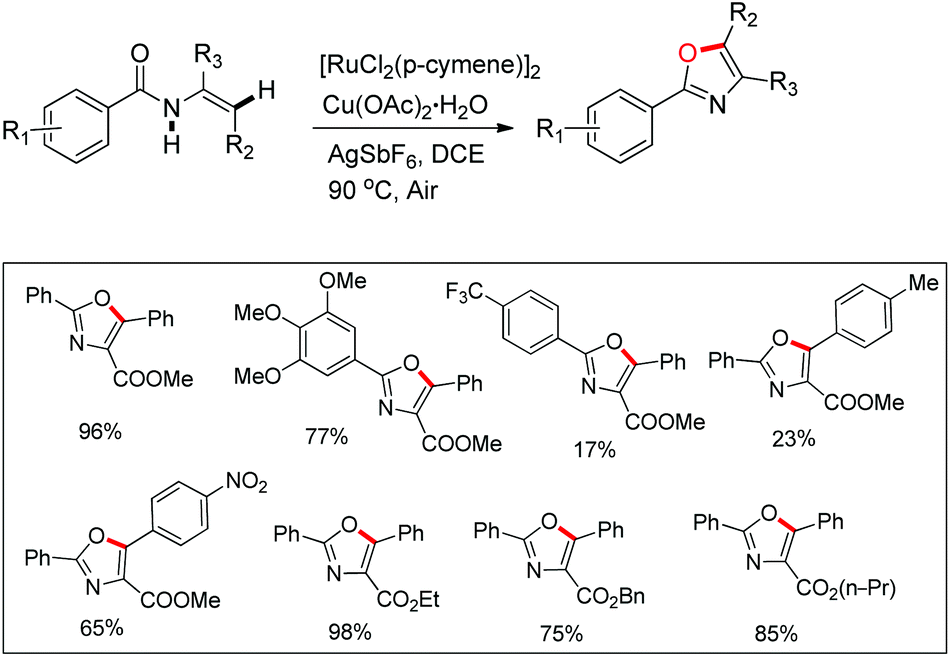
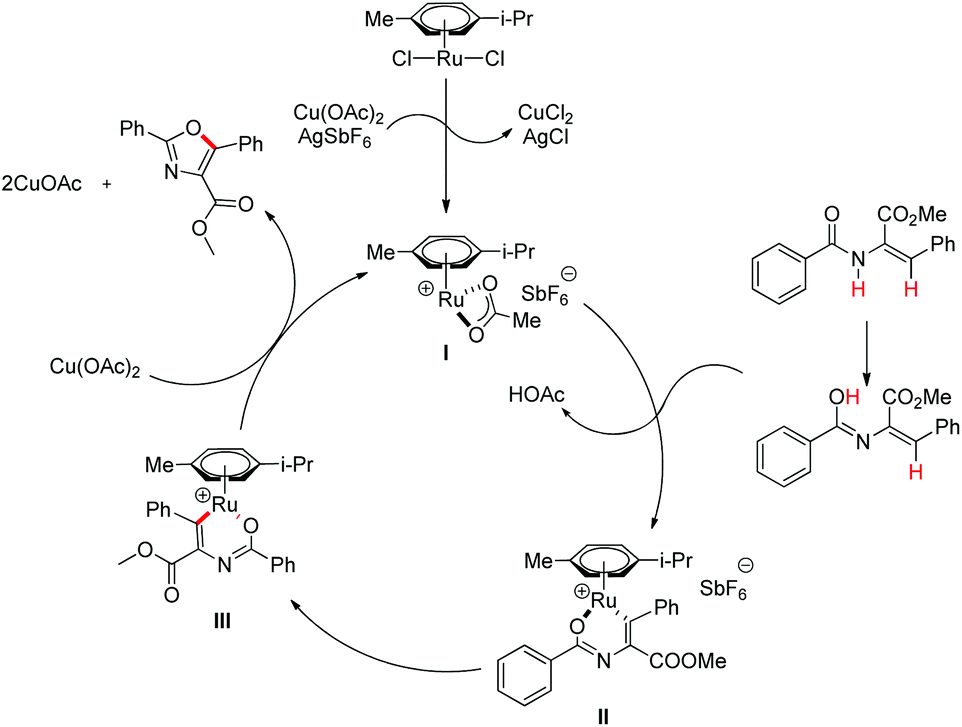
Hu and co-workers recently disclosed an efficient approach for the synthesis of oxazole derivatives from enamides via a ruthenium-catalyzed C–O cyclization (Scheme 76).58 This strategy has broad substrate scope and thiazole/furan derivatives could also be synthesized using appropriate substrates. Moreover, it has operational simplicity.
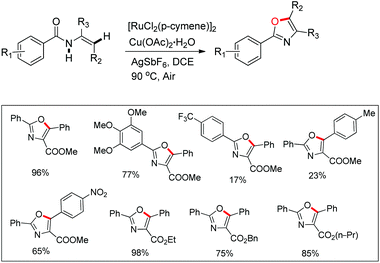 |
| | Scheme 76 Ru(II) catalyzed synthesis of oxazoles. | |
After performing several controlled experiments it was proved that the formation of ruthenacycles was irreversible. Moreover, they established that C–H bond cleavage was not involved in the rate-determining step and therefore, proposed a mechanism for the reaction (Scheme 77).
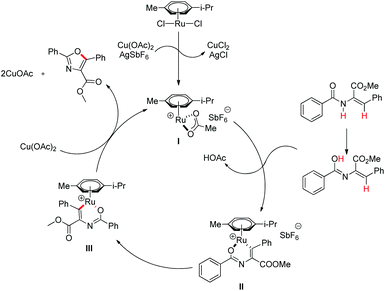 |
| | Scheme 77 Proposed mechanism for the oxazole synthesis. | |
Dong et al. reported a functionalization of unactivated sp3 C–H bonds with internal alcohols as nucleophiles. The reaction was directed by an oxime-masked alcohol and the annulation occurred at the β-position chemoselectively. This led to the formation of aliphatic cyclic ethers with 5- to 7-membered rings (Scheme 78).59 Primary, secondary and tertiary hydroxyl groups can all react to give the corresponding cyclized products. Moreover, benzyl and silyl protected alcohols can also be directly coupled. All the products consist of a mixture of oxime E/Z stereoisomers.
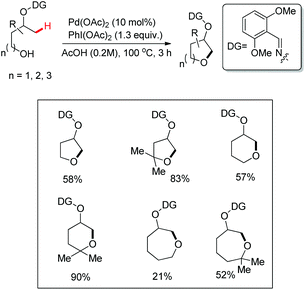 |
| | Scheme 78 Substrate scope for the synthesis of cyclic ethers. | |
They carried out a few controlled experiments which showed that regular benzyl ethers were not deprotected under these reaction conditions. Besides, a significant amount of benzyl acetate was isolated during the reaction. These observations clearly indicated that the ether oxygen might have acted as a nucleophile for the annulation reaction, instead of a free hydroxyl group. Based on these, a mechanism was suggested for the reaction (Scheme 79).
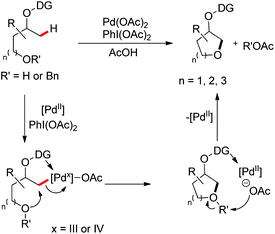 |
| | Scheme 79 Tentative mechanism for the oxime directed cyclization. | |
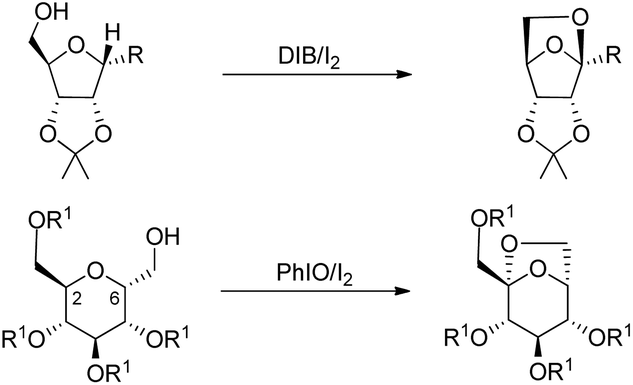
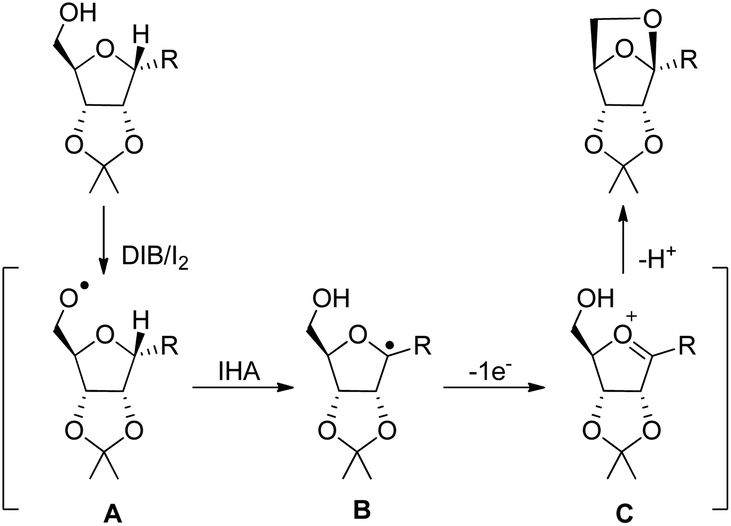
The synthesis of chiral 6,8-dioxabicyclo[3.2.1]octane and 2,7-dioxabicyclo[2.2.1]heptane rings from the reaction of carbohydrates with (diacetoxyiodo)benzene or iodosylbenzene was reported by Suarez et al. using neutral conditions (Scheme 80).60
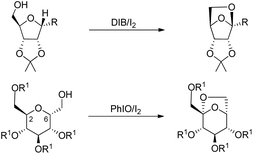 |
| | Scheme 80 Synthesis of 2,7-dioxabicyclo[2.2.1]heptanes and 6,8-dioxabicyclo[3.2.1] octane. | |
The reaction proceeds through the generation of alkoxy radical A which after intramolecular hydrogen abstraction (IHA) generates radical B (Scheme 81). Oxidation of this radical gives the oxycarbenium ion intermediate C which finally forms the cyclised product. The reaction occurs under mild conditions and several bioactive chiral compounds could be synthesized.
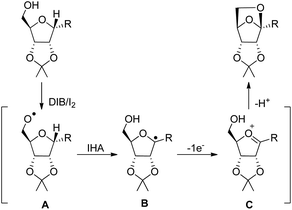 |
| | Scheme 81 Mechanism involving intramolecular hydrogen abstraction. | |
3. Intramolecular C–N bond formation
The C–N bond forming reaction is extremely important because of the ubiquity of nitrogen based compounds in various natural and synthetic products.61 The nitrogen containing heterocycles are used as the building blocks of many pharmaceutical agents. Due to the considerable significance of amide functional groups in biological processes, the construction of the C–N bonds has been extensively explored by chemists. The introduction of an amino group in organic frameworks in a controlled manner was first reported by Ullmann and Goldberg.62 However, wastage of stoichiometric amounts of copper salts and harsh reaction conditions are the main limitations of their process. In recent years, metal complexes are being used for C–N bond formation under mild conditions in a cost effective manner.63a–d The synthesis of nitrogen heterocycles through C–H functionalization using transition metal catalysts was reviewed by Yoshikai et al.63e In this section we have gathered reports related to intramolecular C–N bond formation using C–H functionalization and divided the section according to the nature of the catalyst.
3.1. Pd-Catalyzed intramolecular C–N bond formation
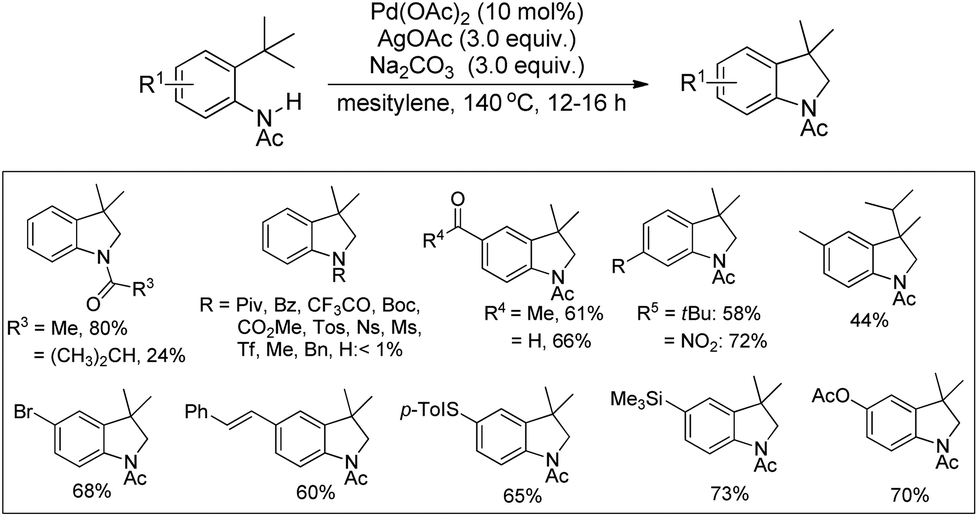
3.1.1 Synthesis of indoline derivatives.
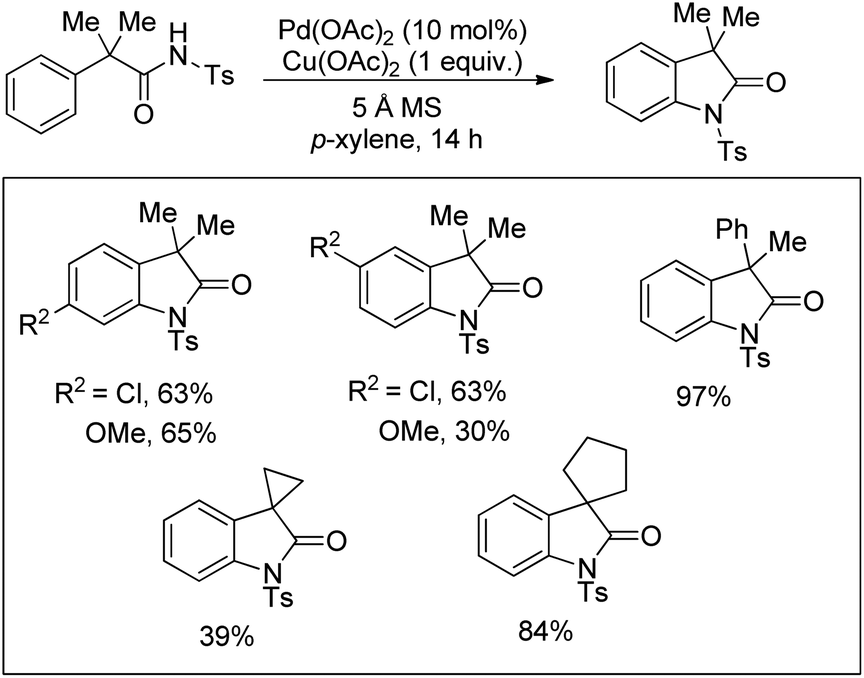
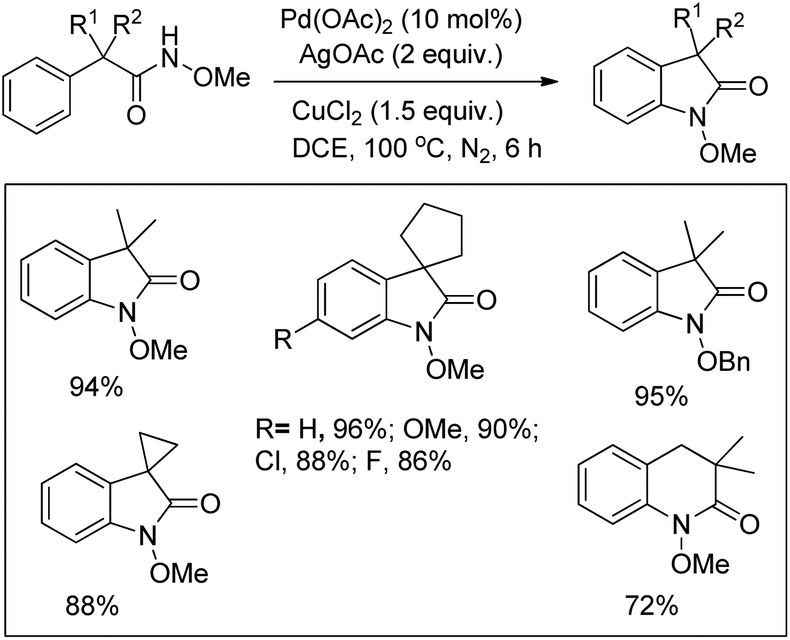
Glorius and co-workers reported the synthesis of indolines by activation of a C(sp3)–H bond of N-acyl anilines using a Pd-catalyst and AgOAc as an oxidant (Scheme 82).64 Substrates having an N-acetyl group provided the best yield. When N-formyl, N-propionyl, and N-isobutyryl substituents were used the desired products were obtained with a reduced yield. Other acyl groups, such as pivaloyl, benzoyl, or trifluoroacetyl, gave no products. No cyclised product was observed when carbamates, sulphonamides, and free primary or secondary amines were used as a substrate.
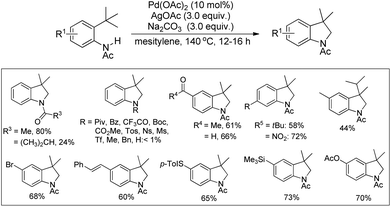 |
| | Scheme 82 Pd-Catalyzed synthesis of indolines. | |
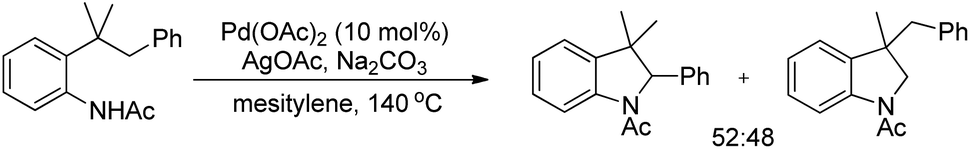
An intramolecular competitive experiment for benzylic and unactivated C(sp3)–H bonds showed that there is negligible preference for the activation of benzylic C–H over unactivated C–H (Scheme 83).
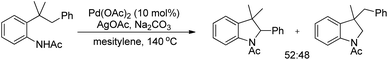 |
| | Scheme 83 Intramolecular competitive reaction for benzylic and unactivated C(sp3)–H. | |
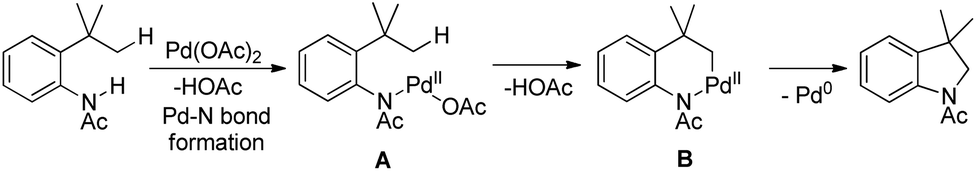
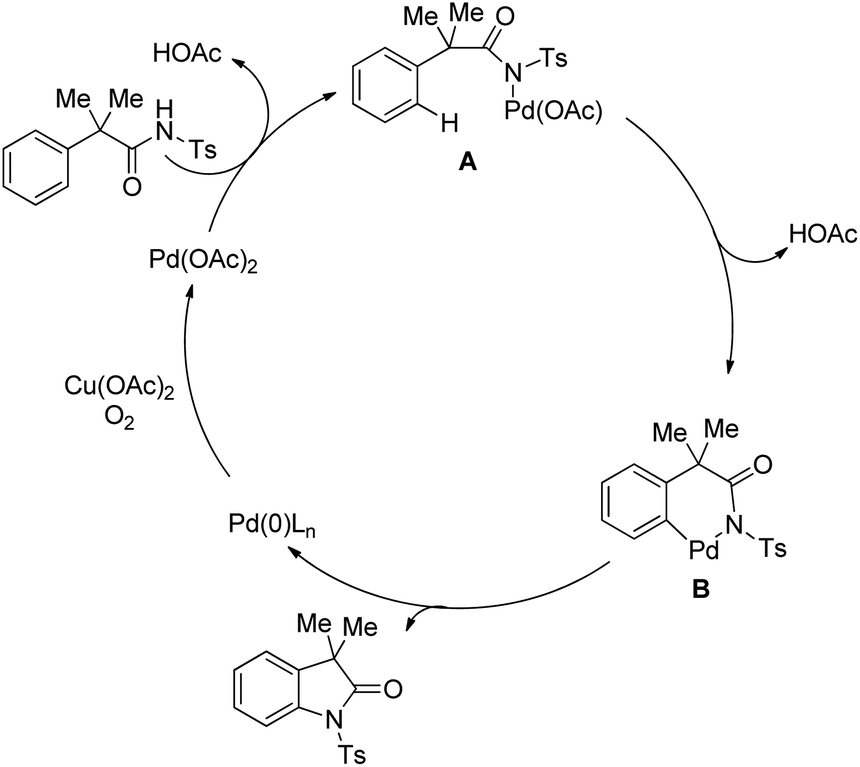
The reaction mechanism involves coordination of a Pd(II) catalyst to an acetanilide moiety giving the intermediate A which after C–H activation gives the palladacycle B. Subsequent reductive elimination gives the product and Pd(0) which can be reoxidized to Pd(II) by the Ag(I) salt present. Two other alternative mechanisms for the formation of the product from the intermediate B were also proposed (Scheme 84).
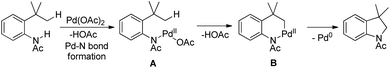 |
| | Scheme 84 Plausible mechanism for cyclization of N-(2-tert-butylphenyl)acetamide to indolines. | |
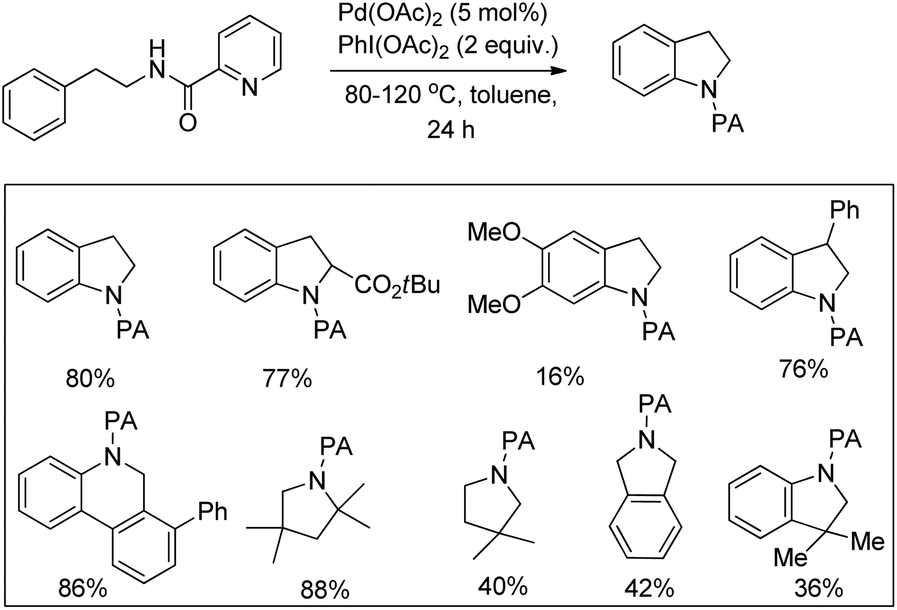
Daugulis et al. used picolinamide (PA) as a directing group in C–H functionalization reactions.65 They further reported the synthesis of indolines and tetrahydroquinolines by Pd-catalyzed C–N bond formation using PA as a directing group. Using alkyl picolinamide substrates pyrrolidine derivatives could also be synthesized in good yields. The reaction is carried out in the presence of the PhI(OAc)2 oxidant and toluene solvent at 80–120 °C (Scheme 85).66
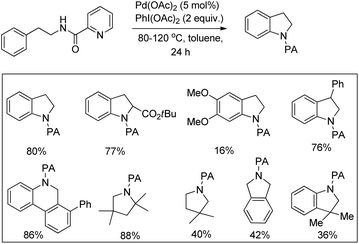 |
| | Scheme 85 DG assisted synthesis of indolines by C–H amination. | |
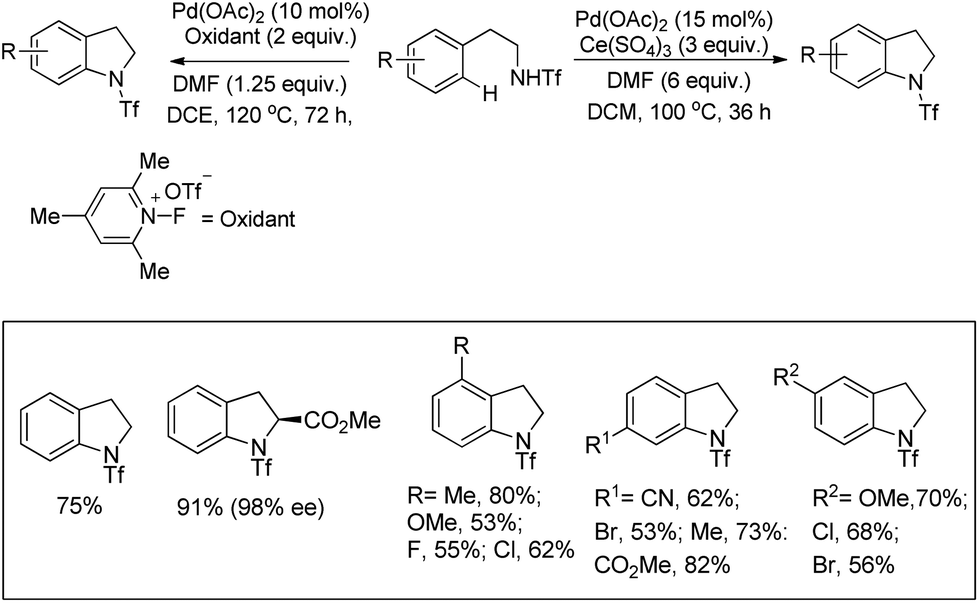
Yu et al. developed a Pd-catalyzed C–H amination reaction for the synthesis of an N-Tf indoline derivative using either Ce(SO4)2 as a one-electron oxidant or a F+ two-electron oxidant (Scheme 86).67 When Ce(SO4)2 was used as an oxidant the yield of the product was reduced which may be due to the formation of unreactive PdSO4. This problem could be overcome by using a F+ oxidant.
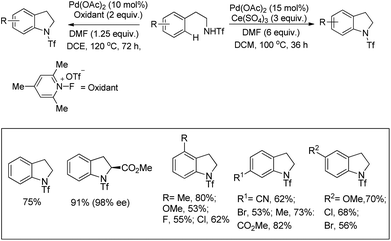 |
| | Scheme 86 Pd-Catalyzed C–H amination reaction by using a one- or two-electron oxidant. | |
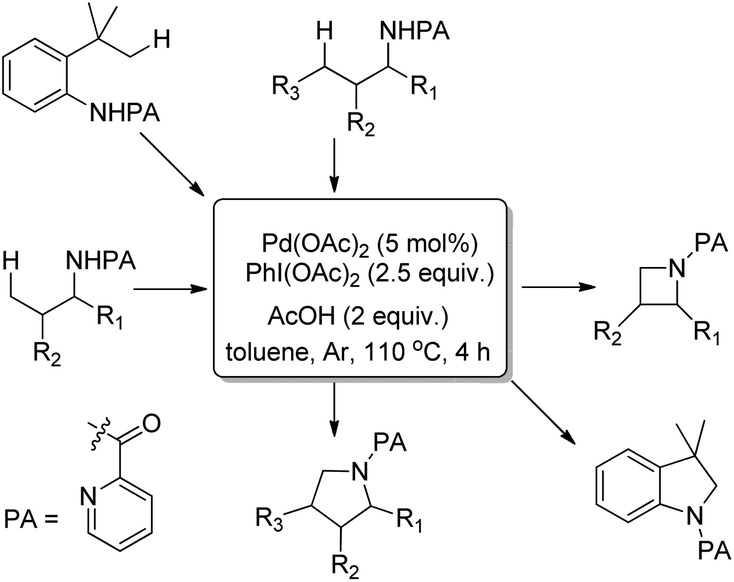
Using the same F+ oxidant and the Pd(OAc)2 catalyst Yu et al. further reported the synthesis of indolines from NH-Tf phenylalanine.68 Chen et al. developed the synthesis of indoline compounds from PA-protected β-arylethylamine substrates using Pd(OAc)2 as a catalyst and PhI(OAc)2 as an oxidant through intramolecular amination of ortho-C(sp2)–H bonds.69 They further developed Pd-catalyzed methods for the synthesis of different heterocyclic compounds such as azetidines, pyrrolidines, and indolines via C–H amination at the γ- and δ-positions of picolinamide (PA) protected amine substrates (Scheme 87).70 Indolines were also synthesized by this method but with or without the use of AcOH in the reaction.
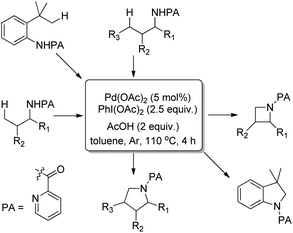 |
| | Scheme 87 Synthesis of indoline and other heterocyclic compounds using a Pd-catalyzed reaction. | |
Various other groups have used the Pd-catalyzed atom economical C–H amination method for the synthesis of indoline compounds from β-arylamine substrates having different amine protecting groups.71
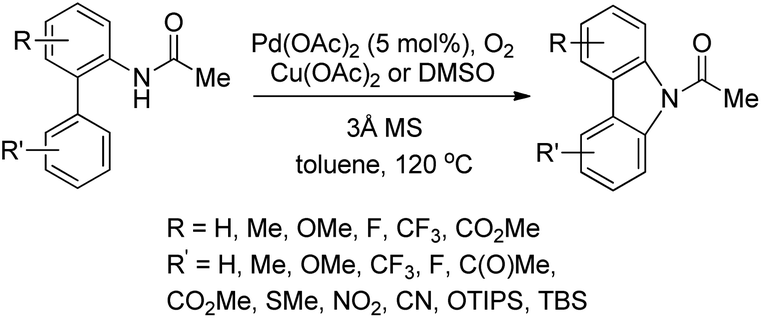
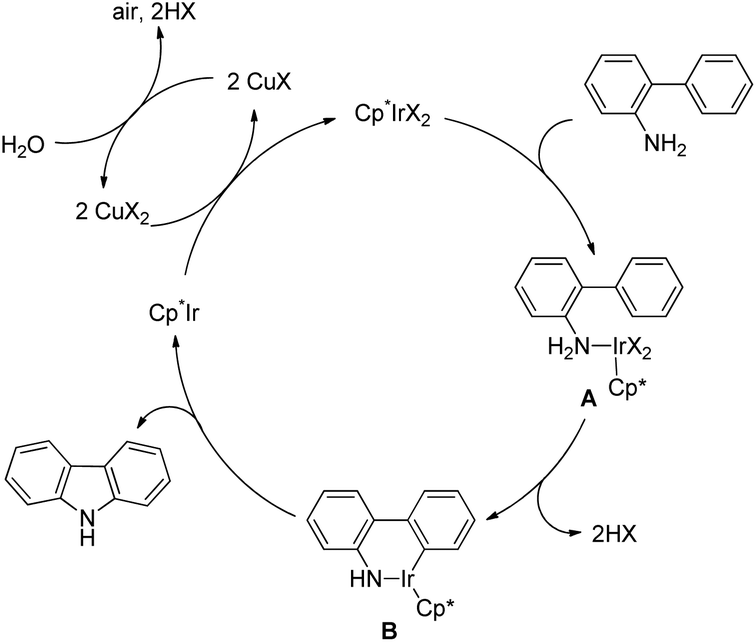
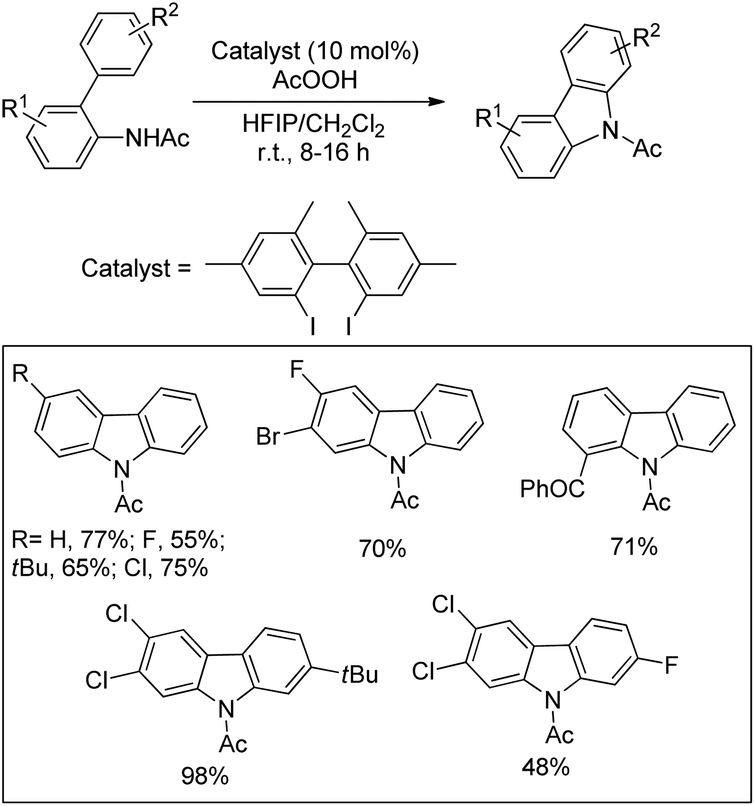
3.1.2 Synthesis of carbazole derivatives.
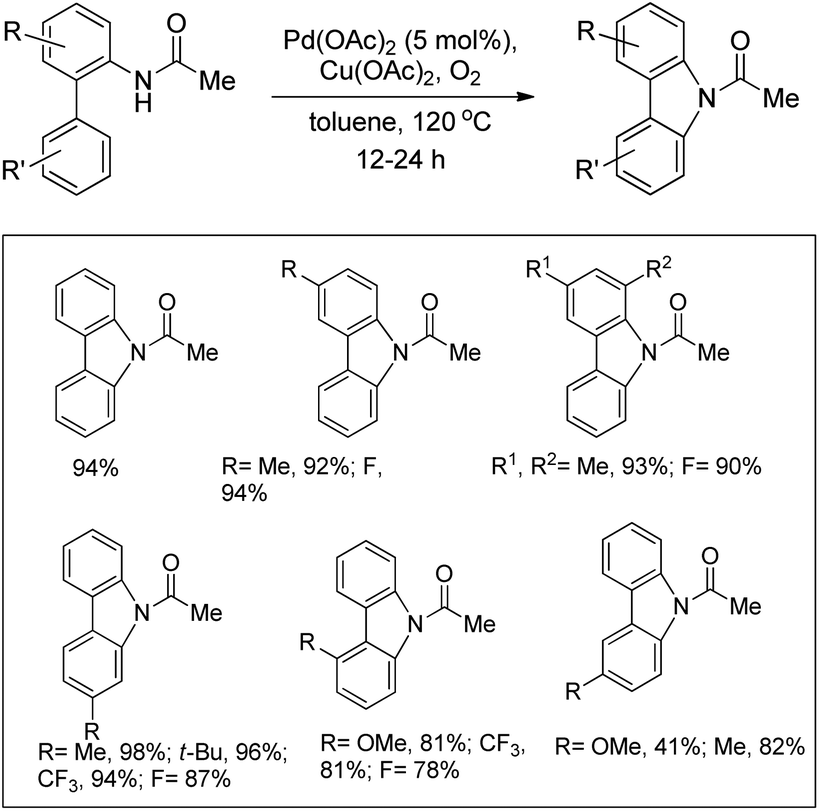
The synthesis of carbazoles by C–H functionalization and C–N bond formation was reported by Buchwald and co-workers.72 2-Phenylacetylides undergo cyclization in the presence of a Pd-catalyst under an oxygen atmosphere to give carbazoles (Scheme 88).
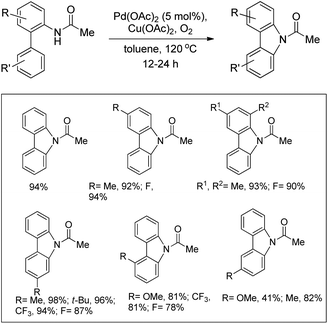 |
| | Scheme 88 Formation of carbazoles by C–N bond formation. | |
The amide substrate adds to the Pd(OAc)2 catalyst which helps the ortho-palladation process and an AcOH molecule is released. Formation of the 6-membered palladacycle and reductive elimination give the carbazole and Pd(0) intermediate. Pd(II) is formed again by the re-oxidation of the Pd(0) species by Cu(OAc)2 (Scheme 89).
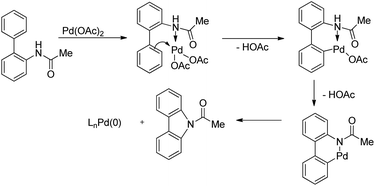 |
| | Scheme 89 Tentative mechanism for the formation of carbazoles. | |
They further developed a method for the synthesis of unsymmetrical carbazoles by a Pd-catalyzed reaction (Scheme 90).73 Different carbazoles could be synthesized by using suitable biaryl amide substrates. The use of Cu(OAc)2 was not necessary when the reaction was carried out in DMSO as a solvent.
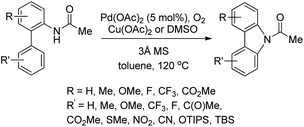 |
| | Scheme 90 Synthesis of unsymmetrical carbazoles. | |
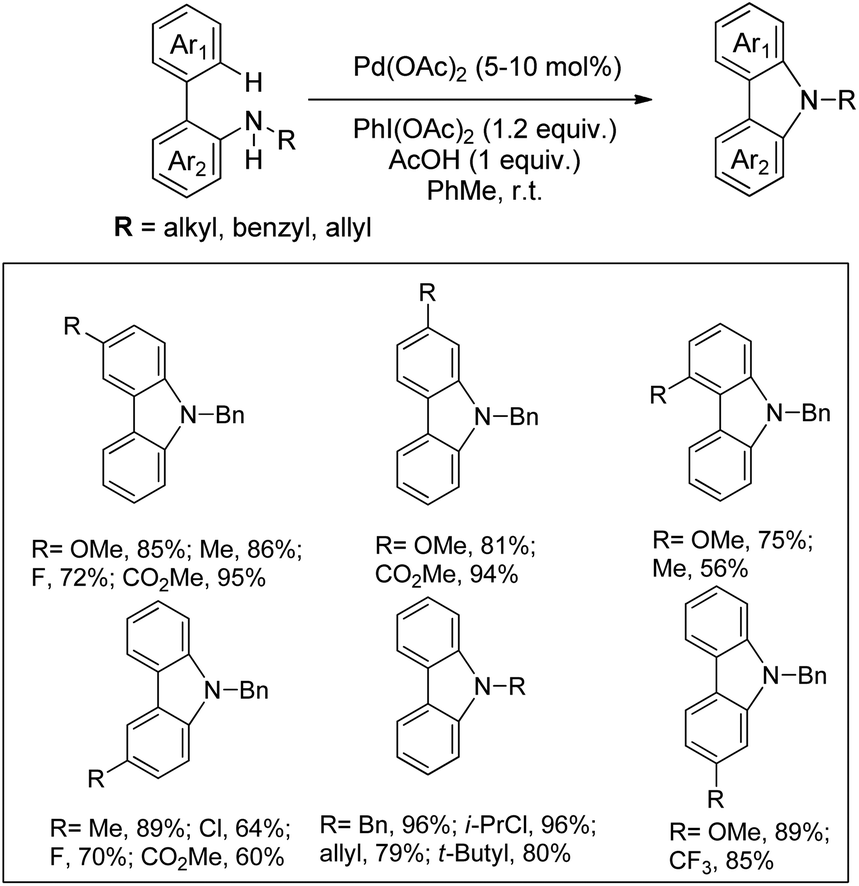
Gaunt and co-workers reported the synthesis of carbazole derivatives from N-alkyl biphenyl using Pd(OAc)2 as a catalyst and PhI(OAc)2 as an oxidant (Scheme 91).74 Substrates having an electron donating group in the Ar1 ring reacted faster than those having an electron withdrawing group which may be due to better coordination of the metal with the more electron-rich C–H bond of the substrate.
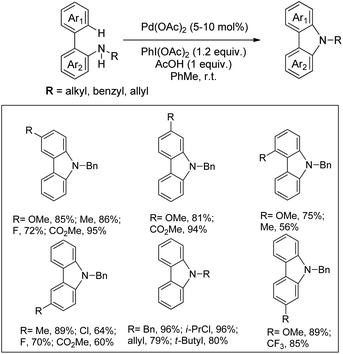 |
| | Scheme 91 Synthesis of carbazoles using PhI(OAc)2 as an oxidant. | |
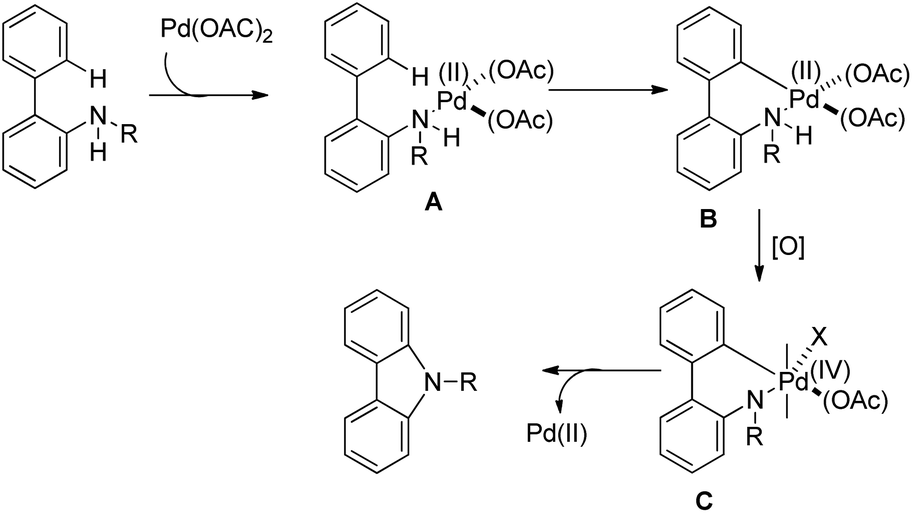
The amine first coordinates with the Pd(II) catalyst resulting in the formation of A which undergoes cyclopalladation to form B. After that, oxidation of the Pd(II) intermediate B to Pd(IV) species C would facilitate C–N bond formation giving the carbazole product (Scheme 92).
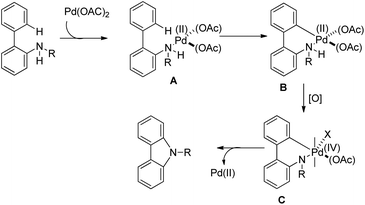 |
| | Scheme 92 Tentative mechanism for Pd-catalyzed C–H amination. | |
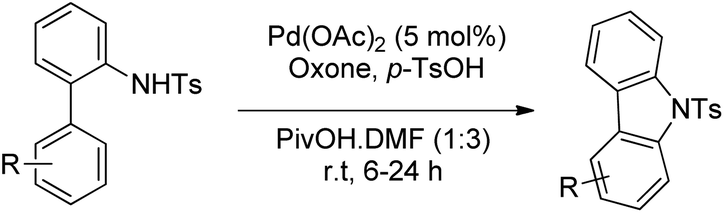
Youn et al. reported a Pd-catalyzed synthesis of carbazoles from N-Ts-2-arylanilines via C–H amination using oxone as the oxidant in the presence of p-toluenesulfonic acid (Scheme 93).75
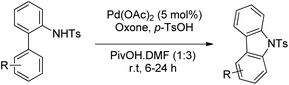 |
| | Scheme 93 Pd-Catalyzed oxidative C–H amination using oxone as an oxidant. | |
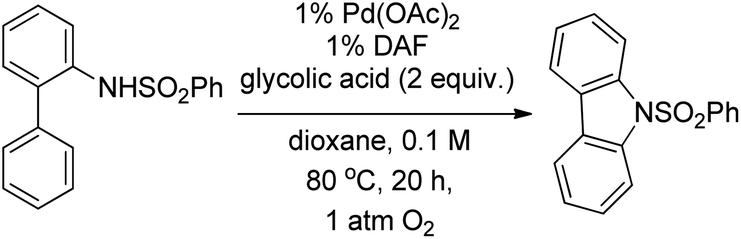
Stahl et al. reported the synthesis of carbazoles using a Pd-catalyzed reaction in the presence of the 4,5-diazafluorenone (DAF) ligand (Scheme 94).76 They observed that the peroxide generated from the decomposition product (1,4-dioxane-2-hydroperoxide and glycolic acid) of 1,4-dioxane promoted the C–H amination reaction.
 |
| | Scheme 94 Carbazole synthesis mediated by in situ formed peroxides from 1,4-dioxane. | |
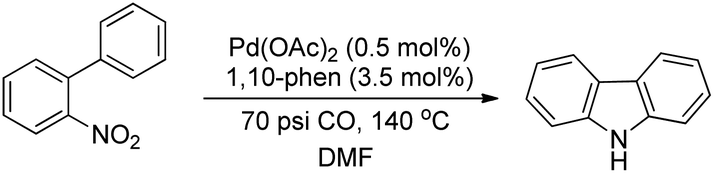
Smitrovich et al. used CO as a stoichiometric reductant for the synthesis of carbazoles from 2-nitro-1,1′-biphenyl derivatives as substrates (Scheme 95). The byproduct of the reaction is CO2 and the products are obtained in a good yield.77
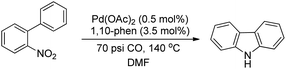 |
| | Scheme 95 C–H functionalization using CO as a stoichiometric reductant. | |
3.1.3 Synthesis of indole and oxiindole derivatives.
Narasaka et al. reported the synthesis of 2,3-disubstituted indoles from 2-aryl-2H-azirines using a Rh(II) catalyst (Scheme 96).78 C2 disubstituted 2H-azirines gave moderate to good yields of the desired products. However, the reaction failed to give any product when C2 monosubstituted 2H-azirine was used as a substrate.
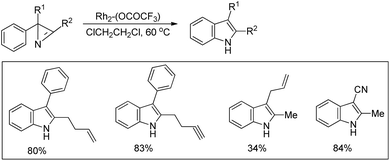 |
| | Scheme 96 Synthesis of 2,3-substituted indoles by a Rh catalyzed reaction. | |
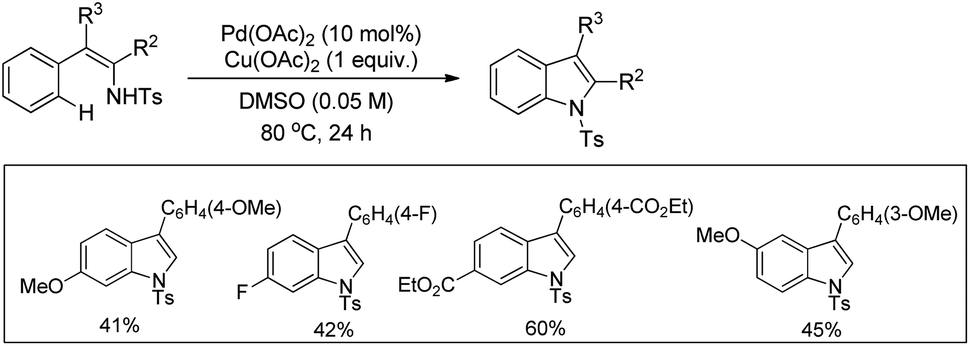
3-Substituted indoles were synthesized by Pd-catalyzed C–H activation followed by an intramolecular C–N bond formation reaction of enamine compounds in the presence of Cu(OAc)2 in DMSO (Scheme 97).79
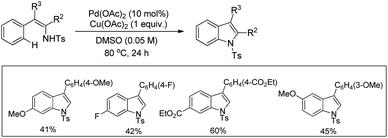 |
| | Scheme 97 Synthesis of 3-substituted indoles by a C–H activation reaction. | |
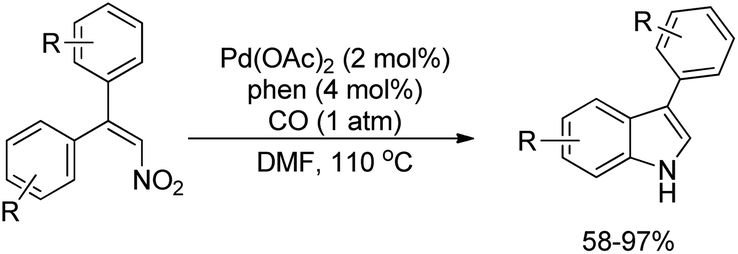
Dong et al. reported the synthesis of indoles using the reductive cyclization of conjugated nitroalkenes through a Pd-catalyzed reaction (Scheme 98).80 Carbon monoxide was used as an inexpensive stoichiometric reductant.
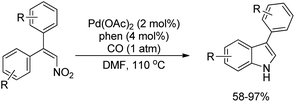 |
| | Scheme 98 Synthesis of indoles using the reduction of nitroalkenes by CO. | |
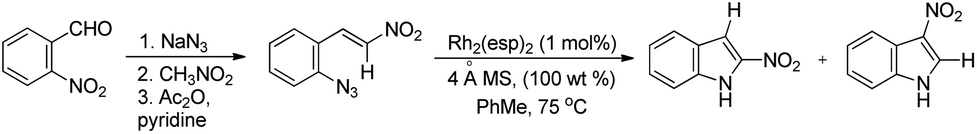
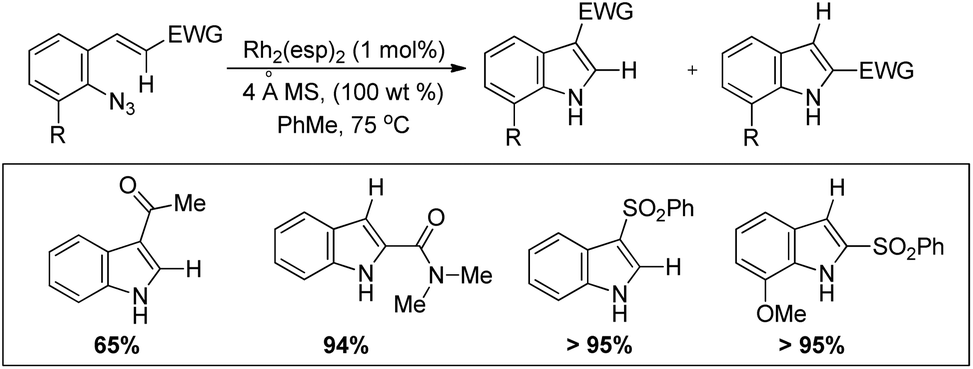
Driver et al. reported the synthesis of 3-substituted indoles from β-substituted styryl azides using the Rh2(esp)2 catalyst, where esp is α,α,α′,α′-tetramethyl-1,3-benzenedipropionate (Scheme 99).81 The reaction involves migration of the electron withdrawing group giving the 3-substituted indole as the major product. For β-nitro styryl azides the presence of a substituent at the ortho-position of the azide group greatly affects the ratio of 2- and 3-nitro indole products. The presence of groups like CF3 gave 95% 2-nitroindole product.
 |
| | Scheme 99 Synthesis of 3-nitro indoles from β-nitro styryl azides. | |
The authors also reported the reaction using different substrates having electron withdrawing groups at the β-position (Scheme 100). 3-Substituted indoles were obtained as the main product for substrates having alkyl or aryl ketones. Substrates having ester and amide moieties at the β-position led to the formation of 2-substituted indoles as the major product. 3-Sulfonylindoles were obtained as the major product when sulfones were used as a substrate. The migration of the sulfonyl group to the 3-position can be inhibited by using an ortho-methoxy-substituted azide substrate. The migratory ability of the –NO2 group is the highest and that of the alkyl group is the lowest.
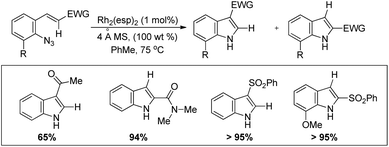 |
| | Scheme 100 Synthesis of 3-substituted indoles using substrates having different EWGs by a Rh catalyzed reaction. | |
Hartwig et al. reported the synthesis of arylindoles from oxime acetates using a Pd-catalyzed reaction (Scheme 101).82 The reaction occurs under redox neutral conditions.
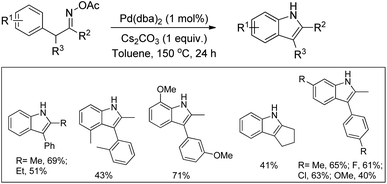 |
| | Scheme 101 Pd-Catalyzed synthesis of substituted indoles. | |
Initially oxidative addition of the N–O bond of the oxime acetate substrate to the Pd(0) catalyst gives the intermediate I which after tautomerization generates complex II. This intermediate II after C–H bond cleavage gives the palladacycle III. The cyclised indole product could be obtained from III through the reductive elimination regenerating the Pd(0) catalyst (Scheme 102).
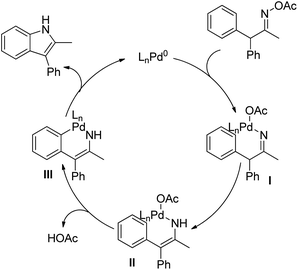 |
| | Scheme 102 Mechanism for the Pd-catalyzed synthesis of indoles. | |
Stahl, Koenig and co-workers reported the synthesis of indole 2-carboxylates from 2-acetamido-3-arylacrylates using a Pd-catalyzed C–H amination reaction (Scheme 103).83 The products could be easily deacetylated using basic hydrolysis giving free indole carboxylates. Some of the suitably substituted indole products could be further functionalized using the Suzuki coupling reaction.
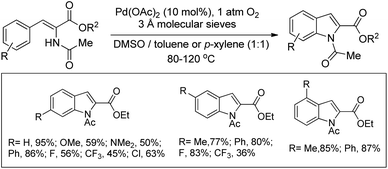 |
| | Scheme 103 Synthesis of indole-2-carboxylates from aryl C–H amination. | |
A Pd-catalyzed reaction for the synthesis of oxindoles from N-tosylphenylacetamide derivatives in the presence of Cu2(OAc)2 was reported by Murakami et al. (Scheme 104).84
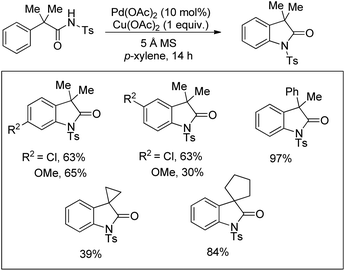 |
| | Scheme 104 Pd-Catalyzed synthesis of 3,3-disubstituted oxindoles. | |
The mechanism starts with the coordination of the Pd(II) catalyst with the N-tosyl substrate to give A which after removal of one AcOH molecule generates the palladacycle B. Intermediate B after reductive elimination gives the product and Pd(0) which gets reoxidized to Pd(II) in the presence of a Cu(II) salt and oxygen (Scheme 105).
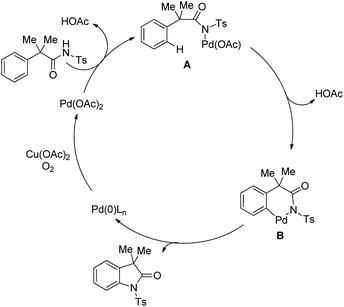 |
| | Scheme 105 Tentative mechanism for the Pd-catalyzed synthesis of oxindoles. | |
Yu et al. used a Pd-catalyzed C–H activation reaction for the synthesis of β-, γ-, and δ-lactams from N-alkoxyamides (Scheme 106).85 No significant difference in the yield was observed when different electron withdrawing and electron donating groups were used in the aryl ring. The reaction could also be applied for allylic N-alkoxyamide substrates.
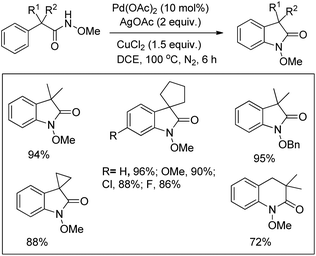 |
| | Scheme 106 Synthesis of lactams by a Pd-catalyzed reaction. | |
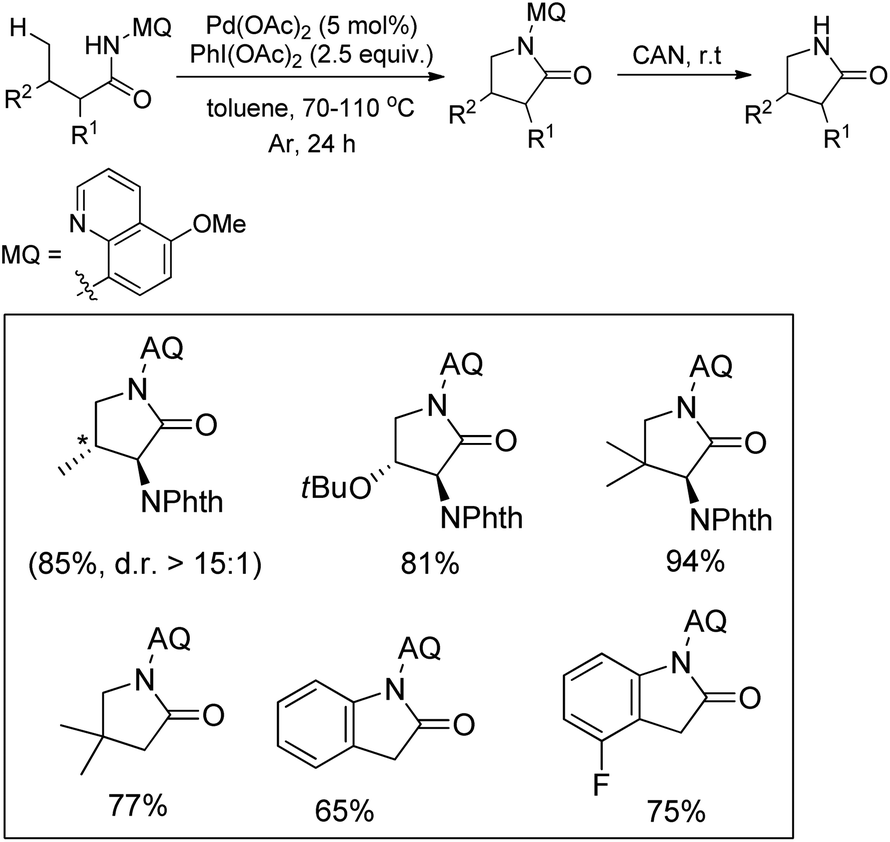
3.1.4 Synthesis of pyrrolidines and pyrrolidones.
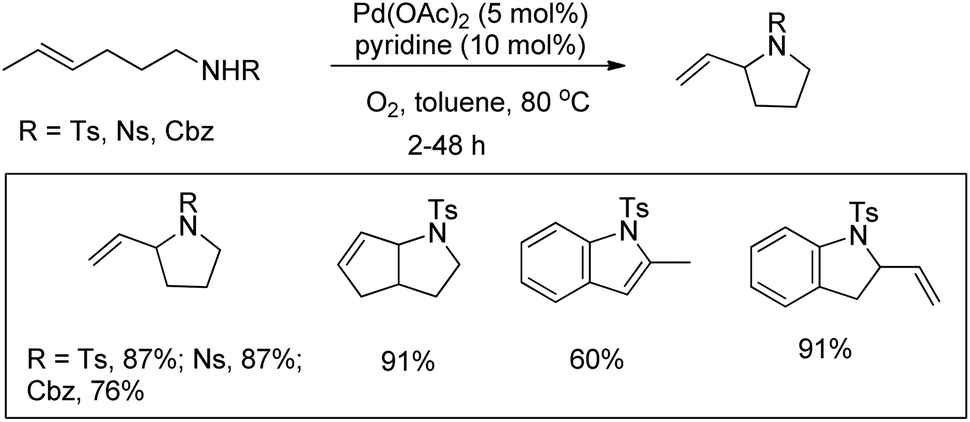
Hegedus in his pioneering work reported a Pd-catalyzed oxidative C(sp2)–H amination reaction using o-allylanilines for the synthesis of indoles and nitrogen heterocycles.86 Stahl et al. used olefinic amines or amides for the synthesis of pyrrolidine compounds using a Pd-catalyzed oxidative amination reaction in toluene as a solvent (Scheme 107).87 Screening of solvents revealed that the reaction works well when nonpolar solvents are used. When the amine protecting group is changed from Ts to Cbz the reaction becomes slower and requires 48 h to give 76% of the product.
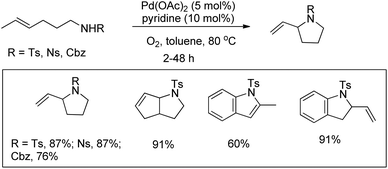 |
| | Scheme 107 Synthesis of pyrrolidines by a Pd-catalyzed reaction. | |
They further reported the synthesis of pyrrolidine derivatives by a Pd-catalyzed intramolecular oxidative C–N bond formation reaction.88
Chen et al. reported the synthesis of pyrrolidones by the directing group assisted Pd-catalyzed intramolecular C–N bond formation reaction (Scheme 108).89 Butanamide substrates having substituents at both α- and β-positions gave a good yield of the products. Indolinones were obtained as a product when aryl acetamides were used as a substrate. The reaction proceeds through the formation of 6-membered palladacycle intermediates giving the γ-lactum products with good selectivity. The directing group could be easily removed by treatment with CAN.
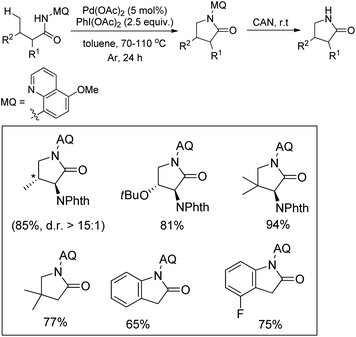 |
| | Scheme 108 Directing group assisted Pd-catalyzed synthesis of pyrrolidones. | |
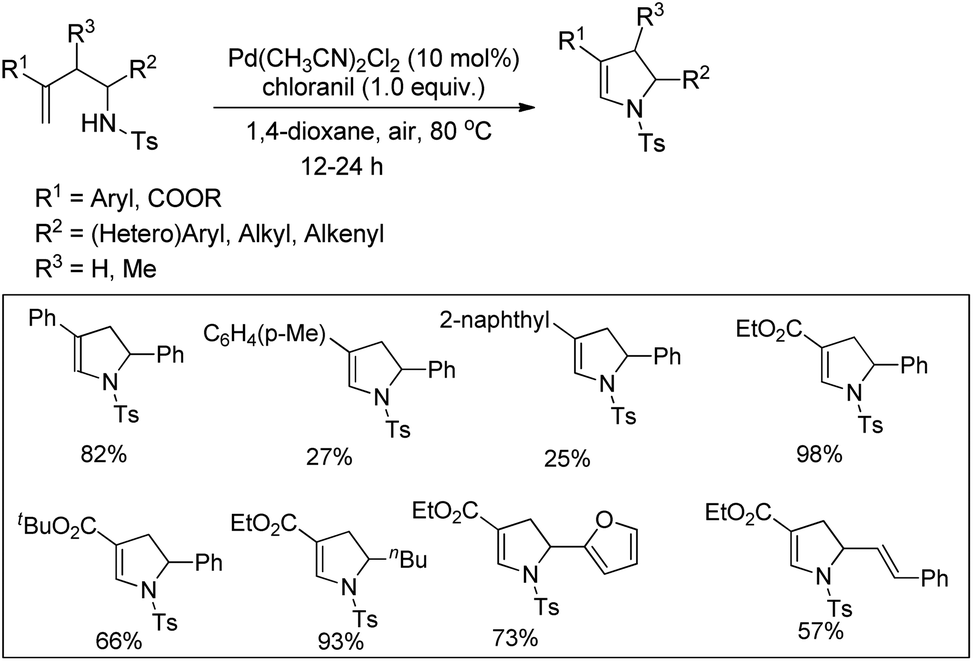
Substituted dihydropyrroles were synthesized by Loh and co-workers from alkenes having a tosyl protected NH group using a Pd-catalyzed reaction under an oxygen atmosphere (Scheme 109).90
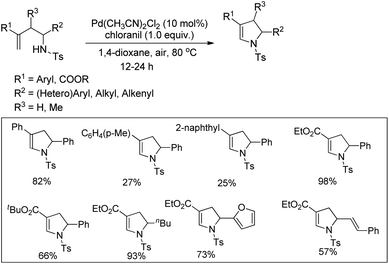 |
| | Scheme 109 Synthesis of substituted dihydropyrroles using a Pd-catalyst. | |
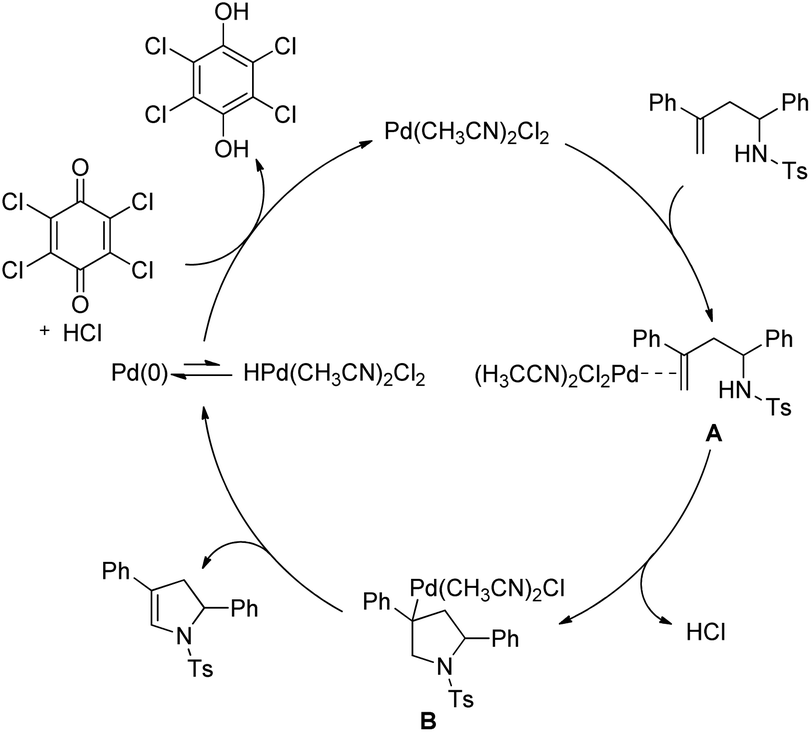
The authors proposed the following mechanism with the help of various controlled experiments (Scheme 110). Initially, the Pd(II) catalyst coordinates with the alkene forming the intermediate A. Nucleophilic attack by the NH-group on the coordinated olefin leads to the formation of B which subsequently gives the product after β-hydride elimination releasing the palladium catalyst. Kinetic isotope studies prove that the β-hydride elimination is not the turnover limiting step in the catalytic cycle.
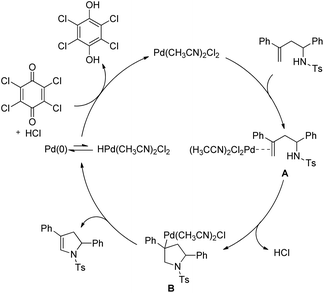 |
| | Scheme 110 Tentative mechanism for the formation of dihydropyrroles. | |
3.1.5 Synthesis of other heterocyclic compounds.
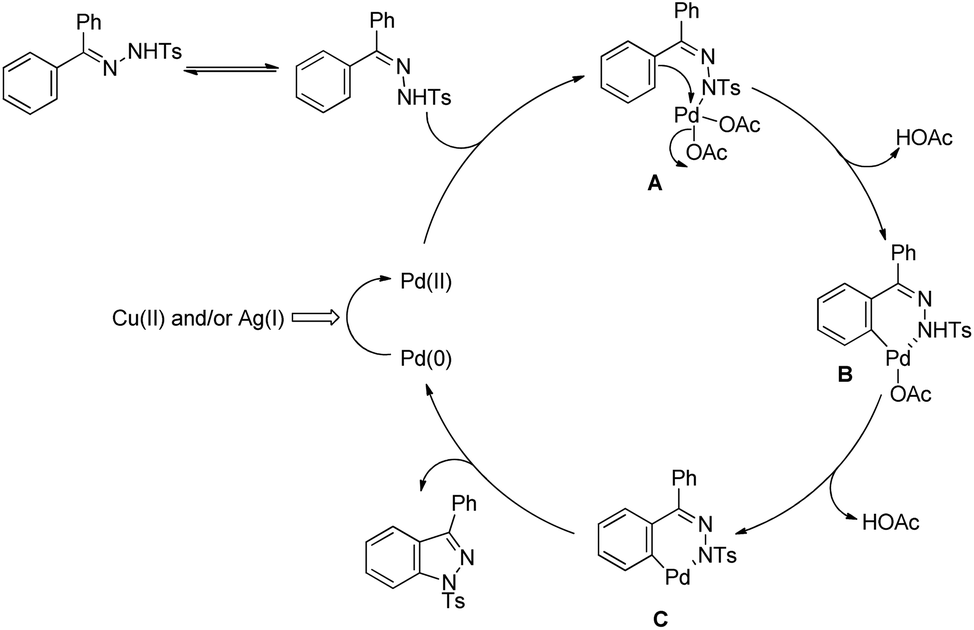
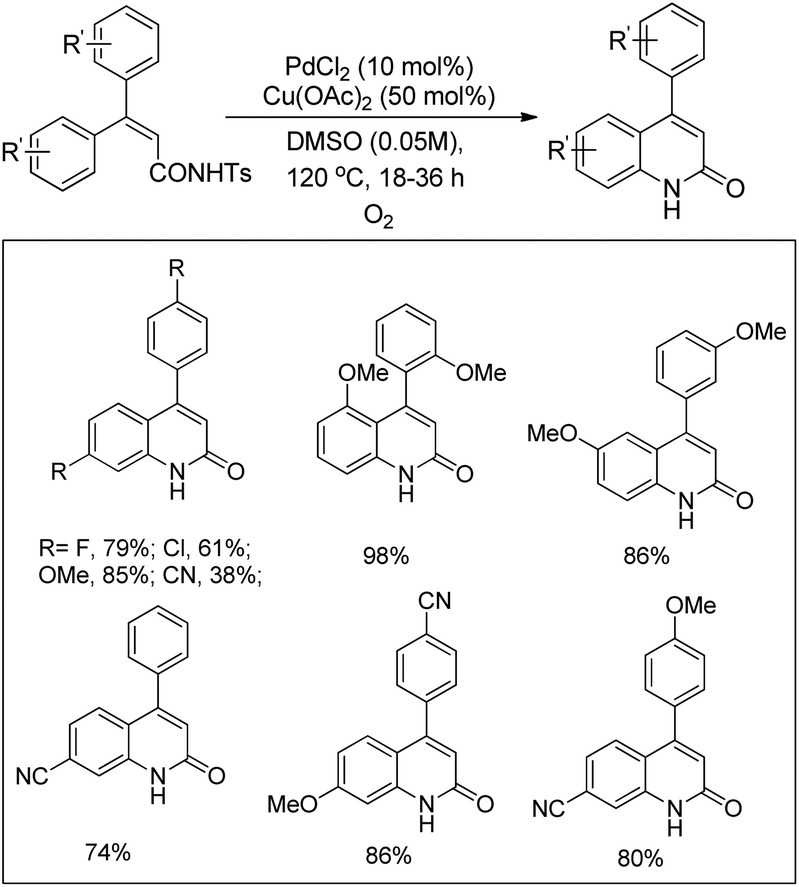
Hiroya and co-workers used C–H activation followed by intramolecular C–N bond formation on diaryl tosylhydrazones for the synthesis of indazoles (Scheme 111).91 The presence of Ag(I) and Cu(II) salts was essential for the cyclization reaction. When tosyl hydrazone substrates with different substituents in both the aryl groups were used the reaction gave a mixture of products depending on the C(sp2)–H amidation bond formed on each aryl ring.
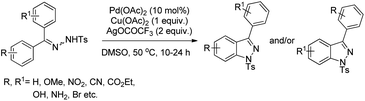 |
| | Scheme 111 Pd-Catalyzed synthesis of indazoles. | |
The reaction starts with coordination of the Pd(II)-catalyst with the substrate which after C–H activation gives the intermediate B. This intermediate after removal of AcOH gives the palladacycle C which subsequently after reductive elimination gives the product (Scheme 112).
 |
| | Scheme 112 Tentative mechanism for the Pd-catalyzed synthesis of indazoles. | |
Doi and co-workers used a Pd-catalyzed intramolecular amidation reaction of N-tosyl-3,3-diarylacrylamides for the synthesis of 4-aryl-2-quinolinones with moderate to good yields (Scheme 113).92 When the unsymmetrical substrates having different R– groups in the aryl rings were used in the reaction, the cyclization occurred in the aryl ring which was in close proximity to the E- or Z-isomer of the amide. By using E- or Z-isomers of the substrates selective C–N bond formation in the products could be achieved.
 |
| | Scheme 113 Pd-Catalyzed 2-quinolinone synthesis via C–N amidation. | |
Fabrizi and co-workers also reported the synthesis of 4-aryl-2-quinolones from o-bromocinnamamide and aryl iodides using Pd(OAc)2 as a catalyst and a molten mixture of Bu4NAc and nBu4NBr as the reaction medium.93
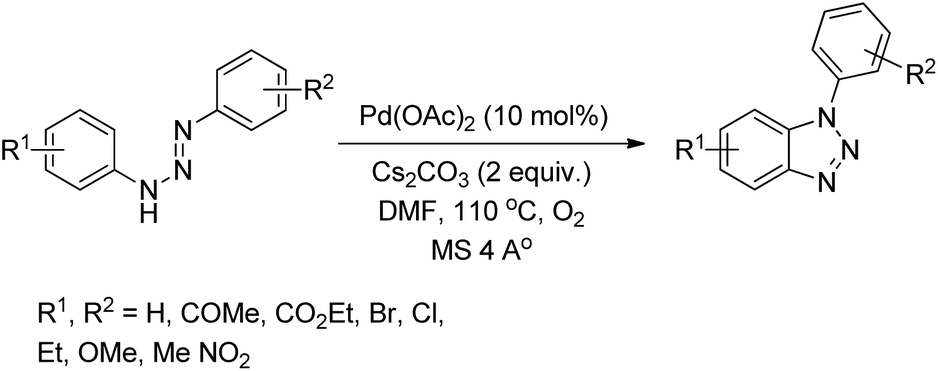
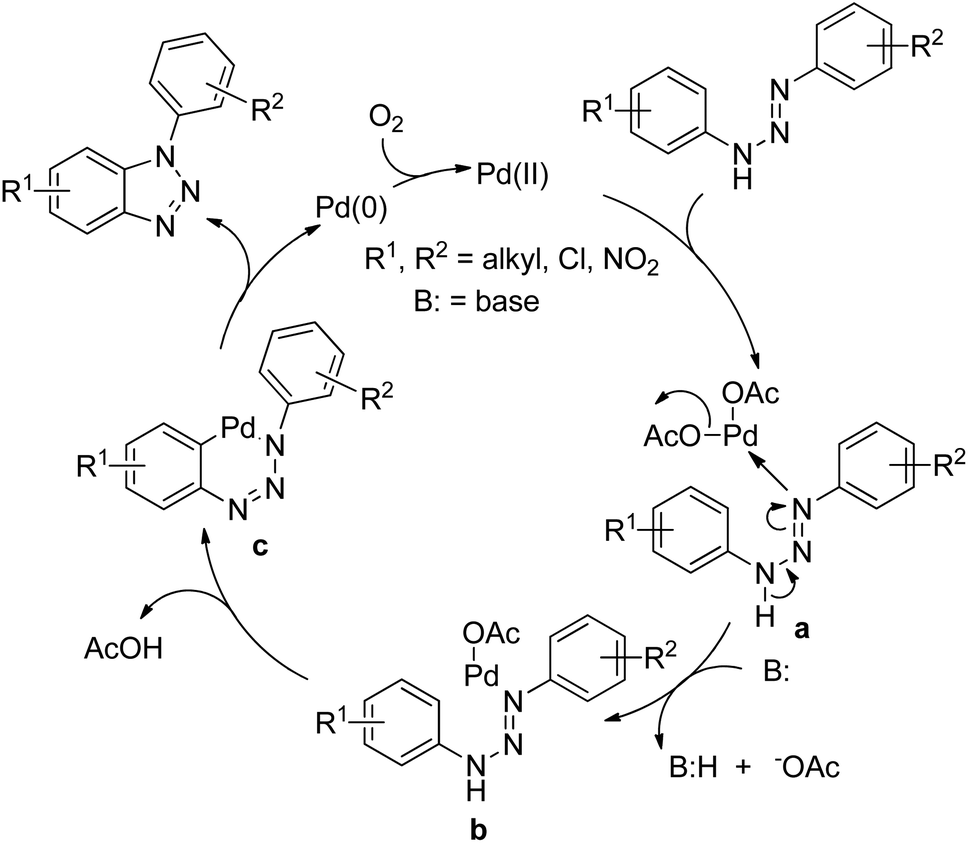
Punniyamurthy et al. reported the synthesis of 1-aryl-1H-benzotriazoles from aryl triazenes using a Pd-catalyzed reaction under an oxygen atmosphere (Scheme 114).94 When substrates having differently substituted aryl rings were used, 2:1 regioisomer formation was observed in which the cyclization occurred preferentially on the non-substituted aryl ring.
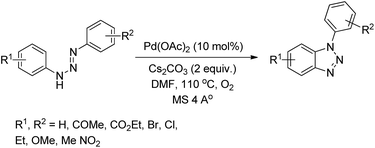 |
| | Scheme 114 Synthesis of 1-aryl-1H-benzotriazoles. | |
The Pd catalyst first coordinates with the aryl triazene substrate which after C–H activation gives the palladacycle c and finally affords the product and reduced Pd(0). The Pd(0) further gets oxidized to Pd(II) in the presence of oxygen (Scheme 115).
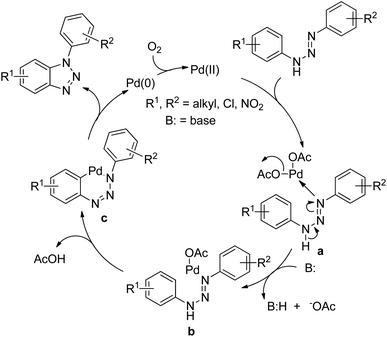 |
| | Scheme 115 Proposed mechanism for the Pd-catalyzed synthesis of 1-aryl-1H-benzotriazoles. | |
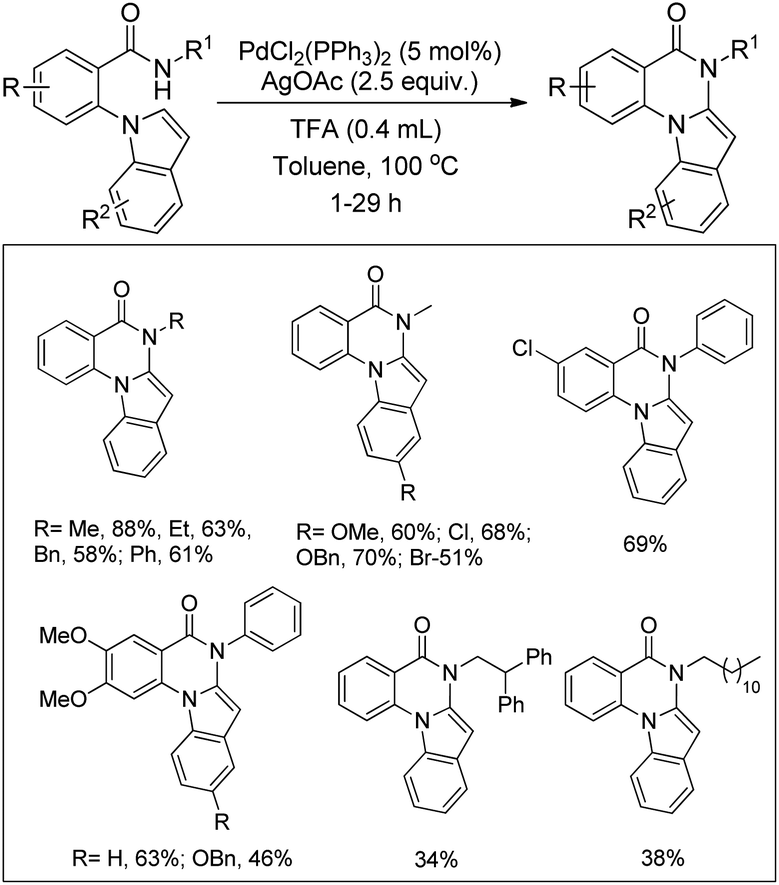
Yao et al. reported the synthesis of indolo[1,2-a]quinazolinones using the Pd(PPh3)2Cl2 catalyst and Ag(OAc)2 oxidant in a stoichiometric amount through the C–H amidation protocol (Scheme 116).95
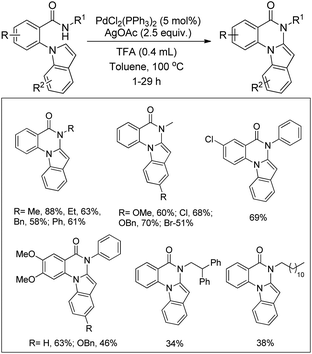 |
| | Scheme 116 Indolo[1,2-a]quinazolinone derivatives through the C–H amidation reaction. | |
3.2. Cu-Catalyzed intramolecular C–N bond formation reaction
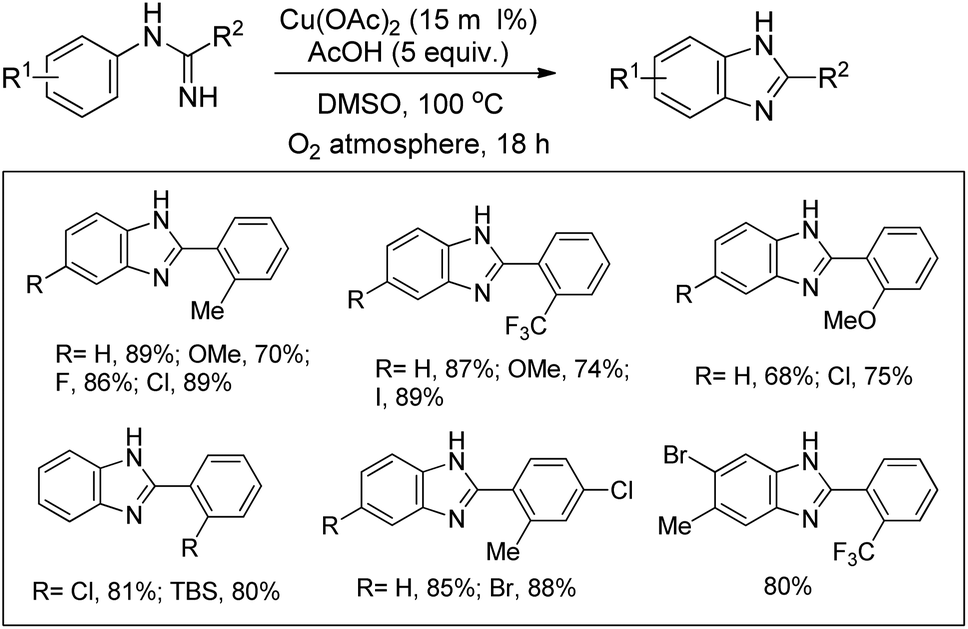
Buchwald et al. reported the synthesis of substituted arylbenzimidazoles from N-arylbenzamidines using Cu-catalyzed C–H amination (Scheme 117).96 High yields of the products were obtained when the substituents were in the 5- and 6-positions of the benzimidazole whereas substitution at the 4- or 4,6-position lowered the product yield.
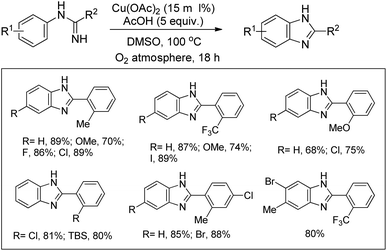 |
| | Scheme 117 Cu-Catalyzed synthesis of benzimidazoles. | |
Shi et al. reported the synthesis of indolines using copper catalyzed amidation of the C(sp3)–H bond (Scheme 118).97 The reaction gave a good yield of the products for substrates having both electron-donating and electron-withdrawing groups in the phenyl ring.
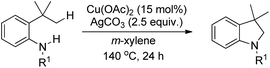 |
| | Scheme 118 Copper catalyzed synthesis of indolines. | |
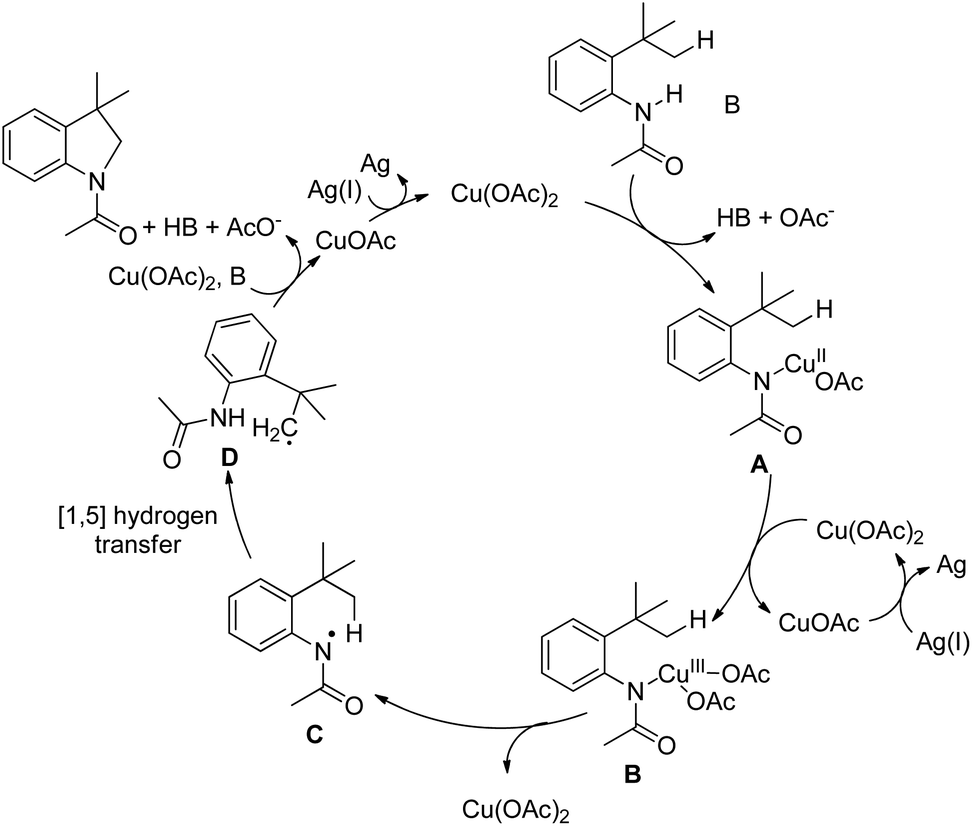
Initially, Cu(OAc)2 coordinates with the amide substrate giving the intermediate A which could be further oxidized to Cu(III) complex B in the presence of Cu(OAc)2 present in the reaction. B then undergoes homolysis producing the nitrogen radical C which after 1,5-hydrogen transfer gives the carbon radical D. Finally, intramolecular C–N cyclization gives the product (Scheme 119).
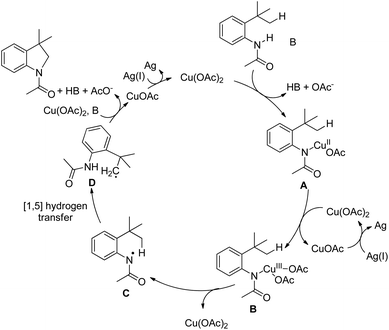 |
| | Scheme 119 Mechanism for Cu-catalyzed intramolecular amidation of the C(sp3)–H bond. | |
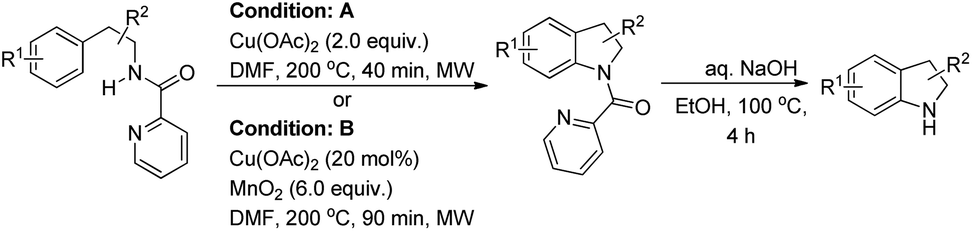
Miura et al. reported a directing group assisted copper mediated or Cu-catalyzed synthesis of indolines under microwave irradiation in the presence of MnO2 as an oxidant (Scheme 120).98 The picolinylamide moiety acts as a directing group. The reaction gave a good yield of heterocycle fused indoline products also. The DG could be easily removed under basic conditions to give the free indoline products.
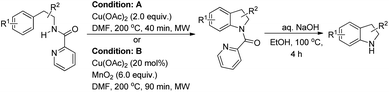 |
| | Scheme 120 Cu-Mediated/-catalyzed intramolecular C–N bond formation. | |
The directing group in the presence of the Cu catalyst helps in forming coordinated intermediate A which after C–H activation yields B. Subsequently one-electron oxidation of Cu(II) into Cu(III) with additional Cu(OAc)2 generates the intermediate C with Cu(III) species. Reductive elimination of C finally gives the product (Scheme 121).
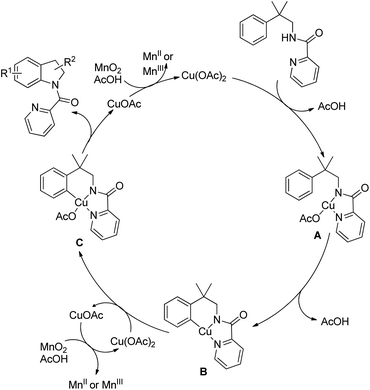 |
| | Scheme 121 Mechanism for DG assisted Cu-catalyzed synthesis of indolines. | |
They also reported a Cu-catalyzed intramolecular C–H amination strategy for the synthesis of carbazoles from picolinamide biaryls (Scheme 122).99 The directing group was removed after the completion of the reaction thus giving access to free carbazoles. The mild conditions allowed the synthesis of pyrrole, thiophene, and furan-based N,S-, N,O-, and N,N-heteroanthracene and tetracene in good yields.
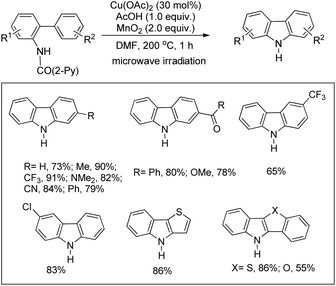 |
| | Scheme 122 Directing group assisted synthesis of carbazoles. | |
Chang and co-workers used PhI(OAc)2 as an oxidant in the presence of Cu(OTf)2 as a catalyst and TFA for the synthesis of carbazoles from N-substituted 2-amidobiaryls.100 Goggiamani et al. reported the synthesis of 4-aryl-2-quinolones from 3,3-diarylacrylamides using CuI as a catalyst (Scheme 123).101 Both symmetrical and unsymmetrical quinolones could be obtained by this method. For unsymmetrical substrates a mixture of products was obtained; however, high selectivity was obtained when o-Me, o-Cl or o-Br groups were present in the aromatic ring trans to the amide group of substrates.
 |
| | Scheme 123 Symmetrical and unsymmetrical quinolones from acrylamides. | |
An aerobic oxidative intramolecular C–H amination protocol was developed by Fu et al. for the synthesis of imidazobenzimidazoles from substituted 2-(1H-imidazol-1-yl)-N-alkylbenzenamine substrates (Scheme 124).102
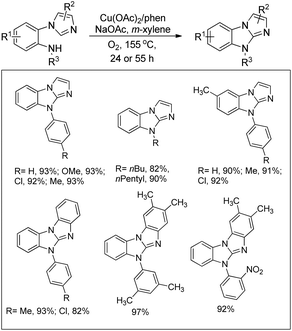 |
| | Scheme 124 Imidazobenzimidazoles from a Cu-catalyzed reaction. | |
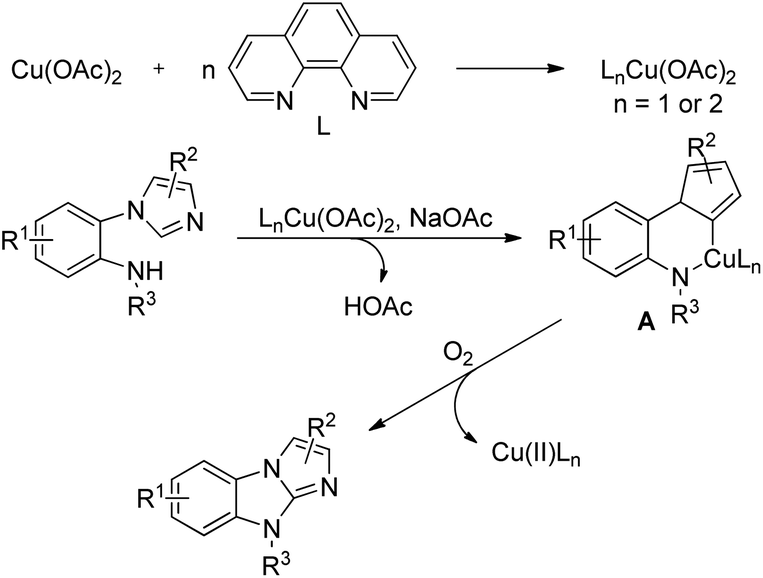
The reaction mechanism involves coordination of the Cu-catalyst with the 1,10-phenanthroline (phen) ligand forming the complex LnCu(OAc)2 which reacts with the hydrazone substrate in the presence of a base (NaOAc) giving the intermediate A. Reductive elimination of A affords the product in the presence of oxygen (Scheme 125).
 |
| | Scheme 125 Mechanism for the synthesis of imidazobenzimidazoles. | |
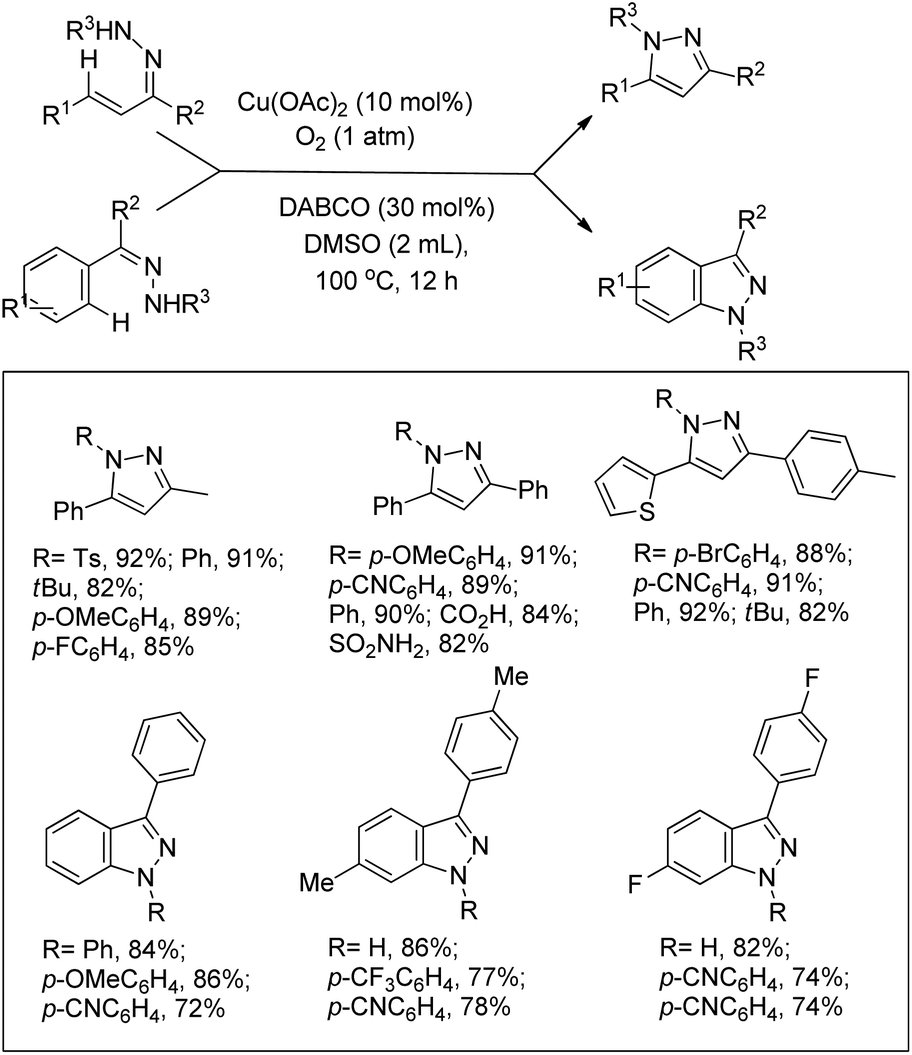
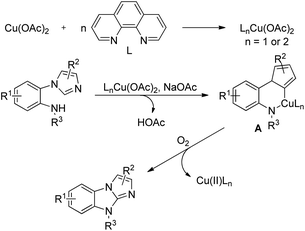
Jiang et al. reported the synthesis of pyrazoles and indazoles using Cu-catalyzed aerobic C(sp2)–H functionalization of the corresponding hydrazone substrates (Scheme 126).103 The products are obtained in good yields and this method works for several functional groups in the substrate.
 |
| | Scheme 126 Cu-Catalyzed aerobic C(sp2)–H functionalization of hydrazones. | |
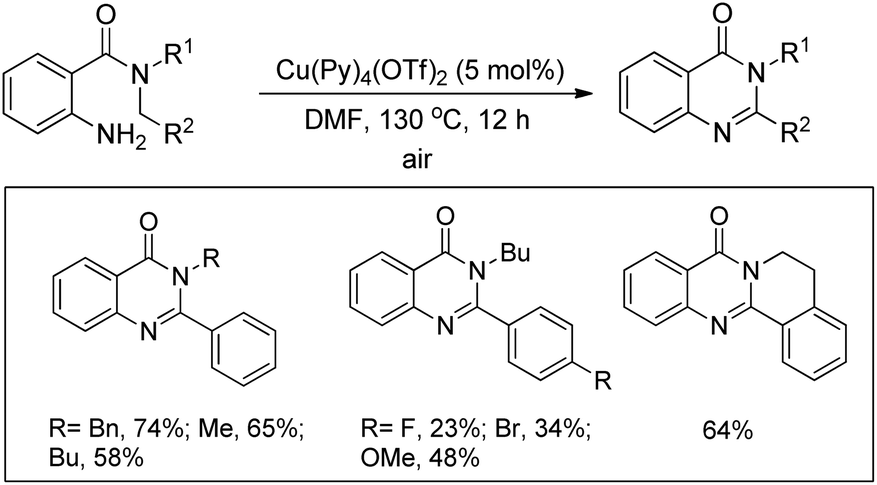
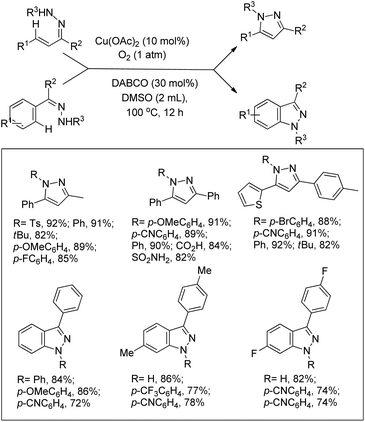
Maiti and co-workers reported the synthesis of 2,3-disubstituted-4(3H)-quinazolinones from 2-amino-N,N-dialkylbenzamides via a Cu-catalyzed C(sp3)–H cyclization (Scheme 127).104 The efficiency of the reaction was largely influenced by the aryl substituent R2. Electron donating groups on the aryl ring slightly lowered the yield whereas electron withdrawing groups provided poorer yields. When secondary aryl amine substrates were used for the reaction dihydroquinazolinones were obtained as the product. A free radical mechanism was proposed for the reaction.
 |
| | Scheme 127 Cu-Catalyzed synthesis of quinazolinones. | |
3.3. Fe-Catalyzed intramolecular C–N bond formation reaction
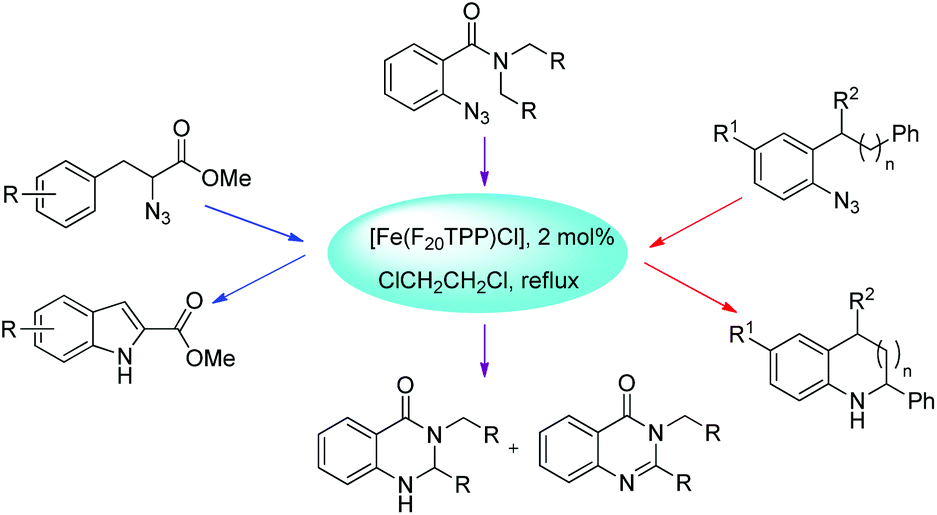
Che et al. used [Fe(F20TPP)Cl] as a catalyst for the synthesis of indoles, indolines, tetrahydroquinolines, dihydroquinazolinones and quinazolinones via intramolecular amination of sp2 and sp3 C–H bonds with aryl/alkyl azides as the nitrogen source (Scheme 128).105 The reaction gave the products in good to moderate yields.
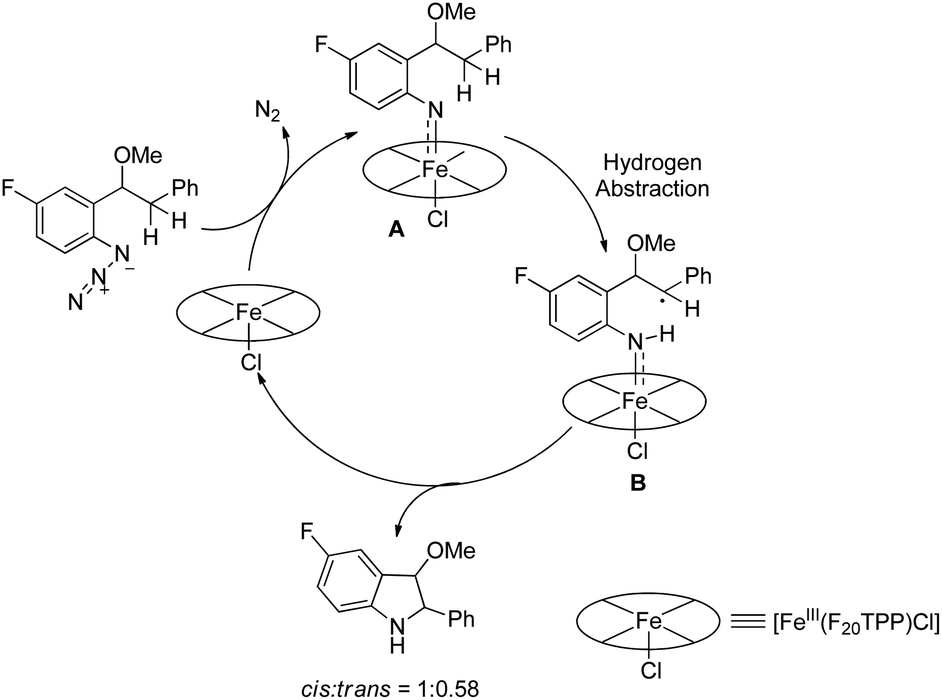
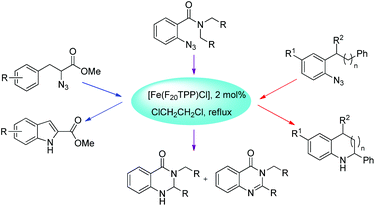 |
| | Scheme 128 Formation of different products catalyzed by [Fe(F20TPP)Cl]. | |
The decomposition of the aryl azide catalyzed by [Fe(F20TPP)Cl] produces the iron-nitrene/imido complex A, which after benzylic hydrogen abstraction gives the radical intermediate B. This radical intermediate undergoes C–N bond formation to give the cis- and trans-isomer product (cis:trans = 1:0.58) (Scheme 129).
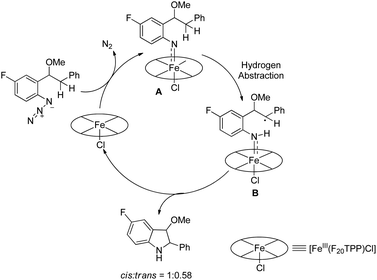 |
| | Scheme 129 Reaction mechanism for the formation of indolines. | |
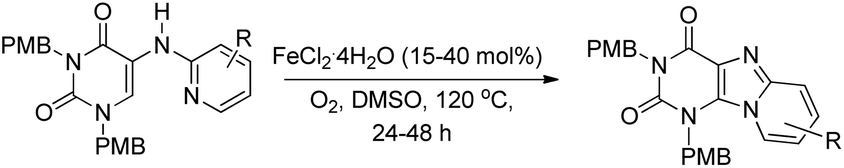
A Fe-catalyzed oxidative C–H amination reaction for the synthesis of purine compounds was reported by Maes et al. (Scheme 130).106 Substrates having electron donating and electron withdrawing groups on the pyridine ring gave products with moderate to good yields.
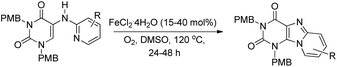 |
| | Scheme 130 Synthesis of purines by a Fe-catalyzed reaction. | |
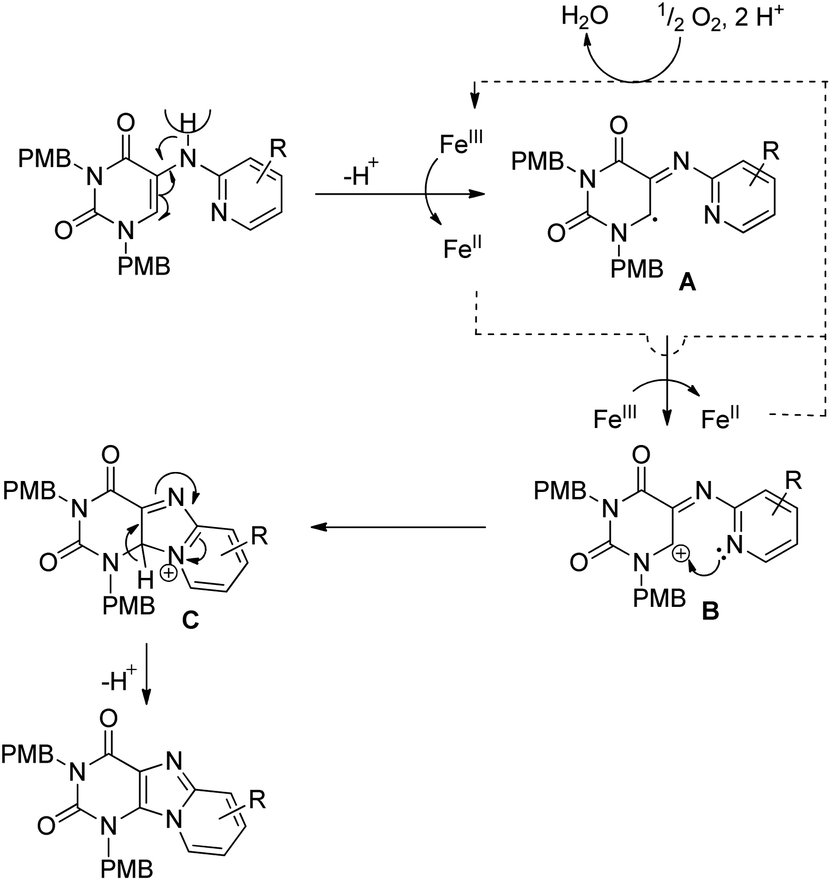
A radical mechanism was proposed for the reaction (Scheme 131). FeIII initiates the catalytic cycle by removing a proton from the substrate generating the radical intermediate A, which after one electron transfer furnishes the carbocation intermediate B. Nucleophilic attack by the pyridine nitrogen atom gives the cyclised intermediate C which after aromatization gives the product.
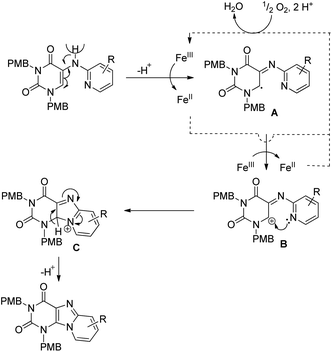 |
| | Scheme 131 Free radical mechanism for the Fe-catalyzed reaction of purine synthesis. | |
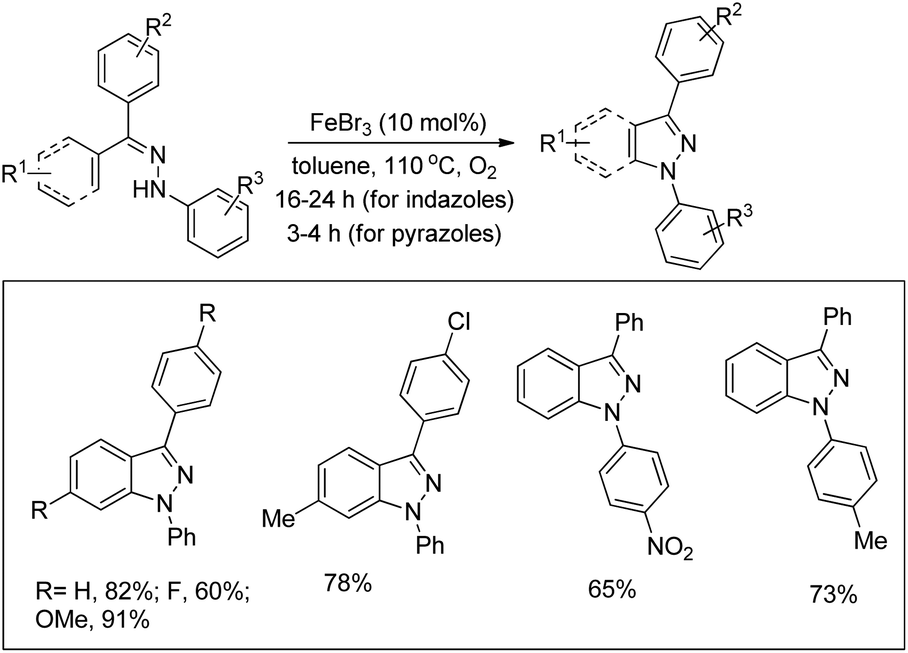
The synthesis of indazoles and pyrazoles from arylhydrazones through Fe-catalyzed C–H amination was reported by Bao et al. in moderate to excellent yields (Scheme 132).107 The phenylhydrazones disubstituted by electron-donating groups gave the indazole products in good yields, whereas the presence of electron-withdrawing groups afforded moderate yields. Hydrazones obtained from unsymmetrical diary ketones gave a mixture of products.
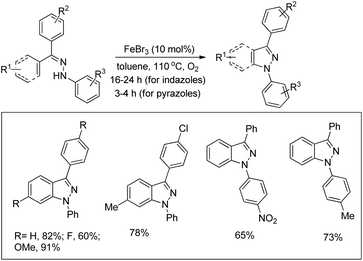 |
| | Scheme 132 Synthesis of indazoles and pyrazoles by the Fe-catalyzed reaction. | |
Pyrazoles could also be synthesized using this method when the substrates derived from α,β-unsaturated ketones and phenylhydrazine were used. Substituents have no significant effect on the yields of the product in the case of pyrazole synthesis.
3.4. Rh-, Ir-, and Ag-catalyzed intramolecular C–N bond formation reaction
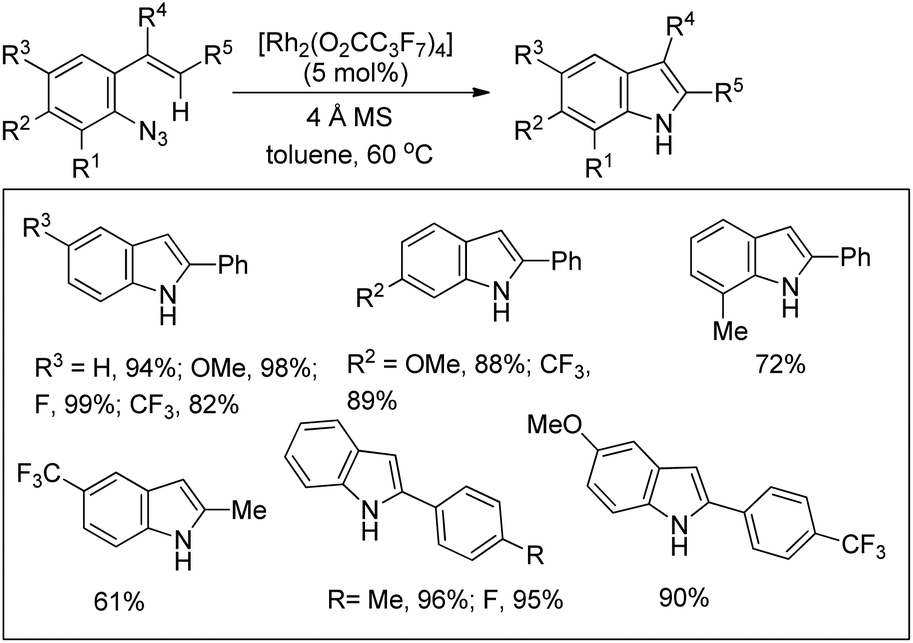
Indoles could be synthesized from aryl azides through a rhodium(II)-catalyzed intramolecular C–N bond formation reaction (Scheme 133).108 In the absence of a molecular sieve the yield decreased substantially. When the R5 group was changed from an aryl group to an alkyl group the yield of the reaction decreased.
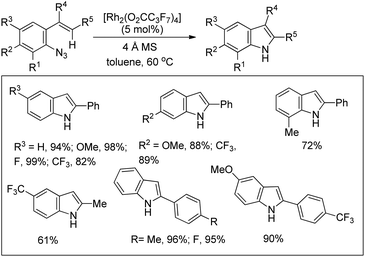 |
| | Scheme 133 Rh(II)-Catalyzed synthesis of indoles. | |
The exact mechanism is uncertain and rhodium is expected to function as a nitrogen atom transfer reagent. Coordination of the Rh(II)-catalyst to the azide would generate A which after removal of one N2 molecule gave intermediate B or D. Functionalization of C–H near the Rh centre would give C through C–N bond formation or through a concerted transition state E. Otherwise, simultaneous formation of a C–N bond with the removal of N2 from A would give the intermediate C which after a 1,5-hydride shift would give the indole product (Scheme 134).
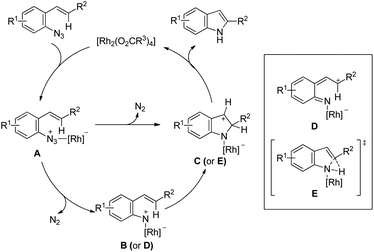 |
| | Scheme 134 Tentative mechanism for the rhodium catalyzed synthesis of indoles. | |
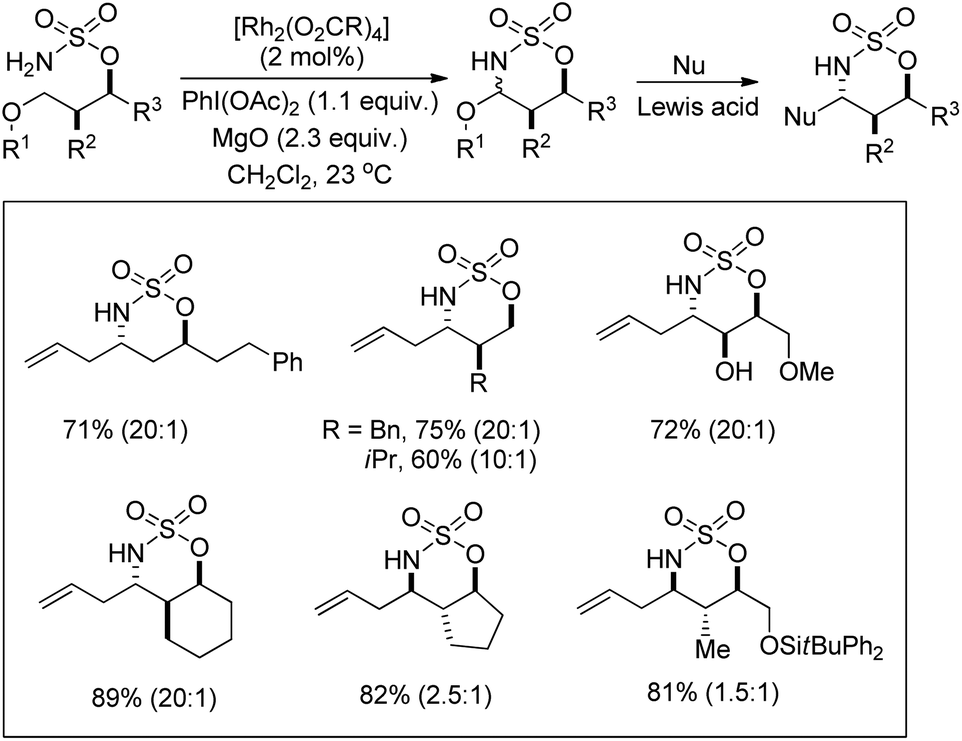
A rhodium catalyzed synthesis of 1,2,3-oxathiazinane-2,2-dioxide heterocycles from sulfamate esters was reported by Du Bois et al. through amination of ethereal α-C–H bonds (Scheme 135).109 The N,O-acetal compounds could be further functionalized with various nucleophiles in the presence of Lewis acids. The regioselectivity of α-C–H bond insertion was improved substantially when [Rh2(O2CCPh3)4] was used as a catalyst instead of the [Rh2(OAc)4] complex. It was observed that the bulky Rh-triphenylacetate catalyst did not favour the insertion into sterically crowded C–H bonds and benzylic sites of substrates offered selectivity.
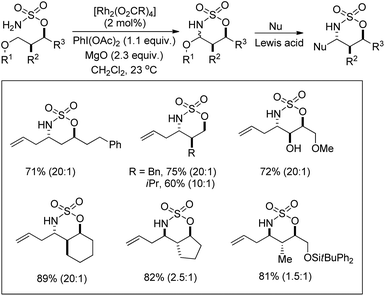 |
| | Scheme 135 Synthesis of oxathiazinane N,O-acetals. The values in parentheses indicates the diastereomeric ratio. | |
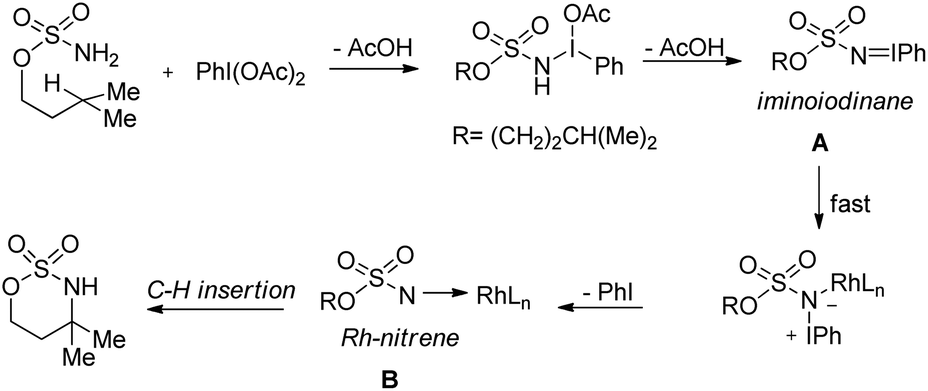
Du Bois conducted a detailed mechanistic study for the synthesis of similar N,O-acetals using various rhodium catalysts (Scheme 136).110 The reaction of PhI(OAc)2 with the sulfamate ester and removal of one AcOH molecule generate the iminoiodinane intermediate A which after elimination of PhI forms the nitrene intermediate B. Finally C–H insertion of this intermediate affords the product.
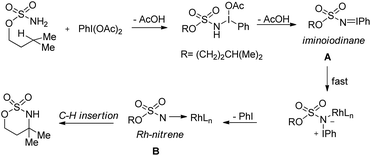 |
| | Scheme 136 Plausible mechanism for the Rh-catalyzed C–N bond formation. | |
Shi et al. reported Ag-catalyzed C–N bond formation for the synthesis of different heterocycles using 4,4′-di-t-butyl-2,2′-bipyridine as a ligand (Scheme 137).111 Various pyrrolidine and tetrahydroquinoline derivatives could be synthesized in good yields by this method which has a preference for primary C(sp3)–H amination.
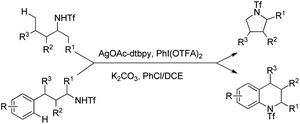 |
| | Scheme 137 Synthesis of 5- and 6-membered N-heterocycles by a Ag-catalyzed reaction. | |
The Ag-catalyst in the presence of strong σ-donor chelating N-ligands forms the Ag complex A which could be oxidized to Ag(III) in the presence of AgI(OTFA)2. The electrophilic Ag(III) centre then attacks the less sterically hindered primary C–H bonds giving the intermediate C which after removal of CF3COOH by deprotonation affords D. Reductive elimination of D gives the product and the Ag(I) catalyst is regenerated (Scheme 138).
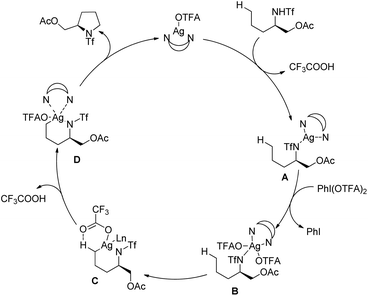 |
| | Scheme 138 Mechanism involving a concerted metallation/deprotonation process. | |
3.5. Intramolecular C–N bond formation reaction using transition metal co-catalysts
Zhu et al. used a Cu(OAc)2 and Fe(NO3)3·9H2O co-catalyst system for the synthesis of pyrido[1,2-a]benzimidazoles from N-aryl-2-aminopyridines via intramolecular C–N bond formation (Scheme 139).112 The reaction was dependent on the substitution on both the pyridine and phenyl rings. When the R2 group was in the p-position and electron withdrawing in nature the reaction took a longer time (up to 66 h in the case of F) to complete compared to the alkyl group (23 h for OMe).
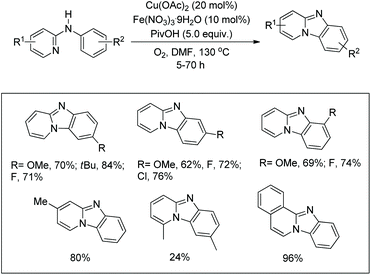 |
| | Scheme 139 Cu(II)/Fe(III) co-catalyzed intramolecular C–H amination. | |
The N-phenyl-2-aminopyridine substrate first forms the Cu(II) complex A which is oxidized to a more electrophilic Cu(III) intermediate E in the presence of Fe(III). E after C–H activation forms the intermediate F which after losing one H+ generates D. Reductive elimination finally gives the product. The reaction can also furnish the product without the presence of an iron catalyst; however, Fe(III) facilitates the generation of the more electrophilic Cu(III) species making the catalytic process much faster (Scheme 140).
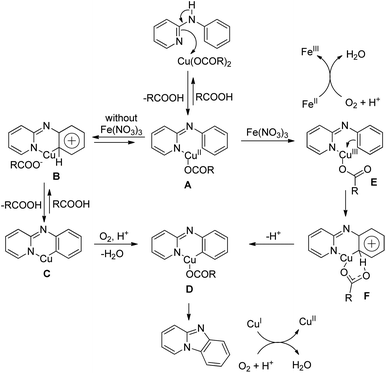 |
| | Scheme 140 Mechanism for the formation of pyrido[1,2-a]benzimidazoles. | |
Roy et al. reported the synthesis of indoloisoquinolines from C3-arylated indoles having an aldoxime group using a Pd and Cu co-catalyst system (Scheme 141).113 It was observed that when only Cu(OTf)2 was used as a catalyst the reaction gave a mixture of cyanide and indoloisoquinoline from the corresponding aldoximes. However, the use of the Pd(OAc)2 catalyst in combination with a copper catalyst led to the selective formation of indoloisoquinolines.
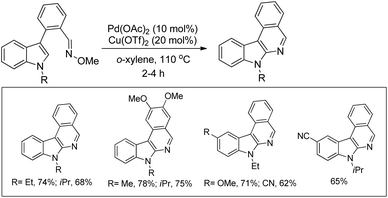 |
| | Scheme 141 Synthesis of indoloisoquinolines using a Cu-/Pd-co-catalyst. | |
The iminyl-Cu intermediate I is generated by cleavage of the N–O bond of the substrate in the presence of the Cu(OTf)2 catalyst. The intermediate I leads to the formation of the product via two pathways (path-b and path-c) presumably through the metallacycles III and IV. The cyanide compound II is obtained through demetallation of the iminyl-Cu species through path-a (Scheme 142).
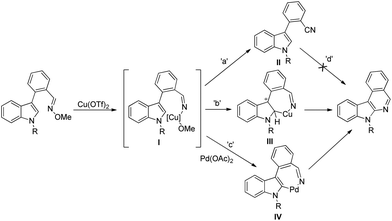 |
| | Scheme 142 Mechanism for the formation of indoloisoquinolines. | |
Cho et al. used a combination of iridium photoredox and Pd-catalysts for the synthesis of carbazoles (Scheme 143).114 The reaction was carried out using visible light under an oxygen atmosphere in the presence of catalytic amounts of Pd(OAc)2 and [Ir(dFppy)2phen]PF6 (dFppy = 2-(2,4-difluorophenyl)pyridine; phen = 1,10-phenanthroline).
 |
| | Scheme 143 Synthesis of carbazoles from N-benzenesulfonyl amidobiaryls. | |
The process utilized the combined visible light photoredox and Pd catalysis and thus did not require strong oxidants for the regeneration of the Pd catalyst. With the help of electrochemical and transient photoluminescence spectroscopy the authors suggest that the catalytic process is initiated by single electron transfer from a palladacyclic intermediate to the photoexcited Ir complex. The photocatalyst is regenerated by molecular oxygen promoted oxidation of its reduced form, and the active catalyst Pd(II) species is regenerated by SET from Pd(I) to O2 or the excited photocatalyst.
Miura et al. further developed a method for the synthesis of carbazole with free NH from 2-aminobiphenyls (without the DG) using Ir-catalyzed C–N bond formation in the presence of a Cu(OAc)2 as a co-catalyst under an air atmosphere (Scheme 144).115
 |
| | Scheme 144 Ir/Cu-Cocatalyzed synthesis of carbazoles. | |
Coordination of an Ir catalyst to the nitrogen atom of the substrate gives the intermediate A which after C–H activation gives B. Reductive elimination of a metal complex from intermediate B furnishes the product. The Cp*Ir(I) species thus formed in the reaction is oxidized by the Cu(II) co-catalyst to regenerate the iridium(III) catalyst (Scheme 145).
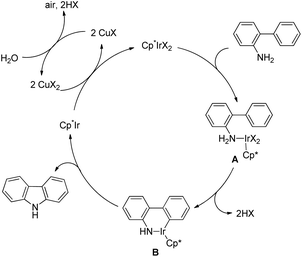 |
| | Scheme 145 Mechanism for the Ir/Cu co-catalyzed C–N bond formation. | |
3.6. Intramolecular C–N bond formation by metal-free and other methods
Antonchick and co-workers used 2,2′-diiodo-4,4′,6,6′-tetramethylbiphenyl as an organocatalyst and peracetic acid as an oxidant in the HFIP solvent for the synthesis of carbazoles through intramolecular C–N bond formation from 2-acetamidobiaryls (Scheme 146).116
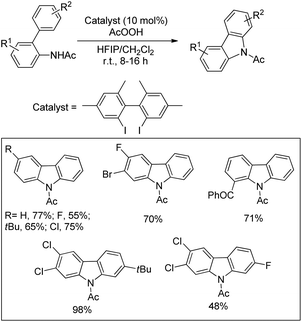 |
| | Scheme 146 Synthesis of carbazoles using an organocatalyst. | |
Muniz et al. reported an iodine catalyzed reaction of sulfamide using a modified hypervalent iodine(III) reagent PhI(mCBA)2 (mCBA = 3-chlorobenzoate) in the presence of visible light for the synthesis of pyrrolidine derivatives (Scheme 147).117
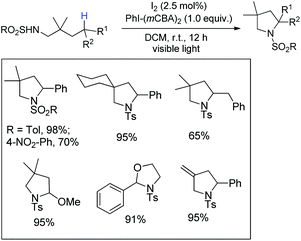 |
| | Scheme 147 Iodine catalyzed synthesis of pyrrolidines. | |
Initially, the reaction of PhI(mCBA)2 with I2 generates the active catalyst I(mCBA) which reacts with the substrate to form the intermediate A with a N–I bond. Irradiation with visible light helps in forming the intermediate B. 1,5-Hydrogen atom abstraction from the benzylic position generates the intermediate C which abstracts an iodine atom from the intermediate A to form the iodinated D. Oxidation of the intermediate D to the alkyl iodine(III) intermediate E and nucleophilic amination result in the formation of a pyrrolidine product (Scheme 148).
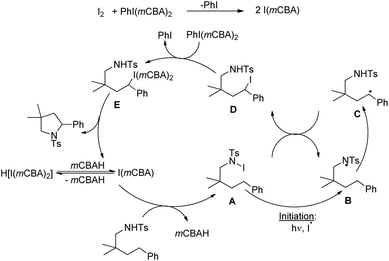 |
| | Scheme 148 Plausible mechanism for the visible light catalyzed synthesis of pyrrolidines. | |
Muniz et al. also reported the NIS promoted synthesis of substituted pyrrolidines under visible light exposure in 53–99% yield (Scheme 149).118 The products obtained did not require any purification by column chromatography as an aqueous workup completely removes the succinimide formed in the reaction. The use of other halide promoters, such as dibrominated hydantoin, led to the formation of unexpected dibrominated imine products.
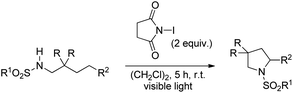 |
| | Scheme 149 NIS promoted synthesis of pyrrolidine sulfonamides. | |
A triiodide mediated strategy for the synthesis of substituted pyrrolidines from sulphonamides through δ-amination of secondary C–H bonds was reported by Nagib et al. (Scheme 150).119 This method uses in situ generated I3− species having the advantage over methods using I2 and in situ generated acetyl hypoiodite (AcOI) which undergoes homolysis in the presence of light resulting in undesired weaker C–H (tertiary, benzylic or α-oxy) oxidations.
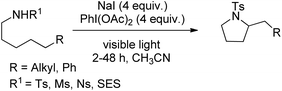 |
| | Scheme 150 Triodide mediated amination of secondary C–H bonds. | |
The reaction starts with the in situ formation of the I3− species which reacts with the substrate to form the haloamine A with a N–I bond. Subsequently, 1,5-HAT and trapping of the I radical generate the intermediate B with a C–I bond which after intramolecular cyclization gives the product. The use of other halide salts (NaBr and NaCl) does not afford the cyclized product (Scheme 151).
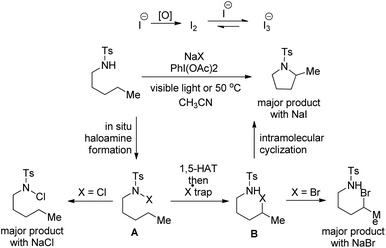 |
| | Scheme 151 Mechanism for triiodide mediated synthesis of pyrrolidines. | |
Muniz et al. further developed a dual light-activated cooperative iodine and photoredox catalysis for intramolecular C–N bond formation by amination of remote C(sp3)–H bonds (Scheme 152).120
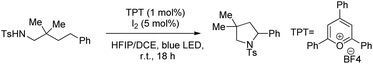 |
| | Scheme 152 Synthesis of pyrrolidines by light-activated I2 and TPT photoredox catalysts. | |
The iodine acts as the primary catalyst giving HOI which reacts with the substrate to form the N–I bond. The visible light induces homolytic cleavage of the N–I bond which is followed by a 1,5-HAT process which activates the remote C(sp3)–H bond. After radical iodine transfer and intramolecular C–N bond formation, the I2 catalyst is reoxidized by photoredox catalysis in a second light-induced process (Scheme 153).
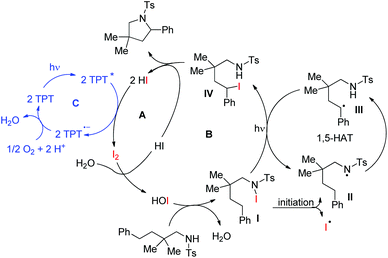 |
| | Scheme 153 Plausible mechanism for the dual light induced I2 catalyzed synthesis of pyrrolidines. | |
Xu et al. reported the synthesis of pyridoimidazoles through an electrochemical method using a solution of Et4NPF6, a reticulated vitreous carbon (RVC) anode and a platinum cathode in refluxing methanol with a constant current of 10 mA (Scheme 154).121 The reaction proceeds through the amidinyl radical intermediate which is involved in the intramolecular C–H/N–H cross-coupling reaction. It is observed that all m-substituted amidines give a 1:1 mixture of regioisomers, whereas the cyclization of p-,m-disubstituted substrates gives benzimidazole regioisomers as the predominant product.
 |
| | Scheme 154 Electrochemical method for the C–H/N–H cross-coupling reaction. | |
Fu et al. reported the visible light mediated synthesis of substituted imidazole derivatives from tertiary aliphatic amines containing β-O-aryl oximes at room temperature (Scheme 155).122 When cyclic amine substrates were used bicyclic imidazole derivatives were obtained.
 |
| | Scheme 155 Visible light mediated synthesis of imidazole derivatives. | |
Reversible binding of both O-2,4-dinitrophenyl and tertiary amine of the starting material converts it into a photosensitizer. The tertiary amine substrate first gets excited by visible light to A which after single electron transfer from the tertiary amine nitrogen atom to the aromatic ring generates the intermediate B. Cleavage of the N–O bond then gives intermediate C having an iminyl radical. Intramolecular hydrogen abstraction in C produces the iminium cation E which after nucleophilic attack gives the product (Scheme 156).
 |
| | Scheme 156 Mechanism for the intramolecular α-C–H imination. | |
A metal-free approach using N-iodosuccinimide for the synthesis of substituted indolo[1,2-a]quinazolinones from functionalized indoles via intramolecular C–N bond formation was reported by Sekar et al. (Scheme 157).123 The reaction did not require any external base, additive and oxidant. The products were obtained in moderate to good yields.
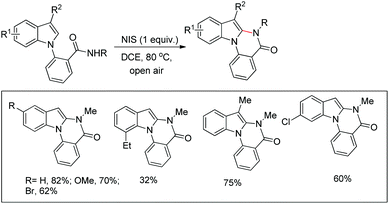 |
| | Scheme 157 Metal-free synthesis of indolo[1,2-a]quinazolinones. | |
Initially the NIS attacks the indole at the C3 position resulting in the formation of iminium ion A. Nucleophilic attack of the succinimide anion on the C2 position of the indole gives intermediate B which after elimination of one molecule of HI gives the indolo[1,2-a]quinazolinone (Scheme 158).
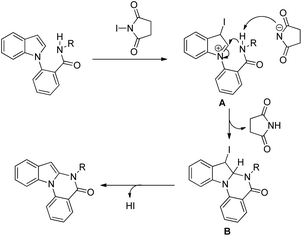 |
| | Scheme 158 Mechanism for the NIS mediated indolo[1,2-a]quinazolinone synthesis. | |
They further developed an I2-mediated intramolecular cyclization through C–H amidation at the C2 position of indoles for the synthesis of various fused cyclic indole compounds.124 This method is used for the synthesis of benzo[4,5]imidazo[1,2-a]indoles, indolo[2,3-b]indoles, dihydroindolo[2,3-b]quinolines and indolo[2,3-b]quinolines from suitably substituted indoles in moderate to good yields.
Our group has developed a method to synthesize α-carboline derivatives from 3-substituted indoles using I2-promoted intramolecular C–H amination and aromatization in open air (Scheme 159).125 The 3-substituted indoles were prepared by a three component reaction of indole, aldehyde and pyrazol-5-amine in the presence of CAN as a catalyst.
 |
| | Scheme 159 Metal-free synthesis of α-carbolines. | |
The reaction starts with the synthesis of the 3-substituted indole A catalyzed by the Ce4+ ion which reacts with I2via a non-free radical process and subsequently gets cyclized to B. As the reaction proceeds via a free-radical pathway, it is believed that in the presence of oxygen, B gets aromatized to the product and generates H2O2 in the process. Once a small amount of H2O2 is generated, it reacts with I2 forming an iodide and a hydroperoxyl radical which initiates the radical chain and compound A gets converted into Bvia [C] to [F] and is finally aromatized to the product (Scheme 160).
 |
| | Scheme 160 Mechanism for the synthesis of α-carbolines. | |
We have further developed an I2 catalyzed C–H amination method for the synthesis of pyrimidine derivatives (Scheme 161).126 The starting material could be easily prepared by a three component reaction of 6-aminouracil, aryl aldehyde and amine. The reaction conditions were mild and several pyrimidines were synthesized in good yields.
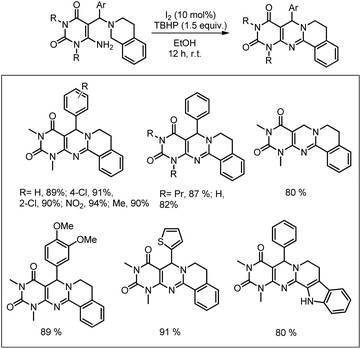 |
| | Scheme 161 I2 catalyzed synthesis of pyrimido[4,5-d]pyrimidines. | |
Quinoxalines could be synthesized from N-(2-acetaminophenyl)enaminones upon treatment with PIDA and benzoic acid as an additive in toluene through an intramolecular C(sp2)–N bond formation reaction (Scheme 162).127 The p-substituted substrates gave slightly higher yields than the substrates with ortho- and meta-substituents presumably due to steric hindrance. The quinoxaline products with keto groups could be further transformed into other products.
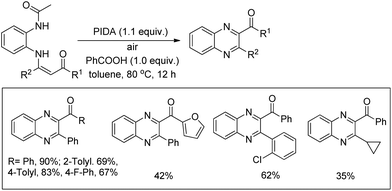 |
| | Scheme 162 PIDA mediated synthesis of quinoxalines. | |
With the help of some controlled experiments the authors proposed a mechanism where initially PIDA reacts with the N-(2-acetaminophenyl)enaminones to give intermediate A. Then, intramolecular cyclization of A gives the product B releasing PhI and CH3COOH in the process. The oxidation of B in the presence of oxygen furnishes C which after aromatization by eliminating one CH3COOH molecule generates the final product (Scheme 163).
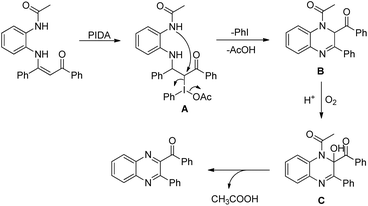 |
| | Scheme 163 Mechanism for the PIDA promoted synthesis of quinoxalines. | |
4. Intramolecular C–S bond formation
The efficient methods for the synthesis of sulphur-based compounds are very much important due to their applications in the pharmaceutical and chemical industries.128 Sulphur is the third most abundant mineral in the human body which plays an important role in the body. It maintains the shape and strength of proteins by mediating electron transfer reactions.129 Some sulphur based organic compounds have important biological activities such as antibacterials, hypoglycemics, anticonvulsants, human immunodeficiency virus (HIV) protease inhibitors, etc.130 For the construction of important sulphur based compounds, an efficient method for the synthesis of the C–S bond is in high demand. Conventionally the C–S bond is formed through the direct coupling of thiols with halides and addition of thiols with unsaturated C–C bonds.131 The reactions such as sulpha-Michael addition reaction,132 Diels–Alder reaction,133 and allylic sulfonation134 are used to synthesize chiral sulphur compounds. But all these reactions have some serious limitations such as waste product formation and require pre-functionalized precursors in many cases. To overcome this, C–S bond formation through C–H activation became an important tool for organic chemists. The study of transition metal catalyzed C–S bond formation compared to C–C and C–N bond formation through C–H activation is less conducted. In this section we described intramolecular C–S bond formation via C–H functionalization and divide the section according to the nature of the catalyst.
4.1. Pd-Catalyzed intramolecular C–S bond formation
In 2008, Doi et al. first reported the Pd(II)-based catalyst C–H activation to synthesise 2-substituted benzothiazoles from thiobenzanilides through C–S bond formation (Scheme 164).135 Different substituted benzothiazoles were obtained in a high yield by using the catalytic system consisting of 10 mol% of Pd(II), 50 mol% of Cu(I), and 2 equiv. of Bu4NBr in DMSO–NMP (1:1). The combination of PdCl2 and CuI has no catalytic activity in the absence of Bu4NBr. Both electron-donating and electron-withdrawing groups furnished good yields. Moreover, functional groups such as alkoxycarbonyl, cyano and halogen were tolerated during the reaction.
 |
| | Scheme 164 Synthesis of substituted 2-arylbenzothiazoles. | |
Due to the advantages of O2, the same group in 2010 reported the synthesis of 2-aryl- and 2-aminobenzothiazoles from thiobenzanilides and thioureas in the presence of PdCl2 (10 mol%) as a catalyst and O2 as an oxidant.136 50 mol% CsF as an additive was required in the DMSO solvent to obtain 2-arylbenzothiazoles whereas NMP was the suitable solvent for 2-aminobenzothiazoles synthesis (Scheme 165).
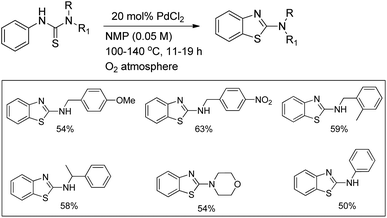 |
| | Scheme 165 Pd-Catalyzed synthesis of 2-aminobenzothiazoles. | |
The reaction is believed to proceed through the coordination of a sulphur atom to Pd(II) and form a 6-membered palladacycle which leads to 2-substituted benzothiazoles and Pd(0) through a reductive elimination process. Molecular oxygen reoxidizes Pd(0) to Pd(II). On the basis of the kinetic isotope experiments, it is suggested that the palladacycle is formed via s-bond metathesis or base-assisted deprotonative metalation instead of electrophilic aromatic palladation or Heck-like insertion (Scheme 166).
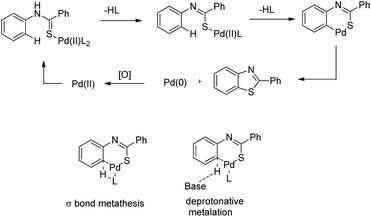 |
| | Scheme 166 Plausible reaction mechanisms for benzothiazole synthesis. | |
In 2008, Doi and co-workers reported another C–S bond formation by using PdCl2 or PdCl2(cod) (Scheme 167).137 They synthesized multi-substituted benzo[b]thiophenes in good to high yields through cyclization of thioenols. Surprisingly, in this reaction the presence of reoxidants decreases the product yield up to 8%.
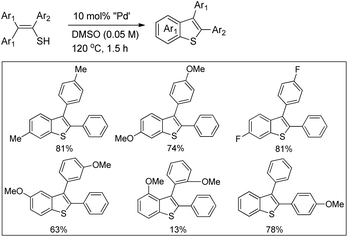 |
| | Scheme 167 Benzo[b]thiophene synthesis from thiols. | |
The reaction proceeds via the formation of disulfides. Palladium plays an important role in the formation of disulfides as well as in the subsequent cyclization (Scheme 168). In the presence of the Pd catalyst, thiols produce disulfides on heating in DMSO. Disulfides then undergo oxidative addition to palladium forming complex X. Then the electrophilic attack of the aryl ring occurs either on the palladium or sulphur atom which gives benzo[b]thiophenes through two possible pathways. It may proceed through the formation of 6-membered palladacycles followed by reductive elimination providing the final product or through the direct formation of the final product along with the starting thioenol.
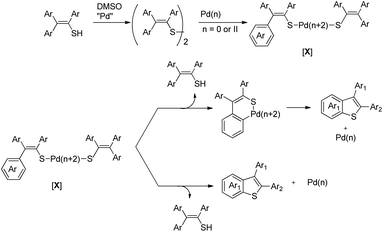 |
| | Scheme 168 Mechanism for the conversion of thiol into benzo[b]thiophenes. | |
Batey et al. used an unusual catalytic system Pd(PPh3)4 and O2/MnO2 as a reoxidant in CH3CN to synthesize 2-aminobenzothiazoles from N-arylthioureas through the formation of an intramolecular C–S bond (Scheme 169).138 Pd(0) with MnO2 furnished a higher yield in comparison with Pd(II). The cyclization process was more rapid in the case of electron-withdrawing groups than the electron-donating groups.
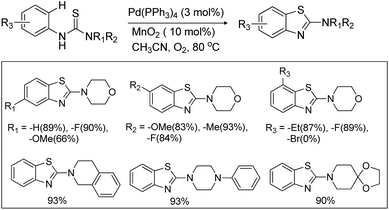 |
| | Scheme 169 Pd catalyzed synthesis of 2-aminobenzothiazoles. | |
On the basis of some controlled experiments and the kinetic isotope effect, they suggested that the cyclization occurs via C–H bond activation and the Pd(II)-peroxo complex formed in the transitional state promoted the proton abstraction. The role of MnO2 is not clear but it may help in the hydrogen peroxide decomposition step (Scheme 170).
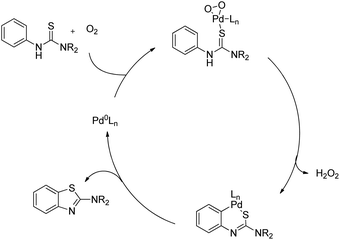 |
| | Scheme 170 Mechanism of aminobenzothiazole formation. | |
Samanta and Antonchick developed a straightforward synthetic method for the regioselective synthesis of dibenzothiophenes from simple benzyl phenyl sulfoxides through double C–H activation directed by the sulfoxide group (Scheme 171).139 Dibenzothiophenes were obtained in good yields by using the PdCl2 catalyst in the presence of AgOAc as an oxidant and p-fluoroiodobenzenes as additives at 110 °C.
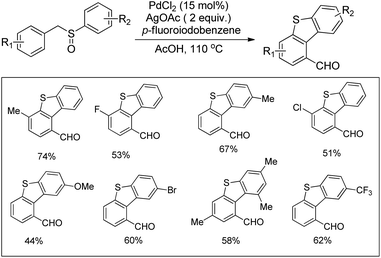 |
| | Scheme 171 Synthesis of dibenzothiophenes. | |
The reaction is believed to proceed via the formation of a dinuclear species by the coordination of Pd(II) to the sulphur atom. Next, oxidative addition of aryl iodides forms a mononuclear complex of Pd(IV) followed by reductive elimination and C–H activation produces a cyclic 6-membered sulfoxide intermediate. Then this intermediate undergoes Pummerer rearrangement in the presence of Pd(OAc)2 and AgOAc and affords dibenzothiophenes through a palladacycle intermediate (Scheme 172).
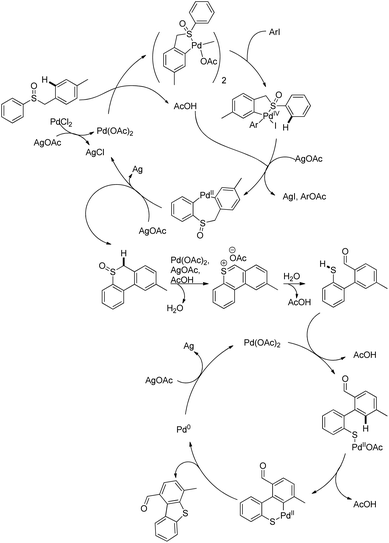 |
| | Scheme 172 Mechanism for the synthesis of dibenzothiophenes. | |
Duan et al. reported a method for the synthesis of dibenzothiophenes and fulvene through a palladium-catalyzed ring opening reaction. They used 2-bromothiophenes and alkynes in the presence of a Pd(II) catalyst which forms a new C–S bond through C–H activation (Scheme 173).140
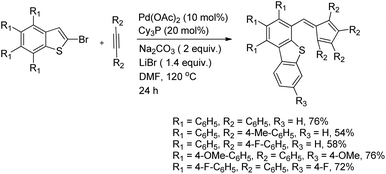 |
| | Scheme 173 Ring opening reaction to synthesize dibenzothiophenes. | |
With the help of some controlled experiments they proposed a mechanism in which oxidative addition of Pd to the starting thiophene gives intermediate A. Intermediate A then reacts with an alkyne to give a vinylic palladium intermediate B followed by C–S bond activation and cycloaddition with another alkyne gives C. Next, intramolecular cyclopalladation with the phenyl group generates a 6-membered palladacycle D, which after subsequent reductive elimination affords the desired product with the regeneration of Pd(0) species (Scheme 174).
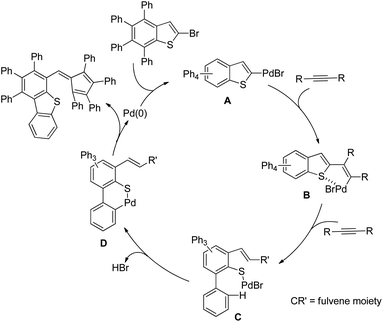 |
| | Scheme 174 Ring opening mechanism for the synthesis of dibenzothiophenes. | |
Liu et al. reported a chemoselective synthesis of sugar-based benzothiazoles from glycosyl thiourea precursors using Pd(COD)Cl2 and Bu4NBr as additives at 100 °C in DMSO under an O2 atmosphere (Scheme 175).141 The reaction was regiospecific at the sterically less-hindered 6-position for 3-substituted thioureas. The reaction did not proceed for non-carbohydrate thiourea and non-pivaloylated glycosyl thiourea.
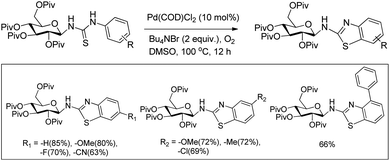 |
| | Scheme 175 Synthesis of sugar-based benzothiazoles. | |
The reaction proceeds via the formation of a Pd(II) arene complex A through coordination to the S-atom of the thiourea and the O-atom of one of the neighbouring pivaloyl carbonyl groups. The Pd(II) arene complex then undergoes cyclopalladation which is stabilized by intramolecular hydrogen bonding (in B and C). Intramolecular hydrogen bonding plays a crucial role in chemoselectivity. The intermediate C then undergoes reductive elimination to furnish the product and Pd(0) species. Molecular oxygen reoxidizes Pd(0) to Pd(II) (Scheme 176).
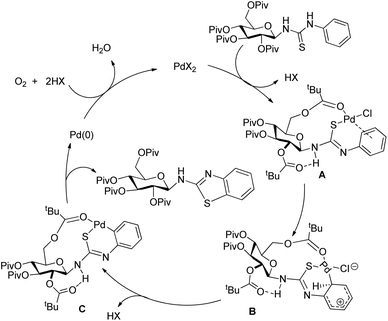 |
| | Scheme 176 Mechanism for the synthesis of sugar-based benzothiazoles. | |
Patel et al. reported a regioselective intramolecular C–S bond formation in the presence of PdCl2 or CuI for the synthesis of 2-aminobenzothiazoles from 2-halothioureas (Scheme 177).142 The specificity of the reaction can be controlled by changing the metal catalyst. In the presence of a Pd(II) catalyst, the 2-aminobenzothiazole was formed through a C–H activation process and a dehalogenative path was favoured in the case of a Cu(I) catalyst.
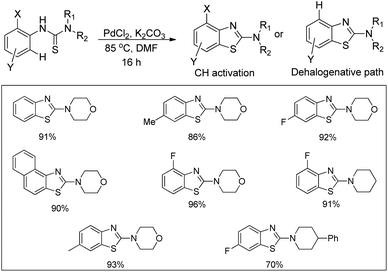 |
| | Scheme 177 Regioselective synthesis of 2-aminobenzothiazoles. | |
In a suggested mechanism, Pd(II) is reduced to Pd(0) in the presence of a base. Two mechanistic pathways are possible depending upon the nature of the substituents present. Either it may follow the oxidative insertion of Pd(0) to the halo group of the thiourea and coordinate with sulphur followed by reductive elimination providing benzothiazoles (path-I) or it may precoordinate with sulphur and form a palladacycle. Then the benzothiazole is formed through C–H activation (path-II) (Scheme 178).
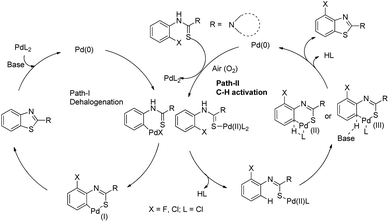 |
| | Scheme 178 Mechanism for the regioselective synthesis of 2-aminobenzothiazoles. | |
4.2. Cu-Catalyzed intramolecular C–S bond formation
Schröder et al. introduced a concept of using disulfides as an oxidant for Cu(I). By employing the C–H activation process they synthesized benzothiazoles from an iminodisulfide via C–S bond formation under an inert atmosphere using Cu(I) (Scheme 179).143
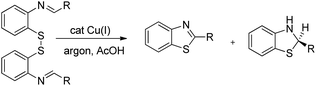 |
| | Scheme 179 Cu(I) catalyzed benzothiazole synthesis. | |
In a plausible mechanism they proposed a Cu(III) complex which is formed from Cu(I) through oxidative addition on the S–S bond followed by hydrogen transfer, thus producing the desired product (Scheme 180).
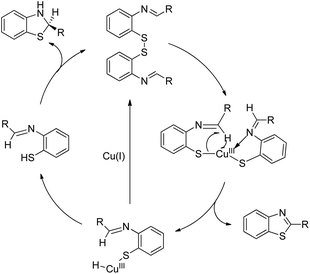 |
| | Scheme 180 Plausible mechanism for the Cu(I) catalyzed benzothiazole synthesis. | |
Patel et al. developed another efficient method of C–H activation to synthesize 2-aminobenzothiazoles via C–S bond formation using environmentally benign Cu(OTf)2 under an oxygen atmosphere. Both electron donating and electron withdrawing groups on the aryl ring of arylthioureas were effective equally in the cyclization process (Scheme 181).144
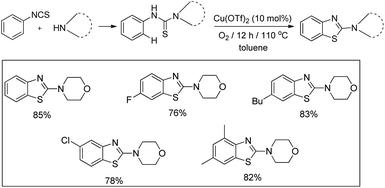 |
| | Scheme 181 Cu(II)-Catalyzed synthesis of 2-aminobenzothiazoles. | |
They also used a ligand assisted Pd-catalyzed protocol for arylthioureas having electron donating groups in the aromatic rings to improve the product yield. 1,10-Phenanthroline was chosen as the ligand in the presence of K2CO3.
4.3. Fe-Catalyzed intramolecular C–S bond formation
Lei et al. described a synthesis of benzothiazoles using FeCl3 through direct C–H functionalization (Scheme 182).145 In this reaction, pyridine plays a vital role to increase the selectivity and yield of the product. When an electron withdrawing group was present on the aryl ring of the aniline moiety the yield decreased significantly.
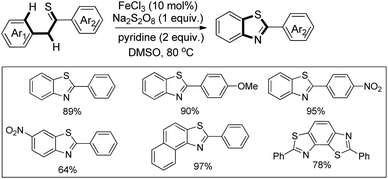 |
| | Scheme 182 Fe(III) catalyzed benzothiazole synthesis. | |
On the basis of some controlled experiments and KIE experiments, it is suggested that the C–H bond cleavage is not the rate determining step. They also suggest that the reaction proceeds via a radical mechanism. First, Fe(III) oxidizes N-phenyl benzothioamide and forms a thioyl radical intermediate. Cyclization of the thioyl radical intermediate followed by oxidation gives 2-phenyl benzothiazoles. Na2S2O8 re-oxidizes the Fe(II) species to regenerate Fe(III) (Scheme 183).
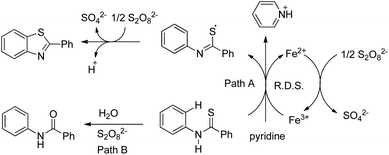 |
| | Scheme 183 Tentative mechanism for Fe(III) catalyzed benzothiazole synthesis. | |
4.4. Au-Catalyzed intramolecular C–S bond formation
Gogoi et al. used an unusual Au-NPs/KMnO4 system to synthesise 2-aminobenzothiazoles forming a C–S bond through C–H activation under an oxygen atmosphere (Scheme 184).146 The reaction of a fruit extract of Kayea assamica with HAuCl4 gave Au-NPs. The catalytic activity remains constant on the addition of KMnO4. They separated and recycled the catalyst eight times without any loss of catalytic activity.
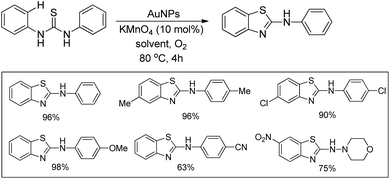 |
| | Scheme 184 Au-NP catalyzed benzothiazole synthesis. | |
The mechanism of the NP-catalyzed reaction is not clear; however, they suggested a plausible mechanism in which gold nanoparticles make the thiourea to bind with molecular oxygen giving A. The intermediate A gives B after eliminating H2O2 which then gives the final product and regenerates the catalyst system (Scheme 185).
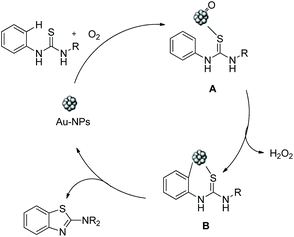 |
| | Scheme 185 Tentative mechanism for Au-NP catalyzed benzothiazole synthesis. | |
4.5. Ru-Catalyzed intramolecular C–S bond formation
Cheng et al. developed a method for the synthesis of 2-substituted benzothiazoles from thioanilides in the presence of Ru(bpy)3(PF6)2, 1,8-diazabicycloundec-7-ene (DBU) and molecular oxygen as the terminal oxidant through C–H activation/C–S bond formation (Scheme 186).147 Based on some controlled experiments and isotope labelling they suggest that the reaction proceeds via a multistep process of radical transformation. Ru(III) accepts a photon and generates an excited Ru(II) species and an O2˙− radical anion. A thioanilide intermediate is formed by deprotonation of the thioanilide precursor which subsequently transforms into a sulphur radical in the presence of Ru(III). Intramolecular cyclization of the sulphur radical intermediate followed by proton abstraction furnishes the benzothiazole product.
 |
| | Scheme 186 Photoredox catalyzed synthesis of benzothiazoles. | |
4.6. I2-Catalyzed intramolecular C–S bond formation
Wu et al. disclosed one I2 promoted intramolecular C–S bond formation through C–H activation. In the presence of I2 and DMSO at 100 °C, aromatic or unsaturated methyl ketones reacted with o-aminobenzenethiols and gave 2-acylbenzothiazoles (Scheme 187).148 The reaction produced moderate to good yields of the product for both electron-withdrawing and electron-donating groups at the ortho-, meta- or para-position of the phenyl ring of aromatic ketones. However, the reaction was not suitable for aliphatic ketones such as acetone, cyclohexanone, and methylethylketone.
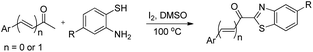 |
| | Scheme 187 I2-Catalyzed synthesis of acylbenzothiazoles. | |
In a plausible mechanism, first, acetophenone converts into phenylglyoxal in the presence of I2 and DMSO. Next, 2-aminobenzenethiol reacts with phenylglyoxal followed by oxidative dehydrogenation to afford the product (Scheme 188).
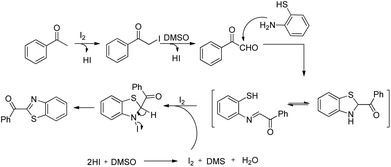 |
| | Scheme 188 Plausible mechanism for I2-catalyzed synthesis of acylbenzothiazoles. | |
5. Conclusions
This review gathered the reports related to intramolecular C–O, C–N and C–S bond formation through the C–H functionalization strategy. Therefore, the synthesis of various heterocycles like furans, pyrans, oxazines, oxazinones, lactones, imidazoles, pyrazoles, pyrimidines, indoles, indolines, carbazoles, carbolines, thiazoles, thiophenes, etc. is included in this review. Intramolecular C–H functionalization reactions are very interesting because of high atom economy and feasibility due to favorable entropy. This is the first review on intramolecular C–H functionalization forming C–O/C–N/C–S bonds and the authors hope that it will encourage organic chemists to work in the area further. Although promising results were obtained in some cases, for example, (i) reactions without using catalysts and external oxidants (section 2.4) and (ii) reactions using visible light with a metal-free catalyst (section 3.6), there are still several important factors which have scope for improvement. For instance, (a) less environmentally benign oxidants can be replaced with air or O2. (b) Expensive and hazardous catalysts, such as Pd, Ir, Rh, Ru, and Os complexes, may be replaced with cheaper and benign Cu, Fe, or I2/iodide salts wherever possible. As intramolecular C–H functionalizations are more feasible than intermolecular reactions, attempts should be made to perform more reactions without using catalysts as one such reaction was reported in section 2.4. (c) Reports of intramolecular asymmetric synthesis for the formation of chiral O-, N-, and S-heterocycles are very few and therefore, it is suggested to develop more intramolecular asymmetric syntheses to prepare chiral heterocycles which would be a more interesting and valuable piece of research. (d) Synthesis of higher membered heterocycles is less reported through this protocol and needs to be further developed. (e) Intramolecular C–H functionalization reactions through directing group assistance are very rare in this field and would be challenging to the researchers.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
MLD is thankful to the Science and Engineering Research Board (SERB), India [Grant No. SB/FT/CS-073/2014] for the financial support under the “Fast Track” Scheme. PJB and BD acknowledge the MHRD, Government of India for research fellowship under the TEQIP-III Project. The authors acknowledge Dr Mridul Hazarika, Vice Chancellor, Gauhati University for constant support and encouragement.
Notes and references
-
(a) D. S. Baldwin, E. H. Reines, C. Guiton and E. Weiller, Ann. Pharmacother., 2007, 41, 1583 CrossRef CAS PubMed;
(b) T. Fukuda, A. Matsumoto, Y. Takahashi, H. Tomoda and S. Omura, J. Antibiot., 2005, 58, 252 CrossRef CAS PubMed;
(c) J. D. Gardner, A. A. Ciociola, M. Robinson and R. L. McIsaac, Aliment. Pharmacol. Ther., 2002, 16, 1317 CrossRef CAS PubMed;
(d) B. Wu, S. He and Y. J. Pan, Planta Med., 2006, 72, 1244 CrossRef CAS PubMed;
(e) A. C. Arai, M. Kessler, G. Rogers and G. Lynch, Mol. Pharmacol., 2000, 58, 802 CrossRef CAS PubMed;
(f) T. M. Böhme, C. E. Augelli-Szafran, H. Hussein, T. Pugsley, K. Serpa and R. D. Schwarz, J. Med. Chem., 2002, 45, 3094 CrossRef PubMed;
(g) K. Cimanga, T. De Bruyne, L. Pieters and A. J. Vlietinck, J. Nat. Prod., 1997, 60, 688 CrossRef CAS PubMed;
(h) T. de Paulis, K. Hemstapat, Y. Chen, Y. Zhang, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332 CrossRef CAS PubMed;
(i) K. Salat, A. Moniczewski and T. Librowski, Mini-Rev. Med. Chem., 2013, 13, 335 CAS.
-
(a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed;
(b) E. B. Murphy and F. Wudl, Polym. Sci., 2010, 35, 223 CAS;
(c) H.-C. Lin, W.-Y. Lin, H.-T. Bai, J.-H. Chen, B.-Y. Jin and T.-Y. Luh, Angew. Chem., Int. Ed., 2007, 46, 897 CrossRef CAS PubMed;
(d) R. C. D. Brown, Angew. Chem., Int. Ed., 2005, 44, 850 CrossRef CAS PubMed;
(e) M. Sonntag and P. Strohriegl, Chem. Mater., 2004, 16, 4736 CrossRef CAS;
(f) N. X. Hu, S. Xie, Z. Popovic, B. Ong and A. M. Hor, J. Am. Chem. Soc., 1999, 121, 5097 CrossRef CAS;
(g) T. Torroba, J. Prakt. Chem., 1999, 341, 99 CrossRef CAS.
-
(a) T. M. Mekhail and M. Markman, Expert Opin. Pharmacother., 2002, 3, 755 CrossRef CAS PubMed;
(b) P. Martins, J. Jesus, S. Santos, L. R. Raposo, C. Roma-Rodrigues, P. V. Baptista and A. R. Fernandes, Molecules, 2015, 20, 16852 CrossRef CAS PubMed;
(c) O. Gidron, Y. Diskin-Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148 CrossRef CAS PubMed;
(d) A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Frechet, J. Am. Chem. Soc., 2012, 134, 2180 CrossRef CAS PubMed;
(e) U. H. F. Bunz, Angew. Chem., Int. Ed., 2010, 49, 5037 CrossRef CAS PubMed;
(f) T. Fallon, A. C. Willis, A. D. Rae, M. N. Paddon-Row and M. S. Sherburn, Chem. Sci., 2012, 3, 2133 RSC;
(g) O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546 CrossRef CAS PubMed;
(h)
M. Kohlmeier, in Nutrient Metabolism, Elsevier, 2nd edn, 2015, pp. 243–263 Search PubMed.
-
(a)
D. J. Brown, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, UK, 1984, vol. 14 Search PubMed;
(b)
R. C. Elderfield, Heterocyclic Compounds, John Wiley & Sons, New York, NY, USA, vol. 6, 1957 Search PubMed;
(c) W. Gul and M. T. Hamann, Life Sci., 2005, 78, 442 CrossRef CAS PubMed;
(d) M. Ishikura and K. Yamada, Nat. Prod. Rep., 2009, 26, 803 RSC;
(e) M. Somei and F. Yamada, Nat. Prod. Rep., 2005, 22, 73 RSC.
-
(a) T. Walsh, Acc. Chem. Res., 2008, 41, 4 CrossRef PubMed;
(b) N. S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257 CrossRef CAS PubMed;
(c) E. Campaigne, D. R. Knapp, E. S. Neiss and T. R. Bosin, Adv. Drug Res., 1970, 5, 1 CAS;
(d) T. R. Bosin and E. Campaigne, Adv. Drug Res., 1977, 11, 191 CAS.
-
(a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed;
(b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed;
(d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed;
(e) N. Yoshikai, Yuki Gosei Kagaku Kyokaishi, 2014, 72, 1198 CrossRef CAS;
(f) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450 CrossRef CAS;
(g) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed;
(h) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS;
(i) D. J. Abrams, P. A. Provenchera and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925 RSC;
(j) J. Liu and L. Zheng, Adv. Synth. Catal., 2019, 361, 1710 CrossRef CAS;
(k) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC.
-
(a) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935 CAS;
(b) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed;
(c) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS PubMed;
(d) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed;
(e) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436 CrossRef CAS PubMed;
(f) W.-J. Yoo and C.-J. Li, Top. Curr. Chem., 2010, 292, 281 CrossRef PubMed;
(g) E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Fare's and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106 CrossRef CAS PubMed;
(h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed;
(i) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC;
(j) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC;
(k) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 1092 CrossRef CAS PubMed;
(l) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769 CrossRef CAS;
(m) B. Schweitzer-Chaput, J. Demaerel, H. Engler and M. Klussmann, Angew. Chem., Int. Ed., 2014, 53, 8737 CrossRef CAS PubMed;
(n) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed;
(o) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459 CrossRef CAS PubMed;
(p) M. L. Deb, C. D. Pegu, P. J. Borpatra and P. K. Baruah, RSC Adv., 2016, 6, 40552 RSC;
(q) M. L. Deb, P. J. Borpatra, C. D. Pegu, R. Thakuria, P. J. Saikia and P. K. Baruah, ChemistrySelect, 2017, 2, 140 CrossRef CAS;
(r) M. L. Deb, B.-S. Saikia, G. K. Rastogi and P. K. Baruah, ChemistrySelect, 2018, 3, 1693 CrossRef CAS;
(s) A. Bakhoda, Q. Jiang, Y. M. Badiei, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2019, 58, 3421 CrossRef CAS PubMed.
-
(a) I. B. Krylov, V. A. Vil’ and A. O. Terent'ev, Beilstein J. Org. Chem., 2015, 11, 92 CrossRef PubMed;
(b) B. Liu and B.-F. Shi, Tetrahedron Lett., 2015, 56, 15 CrossRef CAS;
(c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC;
(d) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC.
-
(a) T. Yoneyama and R. H. Crabtree, J. Mol. Catal. A: Chem., 1996, 108, 35 CrossRef CAS;
(b) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed;
(c) R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS PubMed;
(d) Y. H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed;
(e) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912 RSC;
(f) Q. Liu, P. Wu, Y. Yang, Z. Zeng, J. Liu, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666 CrossRef CAS PubMed;
(g) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827 CrossRef CAS PubMed;
(h) H.-Y. Zhang, H.-M. Yi, G.-W. Wang, B. Yang and S.-D. Yang, Org. Lett., 2013, 15, 6186 CrossRef CAS PubMed;
(i) X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed;
(j) D. Shi, H.-T. Qin, C. Zhu and F. Liu, Eur. J. Org. Chem., 2015, 5084 CrossRef CAS;
(k) K. Neog, A. Borah and P. Gogoi, J. Org. Chem., 2016, 81, 11971 CrossRef CAS PubMed;
(l) T. G. Frihed, M. Bols and C. M. Pedersen, Eur. J. Org. Chem., 2016, 2740 CrossRef CAS;
(m) B. Large, F. Bourdreux, A. Damond, A. Gaucher and D. Prim, Catalysts, 2018, 8, 139 CrossRef.
-
(a)
D. M. X. Donnelly and M. J. Meegan, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katrizky and C. W. Rees, Pergamon, Oxford, U.K., 1984, vol. 4, pp. 657–712 Search PubMed;
(b)
T. Aono, S. Ohkawa and T. Doi, EP Patent483772, 1992 Search PubMed;
(c) S. Ohkawa, K. Fukatsu, S. Miki, T. Hashimoto, J. Sakamoto, T. Doi, Y. Nagai and T. Aono, J. Med. Chem., 1997, 40, 559 CrossRef CAS PubMed;
(d) S. Apers, A. Vlietinck and L. Pieters, Phytochem. Rev., 2003, 2, 201 CrossRef CAS.
-
(a) H. Zhang, E. M. Ferreira and B. M. Stoltz, Angew. Chem., Int. Ed., 2004, 43, 6144 CrossRef CAS PubMed;
(b) J. T. Kuethe, A. Wong, M. Journet and I. W. Davies, J. Org. Chem., 2005, 70, 3727 CrossRef CAS PubMed; and references cited therein;
(c) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed;
(d) M. Lafrance, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 14570 CrossRef CAS PubMed.
- Z. Liang, W. Hou, Y. Du, Y. Zhang, Y. Pan, D. Mao and K. Zhao, Org. Lett., 2009, 11, 4978 CrossRef CAS PubMed.
- X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203 CrossRef CAS.
- B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250 CrossRef CAS PubMed.
- Y. Wei and N. Yoshikai, Org. Lett., 2011, 13, 5504 CrossRef CAS PubMed.
- J. Zhao, Y. Wang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2012, 14, 1078 CrossRef CAS.
- B. D. Dangel, J. A. Johnson and D. Sames, J. Am. Chem. Soc., 2001, 123, 8149 CrossRef CAS.
- X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed.
- Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574 CrossRef CAS PubMed.
-
(a) Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646 CrossRef CAS;
(b) Y.-J. Zhang, T. Abe, T. Tanaka, C.-R. Yang and I. Kouno, J. Nat. Prod., 2001, 64, 1527 CrossRef CAS;
(c) B. Pfundstein, S. K. El Desouky, W. E. Hull, R. Haubner, G. Erben and R. W. Owen, Phytochemistry, 2010, 71, 1132 CrossRef CAS PubMed;
(d) M. A. A. Orabi, S. Taniguchi, M. Yoshimura, T. Yoshida, K. Kishino, H. Sakagami and T. Hatano, J. Nat. Prod., 2010, 73, 870 CrossRef CAS PubMed;
(e) W. Disadee, C. Mahidol, P. Sahakitpichan, S. Sitthimonchai, S. Ruchirawat and T. Kanchanapoom, Phytochemistry, 2012, 74, 115 CrossRef CAS. For reviews of the total synthesis of axially chiral natural products via biaryl lactones, see:
(f) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615 CrossRef CAS;
(g) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed.
- C.-L. Sun, J. Liu, Y. Wang, X. Zhou, B.-J. Li and Z.-J. Shi, Synlett, 2011, 883 CAS.
- Y. Wang, A. V. Gulevich and V. Gevorgyan, Chem. – Eur. J., 2013, 19, 15836 CrossRef CAS PubMed.
- X. Wang, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2014, 53, 11084 CrossRef CAS PubMed.
- J.-J. Dai, W.-T. Xu, Y.-D. Wu, W.-M. Zhang, Y. Gong, X.-P. He, X.-Q. Zhang and H.-J. Xu, J. Org. Chem., 2015, 80, 911 CrossRef CAS PubMed.
- N. P. Ramirez, I. Bosque and J. C. Gonzalez-Gomez, Org. Lett., 2015, 17, 4550 CrossRef CAS PubMed.
- H. Im, D. Kang, S. Choi, S. Shin and S. Hong, Org. Lett., 2018, 20, 7437 CrossRef CAS PubMed.
- M. Zhang, R. Ruzi, N. Li, J. Xie and C. Zhu, Org. Chem. Front., 2018, 5, 749 RSC.
- L. Lia, Q. Yanga, Z. Jia and S. Luo, Synthesis, 2018, 50, 2924 CrossRef.
- S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252 CrossRef CAS PubMed.
-
(a) K. Kikugawa, K. Iizuka, Y. Higuchi, H. Hirayama and M. Ichino, J. Med. Chem., 1972, 15, 387 CrossRef CAS PubMed;
(b) D. J. Faulkner, Nat. Prod. Rep., 1987, 4, 539 RSC;
(c) K. Palczewski, N. Kahn and P. A. Hargrave, Biochemistry, 1990, 29, 6276 CrossRef CAS PubMed;
(d) L. Yet, Chem. Rev., 2000, 100, 2963 CrossRef CAS PubMed;
(e) K. Sprogoe, S. Manniche, T. O. Larsen and C. Christophersen, Tetrahedron, 2005, 61, 8718 CrossRef CAS;
(f) Z. Zhu and J. K. Buolamwini, Bioorg. Med. Chem., 2008, 16, 3848 CrossRef CAS PubMed;
(g) K. Narita, K. Nakamura, Y. Abe and T. Katoh, Eur. J. Org. Chem., 2011, 4985 CrossRef CAS; and references therein;
(h)
J. J. Vaquero, A. M. Cuadro and B. Herradón, in Modern Heterocyclic Chemistry, Wiley-VCH, Weinheim, 2011, pp. 1865–1988 Search PubMed;
(i)
D. L. Riley and W. A. L. van Otterlo, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, pp. 537–549 Search PubMed;
(j) X. F. Xu, W. H. Hu, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2011, 50, 11152 CrossRef CAS;
(k) K. C. Nicolaou, R. C. Yu, L. Shi, Q. Cai, M. Lu and P. Heretsch, Org. Lett., 2013, 15, 1994 CrossRef CAS PubMed.
- X. Wu, X. Geng, P. Zhao, Y.-d. Wu and A.-x. Wu, Org. Lett., 2017, 19, 4584 CrossRef CAS PubMed.
- M. Yu, Z. Wang, J. Hu, S. Li and H. Du, J. Org. Chem., 2015, 80, 9446 CrossRef CAS PubMed.
-
(a) L. Benameur, Z. Bouaziz, P. Nebois, M. H. Bartoli, M. Boitard and H. Fillion, Chem. Pharm. Bull., 1996, 44, 605 CrossRef CAS PubMed;
(b) K. Waisser, K. Gregor, L. Kubicova, V. Klimesova, J. Kunes, M. Machacek and J. Kaustova, Eur. J. Med. Chem., 2000, 35, 733 CrossRef CAS PubMed;
(c) B. A. Arthington-Skaggs, M. Motley, D. W. Warnock and C. J. Morrison, J. Clin. Microbiol., 2000, 38, 2254 CAS;
(d) N. Yamazaki, T. Ito and C. Kibayashi, Org. Lett., 2000, 2, 465 CrossRef CAS PubMed;
(e) O. S. Pedersen and E. B. Pedersen, Synthesis, 2000, 479 CrossRef CAS;
(f) A. J. Cocuzza, D. R. Chidester, B. C. Cordova, S. Jeffrey, R. L. Parsons, L. T. Bacheler, S. E. Viitanen, G. L. Trainor and S. S. Ko, Bioorg. Med. Chem. Lett., 2001, 11, 1177 CrossRef CAS PubMed;
(g) T. M. Böhme, C. E. Augelli-Szafran, H. Hussein, T. Pugsley, K. Serpa and R. D. Schwarz, J. Med. Chem., 2002, 45, 3094 CrossRef PubMed;
(h) P. Zhang, E. A. Terefenko, A. Fensome, Z. Zhang, Y. Zhu, J. Cohen, R. Winneker, J. Wrobel and J. Yardley, Bioorg. Med. Chem. Lett., 2002, 12, 787 CrossRef CAS PubMed;
(i) X. Wang, Y. Dong, J. Sun, X. Xu, R. Li and Y. Hu, J. Org. Chem., 2005, 70, 1897 CrossRef CAS PubMed;
(j) B. P. Mathew, A. Kumar, S. Sharma, P. K. Shula and M. Nath, Eur. J. Med. Chem., 2010, 45, 1502 CrossRef CAS PubMed;
(k) E. Petrlikova, K. Waisser, H. DiviSova, P. Husakova, P. Vrabcova, J. Kunes, K. Kolar and J. Stolarikova, Bioorg. Med. Chem., 2010, 18, 8178 CrossRef CAS PubMed;
(l) G. Cheng, X. Wang, D. Su, H. Liu, F. Liu and Y. Hu, J. Org. Chem., 2010, 75, 1911 CrossRef CAS PubMed;
(m) I. D. Jurberg, B. Peng, E. Wçstefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950 CrossRef CAS PubMed.
- M. L. Deb, S. S. Dey, I. Bento, M. T. Barros and C. D. Maycock, Angew. Chem., Int. Ed., 2013, 52, 9791 CrossRef CAS PubMed.
- S. Mahato, S. Haldar and C. K. Jana, Chem. Commun., 2014, 50, 332 RSC.
- A. Shahrisa, R. Teimuri-Mofrad and M. Gholamhosseini-Nazari, Synlett, 2015, 26, 1031 CrossRef CAS.
- N. A. Waghmode, A. H. Kalbandhe, P. B. Thorat and N. N. Karade, Tetrahedron Lett., 2016, 57, 680 CrossRef CAS.
- M. L. Deb, C. D. Pegu, P. J. Borpatra and P. K. Baruah, Tetrahedron Lett., 2016, 57, 5479 CrossRef CAS.
- M. L. Deb, P. J. Borpatra, P. J. Saikia and P. K. Baruah, Synlett, 2017, 28, 461 CrossRef CAS.
- X.-J. Shang and Z.-Q. Liu, Synthesis, 2017, 49, 4693 CrossRef CAS.
- P. J. Borpatra, M. L. Deb and P. K. Baruah, Tetrahedron Lett., 2017, 58, 4006 CrossRef CAS.
- M. L. Deb, C. D. Pegu, P. J. Borpatra, P. J. Saikia and P. K. Baruah, Green Chem., 2017, 19, 4036 RSC.
- P. J. Borpatra, M. L. Deb and P. K. Baruah, Synlett, 2018, 29, 1171 CrossRef CAS.
- N. Zhang, R. Cheng, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2014, 79, 10581 CrossRef CAS PubMed.
- L. Yang, D. Zhang-Negrerie, K. Zhao and Y. Du, J. Org. Chem., 2016, 81, 3372 CrossRef CAS.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem. – Eur. J., 2013, 19, 5654 CrossRef CAS.
- A. Modak, U. Dutta, R. Kancherla, S. Maity, M. Bhadra, S. M. Mobin and D. Maiti, Org. Lett., 2014, 16, 2602 CrossRef CAS PubMed.
- K. Chen, B. Gao, Y. Shang, J. Du, Q. Gu and J. Wang, Org. Biomol. Chem., 2017, 15, 8770 RSC.
- X.-J. Shang and Z.-Q. Liu, Tetrahedron Lett., 2015, 56, 482 CrossRef CAS.
- A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef CAS.
- M. L. Deb, C. D. Pegu, B. Deka, P. Dutta, A. S. Kotmale and P. K. Baruah, Eur. J. Org. Chem., 2016, 3441 CrossRef CAS.
- V. Satheesh, M. Sengoden and T. Punniyamurthy, J. Org. Chem., 2016, 81, 9792 CrossRef CAS PubMed.
- T. Kamei, M. Uryu and T. Shimada, Org. Lett., 2017, 19, 2714 CrossRef CAS.
- G. Pandey, S. Pal and R. Laha, Angew. Chem., Int. Ed., 2013, 52, 5146 CrossRef CAS PubMed.
- Y. Kim, S.-T. Kim, D. Kang, T.-I. Sohn, E. Jang, M.-H. Baik and S. Hong, Chem. Sci., 2018, 9, 1473 RSC.
- M. Wang, L. Kong, Y. Wang, B. Song, Y. Sun, R. Tang and Y. Li, Org. Lett., 2018, 20, 6130 CrossRef CAS PubMed.
- N. Takemura, Y. Kuninobu and M. Kanai, Org. Biomol. Chem., 2014, 12, 2528 RSC.
- J. Liu, Y. Wen, F. He, L. Gao, L. Gao, J. Wang, X. Wang, Y. Zhang and L. Hu, Org. Chem. Front., 2019, 6, 846 RSC.
- S. J. Thompson, D. Q. Thach and G. Dong, J. Am. Chem. Soc., 2015, 137, 11586 CrossRef CAS PubMed.
- C. G. Francisco, A. J. Herrera and E. Suarez, J. Org. Chem., 2002, 67, 7439 CrossRef CAS PubMed.
-
(a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS;
(b) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
-
(a) F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382 CrossRef;
(b) I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 1691 CrossRef.
-
(a) K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562 CrossRef CAS PubMed;
(b) P. R. Ravisekhara and H. M. L. Davies, Org. Lett., 2006, 8, 5013 CrossRef PubMed;
(c) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652 CrossRef CAS PubMed;
(d) I. Sokolovs, D. Lubriks and E. Suna, J. Am. Chem. Soc., 2014, 136, 6920 CrossRef CAS PubMed;
(e) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS.
- J. J. Neumann, S. Rakshit, T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 6892 CrossRef CAS PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed.
- E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS.
- T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806 CrossRef CAS PubMed.
- C. J. Vickers, T.-S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511 CrossRef CAS PubMed.
- G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS PubMed.
- G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed.
-
(a) T.-S. Mei, D. Leow, H. Xiao, B. N. Laforteza and J.-Q. Yu, Org. Lett., 2013, 15, 3058 CrossRef CAS PubMed;
(b) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712 RSC;
(c) C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884 CrossRef CAS PubMed.
- W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560 CrossRef CAS PubMed.
- W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603 CrossRef CAS PubMed.
- J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184 CrossRef CAS.
- S. W. Youn, J. H. Bihn and B. S. Kim, Org. Lett., 2011, 13, 3738 CrossRef CAS PubMed.
- A. B. Weinstein and S. S. Stahl, Catal. Sci. Technol., 2014, 4, 4301 RSC.
- J. H. Smitrovich and I. W. Davies, Org. Lett., 2004, 6, 533 CrossRef CAS PubMed.
- S. Chiba, G. Hattori and K. Narasaka, Chem. Lett., 2007, 36, 52 CrossRef CAS.
- K. Inamoto, T. Saito, K. Hiroya and T. Doi, Synlett, 2008, 3157 CrossRef CAS.
- T. H. H. Hsieh and V. M. Dong, Tetrahedron, 2009, 65, 3062 CrossRef CAS.
- B. J. Stokes, S. Liu and T. G. Driver, J. Am. Chem. Soc., 2011, 133, 4702 CrossRef CAS.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed.
- K. Clagg, H. Hou, A. B. Weinstein, D. Russell, S. S. Stahl and S. G. Koenig, Org. Lett., 2016, 18, 3586 CrossRef CAS PubMed.
- T. Miura, Y. Ito and M. Murakami, Chem. Lett., 2009, 38, 328 CrossRef CAS.
- M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058 CrossRef CAS PubMed.
-
(a) L. S. Hegedus, G. F. Allen, J. J. Bozell and E. L. Waterman, J. Am. Chem. Soc., 1978, 100, 5800 CrossRef CAS;
(b) L. S. Hegedus and J. M. Mckearin, J. Am. Chem. Soc., 1982, 104, 2444 CrossRef CAS.
- S. R. Fix, J. L. Brice and S. S. Stahl, Angew. Chem., Int. Ed., 2002, 41, 164 CrossRef CAS PubMed.
-
(a) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328 CrossRef CAS PubMed;
(b) P. B. White and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 18594 CrossRef CAS PubMed.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS.
- B. Jiang, F.-F. Meng, Q.-J. Liang, Y.-H. Xu and T.-P. Loh, Org. Lett., 2017, 19, 914 CrossRef CAS PubMed.
- K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS PubMed.
- K. Inamoto, T. Saito, K. Hiroya and T. Doi, J. Org. Chem., 2010, 75, 3900 CrossRef CAS.
- G. Battistuzzi, R. Bernini, S. Cacchi, I. De Salve and G. Fabrizi, Adv. Synth. Catal., 2007, 349, 297 CrossRef CAS.
- R. K. Kumar, M. A. Ali and T. Punniyamurthy, Org. Lett., 2011, 13, 2102 CrossRef CAS.
- T. Kotipalli, V. Kavala, D. Janreddy, C.-W. Kuo, T.-S. Kuo, H.-N. Huang, C.-H. He and C.-F. Yao, RSC Adv., 2014, 4, 2274 RSC.
- G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed.
- F. Pan, B. Wu and Z.-J. Shi, Chem. – Eur. J., 2016, 22, 6487 CrossRef CAS PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 3242 CrossRef CAS PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892 CrossRef CAS.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996 CrossRef CAS PubMed.
- R. Berrino, S. Cacchi, G. Fabrizi and A. Goggiamani, J. Org. Chem., 2012, 77, 2537 CrossRef CAS.
- X. Wang, Y. Jin, Y. Zhao, L. Zhu and H. Fu, Org. Lett., 2012, 14, 452 CrossRef CAS PubMed.
- X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636 CrossRef CAS PubMed.
- A. V. A. Gholap, S. Maity, C. Schulzke, D. Maiti and A. R. Kapdi, Org. Biomol. Chem., 2017, 15, 7140 RSC.
- Y. Liu, J. Wei and C.-M. Che, Chem. Commun., 2010, 46, 6926 RSC.
- J. Maes, T. R. M. Rauws and B. U. W. Maes, Chem. – Eur. J., 2013, 19, 9137 CrossRef CAS PubMed.
- T. Zhang and W. Bao, J. Org. Chem., 2013, 78, 1317 CrossRef CAS PubMed.
- M. Shen, B. E. Leslie and T. G. Driver, Angew. Chem., Int. Ed., 2008, 47, 5056 CrossRef CAS.
- K. W. Fiori, J. J. Fleming and J. Du Bois, Angew. Chem., Int. Ed., 2004, 43, 4349 CrossRef CAS PubMed.
- K. W. Fiori, C. G. Espino, B. J. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042 CrossRef CAS.
- M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS PubMed.
- H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217 CrossRef CAS.
- S. Hazra, B. Mondal, H. Rahaman and B. Roy, Eur. J. Org. Chem., 2014, 2806 CrossRef CAS.
- S. Choi, T. Chatterjee, W. J. Choi, Y. You and E. J. Cho, ACS Catal., 2015, 5, 4796 CrossRef CAS.
- C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 1597 CrossRef CAS PubMed.
- A. P. Antonchick, R. Samanta, K. Kulikov and J. Lategahn, Angew. Chem., Int. Ed., 2011, 50, 8605 CrossRef CAS PubMed.
- C. Martinez and K. Muniz, Angew. Chem., Int. Ed., 2015, 54, 8287 CrossRef CAS PubMed.
- C. Q. O'Broin, P. Fernandez, C. Martinez and K. Muniz, Org. Lett., 2016, 18, 436 CrossRef PubMed.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974 CrossRef CAS PubMed.
- P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muniz, Angew. Chem., Int. Ed., 2017, 56, 8004 CrossRef CAS PubMed.
- H.-B. Zhao, Z.-W. Hou, Z.-J. Liu, Z.-F. Zhou, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 587 CrossRef CAS PubMed.
- J. Li, P. Zhang, M. Jiang, H. Yang, Y. Zhao and H. Fu, Org. Lett., 2017, 19, 1994 CrossRef CAS PubMed.
- S. Badigenchala and G. Sekar, J. Org. Chem., 2017, 82, 7657 CrossRef CAS.
- S. Badigenchala, V. Rajeshkumar and G. Sekar, Org. Biomol. Chem., 2016, 14, 2297 RSC.
- B. Deka, P. K. Baruah and M. L. Deb, Org. Biomol. Chem., 2018, 16, 7806 RSC.
- P. J. Borpatra, G. K. Rastogi, B.-S. Saikia, M. L. Deb and P. K. Baruah, ChemistrySelect, 2019, 4, 3381 CrossRef CAS.
- H. Zhang, J. Shen, Z. Yang and X. Cui, RSC Adv., 2019, 9, 7718 RSC.
-
(a) A. Casini, J.-Y. Winum, J.-L. Montero, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2003, 13, 837 CrossRef CAS PubMed;
(b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133 CrossRef CAS PubMed.
- S. S. Mansy and J. A. Cowan, Acc. Chem. Res., 2004, 37, 719 CrossRef CAS PubMed.
-
(a) A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979 CrossRef CAS PubMed;
(b) D. C. Cole, W. J. Lennox, S. Lombardi, J. W. Ellingboe, R. C. Bernotas, G. J. Tawa, H. Mazandarani, D. L. Smith, G. Zhang, J. Coupet and L. E. Schechter, J. Med. Chem., 2005, 48, 353 CrossRef CAS PubMed;
(c) M. Banerjee, A. Poddar, G. Mitra, A. Surolia, T. Owa and B. Bhattacharyya, J. Med. Chem., 2005, 48, 547 CrossRef CAS PubMed;
(d) A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- D. Uraguchi, N. Kinoshita, D. Nakashima and T. Ooi, Chem. Sci., 2012, 3, 3161 RSC.
- A. Sakakura, H. Yamada and K. Ishihara, Org. Lett., 2012, 14, 2972 CrossRef CAS PubMed.
- X. Wu, Y. Chen, M. Li, M. Zhou and S. K. Tian, J. Am. Chem. Soc., 2012, 134, 14694 CrossRef CAS PubMed.
- K. Inamoto, C. Hasegawa, K. Hiroya and T. Doi, Org. Lett., 2008, 10, 5147 CrossRef CAS PubMed.
- K. Inamoto, C. Hasegawa, J. Kawasaki, K. Hiroya and T. Doi, Adv. Synth. Catal., 2010, 352, 2643 CrossRef CAS.
- K. Inamoto, Y. Arai, K. Hiroya and T. Doi, Chem. Commun., 2008, 5529 RSC.
- L. L. Joyce and R. A. Batey, Org. Lett., 2009, 11, 2792 CrossRef CAS PubMed.
- R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217 CrossRef CAS PubMed.
- H. Huang, J. Li, C. Lescop and Z. Duan, Org. Lett., 2011, 13, 5252 CrossRef CAS PubMed.
- C. Shen, H. Xia, H. Yan, X. Chen, S. Ranjit, X. Xie, D. Tan, R. Lee, Y. Yang, B. Xing, K.-W. Huang, P. Zhang and X. Liu, Chem. Sci., 2012, 3, 2388 RSC.
- S. K. Sahoo, A. Banerjee, S. Chakraborty and B. K. Patel, ACS Catal., 2012, 2, 544 CrossRef CAS.
- J. Srogl, J. Hỳvl, Á. Révész and D. Schröder, Chem. Commun., 2009, 3463 RSC.
- A. Banerjee, S. K. Santra, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 9096 CrossRef CAS.
- H. Wang, L. Wang, J. Shang, X. Li, H. Wang, J. Guia and A. Lei, Chem. Commun., 2012, 48, 76 RSC.
- P. Gogoi, B. Paul, S. Hazarika and P. Barman, RSC Adv., 2015, 5, 57433 RSC.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98 CrossRef CAS PubMed.
- Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086 RSC.
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Bhaskar
Deka
,
Bhaskar
Deka