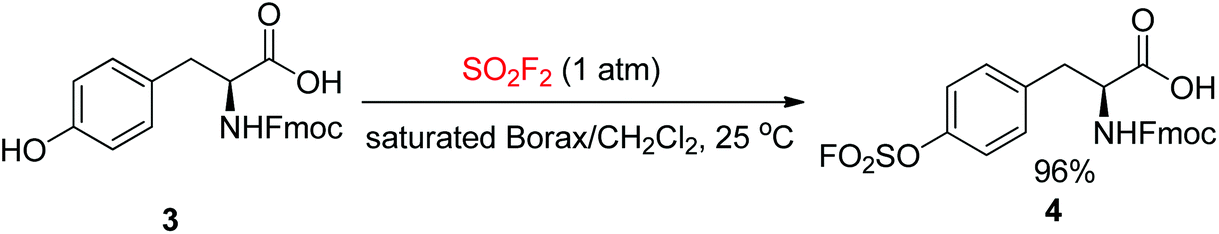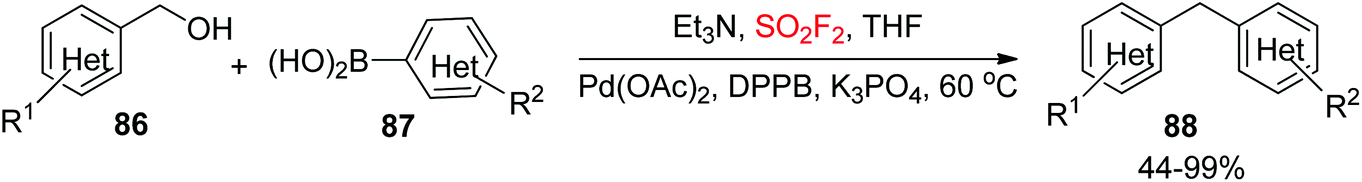DOI:
10.1039/C9QO00747D
(Review Article)
Org. Chem. Front., 2019,
6, 3490-3516
Applications of sulfuryl fluoride (SO2F2) in chemical transformations
Received
10th June 2019
, Accepted 1st September 2019
First published on 17th September 2019
Abstract
A number of novel methodologies concerning the chemical, biological and medicinal applications of sulfuryl fluoride (SO2F2) gas have dramatically improved year by year. SO2F2 is a cheap, abundant and relatively inert electrophile, and also has been widely used as a fumigant for over five decades. Recently, it has gained significant attention as a reagent in organic synthesis. Herein, we summarize chemical transformations using the readily available feedstock SO2F2 gas.
 Ravindar Lekkala | Ravindar Lekkala received his B.Sc. in 2003 from Kakatiya University, India. He is currently pursuing his M.Sc. under the supervision of Prof. Hua-Li Qin at Wuhan University of Technology, P. R. China. His research focuses on synthetic, bioorganic, and medicinal chemistry. |
 Revathi Lekkala | Revathi Lekkala received her M.Sc. in 2011 from Kakatiya University, India. She is currently pursuing her Ph.D. under the supervision of Prof. Hua-Li Qin at Wuhan University of Technology, P. R. China. Her research focuses on the development of new methods in organic synthesis and medicinal chemistry. |
 Balakrishna Moku | Balakrishna Moku grew up in Telangana, India. He obtained his M.Sc. in organic chemistry from Osmania University in 2006 and his Ph.D. in 2014 from the Indian Institute of Chemical Technology (degree obtained from Kakatiya University) under the supervision of Dr Ahmed Kamal. After that he worked for Intonation Research Laboratories Pvt. Ltd, India. Then he moved to Wuhan University of Technology, China to undertake postdoctoral research with Prof. Hua-Li Qin in 2017. Now his area of research is transition metal catalyzed conjugate additions. |
 K. P. Rakesh | Dr K. P. Rakesh received his M.Sc. (2011) and Ph.D. (2015) in chemistry from the University of Mysore, India. He is currently undertaking postdoctoral research under the guidance of Prof. Hua-Li Qin at Wuhan University of Technology, P. R. China. His research focuses on the development of new synthetic methods, amino acid and peptide chemistry, and medicinal chemistry. |
 Hua-Li Qin | Professor Hua-Li Qin received his B.S. in analytical chemistry from Anhui University, P. R. China; his M.S. in organic chemistry from the University of Science and Technology of China, P. R. China (adviser: Professor Z.-X. Wang); and his Ph.D. in organic and medicinal chemistry from Boston University, MA, USA (adviser: Professor J. S. Panek). Then, he was a visiting professor at The Scripps Research Institute, La Jolla (adviser: K. B. Sharpless), before taking up a professorship at Wuhan University of Technology in 2013, after spending some years working in the pharmaceutical industry. His research interests include the design and synthesis of new molecules for the discovery of multitargeted drugs to overcome the multidrug resistance of diseases; the development of robust synthetic methods; and the transformations and applications of the “new click chemistry”, sulfur(VI) fluoride exchange (SuFEx), in the discovery and development of new medicines. |
1. Introduction
The synthesis of sulfuryl fluoride (SO2F2) gas was first reported by Moissan in 19011 and afterward developed in the 1950s by Dow Chemical as the pest control agent Vikane.2 SO2F2 is a colourless, odourless gas, which is 3.5 times heavier than air at normal temperature and pressure (Table 1). For more than five decades, gaseous SO2F2 has been broadly used as a fumigant2a,3 and recently it has gained significant attention in organic synthesis as a reagent. SO2F2 has been produced at more than 3 million kilograms per year since 2000, with low cost,3a and it is a relatively inert electrophile (stable up to 400 °C when dry).4 Under neutral conditions, SO2F2 hydrolyzes slowly in water, and also hydrolyzes more rapidly under basic conditions to provide fluoride and fluorosulfate ions.5 SO2F2 has comparatively small quadrupole and magnetic moments;6 it does not undergo photolysis in the actinic region of solar radiation and is stable toward ozone and the active radicals of the atmosphere (Cl˙, OH˙).3a,b
Table 1 Physical properties of SO2F2
| CAS |
2699-79-8 |
| Molecular weight |
102.1 g mol−1 |
| Density (25 °C, 1 atm) |
4.18 mg mL−1 (air: 1.18 mg mL−1) |
| Boiling point |
−55 °C |
| Vapor pressure |
1611.47 kPa at 20 °C |
| Odor |
Odorless |
| Appearance |
Colorless gas |
| Flammability |
Non-flammable |
| Solubility (25 °C, g L−1) |
Water: 0.75; 1-octanol: 14; heptanes: 22; 2,2-dichloroethane: 25; MeOH: 33; EtOAc: 59; acetone: 71 |
The reaction of phenols with SO2F2 gas reaches its zenith for aromatic compounds, as the generated aryloxy fluorosulfate derivatives are highly stable. In particular, aliphatic amines and phenols undergo selective modification with SO2F2 gas, aromatic amines, and carboxylates, leaving aliphatic alcohols untouched. Since the report by the Sharpless group in 2014,7 SO2F2 gas has been utilized by a number of researchers to synthesize fluorosulfates from a diversity of phenols and alcohols.8 In 2014, the research group of Sharpless reported a powerful synthetic methodology of the sulfur(VI) fluoride exchange (SuFEx) reaction, a class of new click chemistry, signifying that the sulfur(VI) fluoride functional group could be used in a targeted and controllable manner for biological, material, and medicinal applications.9 For the SuFEx chemistry and versatile manipulations, SO2F2 is one of the key reagents.10
Because of the industrial scale of SO2F2 manufacture and utilization, several reports on the assessment of its environmental and physiological toxicity impacts have appeared.11 Gaseous SO2F2 is commonly introduced through a balloon, packed from a stressed lecture bottle. Even though SO2F2 gas is synthesized on a large scale and broadly utilized as a fumigant, its impact on the environment and the human body should not be neglected.12 Symptoms of extended SO2F2 exposure include throat, eye and nose, and coughing, seizures, nausea, muscle twitching, respiratory irritation, vomiting, pulmonary edema, and abdominal pain. Continuous exposure to elevated concentrations of SO2F2 may cause damage to kidneys and lungs.11a Serious long-term effects on reproduction and carcinogenic and mutagenic effects of SO2F2 have not been identified.
Note: SO2F2 is a neurotoxic and strong greenhouse gas; please handle with care in fume hoods.
2. Chemical applications of sulfuryl fluoride (SO2F2)
A number of novel methodologies concerning the chemical applications of sulfuryl fluoride have dramatically improved year by year. Herein, we summarize chemical transformations using the readily available feedstock SO2F2 gas.
2.1. Chemical transformations of phenols
2.1.1. Phenols to aryl fluorosulfates.
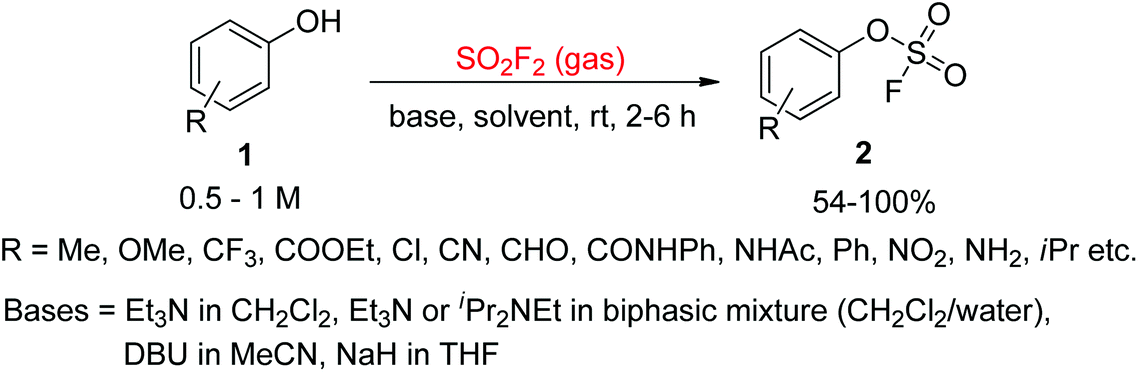
In 1930, for the first time, Lange and Muller reported the synthesis of aryl fluorosulfates from arenediazonium fluorosulfate salts by pyrolysis.13 Later on, Coffman and Cramer synthesized aryl fluorosulfates from substituted phenols upon reaction with ClSO2F with the aid of pyridine.14 However, both of these procedures had disadvantages of costly starting materials, risky reaction conditions, and low yields. In the 1970s, Firth15 and other researchers16 reported a simple procedure for the assembly of aryl fluorosulfate derivatives from cheap and abundant phenols and SO2F2 gas. A more advanced and efficient synthetic methodology for the production of aryl fluorosulfates 2 from phenol derivatives 1 upon reaction with gaseous SO2F2 in the presence of a base was reported in 2014 by the Sharpless group (Scheme 1).7
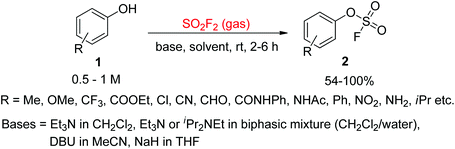 |
| | Scheme 1 Production of aryl fluorosulfate derivatives from phenols treated with gaseous SO2F2. | |
The ease of obtaining the Fmoc-protected Y(OSO2F) solid-phase peptide synthesis (SPPS) building block and the high stability of aromatic fluorosulfates facilitate the construction of peptides having an Ar–O–SO2F side-chain utilizing a strategy rooted in Fmoc chemistry. The Fmoc-Y(OSO2F)-OH amino acid 4 used in protein ligation17 was synthesized in 2016 by the Sharpless group in one step from Fmoc-protected Tyr upon reaction with SO2F2 gas in a biphasic solvent system (saturated aqueous borax buffer/CH2Cl2) (Scheme 2).8c
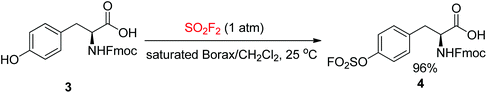 |
| | Scheme 2 Synthesis of Fmoc-fluorosulfated tyrosine 4. | |
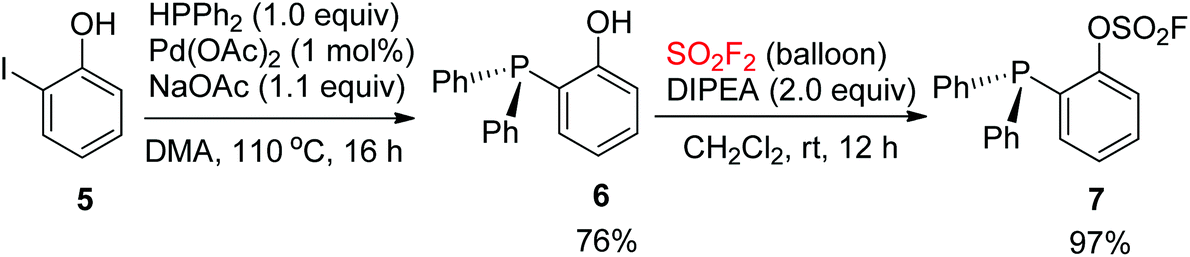
In 2017, Wang and co-workers synthesized 2-(diphenylphosphanyl)phenyl fluorosulfate 7. The Pd-catalyzed coupling reaction between diphenylphosphine and 2-iodophenol 5 provided 2-(diphenyl)phosphanylphenol 6, which then reacted with SO2F2 gas with the aid of N,N-diisopropylethylamine (DIPEA) to furnish 2-(diphenylphosphanyl)phenyl fluorosulfate 7 as a white crystalline compound in 97% yield (Scheme 3).8d
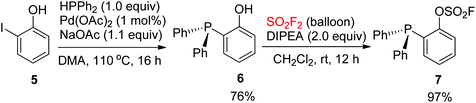 |
| | Scheme 3 Synthesis of 2-(diphenyl)phosphanyl phenyl fluorosulfate. | |
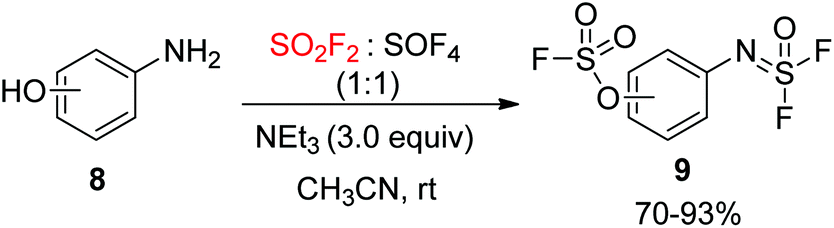
A mixture of SO2F2 and SOF4 was utilized by the Sharpless group18 for the chemoselective SuFEx reaction of aminophenol 8. It was found that the fluorosulfate was generated from the phenol group upon treatment with SO2F2. The aid of Et3N as a base improved the rate of reaction and yield of the transformation with SOF4. This outcome was elucidated by the preferential reaction pairing of phenols with SO2F2 and amines with SOF4, along with a decrease in the chances for a cross-over reaction of each gas as the reaction proceeded, and, even in the case of F2OS![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Ar–OH, the improved acidity of the phenol group (Scheme 4).
N–Ar–OH, the improved acidity of the phenol group (Scheme 4).
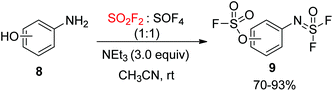 |
| | Scheme 4 Selectivity of SOF4 and SO2F2 towards aromatic amino and hydroxy groups respectively. | |
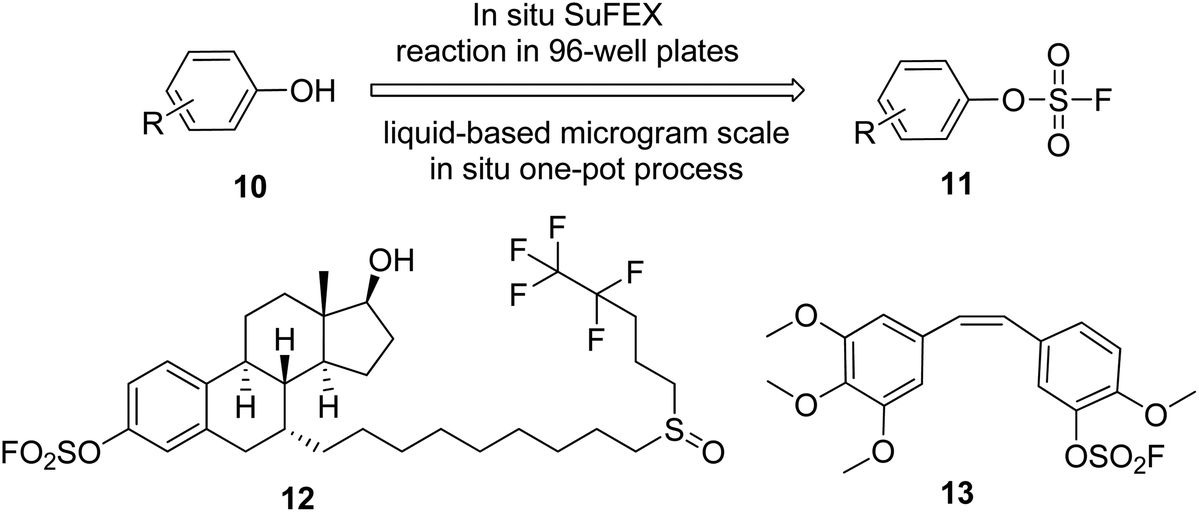
Recently, Wu et al. demonstrated the utilization of the SuFEx method for the late-stage functionalization of phenolic anticancer drugs and transforming them into their corresponding aryl fluorosulfonates in situ in a 96-well plate (Scheme 5).19 Three in situ generated aryl fluorosulfonates exhibited stronger anticancer cell proliferation activity than their phenol precursors. Among them, fulvestrant 12 showed a significantly enhanced activity of estrogen receptor (ER) expression down-regulation on the ER+ breast cancer cell line MCF-7, whereas combretastatin A4 13 showed an effect 70 times that of the original drug in the anticancer activity test against the drug-resistant colon cancer cell line HT-29.
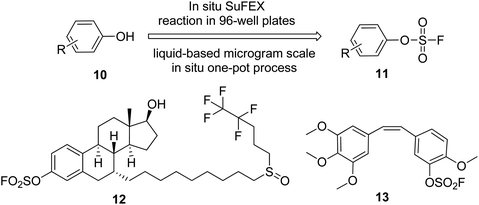 |
| | Scheme 5 Late-stage drug functionalization by SuFEx click chemistry. | |
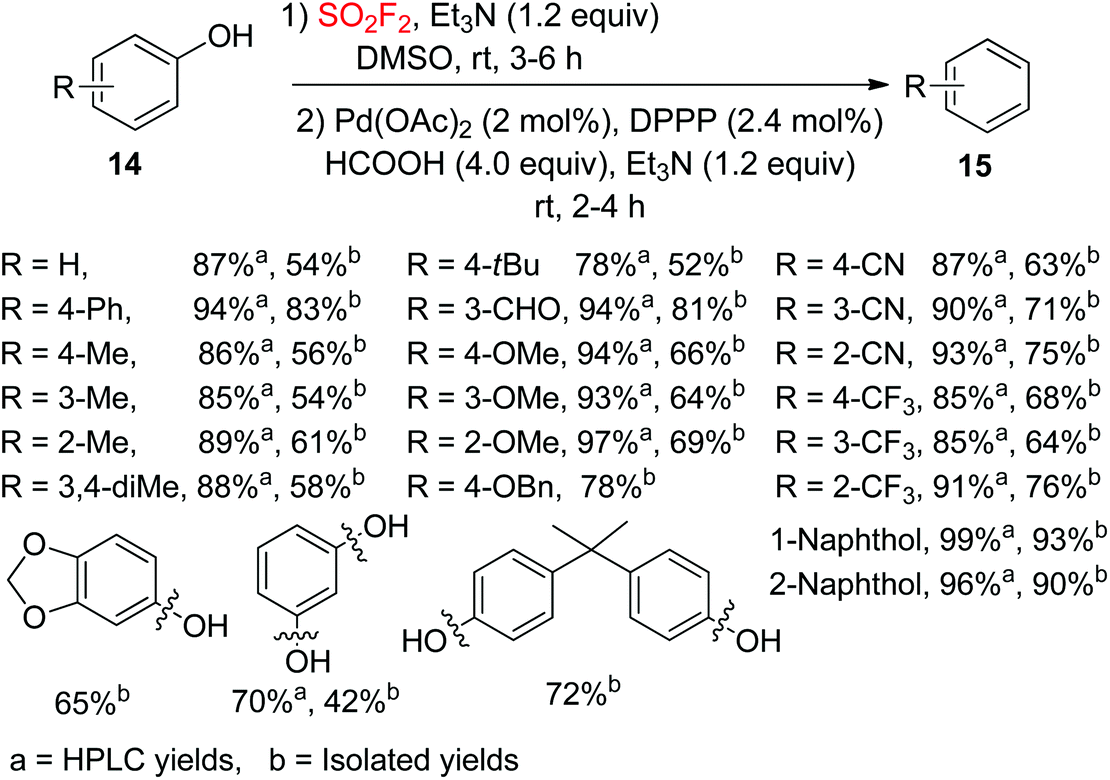
2.1.2. Phenols to arenes.
The development of novel methodologies for the hydrodeoxygenation (HDO) of phenolic compounds is of great significance for the development of bio-oils.20 In 2017, our laboratory developed a facile one-pot procedure for the palladium catalyzed hydrodeoxygenation of phenols 14 to arenes 15 during an ArOSO2F intermediate transformation under a SO2F2 atmosphere by employing Et3N/HCOOH as the transfer hydrogen reductant (Scheme 6).21 The substrates containing alkyl, aryl, formyl, ether and cyano groups were efficiently transformed into their hydrodeoxygenation products in moderate to good yields. Furthermore, also multifunctionalized substrates were transformed into the corresponding products with slightly lower yields than monofunctionalized phenols. Notably, compounds having two phenolic hydroxyl groups on two different aryl rings or a single aryl ring were well tolerated by the hydrodeoxygenation system and furnished the corresponding arenes in good yields, which confirmed the huge benefit of this developed procedure for the development of bio-oil with the survival of multi-phenolic hydroxyl groups.
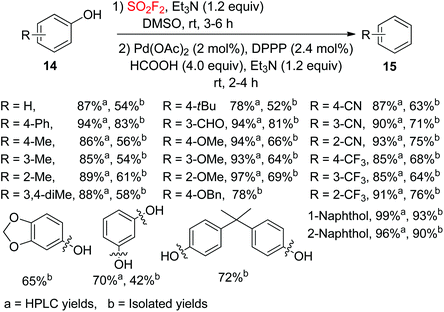 |
| | Scheme 6 SO2F2 mediated hydrodeoxygenation of phenols. | |
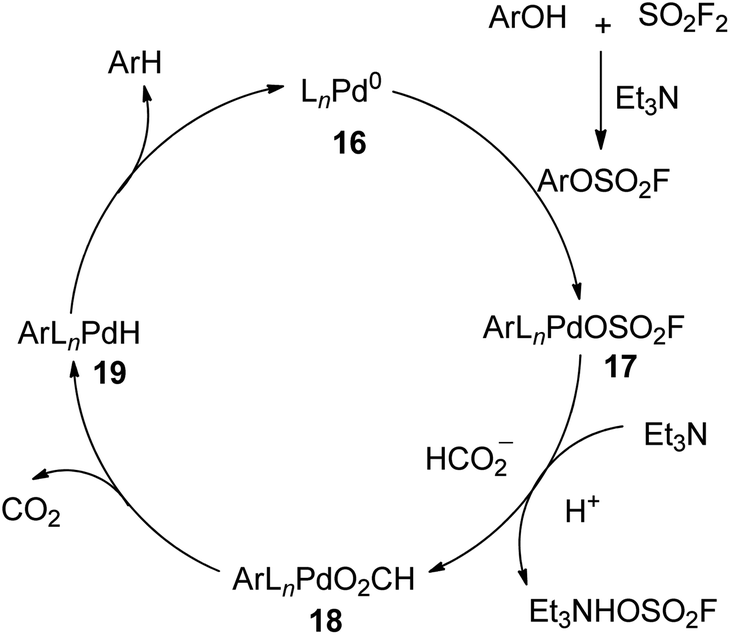
A plausible mechanism for SO2F2 mediated hydrodeoxygenation of phenols was described (Scheme 7). Initially, phenols reacted with SO2F2 gas with the aid of Et3N and generated the aryl fluorosulfate intermediate, which subsequently underwent an oxidative addition with [LnPd0] complex 16 to furnish ArLnPdOSO2F 17. Subsequently, the base-assisted ligand exchange of fluorosulfonate of intermediate 17 with the formate ion generated intermediate 18, which then provided intermediate 19 by releasing carbon dioxide. Finally, the reductive elimination of intermediate 19 yielded the arene while regenerating the Pd0Ln complex 16. During this conversion, an excess amount of Et3N was needed for the neutralization of the yielded acid complex and to speed up the dissociation of HOSO2F from intermediate 17.
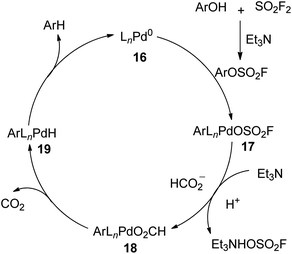 |
| | Scheme 7 Proposed mechanism for SO2F2 mediated hydrodeoxygenation of phenols. | |
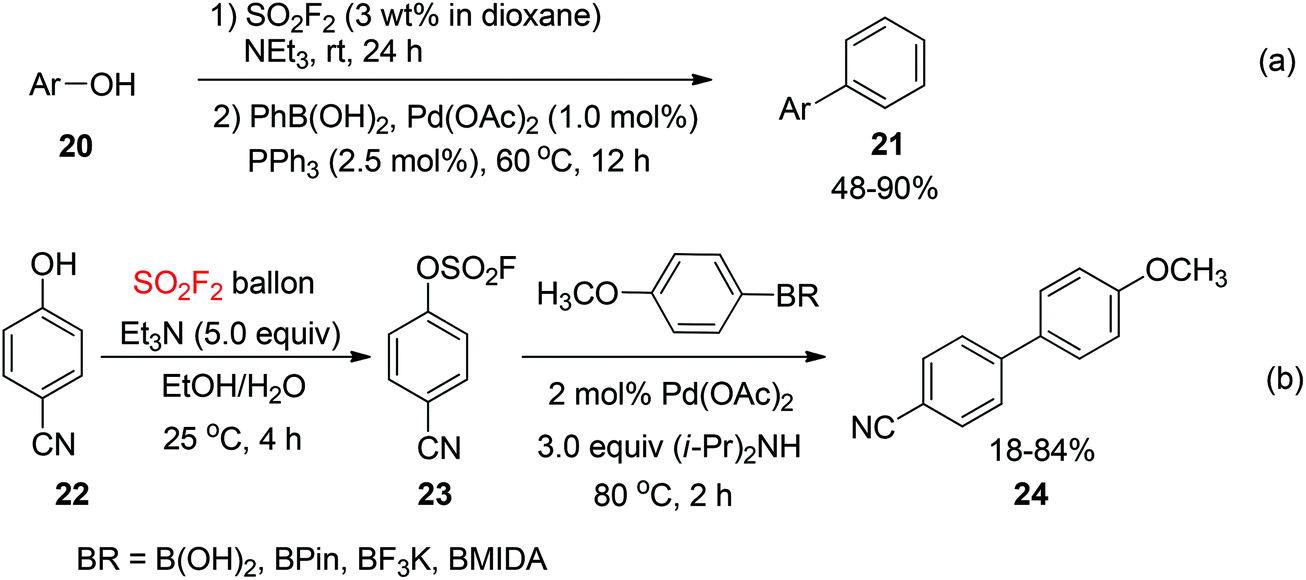
2.1.3. Phenols to biaryls.
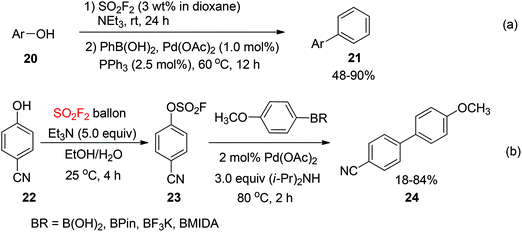
In 2015, Hanley and co-workers reported a SO2F2-mediated one-pot procedure for the transformation of phenols 20 into the corresponding biaryls 21 through the Pd-catalyzed Suzuki–Miyaura coupling with phenylboronic acid (Scheme 8a).22 The reactions were conducted in a fume hood by addition of SO2F2 in 1,4-dioxane to a phenol derivative and NEt3. Compared to the coupling reaction of isolated fluorosulfates, the electronic and steric properties of phenol derivatives had a greater influence on the outcome of the reaction. The coupling reactions of electron-rich phenols furnished lower yields than those of neutral or electron-deficient phenols. Higher yields were furnished with an excess amount of SO2F2. However, a nitrogen atmosphere and a phosphine ligand were required in this procedure. Recently, Yuan and co-workers reported a SO2F2-mediated one-pot transformation of phenols into biaryls catalyzed by palladium without a phosphine ligand under standard reaction conditions (Scheme 8b).23 The conversion of 4-hydroxybenzonitrile 22 into 4-cyanobiphenyl 24 was furnished in 83% and 84% yields respectively when phenylboronic acid pinacol ester and phenylboronic acid were used as coupling partners. However, only 18% yield was obtained with N-methyliminodiacetic acid (MIDA) boronate under the same reaction conditions.
 |
| | Scheme 8 (a) and (b) SO2F2-mediated one-pot transformations of phenols into the corresponding biaryls. | |
2.1.4. Phenols to diarylamines.
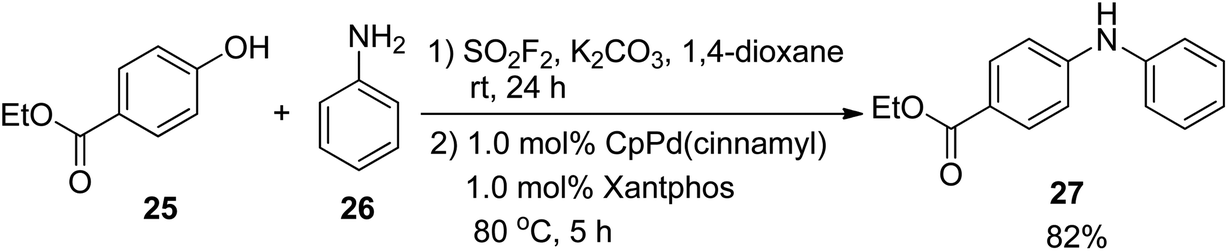
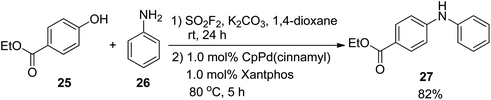
In 2016, the Hanley research group reported a SO2F2-mediated one-pot procedure for the transformation of ethyl 4-hydroxybenzoate 25 into the corresponding biarylamine 27 with 82% isolated yield through the Pd-catalyzed coupling with aniline 26 under the optimal reaction conditions (Scheme 9).10a
 |
| | Scheme 9 SO2F2-mediated one-pot transformations of phenols into biarylamines. | |
2.1.5. Phenols to carbonyl compounds.
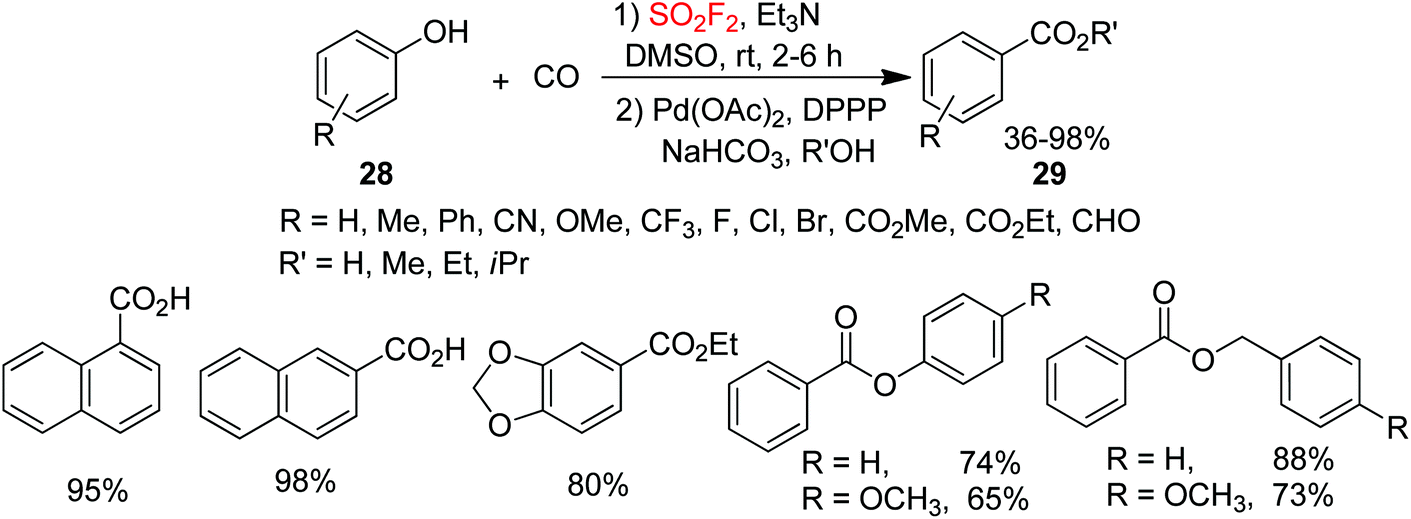
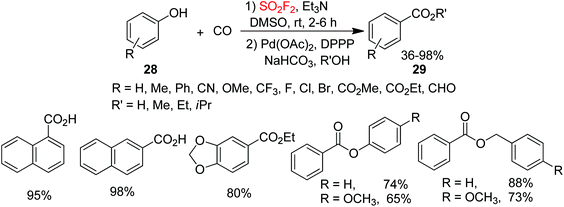
Carboxylic acids and esters are significant motifs in agrochemicals, polymers, pharmaceuticals, biologically active molecules, and natural products.24 Hence, the development of efficient, mild, general and reliable methodologies for the assembly of carboxylic acids or esters is of great importance. In continuation of our interest on SO2F2-mediated chemical transformations, recently, arylcarboxylic acids and esters 29 were synthesized by our laboratory from inexpensive and commercially available phenols 28 upon reaction with carbon monoxide (CO) through Pd-catalysis mediated by SO2F2 gas (Scheme 10).25 Under the optimal reaction conditions, different functionalized phenols were all efficiently transformed into their respective carboxylic acids and esters in moderate to excellent yields through the Pd-catalyzed CO insertion. Interestingly, phenols having electron-withdrawing groups were obtained in better yields than phenols having electron-donating groups. Gratefully, para-electron-rich group substituted substrates or some non-substituted phenols were also transformed into the required products in nearly quantitative yields. Furthermore, bisphenol resorcine was also transformed smoothly into a bisester. In addition, both homo and hetero-esterifications of phenols also efficiently proceeded under identical reaction conditions and afforded the corresponding esters in high yields. Also, benzyl alcohols were capable of furnishing the corresponding products in high yields.
 |
| | Scheme 10 SO2F2-mediated hydroxycarbonylation and alkoxycarbonylation of phenols. | |
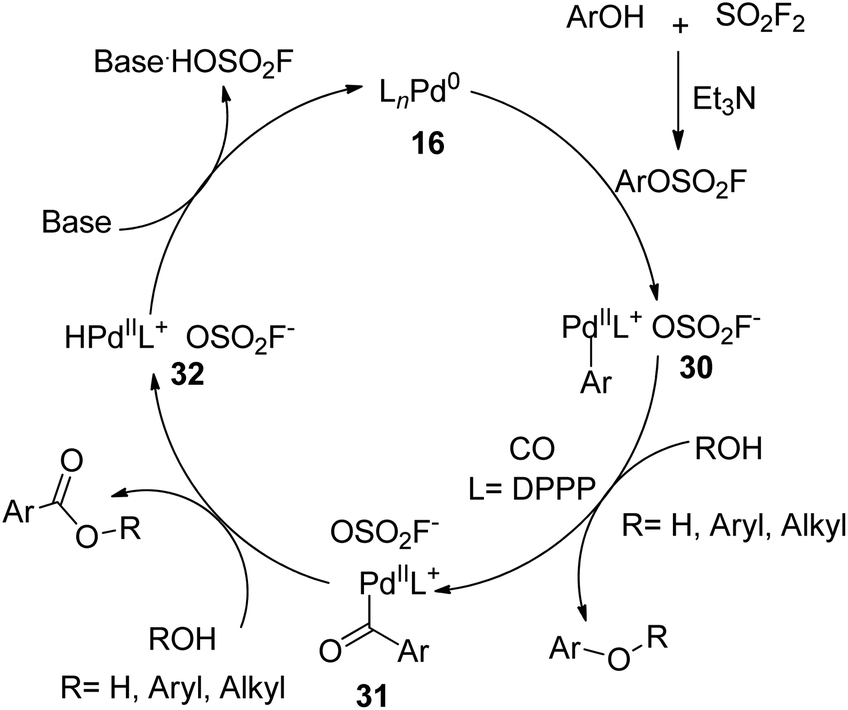
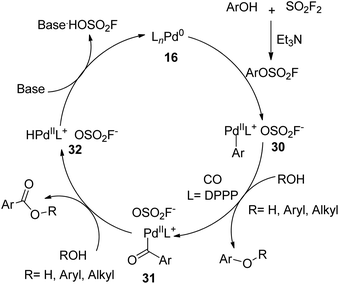
A plausible mechanism for SO2F2-mediated hydroxycarbonylation and esterification of phenols was proposed (Scheme 11). Initially, phenols were treated with SO2F2 gas with the aid of Et3N and generated the aryl fluorosulfate intermediate, which subsequently underwent an oxidative addition with [LnPd0] complex 16 to furnish ArLnPdOSO2F 30. The subsequent insertion of carbon monoxide into ArLnPdOSO2F 30 generated intermediate 31, which was then followed by a nucleophilic attack of ROH to yield the corresponding product with the concurrent generation of intermediate 32. Finally, the base-assisted deprotonation and reductive elimination of intermediate 32 regenerated the active [LnPd0] complex 16.
 |
| | Scheme 11 Proposed mechanism for SO2F2 mediated hydroxycarbonylation and esterification of phenols. | |
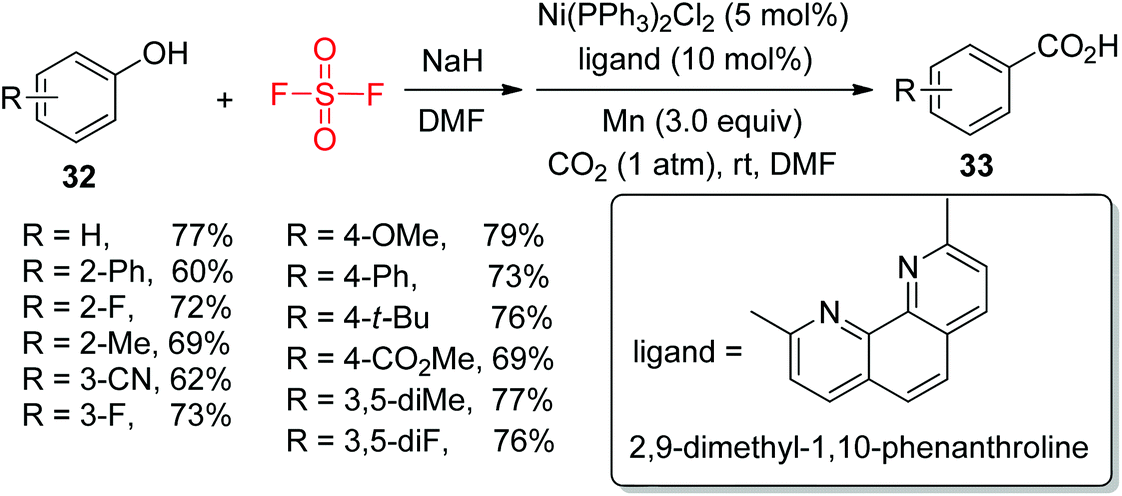
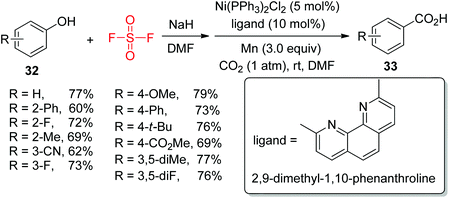
The development of practical and efficient methods for the formation of carboxylic acids utilizing CO2 as a C1 synthon is of great significance. For the first time, the group of Mei very recently demonstrated an efficient procedure for Ni-catalyzed one-pot transformation of phenols 32 into carboxylic acids 33 using CO2 through aryl fluorosulfate intermediates under mild conditions (Scheme 12).26 A wide variety of phenols having both electron-withdrawing and electron-donating groups were efficiently transformed into their respective carboxylic acids in moderate to good yields upon treatment with CO2.
 |
| | Scheme 12 Ni-Catalyzed one-pot transformation of phenols into carboxylic acids. | |
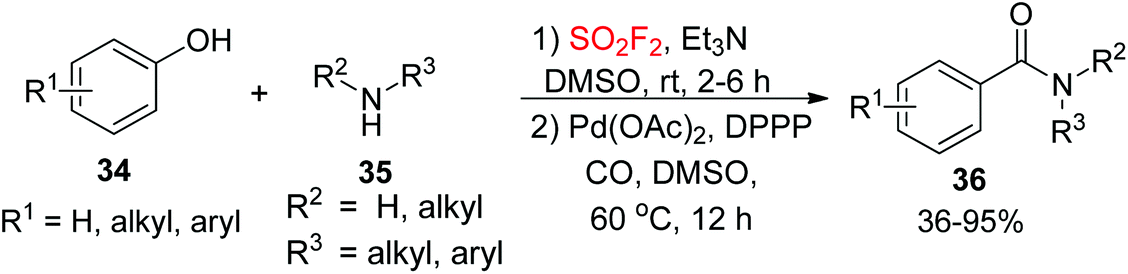
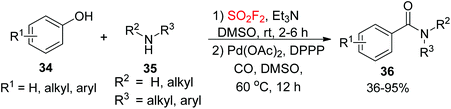
Amides, a class of structurally ubiquitous motifs, are important key intermediates to assemble a wide variety of ligands, peptides, polymers, agrochemicals, pharmaceuticals, and natural products,27 and also significant motifs prevalent in alkaloids, proteins, peptides, pharmaceutical agents, and other naturally occurring molecules.28 Therefore, the development of novel synthetic methodologies for the assembly of amides is of great significance in organic chemistry. Recently, our laboratory developed a convenient one-pot Pd-catalyzed CO insertion process for the assembly of arylcarboxylic amides 36 from their corresponding phenols 34 upon dehydroxylative coupling with amines 35 utilizing SO2F2 gas (Scheme 13).29 The reactions proceeded well under established reaction conditions and tolerated a variety of functional groups of both phenols and amines.
 |
| | Scheme 13 SO2F2 mediated, Pd-catalyzed amidation of phenols. | |
2.1.6. Phenols to aryl nitriles.
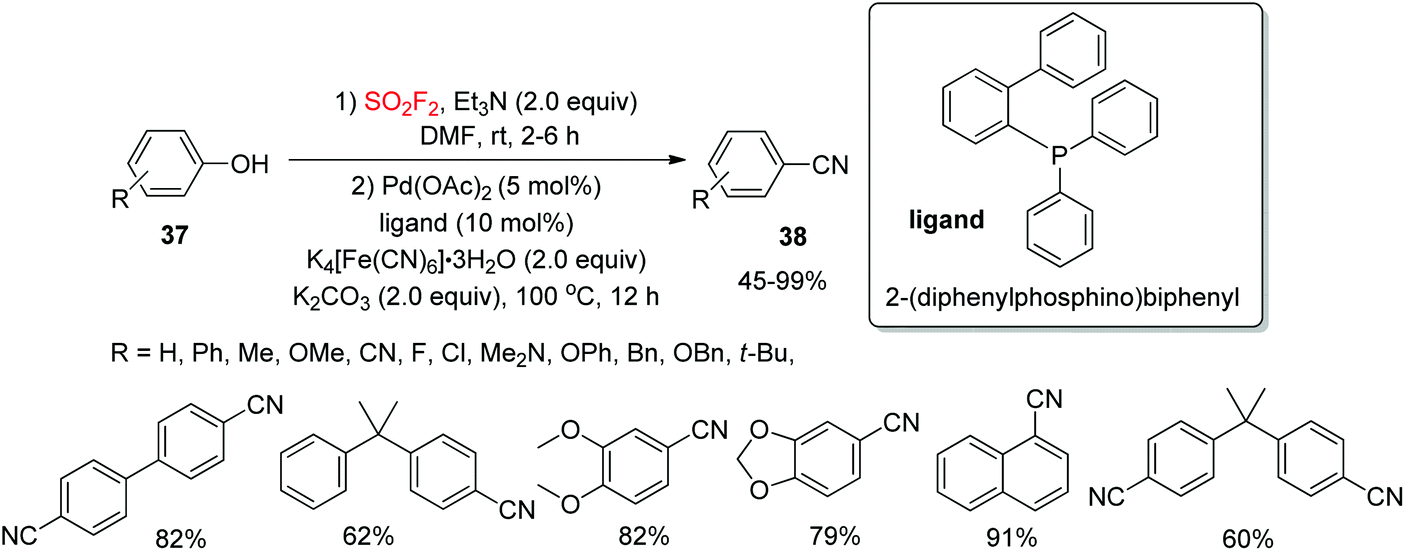
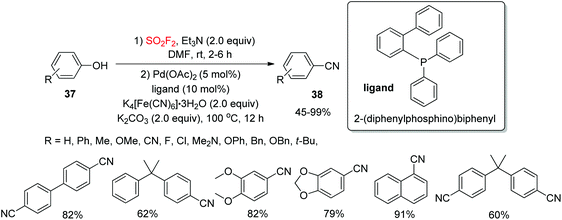
Aryl nitriles, a class of structurally omnipresent moieties, are important patterns in a number of biologically active molecules and have been generally utilized in some functional materials, dyes, agrochemicals, and pharmaceuticals.30 As a result of the broad applicability of aryl nitriles as synthetic intermediates for the construction of many functional groups,31 the development of efficient, mild, general, and reliable synthetic routes for the assembly of aryl nitriles has been a significant and highly desirable challenging target in organic transformations. Recently, our laboratory32 reported a SO2F2 mediated one-pot dehydroxylative conversion of phenols 37 into aryl nitrile derivatives 38via the Pd-catalyzed SN2 coupling reaction employing an inexpensive food additive, K4[Fe(CN)6], as a cyanide source (Scheme 14). Under the standard reaction conditions, a wide variety of functionalized phenols were tolerated smoothly and the targeted aryl nitrile derivatives were furnished in good yields. Furthermore, multi-functionalized phenols and also naphthalenones were efficiently converted into their cyanated products under similar reaction conditions.
 |
| | Scheme 14 SO2F2 mediated Pd-catalyzed dehydroxylative coupling reaction. | |
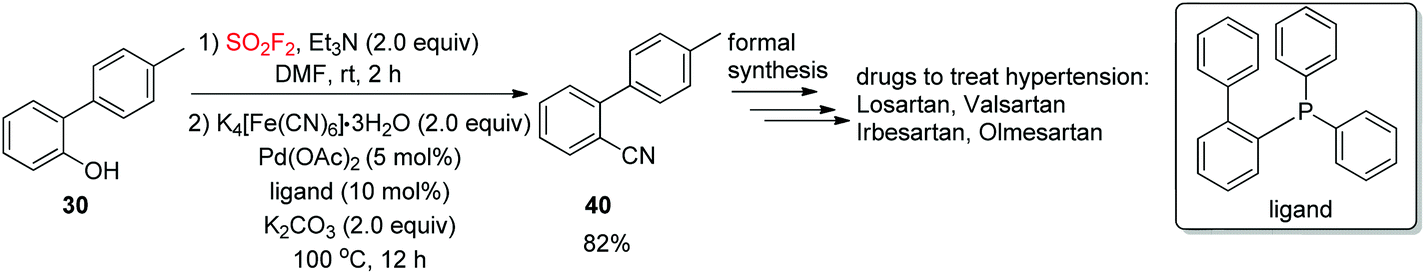
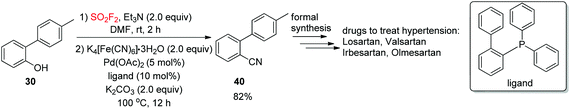
Furthermore, to show the convenience of this new cyanation method for the synthesis of complex drug molecules, the formation of o-tolylbenzonitrile (OTBN, 40),33 the significant pharmaceutical precursor for the formal synthesis of the drugs olmesartan, irbesartan, losartan and valsartan, was achieved smoothly from the respective phenol (4′-methyl-[1,1′-biphenyl]-2-ol) 39 in 82% isolated yield utilizing a newly developed dehydroxylative process (Scheme 15).
 |
| | Scheme 15 Applications of the cyanation method in the formal synthesis of drugs to treat hypertension. | |
2.1.7. Phenols to fluoroarenes.
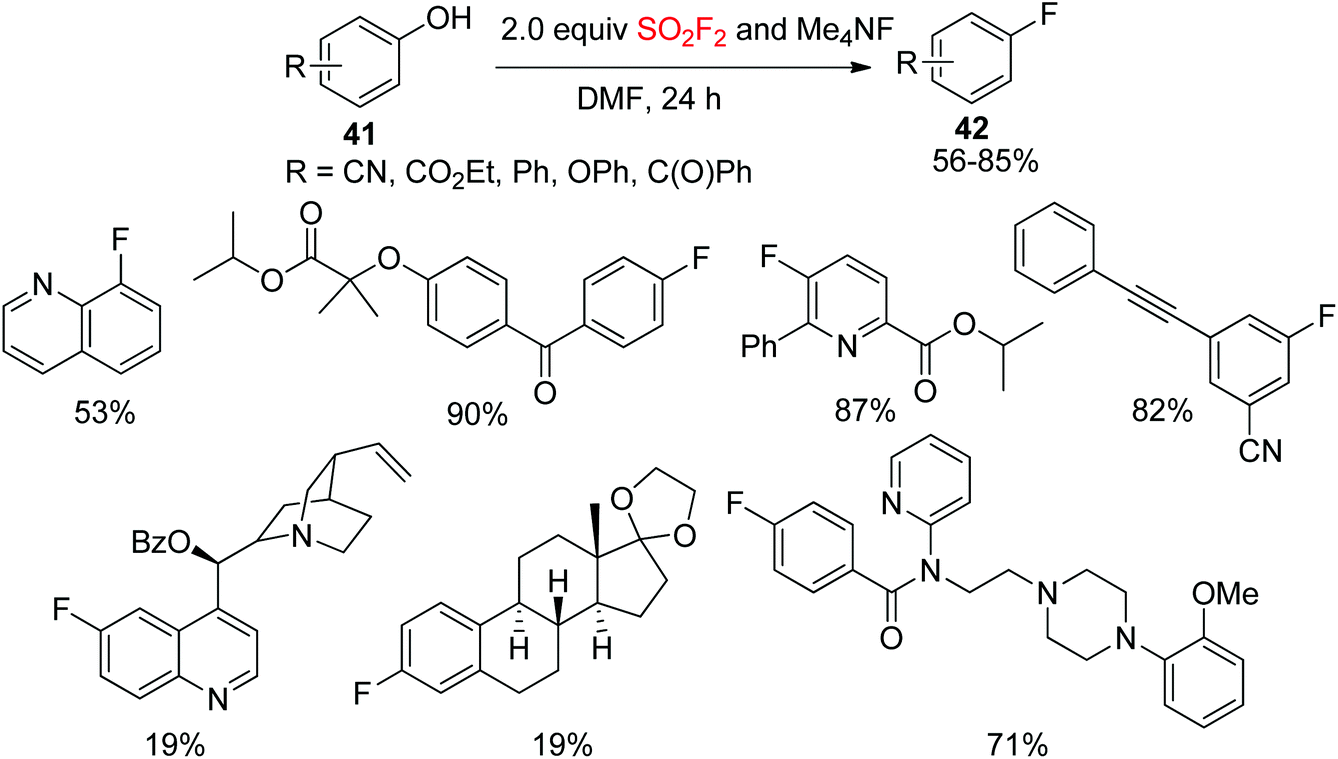
Deoxyfluorination is a powerful strategy for the installation of fluorine into organic compounds.34 The most commonly utilized deoxyfluorination reagent is diethylamino sulfur trifluoride (DAST), which was first reported in 1973.35 Recently, Sanford and co-workers demonstrated that SO2F2 can be utilized for the deoxyfluorination of phenols. Under mild conditions, a wide variety of phenols 41 were directly converted into their corresponding fluoroarenes 42 through the aryl fluorosulfate intermediates utilizing tetramethylammonium fluoride (Me4NF) and SO2F2 as deoxyfluorination reagents (Scheme 16).36 In this protocol, a series of bioactive compounds were converted smoothly into their respective fluorine-containing drugs and ligands in up to 90% yield under established reaction conditions.
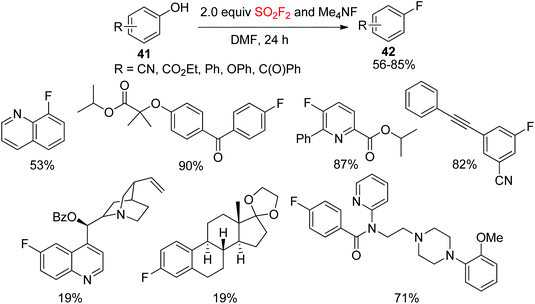 |
| | Scheme 16 Deoxyfluorination of phenols using SO2F2 and Me4NF. | |
2.2. Chemical transformations of silyl ethers to fluorosulfates
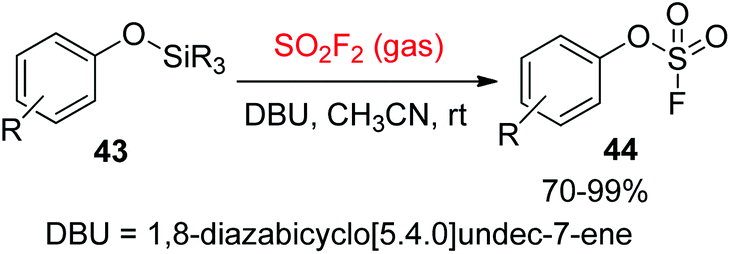
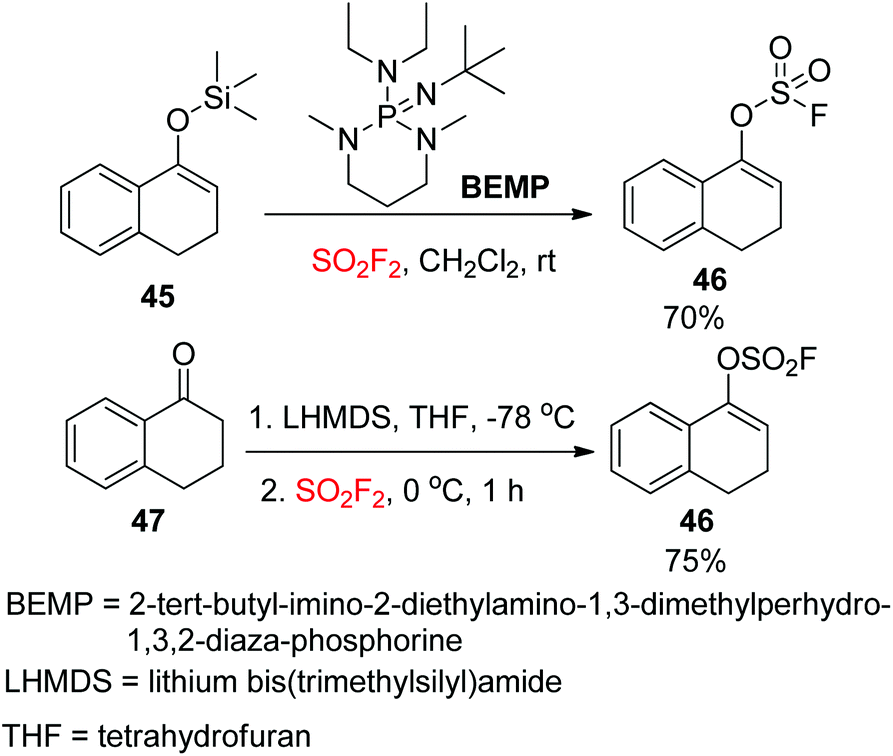
The Sharpless group7a,37 reported another powerful synthetic method for the formation of aryl fluorosulfate derivatives that engaged silicon-based starting materials. In this method, aryl silyl ethers 43 were found to be excellent starting materials for the construction of aryl fluorosulfates 44 upon reaction with SO2F2 gas with the aid of DBU (Scheme 17). In addition, gaseous SO2F2 was found to be an efficient reagent to synthesize enol fluorosulfates 46 from the respective silyl ether 45 or lithium enolate 47 (Scheme 18).
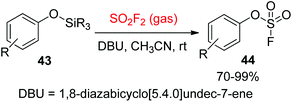 |
| | Scheme 17 The transformation of aryl silyl ethers into aryl fluorosulfates. | |
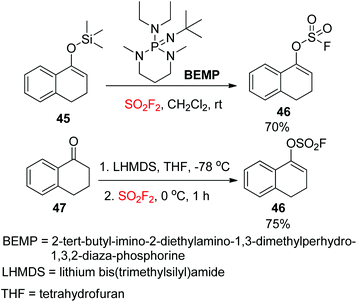 |
| | Scheme 18 Synthesis of enol fluorosulfates. | |
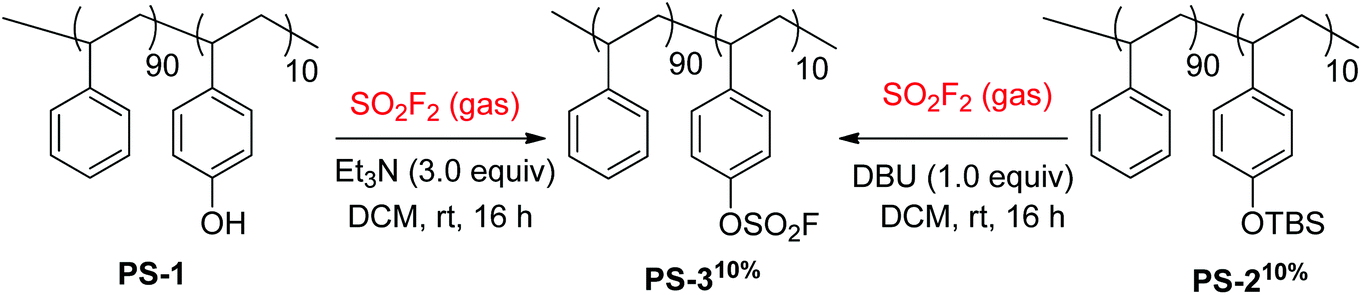
The fluorosulfate-derivatized styrene copolymer PS-310% was synthesized in two different ways utilizing sulfuryl fluoride gas by Fokin and co-workers (Scheme 19).38 Phenol-containing styrene co-polymer PS-1 was directly converted into fluorosulfate-containing co-polystyrene PS-310% upon reaction with SO2F2 gas and Et3N base. Likewise, the silyl ether group of PS-210% was also transformed into the fluorosulfate group of PS-310% upon reaction with SO2F2 gas with the aid of the base DBU.
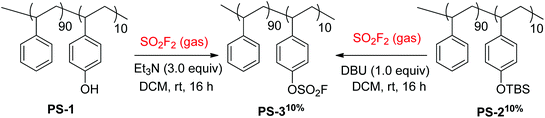 |
| | Scheme 19 Synthesis of fluorosulfate-containing co-polystyrenes using SO2F2 gas. | |
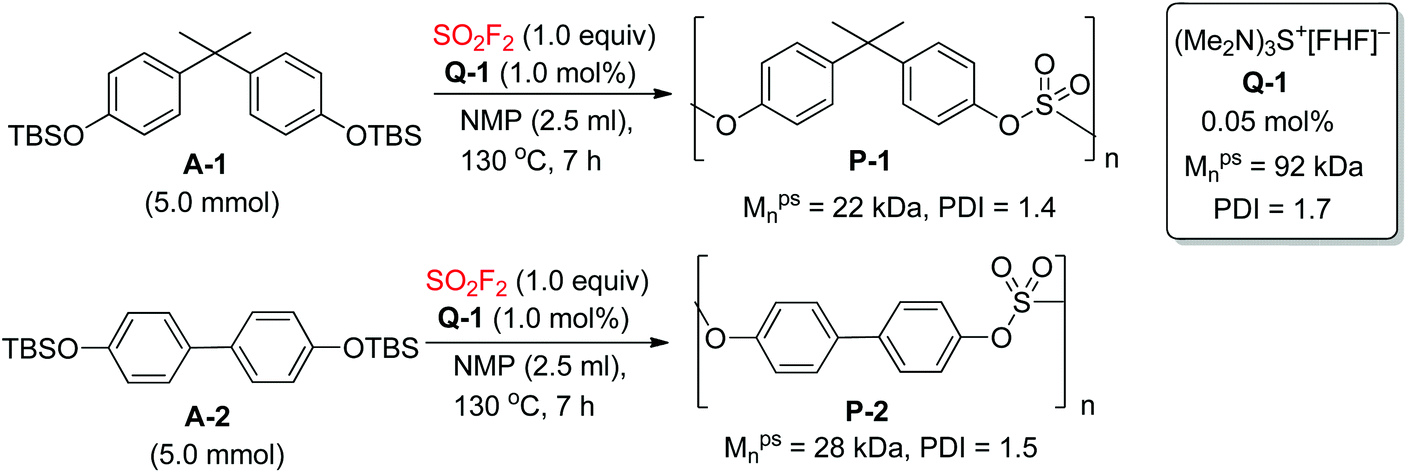
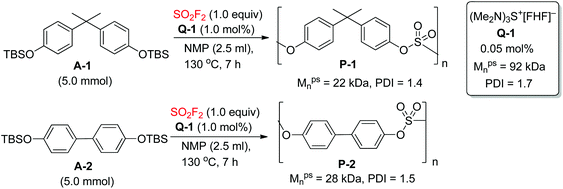
Polysulfates have exceptional mechanical properties making them potentially valuable engineering polymers. Recently, the Sharpless group reported a new set of catalysts for SuFEx-based polysulfate construction, namely the bifluoride salts (Q+[FHF]−, where Q+ represents a diversity of inorganic and organic cations) (Scheme 20).9b The Q-1 catalyzed direct polycondensation of monomer A-1 with SO2F2 yielded polysulfate P-1 with a moderate molecular weight (Mpsn = 22 kDa, PDI = 1.4). In addition, polysulfate P-2 was obtained in near quantitative conversion (Mpsn = 28 kDa, PDI = 1.5) from the monomer A-2 catalyzed by Q-1 under identical reaction conditions.
 |
| | Scheme 20 Synthesis of polysulfates via the bifluoride-catalysed SuFEx reaction. | |
2.3. Chemical transformations of heterocycles
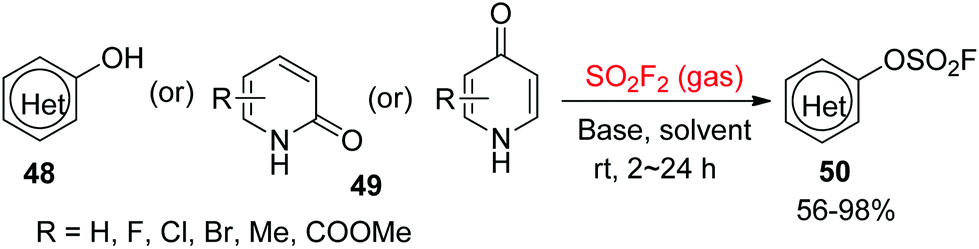
2.3.1. Pyridinols and pyridones to heteroaryl fluorosulfates.
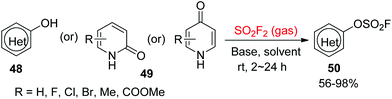
In 2016, Sharpless and co-workers39 utilized commercially available SO2F2 gas to synthesize the heteroaryl fluorosulfates 50 from the corresponding pyridinol 48 or pyridone substrates 49 under the standard reaction conditions (Scheme 21). Pyridinol substrates were converted smoothly into their respective heteroaryl fluorosulfates in the presence of Et3N (1.5 equiv.) in CH2Cl2, whereas N,N-diisopropylethylamine (DIPEA, 1.5–3.0 equiv.) in CH3CN was required for the transformation of pyridones into their respective heteroaryl fluorosulfates. The N- and S-containing hydroxyheterocycles, such as methyl 3-hydroxythiophene-2-carboxylate, quinolin-8-ol, 1H-indol-5-ol and benzo[b]thiophen-4-ol, were also converted efficiently into their corresponding heteroaryl fluorosulfates.
 |
| | Scheme 21 Conversions of pyridinols and pyridones into heteroaryl fluorosulfates. | |
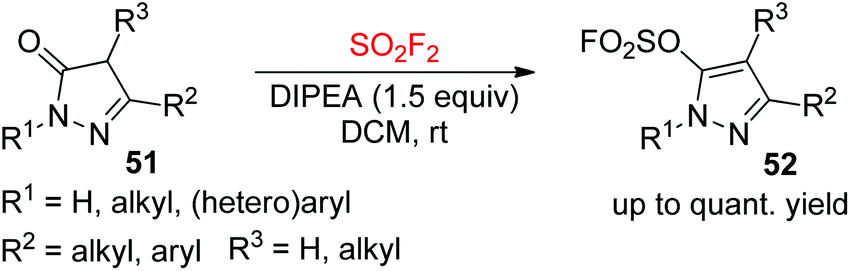
2.3.2. Pyrozolones to heteroaryl fluorosulfates.
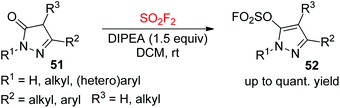
Functionalized pyrazole derivatives as core moieties are present in various biologically active molecules with broad applications in some functional materials, agrochemicals, and pharmaceuticals.40 On the other hand, the fluorosulfate moieties have played a significant role in biology and medicinal chemistry.41 On the basis of the importance of both fluorosulfates and pyrazoles, our laboratory very recently synthesized N-heterocyclic compounds 52 having both fluorosulfate and pyrazole functionalities from pyrazolones 51 upon reaction with SO2F2 gas under very mild conditions (Scheme 22).42 A wide variety of both 2,5-disubstituted and 2,4,5-trisubstituted pyrazolones were all effectively transformed into their respective pyrazolyl fluorosulfates in up to quantitative yields under optimal reaction conditions.
 |
| | Scheme 22 SO2F2 mediated conversion of pyrozolones into pyrazolyl fluorosulfates. | |
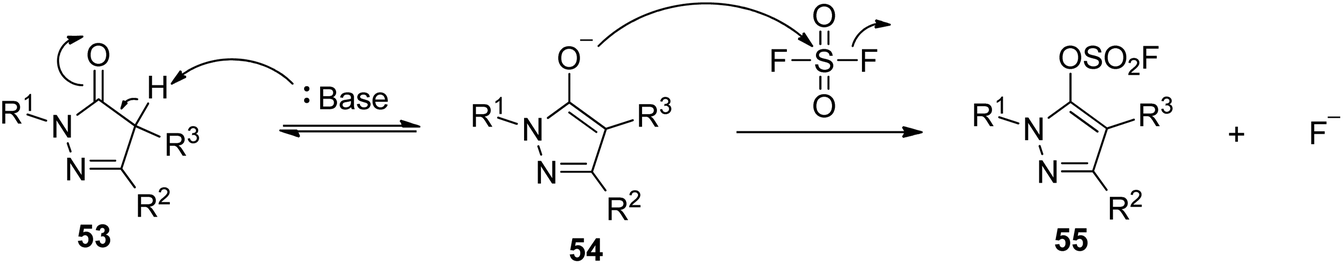
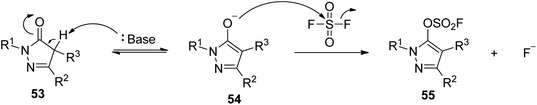
A possible reaction mechanism for SO2F2 mediated conversion of pyrozolones into pyrazolyl fluorosulfates was proposed (Scheme 23). The reaction was initiated by the base-assisted transformation of pyrozolones 53 into their enol forms 54 (which was in equilibrium with its own ketone), which then reacted with SO2F2 to yield the final products pyrazolyl fluorosulfates 55 while generating a fluoride ion. Due to the aromaticity and aromatic character of the product, the conversion of non-aromatic pyrazolones into fluorosulfated pyrazones is hypothetically favoured.
 |
| | Scheme 23 Plausible reaction mechanism. | |
2.4. Chemical transformations of alcohols
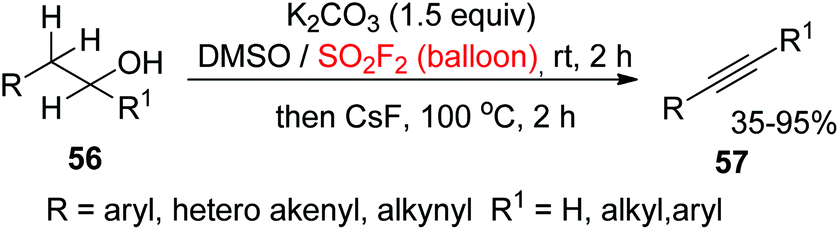
2.4.1. Alcohols to alkynes.
Alkynes are versatile building blocks in the areas of drug discovery,43 chemical biology,44 organic chemistry,45 and materials science46 and are also advantageous motifs prevalent in ligands, natural products, pharmaceuticals, catalyst architectures, materials, and agrochemicals.47 Hence, the development of novel synthetic methodologies for the assembly of alkynes is of great significance. With the intention of developing cost-effective, stable, and powerful strategies for chemical transformations, recently, our laboratory utilized SO2F2 gas for the construction of alkynes 57 from alcohols 56 through a dehydration and dehydrogenation process without any transition metals (Scheme 24).48 We envisioned that SO2F2 would perform the role of an electrophilic activator similar to oxalyl chloride under basic conditions, leading to oxidation of the alcohols to aldehydes or ketones by DMSO (Swern oxidation).49
 |
| | Scheme 24 SO2F2 mediated transformation of alcohols into alkynes. | |
Under mild conditions, only cheap and abundant reagents were utilized for the conversion of alcohols into alkynes. A wide variety of electronically and structurally varied homobenzylic 1° alcohols having both electron-withdrawing and electron-donating groups were obtained efficiently and furnished the final products in up to 95% yield. Notably, naphthyl substrates, alcohols containing heteroarenes and two ethanol groups were all well tolerated. In addition, the transformation of homoallylic 1° alcohols, homopropargyl, and 2° alcohols into their corresponding alkynes also occurred smoothly under similar reaction conditions.
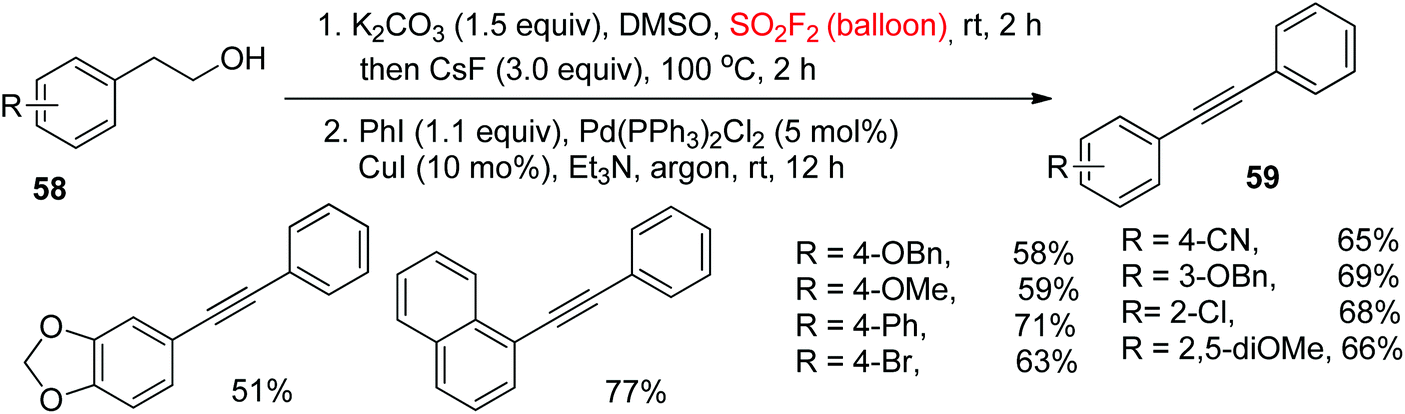
The Sonogashira reaction for the synthesis of diarylalkynes utilizing terminal alkynes is the most useful and powerful synthetic method.50 For additional significance of the utility of our process, the Sonogashira coupling reaction following the Krause et al.51 procedure, utilizing the crude alkynes generated by direct dehydration/dehydrogenation of alcohols, was effectively incorporated into a one-pot method (Scheme 25). Under the standard reaction conditions, the unsymmetrical diarylalkynes 59 were synthesized in 51%–71% yields from a variety of 2-arylethanols 58 having both electron-withdrawing (CN, Cl, Br) and electron-donating groups (Ph, OMe, OBn) at different positions of the aromatic ring upon reaction with iodobenzene. Remarkably, 2-(1-naphthyl)ethanol was also well tolerated with 77% yield of the Sonogashira coupling product.
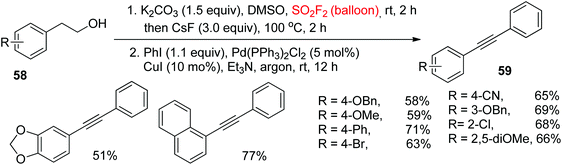 |
| | Scheme 25 Direct conversion of 2-arylethanols into diarylalkynes by one-pot dehydration/dehydrogenation and Sonogashira coupling. | |
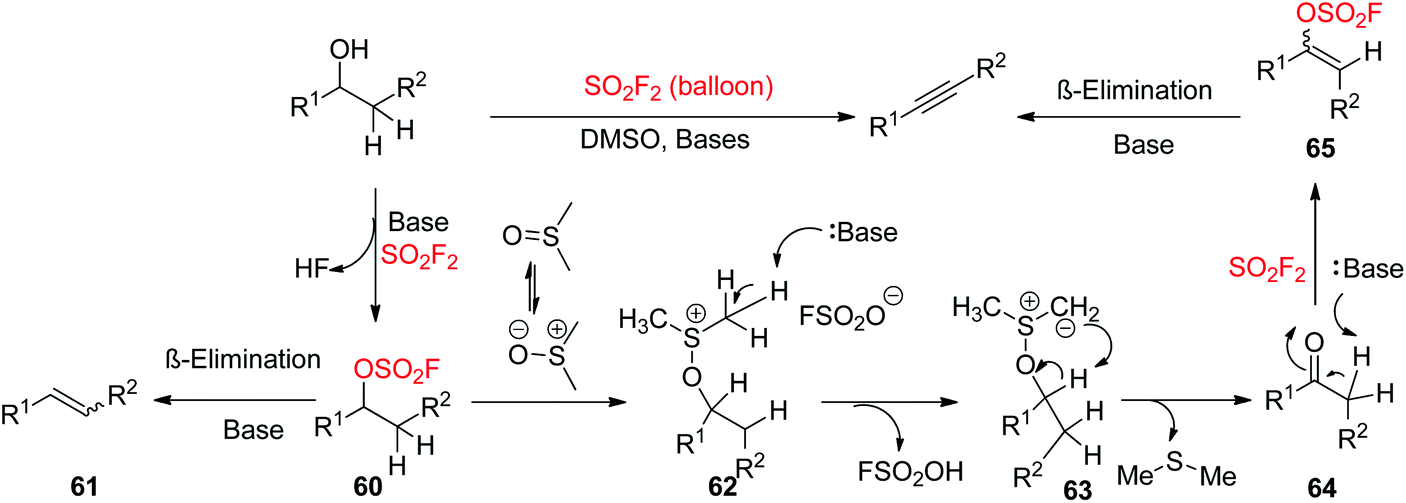
A possible reaction mechanism for SO2F2 mediated transformation of 2-arylethanols into diarylalkynes was proposed (Scheme 26). The reaction was initiated by the base-assisted construction of intermediate 60 from alcohols upon treatment with SO2F2 gas. The consequent SN2 displacement by DMSO acting as a nucleophile generated the cationic intermediate 62, which was then deprotonated by the base to give sulphur ylide 63. The consequent intramolecular deprotonative elimination of sulphur ylide 63 provided Me2S and aldehyde (ketone) 64, which then generated vinyl sulfurofluoridate 65 upon treatment with a base and SO2F2 gas. Finally, vinyl sulfurofluoridate 65 underwent β-elimination with the aid of a base to yield the desired alkyne as the major product. However, the alkene 61 by-product was also furnished when the intermediate 60 underwent β-elimination directly earlier to SN2 displacement by DMSO.
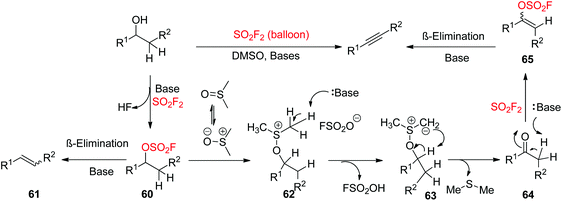 |
| | Scheme 26 Proposed mechanism for SO2F2-mediated transformation of 2-arylethanols into biarylalkynes. | |
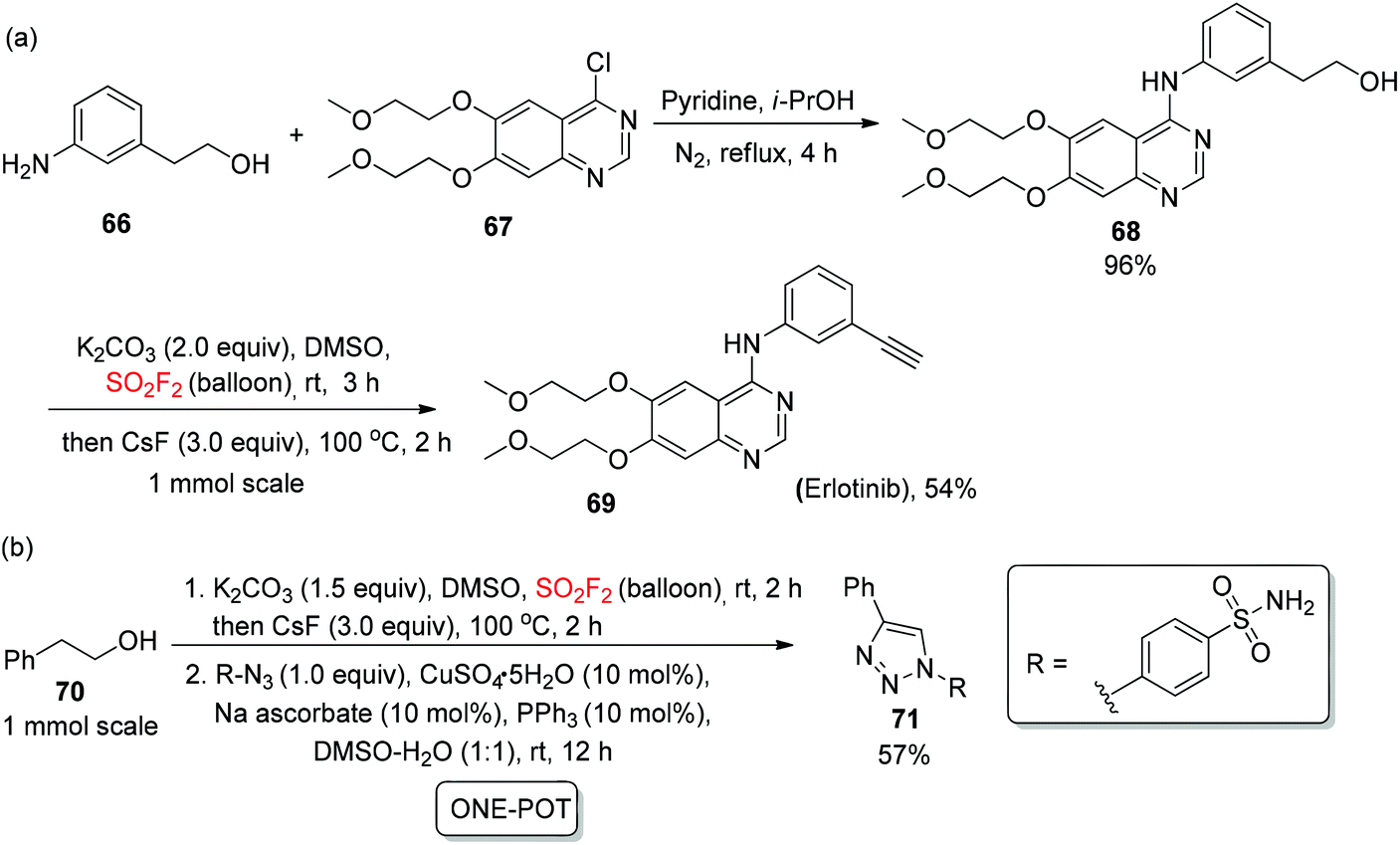
In order to show the convenience of this new protocol in drug synthesis, natural products, biologically active molecules, and the drug erlotinib 69 (a tyrosine kinase inhibitor) was synthesized from alcohol 66 during a two-step process of nucleophilic addition and an oxidatively dehydrative dehydrogenation method in 54% yield (Scheme 27a). In addition, with the production of a carbonic anhydrase (CA) inhibitor, the 1,4-substituted triazole 71 was also obtained52 utilizing the developed “one-pot” procedure of oxidation, enolization, elimination, and 1,3-dipolar cycloaddition in 57% yield (Scheme 27b).
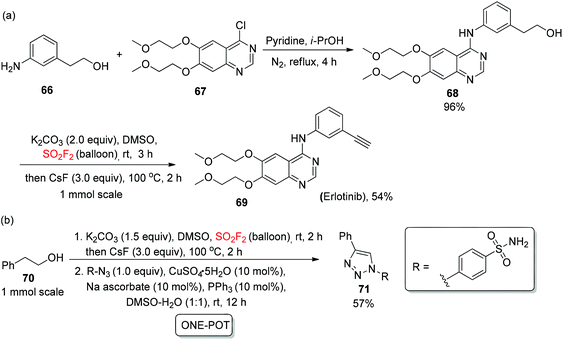 |
| | Scheme 27 (a) and (b) Applications of a novel cascade process in the assembly of biologically active molecules. | |
2.4.2. Alcohols to carbonyl compounds.
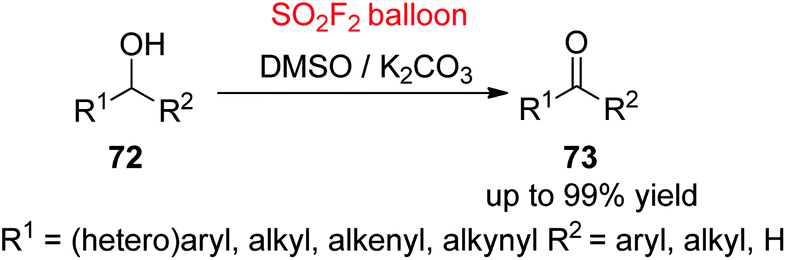
Ketones and aldehydes are important key intermediates for the preparation of a broad range of versatile pharmaceuticals, materials, fragrances, vitamins, and fine chemicals, and other chemical conversions.53 Therefore, the development of novel methodologies for the construction of ketones or aldehydes from alcohols through selective oxidation while preventing unwanted over-oxidation to by-products, such as esters and carboxylic acids, is extremely significant in both industry and laboratory.54 Over the past decades, different transition metals (Pd, Ir, Cu, Fe, Pt, Ru, Rh, Au, etc.) have been utilized as a catalyst for the selective oxidation of alcohols to carbonyl compounds; the utilization of non-catalytic oxidation methods is a highly predominating protocol in both industry and laboratory.55 For the first time, our laboratory utilized SO2F2 gas as a “perfect” activator for a general selective oxidation of alcohols 72 to carbonyl groups 73 with the aid of a cheap and easily removable inorganic base in DMSO as both solvent and oxidant at room temperature (Scheme 28).56 Under the developed reaction conditions, a wide variety of functionalized 1° and 2° benzylic alcohols, and also aliphatic, allylic and propargylic alcohols were all oxidized smoothly to their respective aldehydes and ketones in up to 99% yields. The reaction mechanism was similar to Scheme 26. A significant result that occurred from the utilization of the “perfect electrophile” SO2F2 was the formation of 1 equiv. fluoride after the consumption of alcohols. The utilization of KF by itself leads to excellent yields of products, suggesting that fluoride was capable (pKa of HF in DMSO is 15 ± 2)57 of generating sulphur ylide in the view of Swern-type oxidation – the first time this was observed.
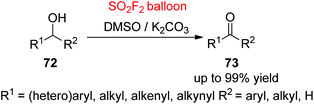 |
| | Scheme 28 Oxidation of alcohols. | |
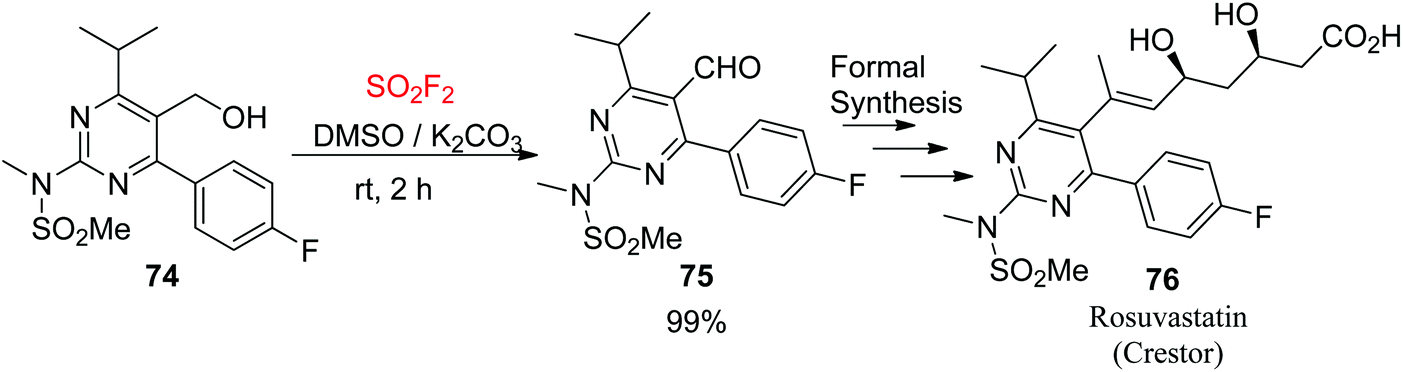
The synthesis of the pyrimidinyl aldehyde 75, the substrate for rosuvastatin 76 (Crestor™, an epic HMG-CoA reductase inhibitor),58 from the functionalized alcohol 74 was achieved efficiently to give the aldehyde with a nearly quantitative yield without chromatography (Scheme 29). This formal synthesis59 of the drug demonstrated well the efficiency of the SO2F2/DMSO oxidation in the target-oriented construction of complex molecules.
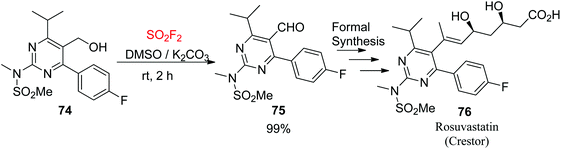 |
| | Scheme 29 Application of SO2F2/DMSO oxidation in the formal synthesis of rosuvastatin. | |
2.4.3. Alcohols to nitriles.
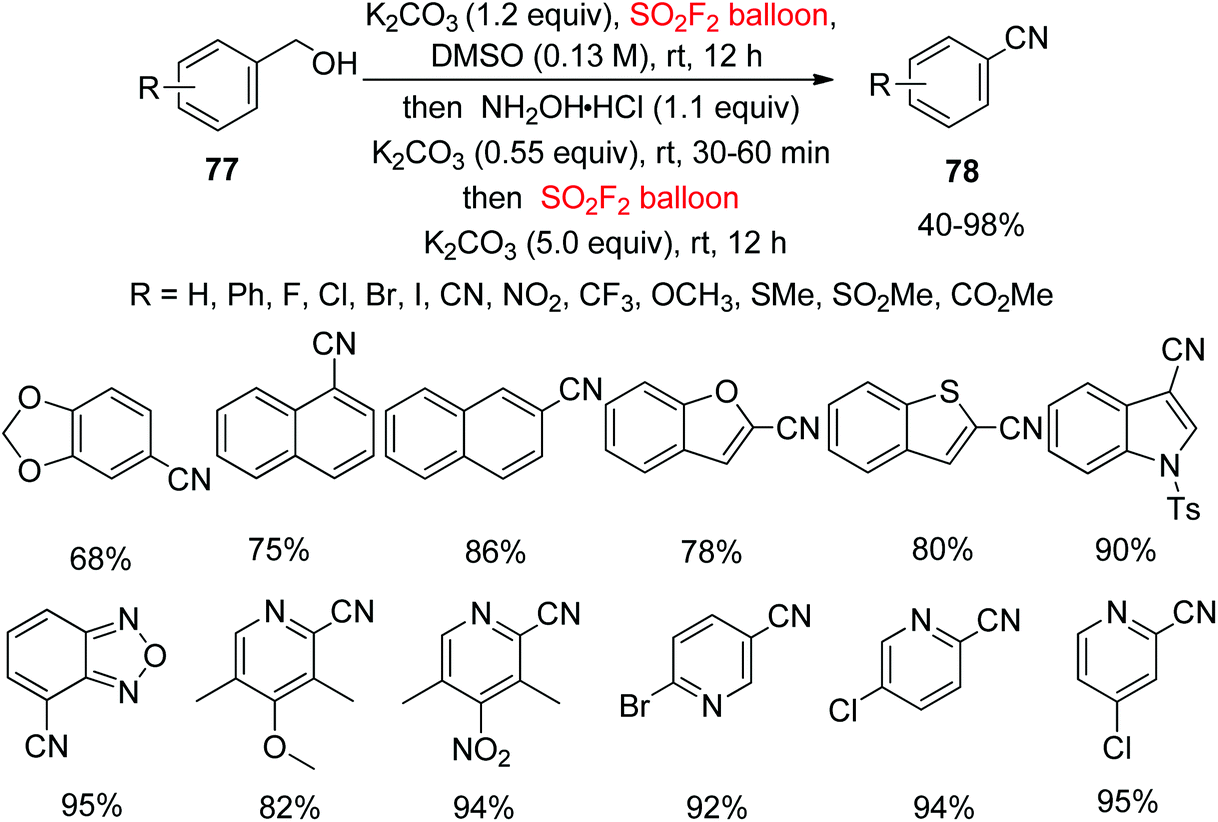
The common strategies for the conversion of alcohols into nitriles are via transition-metal (Cu, Ni, Pd, etc.) catalyzed SN2 substitution, or regular SN2-type reactions utilizing more noxious metal cyanides such as Zn(CN)2, KCN, NaCN, CuCN, etc. with organo-halides, triflates or mesylates and other activated derivatives from hydroxyl groups.60 In continuation of our interest on SO2F2 mediated transformations of hydroxyl groups into nitriles, a new one-pot method for the transformation of readily-available alcohols 77 into highly valuable nitriles 78 using cheaper SO2F2 gas through the transition-metal free cascade method was developed very recently in our laboratory without introducing an “additional carbon atom” (Scheme 30).61 Under the optimal reaction conditions, different functionalized alcohols were converted smoothly into their cyanated products in good yields. Furthermore, di-substituted phenyl methanols and also naphthalene methanols were tolerated smoothly under similar conditions. Remarkably, nitriles having O-heterocycles, N-heterocycles and S-heterocycles were furnished in excellent to quantitative yields from their corresponding alcohols. Most importantly, nitriles containing a pyridine ring were also successfully obtained in quantitative yields from their corresponding alcohols with this protocol.
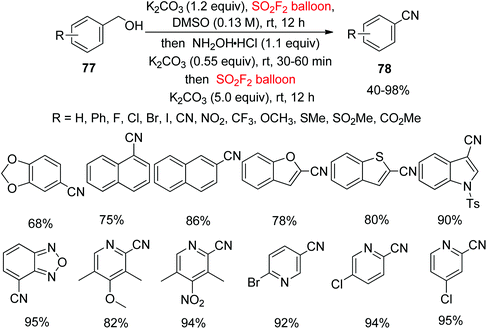 |
| | Scheme 30 SO2F2 mediated transformations of alcohols into nitriles. | |
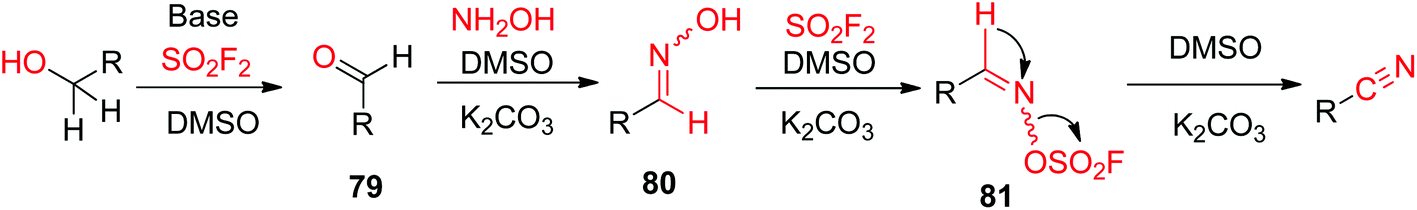
A possible reaction mechanism for SO2F2 mediated transformations of alcohols into nitriles was proposed (Scheme 31). Initially, the alcohol was converted into aldehyde 79 (similar to Scheme 26), which then underwent dehydration to generate aldoxime intermediate 80 upon reaction with NH2OH. Subsequently, aldoxime intermediate 80 reacted with SO2F2 to generate sulfonyl ester 81, which finally underwent β-elimination with the aid of a base to yield the respective nitrile.
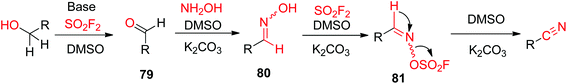 |
| | Scheme 31 Proposed mechanism for SO2F2 mediated transformations of alcohols into nitriles. | |
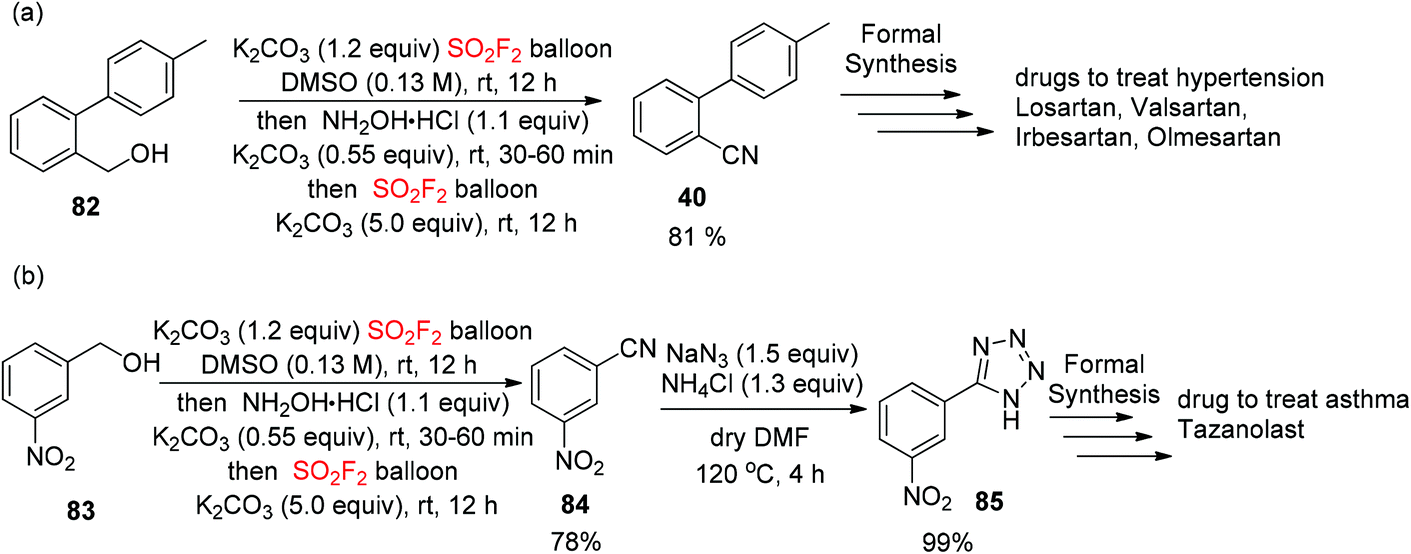
In addition, to show the convenience of this new cascade method for the assembly of complex drug molecules, the construction of o-tolylbenzonitrile (OTBN, 40), the significant pharmaceutical precursor for the formal synthesis of the drugs olmesartan, irbesartan, losartan and valsartan, was achieved smoothly from alcohol ((4′-methyl-[1,1′-biphenyl]-2-yl)methanol) 82 in 81% isolated yield utilizing this developed procedure (Scheme 32a). Furthermore, starting from the alcohol (3-nitrophenyl)methanol 83, the formal synthesis of tetrazole 85 for the asthma medicine tazanolast62 was accomplished in two steps with 99% yield by utilizing this developed novel cascade method (Scheme 32b).
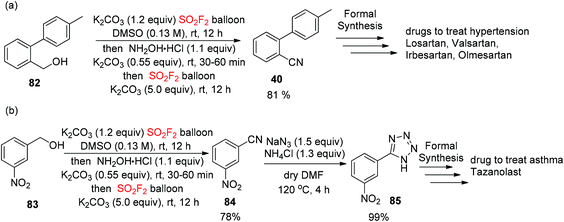 |
| | Scheme 32 (a) and (b) Applications of the novel cascade method in the formal synthesis of drugs. | |
2.4.4. Alcohols to di(hetero)arylmethanes.
Two main strategies have been widely utilized for the synthesis of di(hetero)arylmethanes. The first one is a transition-metal-catalyzed coupling reaction63 and the second one is a Friedel–Crafts type of reaction.64 Because of the steric and electronic limitations of Friedel–Crafts reactions, the transition-metal-catalyzed coupling reaction, particularly the Suzuki type of cross-coupling reaction between organohalides and organoboronic acids (or esters), has been significantly utilized for the assembly of di(hetero)arylmethanes effectively.65 Very recently, our laboratory utilized SO2F2 gas for the synthesis of di(hetero)arylmethanes 88 from (hetero)benzylic alcohols 86 upon dehydroxylative coupling with (hetero)arylboronic acids 87 catalyzed by the transition metal Pd (Scheme 33).66 Both the arylboronic acids and benzylic alcohols containing different substituent groups were efficiently tolerated and furnished the desired diarylmethanes in good to quantitative yields. Furthermore, also bis-substituted benzylic alcohols were transformed easily into their respective diarylmethane derivatives in good yields. Remarkably, naphthalene boronic acids and 1-naphthylmethanol were all well tolerated with good yields of the corresponding products. Most importantly, both heteroaryl boronic acids and heteroaryl methanols also furnished their corresponding diarylmethanes having an aromatic heterocycle such as pyridine, thiophene, and furan in reasonable to good yields, which pointed out the importance of this protocol for the assembly of bioactive heterocycle-containing molecules.
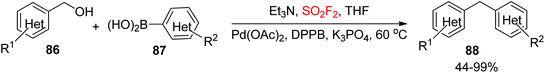 |
| | Scheme 33 Dehydroxylative coupling reaction between benzylic alcohol and aryl boronic acid. | |
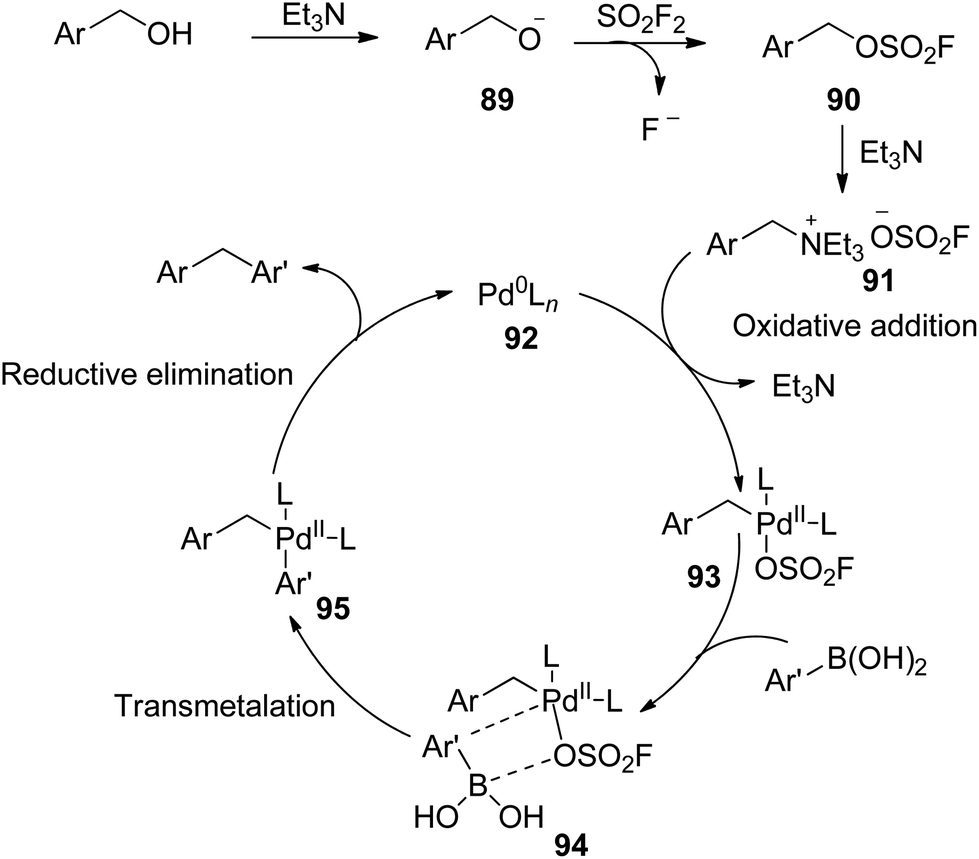
A possible reaction mechanism proposed in Scheme 34 was initiated by the base-promoted deprotonation of benzylic alcohols to give the alkoxide 89, which was then treated with SO2F2 gas to furnish fluorosulfonate ester 90. Subsequently, the resultant intermediate 90 generated the benzyltriethylammonium salts 91 upon treatment with the base Et3N. The subsequent oxidative addition of benzyltriethylammonium salts 91 with Pd0Ln complex 92 generated the intermediate 93,67 which then coordinated with boronic acid to give complex 94. The transmetalation of complex 94 then generated the intermediate 95,68 which finally underwent reductive elimination to yield the diarylmethane while regenerating the Pd0Ln complex 92.
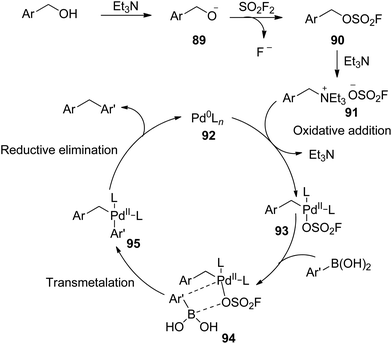 |
| | Scheme 34 Proposed mechanism for the dehydroxylative coupling reaction between benzylic alcohol and aryl boronic acid. | |
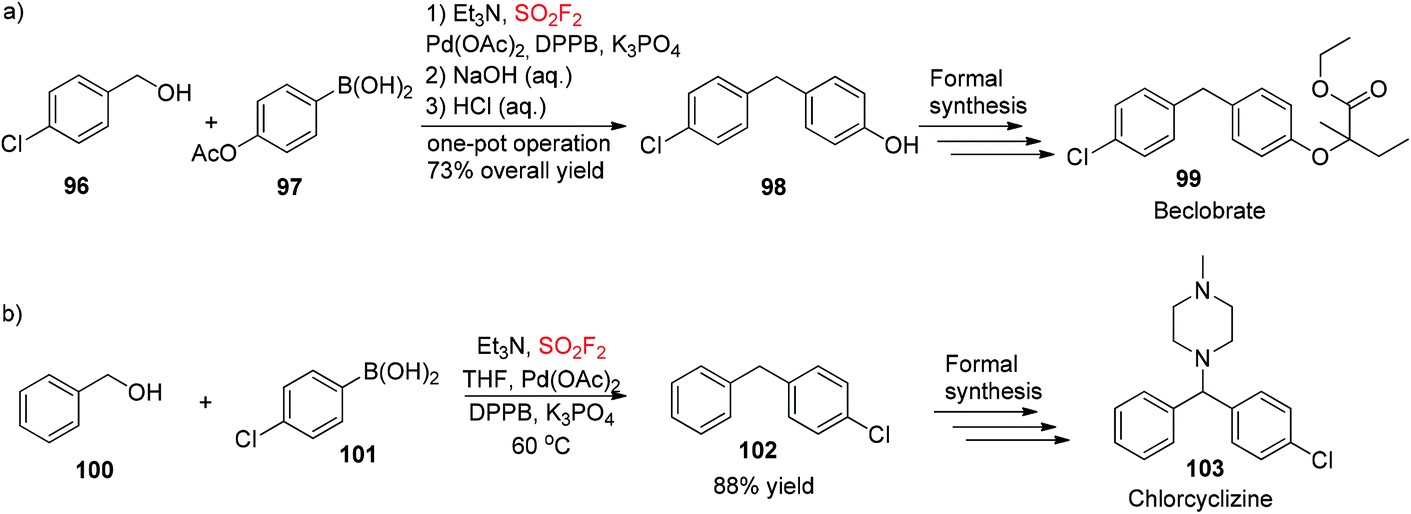
To show the synthetic applicability of our developed dehydroxylative coupling procedure, 4-(4-chlorobenzyl)phenol 98, the key intermediate for the formal synthesis of beclobrate6999, was obtained during a one-pot procedure from (4-acetoxyphenyl)boronic acid 97 and (4-chlorophenyl)methanol 96 in 73% overall yield (Scheme 35a). Furthermore, 1-benzyl-4-chlorobenzene 102, the starting material for formal synthesis of chlorcyclizine70103, was synthesized during dehydroxylative coupling between (4-chlorophenyl)boronic acid 101 and benzyl alcohol 100 utilizing this newly developed protocol in good yield (Scheme 35b).
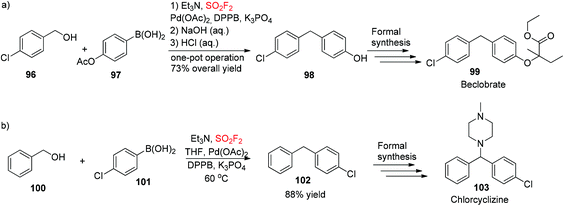 |
| | Scheme 35 Applications of a newly developed protocol in the formal synthesis of Beclobrate (a) and Chlorcyclizine (b). | |
2.4.5. Alcohols to 1,3-dienes.
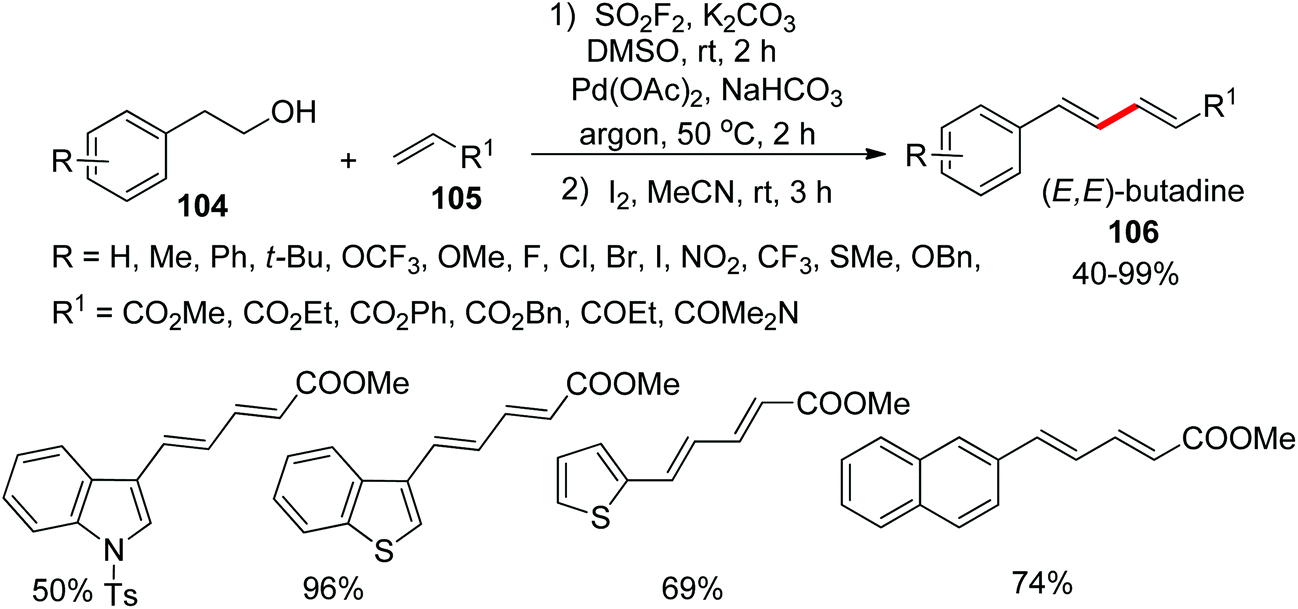
1,3-Dienes represent the core motif of several complex natural products71 and biologically active molecules.72 Recently, many researchers have developed synthetic routes for the preparation of 1,3-dienes through alkene–alkene and alkene–alkyne cross-coupling reactions.73 For the first time, from our research group, 1,3-dienes 106 were synthesized very recently from commercially available homobenzylic alcohols 104 upon treatment with olefins 105 through Pd-catalysis mediated by SO2F2 gas (Scheme 36).74 Under established reaction conditions, the homobenzylic alcohols functionalized with both electron-withdrawing and electron-donating groups were smoothly transformed into their respective 1,3-dienes in moderate to good yields upon treatment with acrylates. Furthermore, naphthalene alcohols and heterocyclic alcohols were also tolerated smoothly in 50–96% yields. Under identical reaction conditions, olefin coupling partners were also examined and furnished 1,3-dienes in 77–88% yields.
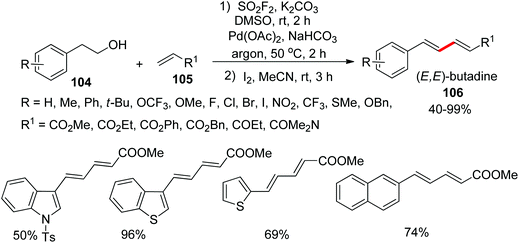 |
| | Scheme 36 Pd-Catalyzed conversion of homobenzylic alcohols into 1,3-dienes mediated by SO2F2 gas. | |
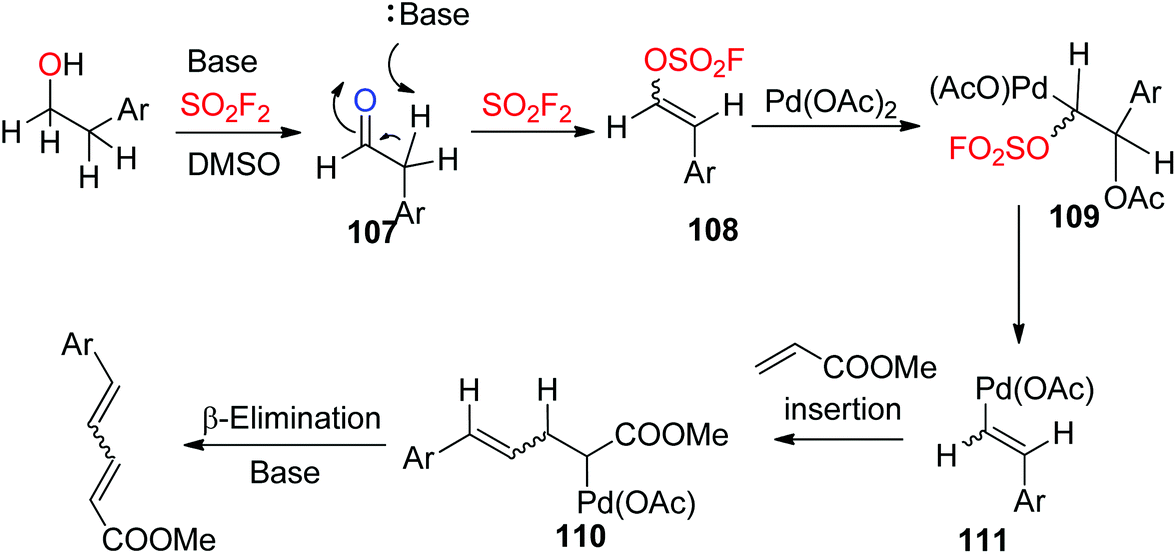
A plausible mechanism for Pd-catalyzed, SO2F2 mediated transformation of homobenzylic alcohols into 1,3-dienes was proposed (Scheme 37). Initially, the alcohol was converted into aldehyde 107 (similar to Scheme 26), which then generated vinyl sulfurofluoridate 108 upon treatment with a base and SO2F2 gas. Subsequently, vinyl sulfurofluoridate 108 was captured by the Pd catalyst and generated intermediate 109, which then underwent an elimination to furnish intermediate 110. The coordination of methyl acrylate to the metal center followed by insertion into the Pd-vinyl bond generated an intermediate 111, which finally underwent β-elimination with the aid of a base to furnish the 1,3-diene.
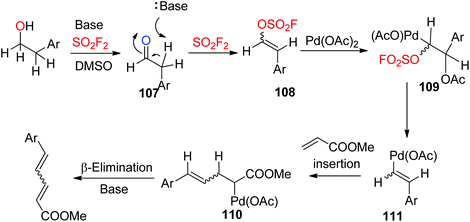 |
| | Scheme 37 Proposed reaction mechanism. | |
2.4.6. Alcohols to diaryltriazoles.
Synthesis of substituted 1,2,3-triazoles was an exceptional, influential method for drug discovery, medicinal chemistry, chemical biology, and materials science.75 Substituted 1,2,3-triazole derivatives are significant classes of chemicals ubiquitously present in agrochemicals, dyes, functional materials, catalysts, ligands, biologically active molecules, and natural products.76 The Huisgen alkyne-azide cycloaddition reaction for the assembly of 1,4-disubstituted 1,2,3-triazole derivatives77 is one of the most useful and powerful alkyne conversions in different research areas. Recently, our laboratory followed the Feringa procedure78 utilizing SO2F2 gas for a one-pot production of 1,4-diaryl-1,2,3-triazoles 113 directly from homobenzylic 1° alcohols 112 using CuSO4·5H2O and PPh3 as a ligand (Scheme 38).48 Homobenzylic 1° alcohols having both electron-poor and electron-rich substituents, 2-(1-naphthyl)ethanol, 2-heteroaryl ethanols, homoallylic alcohol, and homopropargyl alcohol, were all smoothly converted into their corresponding 1,2,3-triazoles in up to 92% yield under the optimal reaction conditions.
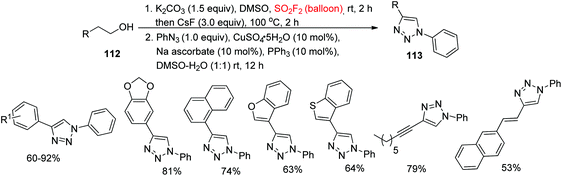 |
| | Scheme 38 SO2F2-mediated transformation of 2-substituted ethanols into 1,4-diaryl-1,2,3-triazoles. | |
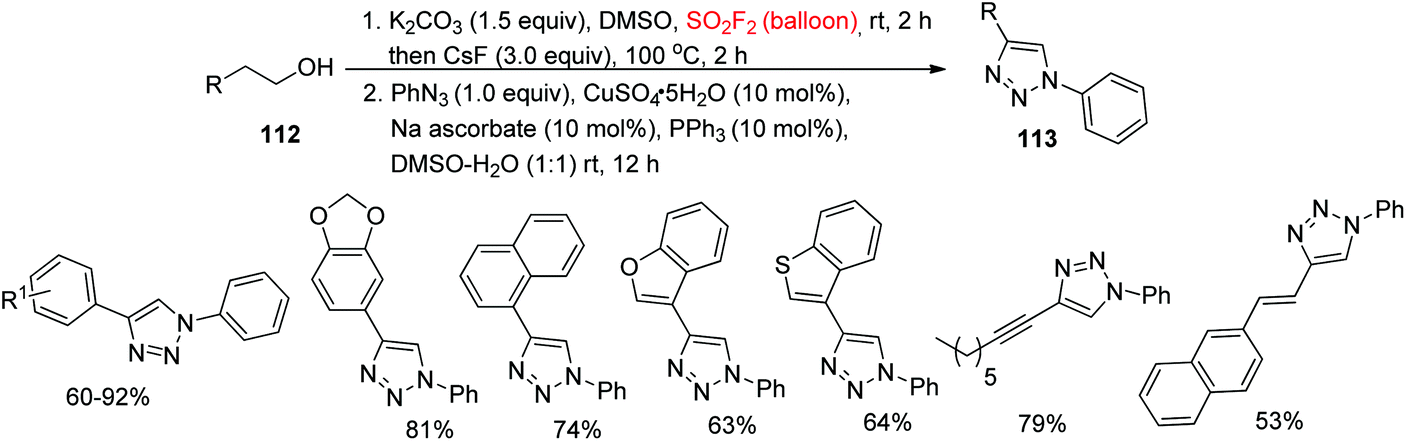
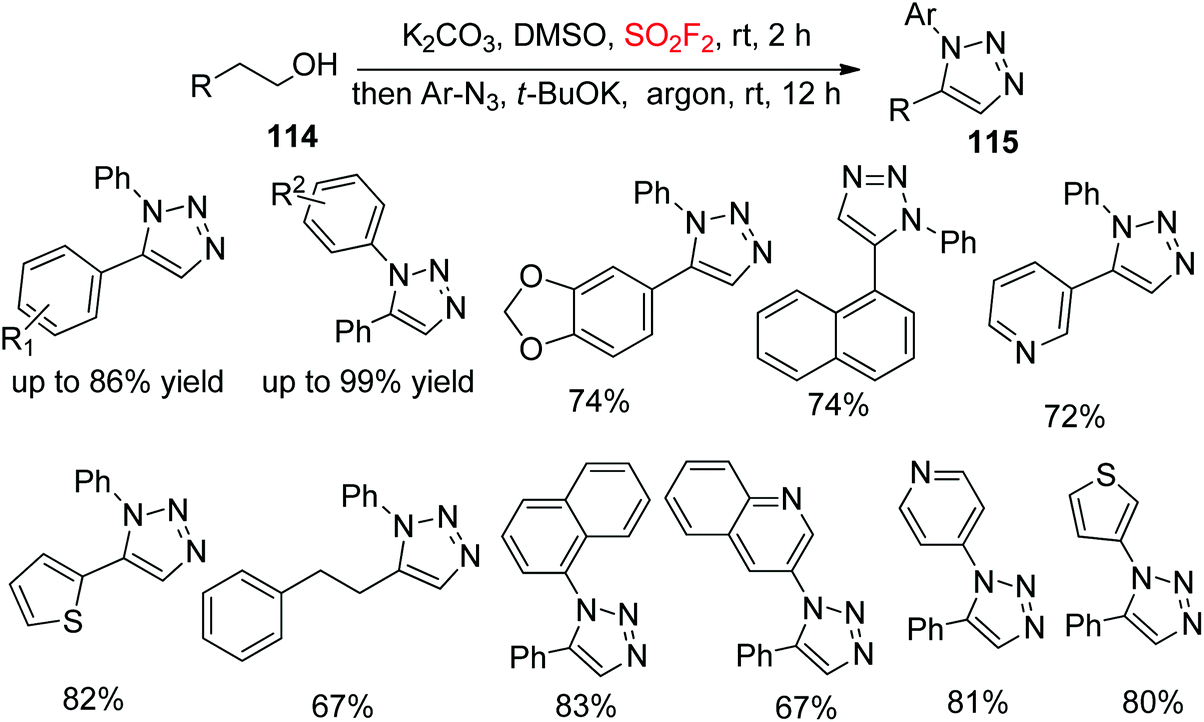
In continuation of our interest on diaryltriazole synthesis utilizing gaseous SO2F2, very recently, our laboratory synthesized 1,5-diaryl-1,2,3-triazoles 115 from cheap and commercially available alcohols 114 upon dehydrative cycloaddition with aryl azides through the transition-metal free cascade method mediated by SO2F2 gas (Scheme 39).79 A variety of functionalized arylethanols were smoothly converted into the desired 1,5-diaryl-1,2,3-triazoles in high yields. Furthermore, also multifunctionalized arylethanols were efficiently transformed into their respective triazoles in good yields regio-selectively. Remarkably, naphthyl substrates, alcohols having heteroarenes and long-chain aliphatic alcohols were all well tolerated with good yields of the corresponding products. In addition, diverse aryl azides having different substituent groups and also heteroaryl azides all furnished their corresponding triazoles in high yields under the same reaction conditions.
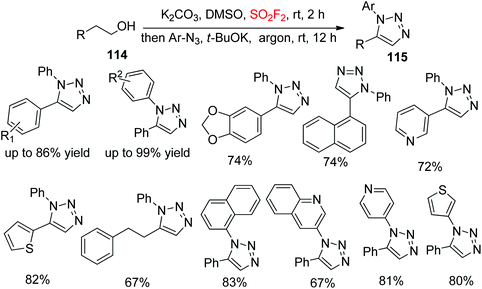 |
| | Scheme 39 SO2F2 mediated transformations of alcohols into diaryltriazoles. | |
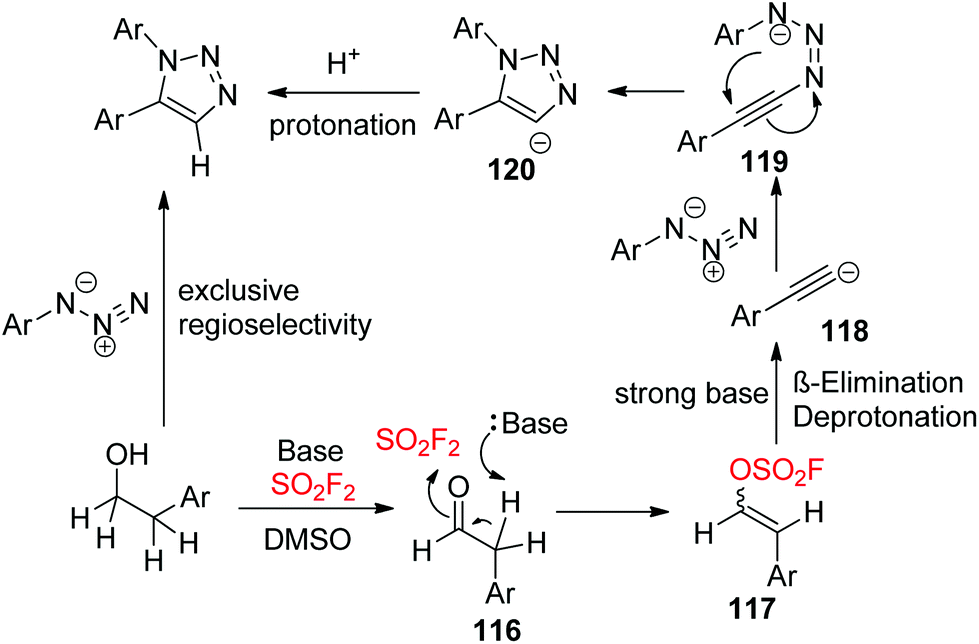
A plausible mechanism for SO2F2 mediated transformations of alcohols into diaryltriazoles was proposed (Scheme 40). Initially, the alcohol was converted into aldehyde 116 (similar to Scheme 26), which then generated vinyl sulfurofluoridate 117 upon treatment with a base and SO2F2 gas. Subsequently, vinyl sulfurofluoridate 117 underwent β-elimination and deprotonation with the aid of a base to give the reactive acetylide 118,80 which then generated the triazenide intermediate 119 by a nucleophilic attack on the terminal nitrogen of the aryl azide. The resultant intermediate 119 then underwent an intramolecular cyclization to give the 1,5-aryl-1,2,3-triazolyl anion 120, which finally furnished the 1,5-diaryl-1,2,3-triazole with exclusive regioselectivity upon reaction with a proton cation.
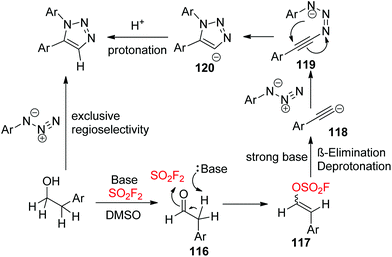 |
| | Scheme 40 Proposed mechanism for SO2F2 mediated transformations of alcohols into diaryltriazoles. | |
2.5. Chemical transformations of amines
2.5.1. Amines to sulfamides.
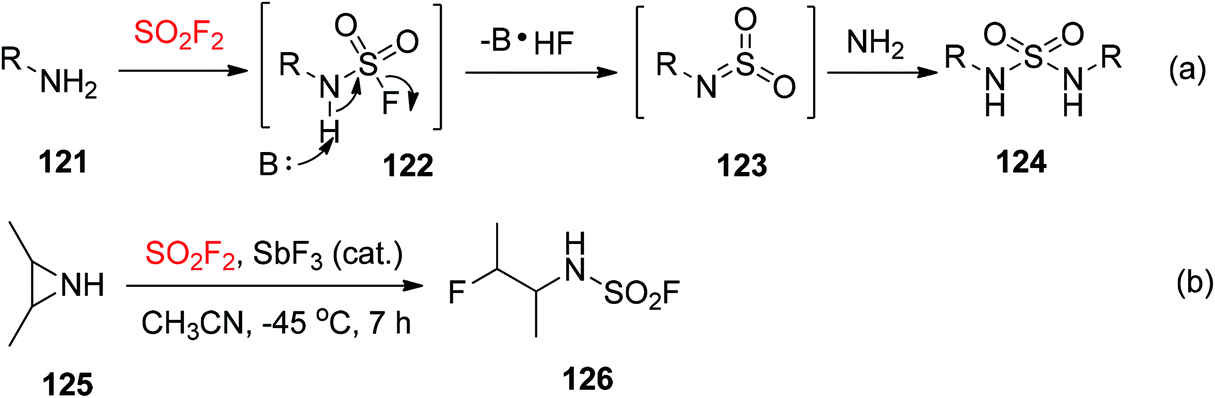
Primary amines 121 were easily reacted with SO2F2 gas and resulted in fluorosulfated products A. Due to the more acidic nature of the N-sulfamoyl proton, the resultant products 122 then underwent facile elimination to yield azasulfene intermediates 123,81 which were captured by amines to furnish symmetrically substituted sulfamides 124 (Scheme 41a).82 A few methods utilized SO2F2 gas for the synthesis of N-monosubstituted sulfamoyl fluorides 126 from secondary amines 125 including ring-opening of an aziridine under the standard conditions (Scheme 41b).83
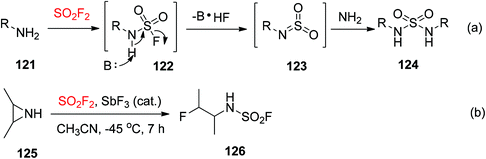 |
| | Scheme 41 Synthesis of sulfamides (a) and sulfamoyl fluorides (b) utilizing SO2F2 gas. | |
2.5.2. Amines to sulfamoyl fluorides.
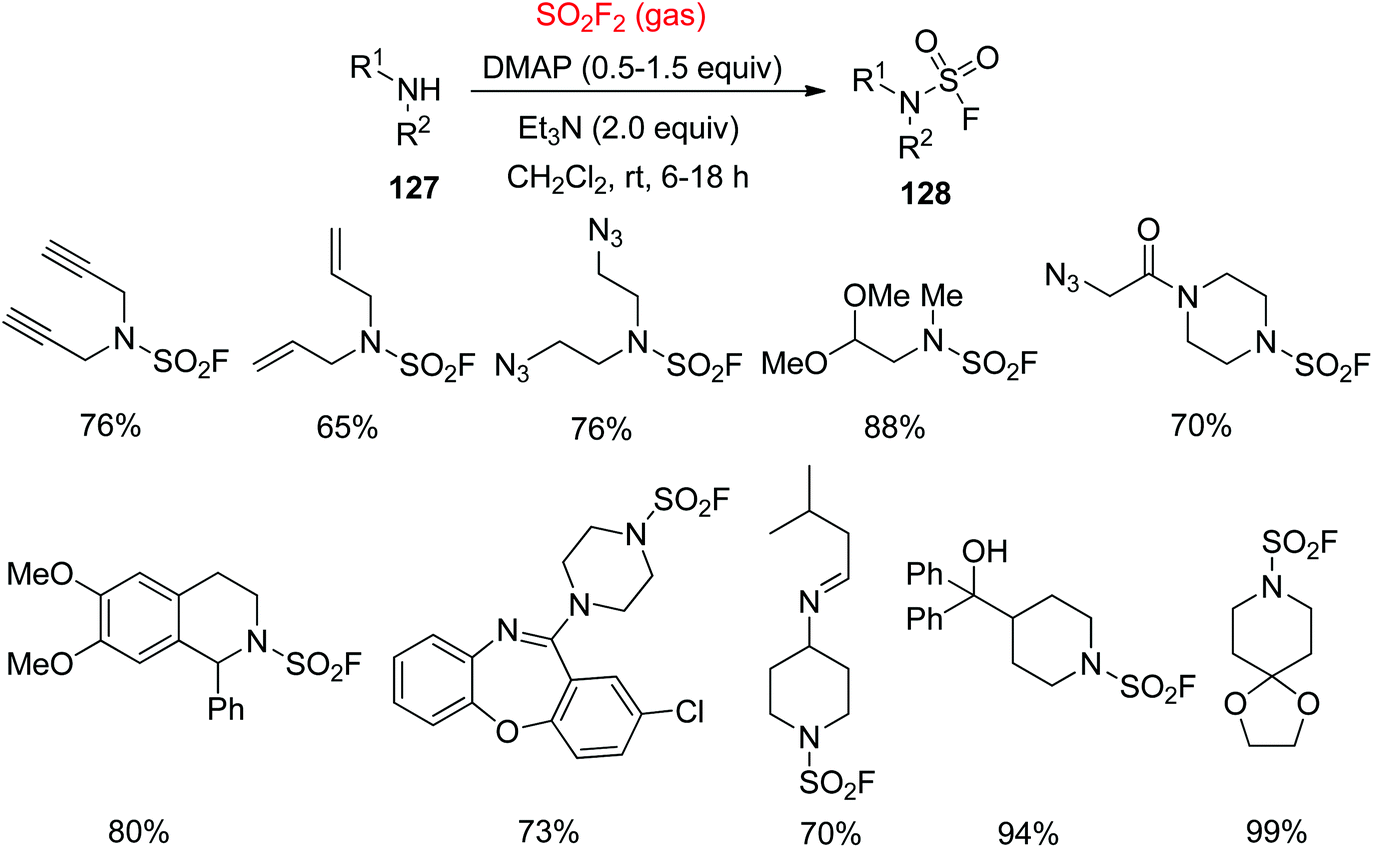
In contrast, remarkably more stable and robust N-disubstituted sulfamoyl fluorides 128 were synthesized smoothly from the secondary amines 127 by treating with SO2F2 gas in the presence of N,N-4-dimethylaminopyridine (DMAP) and Et3N for 6–18 hours at room temperature in CH2Cl2 (Scheme 42).7a Poor nucleophiles like disubstituted anilines were not reacted with SO2F2 gas under these reaction conditions within a reasonable period of time.
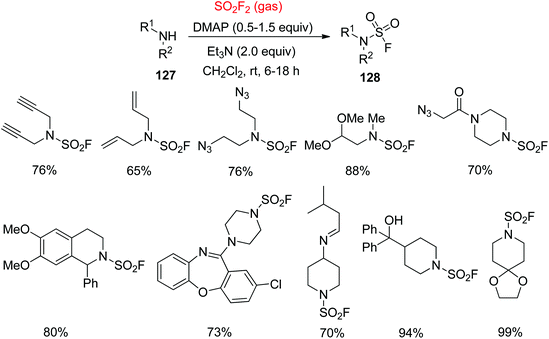 |
| | Scheme 42 Construction of N-disubstituted sulfamoyl fluorides using SO2F2 gas. | |
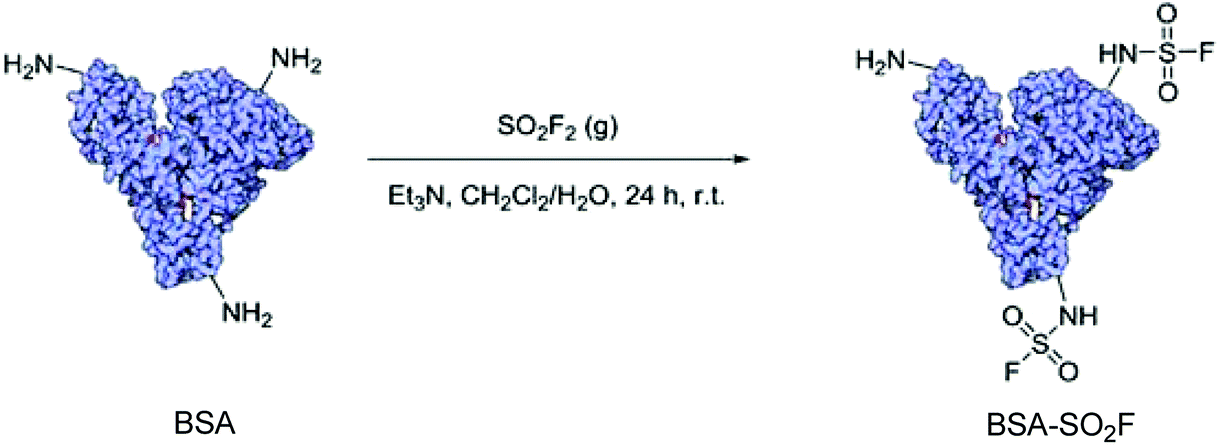
In 2016, for the first time, Averick and co-workers synthesized a novel bioconjugate BSA-SO2F from the model protein BSA (bovine serum albumin) upon treatment with SO2F2 gas under biphasic conditions (Scheme 43).84 Importantly, BSA-SO2F was demonstrated to be biocompatible after 72 h of incubation with A549 lung endothelial cells. Because of the unique and selective reactivity of the –SO2F group with amines, BSA-SO2F could be self-condensed to construct a biocompatible hydrogel that could be utilized in co-culture with HEK 293 cells.
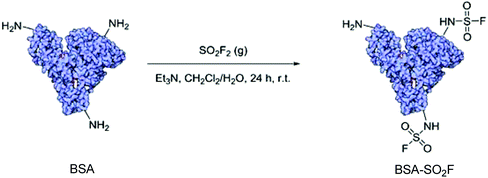 |
| | Scheme 43 SuFEx modification of BSA with SO2F2 gas. | |
2.5.3. Amines to amides.
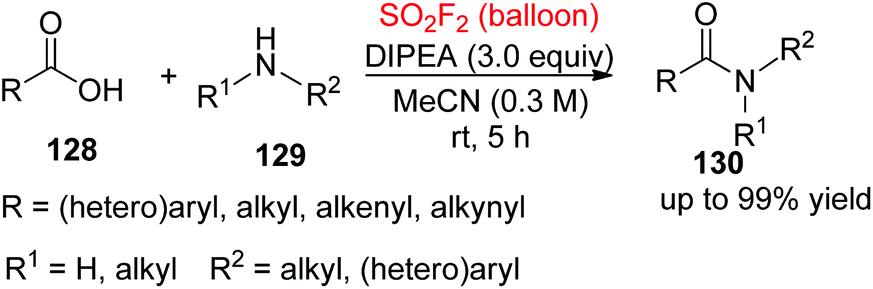
The formation of the amide bond and peptide linkage is one of the basic conversions in all life processes. Very recently, our laboratory developed a mild, simple, practical, robust and highly efficient methodology for the formation of an amide bond and a peptide linkage via a SO2F2-mediated direct clickable coupling of carboxylic acids 128 with amines 129 (Scheme 44).85 The reactions proceeded well under mild conditions and tolerated a range of functional groups of both aliphatic and aromatic amines and carboxylic acids.
 |
| | Scheme 44 SO2F2 mediated coupling of carboxylic acids with amines. | |
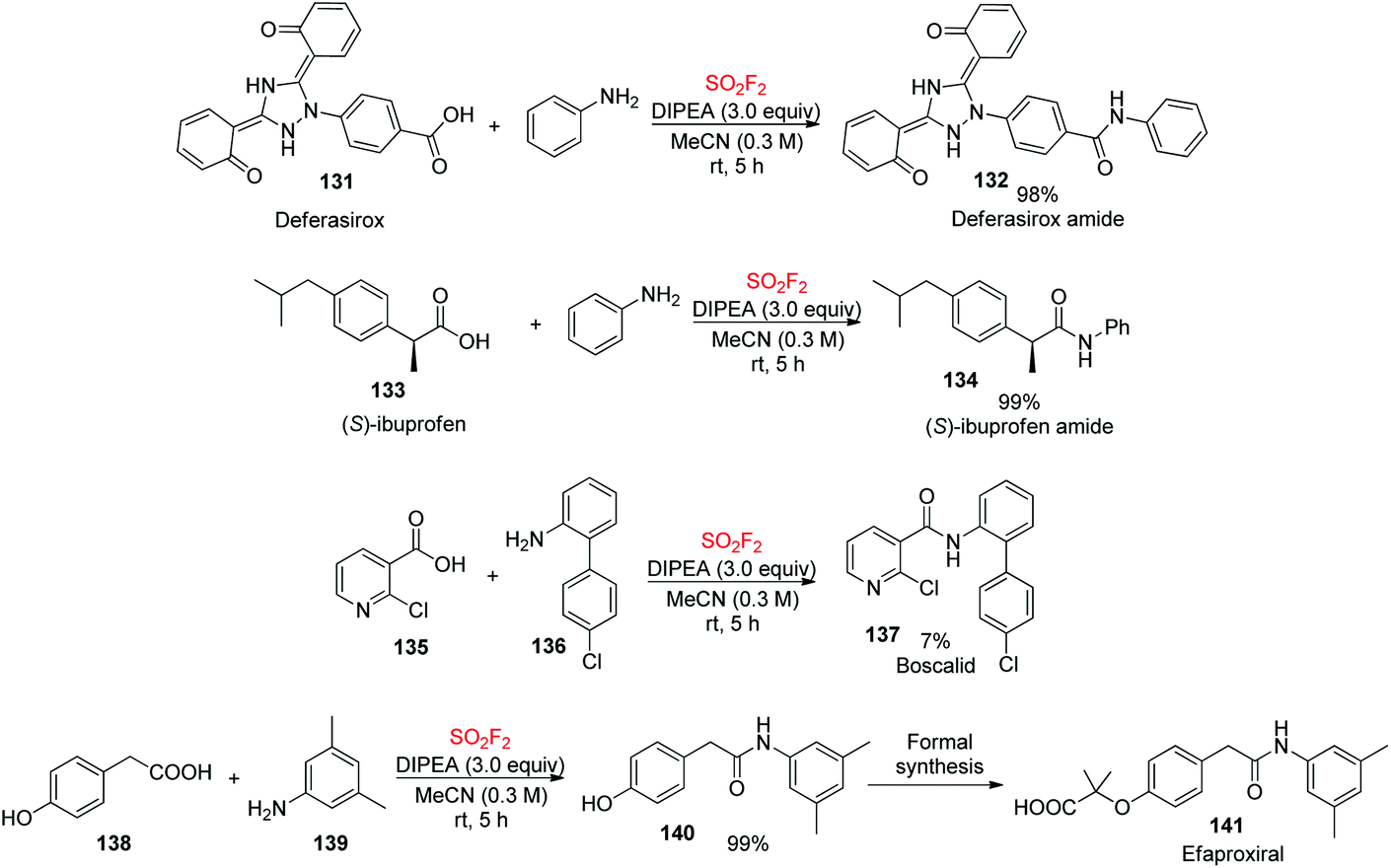
Our newly developed SO2F2-mediated direct clickable coupling process was utilized for the assembly of biologically active molecules (Scheme 45). Deferasirox 131 and ibuprofen 133 were efficiently transformed into their corresponding amides in quantitative yields upon coupling with aniline. Furthermore, the construction of Boscalid 137, a fungicide, was accomplished in good yields during the coupling of the corresponding amine 136 and carboxylic acid 135 utilizing this protocol. In addition, the formation of amide (N-(3,5-dimethylphenyl)-2-(4-hydroxyphenyl)acetamide) 140, a key intermediate for formal synthesis of the cholesterol drug efaproxiral 141,86 was also achieved in a quantitative yield.
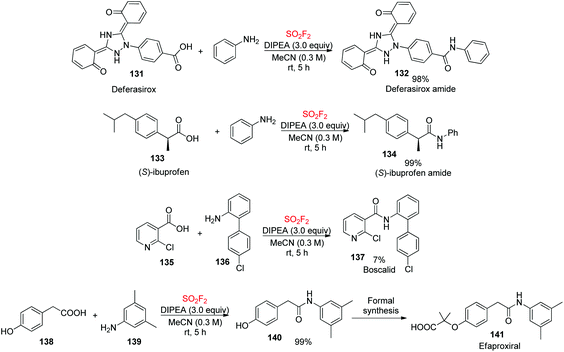 |
| | Scheme 45 Applications of the newly developed method in the synthesis and modification of biologically active molecules. | |
2.5.4. Amines to fluoroalkylated amines.
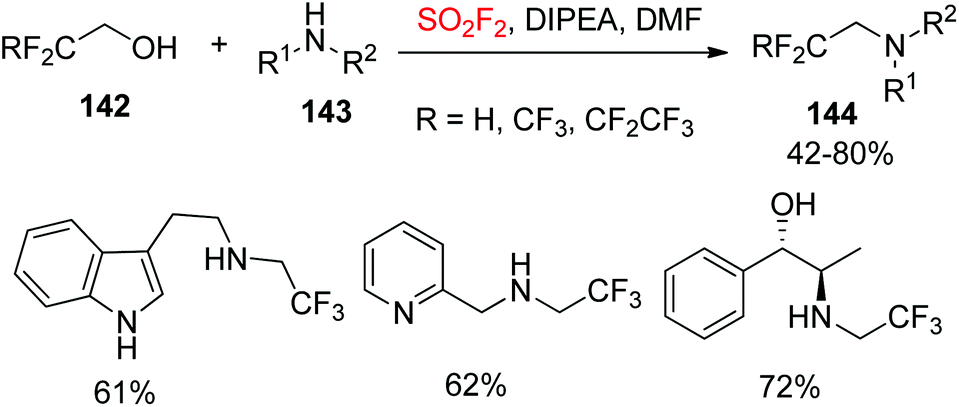
Recently, Sammis and co-workers reported a new application of SO2F2 gas in a novel one-pot 1,1-dihydrofluoroalkylation reaction, which simply involved bubbling SO2F2 through a solution of 1,1-dihydrofluoroalcohol, amine, and diisopropylethylamine (Scheme 46).10b A wide variety of 1,1-dihydrofluoroalcohols 142, and also a range of 1° and 2° amines 143 converted smoothly into their respective 1,1-dihydrofluoroalkylated amines 144 in moderate to good yields. The reaction also showed exceptional functional group tolerance.
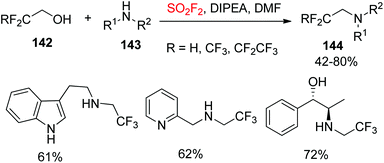 |
| | Scheme 46 SO2F2-mediated one-pot 1,1-dihydrofluoroalkylation. | |
2.6. Chemical transformations of N-arylhydroxylamines into para-amino-arylfluorosulfates
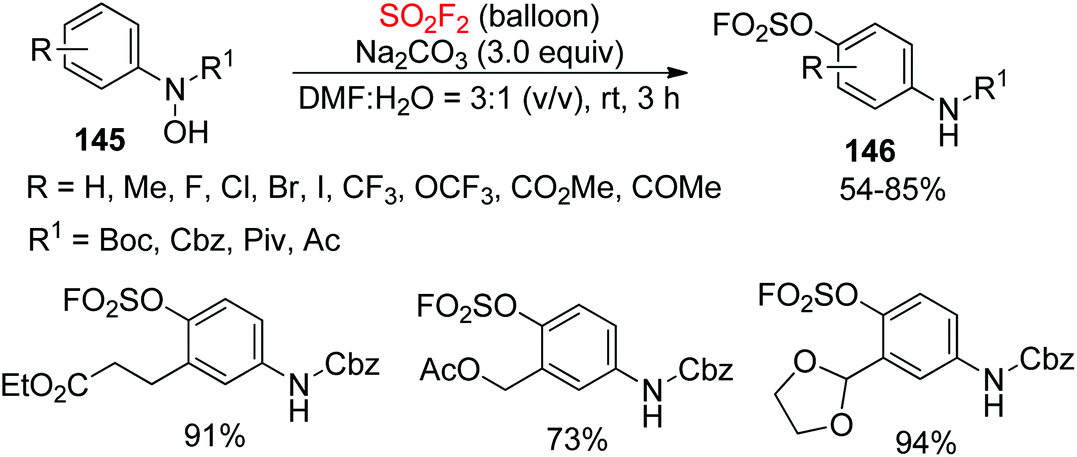
Anilines are widespread in natural materials and pharmaceuticals and have been broadly employed as versatile elements in fine chemicals, synthetic chemistry, and peptide chemistry87 Very recently, our laboratory developed a new and practical methodology for the assembly of aryl fluorosulfates (ArOSO2F) containing anilines 146 from N-arylhydroxylamines 145 upon reaction with SO2F2 gas in the presence of an inorganic base (Scheme 47).88 Under the established reaction conditions, a wide variety of electronically and structurally varied N-arylhydroxylamines protected by tert-butoxycarbonyl(Boc), Cbz and carbonyl groups were all converted efficiently into their respective para-amino-arylfluorosulfates in up to 94% yield.
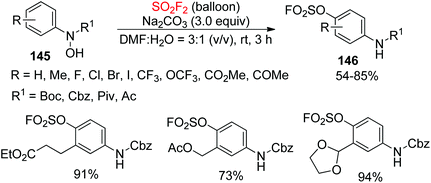 |
| | Scheme 47 SO2F2-mediated rearrangement of N-arylhydroxylamines protected by tert-butoxycarbonyl(Boc), Cbz and carbonyl groups. | |
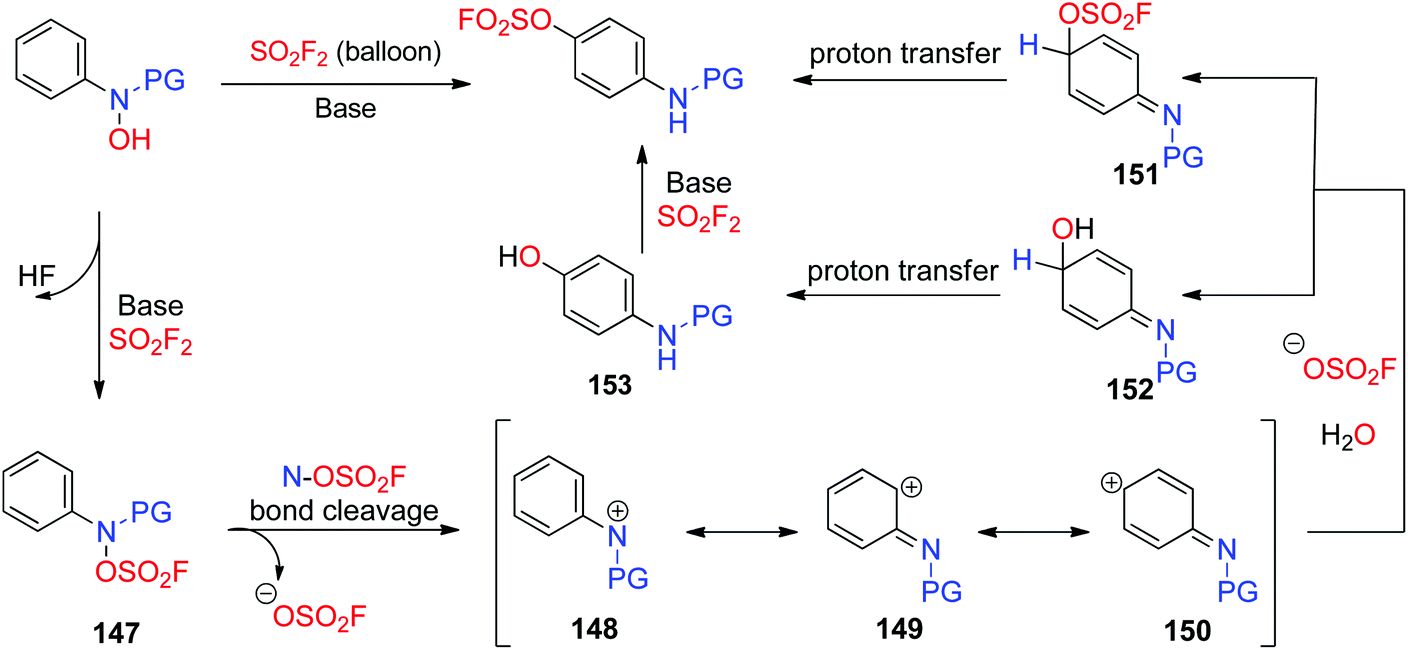
A plausible mechanism for the base-promoted, SO2F2 mediated para-selective rearrangement was proposed (Scheme 48). The reaction was initiated by the base-assisted SuFEx type substitution of the hydroxy group of N-arylhydroxylamine with SO2F2 to give the fluorosulfate 147, which subsequently generated the cationic intermediate 148 after the N–OSO2F bond fission. The resultant cationic intermediate 148 has two other resonance contributors 149 and 150. Subsequently, the more stable carbocation intermediate 150 underwent nucleophilic addition with an OSO2F anion and H2O to afford a mixture of 151 and 152 respectively, which then underwent a proton transfer process to yield the final product fluorosulfate and aminophenol 153, respectively. Finally, aminophenol 153 also furnished the fluorosulfate upon reaction with another equivalent of SO2F2.
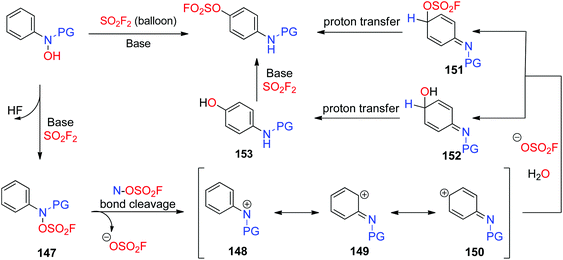 |
| | Scheme 48 Proposed mechanism of the base-promoted, SO2F2 mediated para-selective rearrangement. | |
2.7. Chemical transformations of carbonyl compounds
2.7.1. Aldehydes to nitriles.
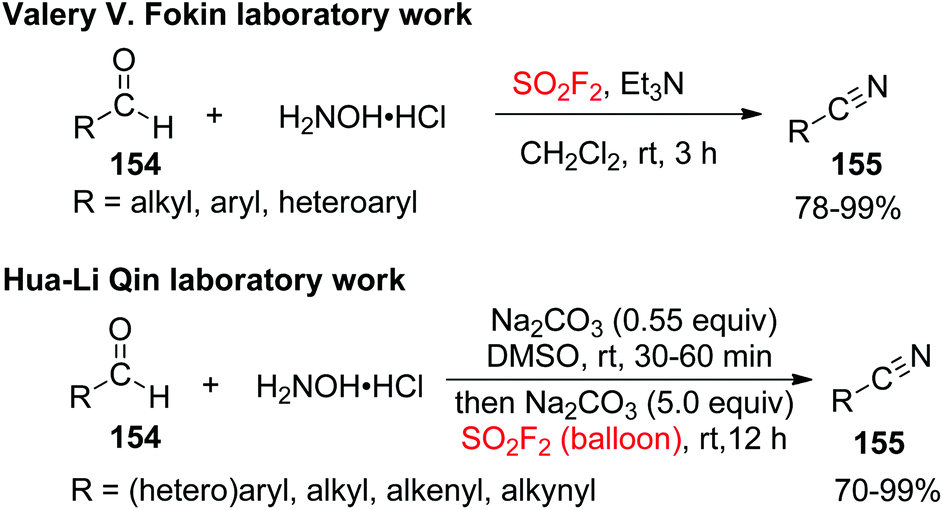
Because of the environment friendliness, low cost and ready availability, aldehydes have been used as substrates for the construction of nitriles through transoximation,89 metal-catalyzed dehydration of aldoximes,90 or fragmentation of the coordinated cyanide anion,91 or by employing N-containing reagents92 for the source of nitrogen. Very recently, another influential and cost-effective transition-metal free one-pot cascade procedure was introduced by both Fokin and Qin laboratories simultaneously for the SO2F2 mediated direct transformations of inexpensive aldehydes 154 into highly valuable nitriles 155 utilizing the advantages of inorganic reagents under mild conditions (Scheme 49).93 A wide variety of electronically and structurally varied arylaldehydes having both electron-donating and electron-withdrawing groups were obtained efficiently and furnished the desired products in excellent to quantitative yields. Remarkably, polycyclic substrates, compounds having two aldehyde groups on two different aryl rings or a single aryl ring, and also heterocyclic compounds all well tolerated the cyanation system and furnished the respective nitriles in excellent to quantitative yields. Furthermore, the transformation of regular aliphatic aldehydes and aliphatic allylic aldehydes into nitriles also occurred smoothly with absolute retention of the original E configuration of the double bonds under identical reaction conditions.
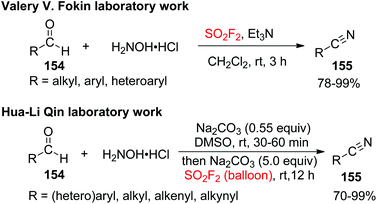 |
| | Scheme 49 SO2F2 mediated transformations of aldehydes into nitriles. | |
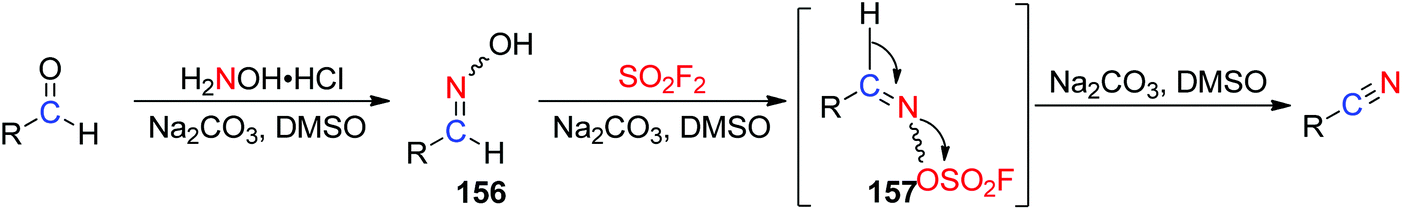
A possible reaction mechanism for SO2F2 mediated conversion of aldehydes into carbon–nitrogen triple bonds of nitriles was proposed (Scheme 50). The reaction was initiated by the formation of aldoxime intermediate 156 after dehydration of aldehyde in DMSO upon treatment with NH2OH in the presence of a Na2CO3 base. The subsequent treatment of 156 with SO2F2 in the presence of the same base generated the desired sulfonyl ester intermediate 157, which finally underwent a base-assisted β-elimination to furnish the corresponding nitrile product.
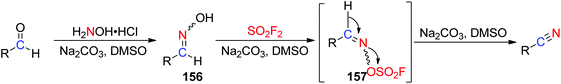 |
| | Scheme 50 A proposed one-pot procedure for SO2F2 mediated transformations of aldehydes into nitriles. | |
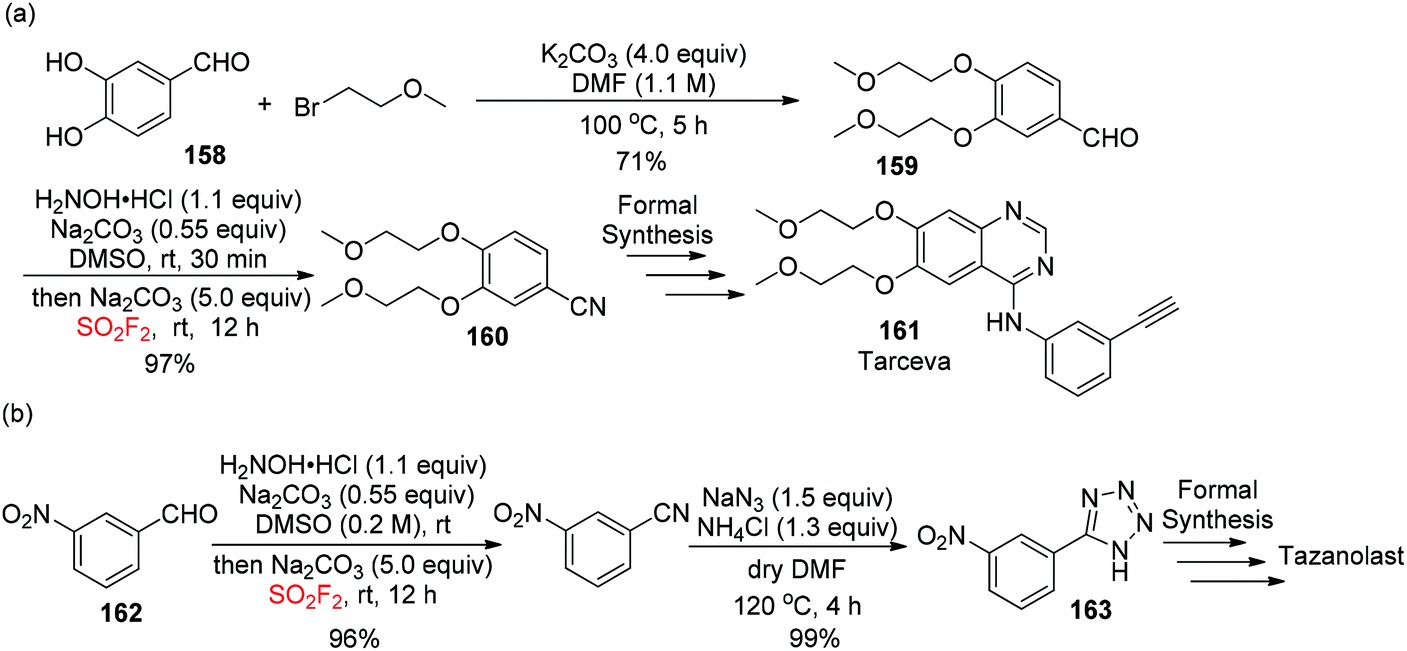
To show the synthetic efficiency of our developed cyanation method for the construction of complex molecules, aldehyde 159 (synthesized from aldehyde 158) was examined under the optimized conditions and afforded the desired nitrile product 160, the substrate for tarceva 161, in 97% yield (Scheme 51a).94 Furthermore, 5-(3-nitrophenyl)-1H-tetrazole 163, a key precursor for tazanolast, was obtained in two steps with 99% yield from aldehyde 162 (Scheme 51b).
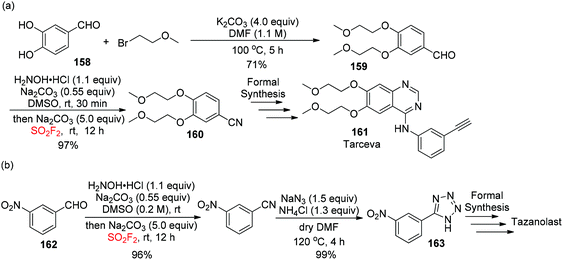 |
| | Scheme 51 Applications of the novel cascade method in the formal synthesis of Tarceva (a) and Tazanolast (b). | |
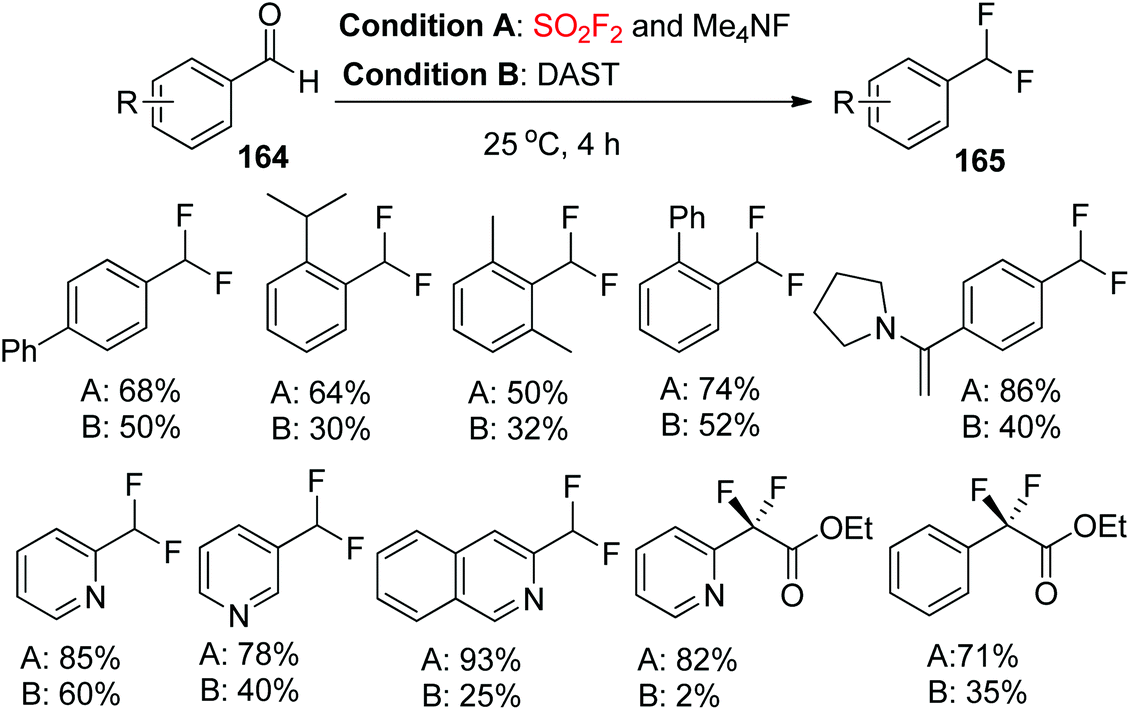
2.7.2. Benzaldehydes to difluoromethylarenes.
Very recently, the Sanford research group reported that the combination of Me4NF and SO2F2 is efficient for the deoxyfluorination of a range of (hetero)aryl aldehydes and α-ketoesters under mild conditions. Moreover, the combination of Me4NF and SO2F2 afforded considerably higher yields than that of DAST in all cases examined (Scheme 52).95
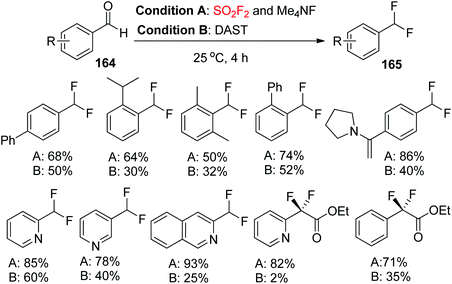 |
| | Scheme 52 Deoxyfluorination of benzaldehydes using SO2F2 and Me4NF. | |
2.7.3. 2′-Hydroxyacetophenones to benzo-oxetes.
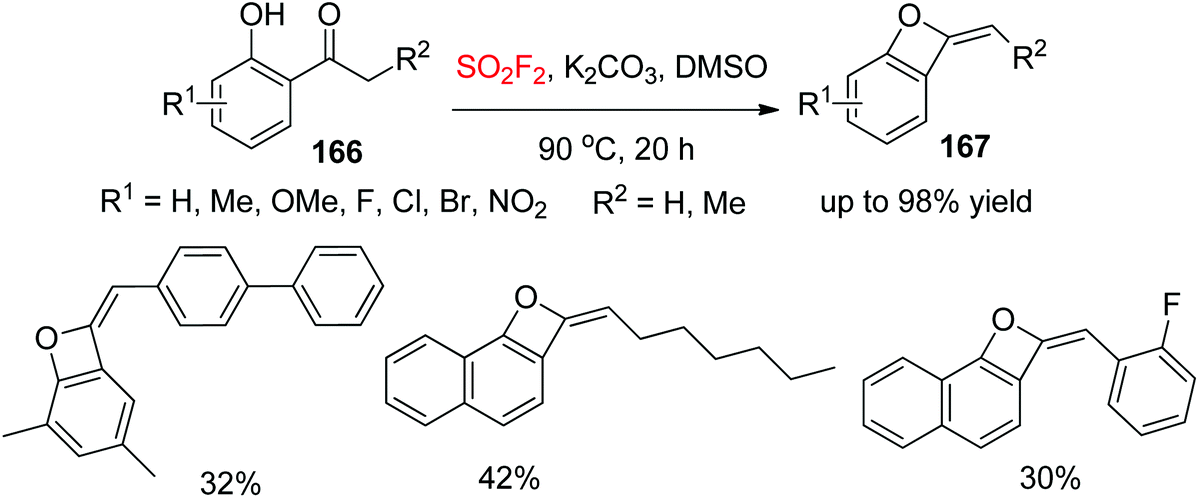
Oxetanes are significant motifs in some biologically active molecules and natural products,96 and also serve as versatile elements in drug discovery.97 In continuation of our interest on SO2F2-mediated chemical transformations, we developed very recently a new strategy for assembling benzo-oxetes 167 from ketones 166, taking advantage of readily available SO2F2 and ketones through a catalyst-free cascade process (Scheme 53).98 A variety of 2′-functionalized hydroxyacetophenones were successfully converted into their respective benzo-oxetes under the established reaction conditions. Furthermore, bis-substituted 2′-hydroxyacetophenones and naphthalenones were also transformed efficiently into their respective benzo-oxetes in up to 95% yield.
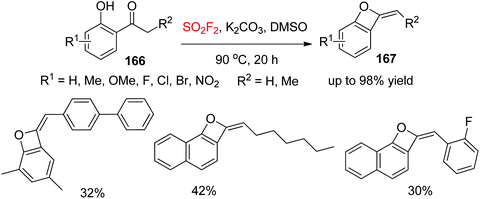 |
| | Scheme 53 SO2F2-mediated conversion of 2′-hydroxyacetophenones into benzo-oxetes. | |
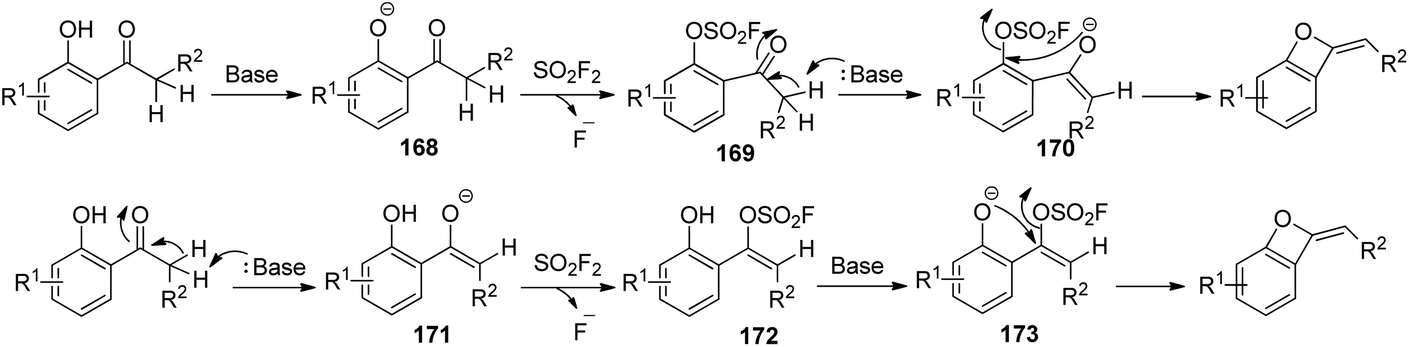
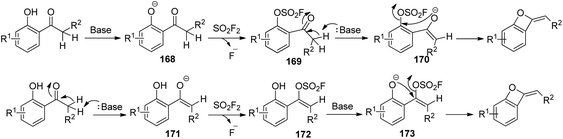
Two plausible reaction mechanisms of SO2F2-mediated conversion of 2′-hydroxyacetophenone into benzo-oxetes were proposed (Scheme 54). In the first one, initially, 2′-hydroxyacetophenone was deprotonated by the base to furnish phenoxide intermediate 168, which then generated an intermediate 169 and a fluoride anion upon treatment with SO2F2. The proton of the acetyl group next to the carbonyl moiety of intermediate 169 was deprotonated by the base to yield the enolated intermediate 170, which finally underwent an intramolecular cyclization to give the corresponding benzo-oxetes. In the second one, initially, the proton of the acetyl group adjacent to the carbonyl moiety of 2′-hydroxyacetophenone was deprotonated by the base to yield the enol intermediate 171, which then generated an intermediate 172 and a fluoride anion upon treatment with SO2F2. Subsequently, the base-assisted deprotonation of intermediate 172 generated the phenol anion 173, which finally underwent an intramolecular cyclization to furnish the corresponding benzo-oxetes.
 |
| | Scheme 54 Two possible reaction mechanisms. | |
2.8. Chemical transformations of arynes into arenesulfonyl fluorides
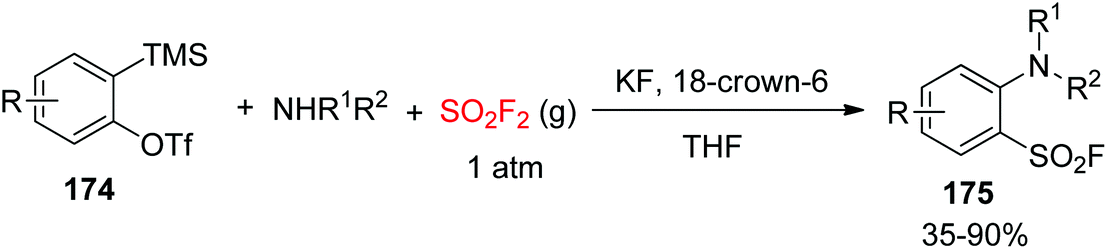
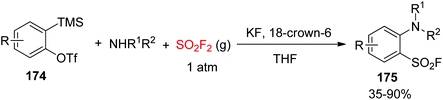
Very recently, the Kim group developed a novel methodology for the synthesis of arenesulfonyl fluorides 175 from the multicomponent reaction (MCR) involving arynes 174, 2° amines, and SO2F2 gas under transition-metal-free conditions (Scheme 55).99 Under mild conditions, zwitterionic intermediates generated from arynes upon reaction with amines captured SO2F2 gas and offered a novel and practical procedure for the synthesis of 2-alkylaryl-, 2-dialkyl-, or 2-diarylamino-substituted arenesulfonyl fluorides in up to 90% yield.
 |
| | Scheme 55 Synthesis of arenesulfonyl fluorides. | |
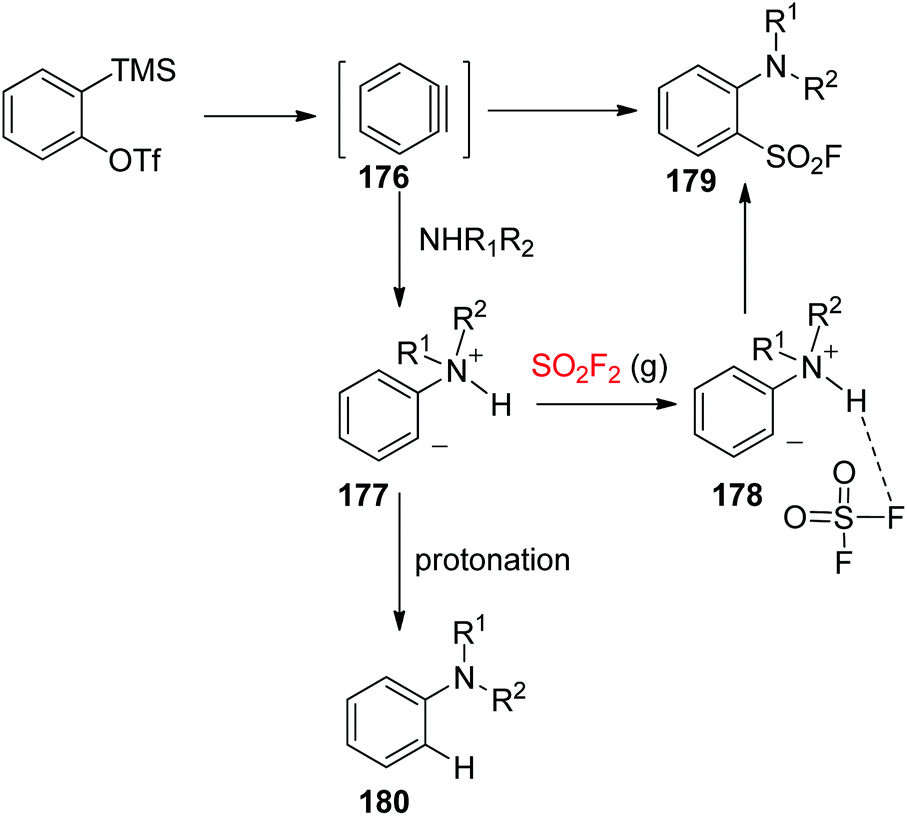
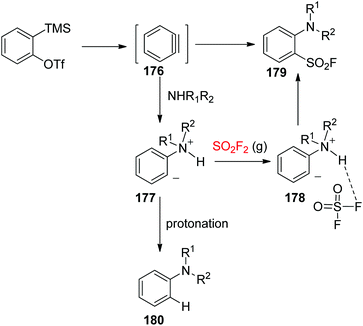
A possible reaction mechanism for the synthesis of arenesulfonyl fluorides was proposed (Scheme 56). Nucleophilic attack of the 2° amine on the aryne, furnished in situ from the starting material with the aid of a fluoride source, generated a zwitterionic intermediate 177. Subsequently, the resultant zwitterionic intermediate 177 was treated with SO2F2 gas to yield the corresponding arenesulfonyl fluoride 179 or with a proton from the ammonium salt to provide the protonated by-product 180. A strong hydrogen bond between SO2F2 and the proton of the ammonium salt as shown in 178 led to the required three-component reaction instead of proton abstraction.
 |
| | Scheme 56 Proposed mechanism for the synthesis of arenesulfonyl fluorides. | |
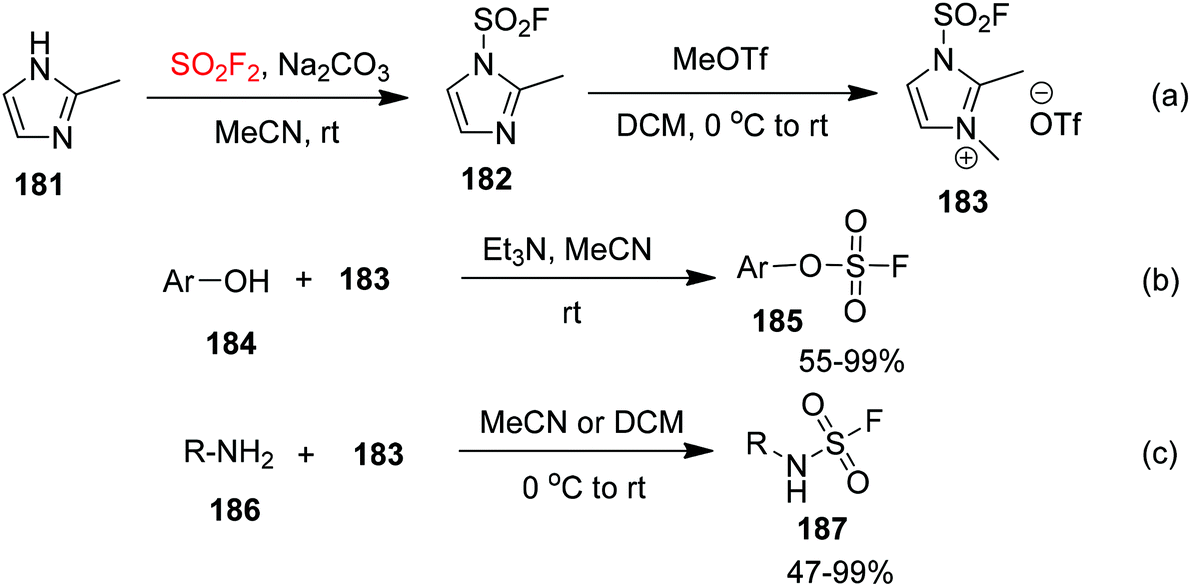
2.9. Chemical transformations of 2-methyl-1H-imidazole into a fluorosulfuryl imidazolium salt
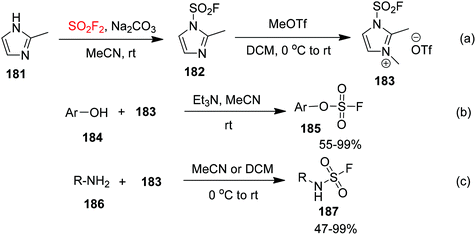
Recently, Sharpless and co-workers10e synthesized a fluorosulfonyl imidazolium salt 183 (a fluorosulfonyl transfer reagent stably stored as a solid salt) from 2-methyl-1H-imidazole 181 upon treatment with SO2F2 gas at room temperature (Scheme 57a). The fluorosulfonyl imidazolium salt 183 has both high reactivity and high selectivity. Under ambient conditions, a diversity of phenols or heteroaryl hydroxides 184 reacted with fluorosulfonyl imidazolium salt 183 with the aid of triethylamine to give fluorosulfates 185 in 55–99% yields (Scheme 57b). More importantly, the reaction between fluorosulfonyl imidazolium salt 183 and primary amines 186 furnished the monosubstituted primary amine sulfonyl fluoride (R-NH-SO2F) 187 in 47–99% yields under mild reaction conditions without any additives (Scheme 57c). If a phenolic hydroxyl group is present in the molecule, the reagent will specifically react with the primary amine without any interaction with the oxygen nucleophilic functional group. The development of this reagent provides a good alternative for the introduction of sulfonyl fluoride groups.
 |
| | Scheme 57 Synthesis of fluorosulfuryl imidazolium salt (a); and (b) and (c) showed applications of fluorosulfuryl imidazolium salt in organic synthesis. | |
3. Conclusion
Sulfuryl fluoride (SO2F2) has been proven to be an important reagent for the formation of new chemical bonds in synthetic chemistry. In recent years, several SO2F2-mediated transformation reactions featuring mild and efficient conditions have been reported. This review has covered the diverse chemical transformations and medicinal applications regarding SO2F2 gas. There are still a number of challenges in the field of SO2F2-mediated transformations, but there is no doubt that the future development of this field will lead to the formation of fine and useful chemicals. Additionally, we hope that the utilization of much more reactive reagents such as triflic anhydride and methyl bromide could also improve the ways for the chemical transformations and lead to the formation of fine and useful chemicals. We believe that this review article will be useful for synthetic organic chemistry researchers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (grant no. 21772150) and Wuhan University of Technology for financial support.
References
- H. Moissan and P. Lebeau, Compt. Rend., 1901, 132, 374 CAS.
-
(a) E. E. Kenaga, J. Econ. Entomol., 1957, 50, 1 CrossRef CAS;
(b) Dow AgroSciences Technical Bulletin, “Sulfuryl Fluoride Gas Fumigant”, April 2002.
-
(a) M. P. Sulbaek Andersen, D. R. Blake, F. S. Rowland, M. D. Hurley and T. J. Wallington, Environ. Sci. Technol., 2009, 43, 1067 CrossRef CAS PubMed;
(b) G. P. Opit, E. Thoms, T. W. Phillips and M. E. Payton, J. Econ. Entomol., 2016, 109, 930 CrossRef CAS PubMed;
(c) A. Cao, M. Guo, D. Yan, L. Mao, Q. Wang, Y. Li, X. Duana and P. Wang, Pest Manage. Sci., 2014, 70, 219 CrossRef CAS PubMed;
(d) S. Abbar, Ö. Sağlam, M. W. Schilling and T. W. Phillips, J. Stored Prod. Res., 2018, 76, 7 CrossRef;
(e) A. Uzunovic, A. Mukherjee, P. Elder and S. W. Myers, For. Prod. J., 2017, 67, 4 CAS;
(f) R. Jagadeesan and M. K. Nayak, Pest Manage. Sci., 2016, 73, 1391 CrossRef PubMed;
(g) F. Sen, K. B. Meyvacı, U. Aksoy and G. Tan, Acta Hortic., 2016, 1120, 241 CrossRef.
-
(a) L. Revathi, L. Ravindar, J. Leng, K. P. Rakesh and H.-L. Qin, Asian J. Org. Chem., 2018, 7, 662 CrossRef CAS;
(b) C. Veryser, J. Demaerel, V. Bieliūnas, P. Gilles and W. M. De Borggraeve, Org. Lett., 2017, 19, 5244 CrossRef CAS PubMed.
- G. E. Cady and S. Misra, Inorg. Chem., 1974, 13, 837 CrossRef CAS.
- R. G. Stone, J. M. Pochan and W. H. Flygare, Inorg. Chem., 1969, 8, 2647 CrossRef CAS.
-
(a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 (
Angew. Chem.
, 2014
, 126
, 9584
) CrossRef CAS PubMed;
(b) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Okadale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466 (
Angew. Chem.
, 2014
, 126
, 9620
) CrossRef CAS PubMed.
-
(a) Q. Liang, P. Xing, Z. Huang, J. Dong, K. B. Sharpless, X. Li and B. Jiang, Org. Lett., 2015, 17, 1942 CrossRef CAS PubMed;
(b) Q. Chen, H. Yu, Z. Xu, L. Lin, X. Jiang and R. Wang, J. Org. Chem., 2015, 80, 6890 CrossRef CAS PubMed;
(c) W. Chen, J. Dong, S. Li, Y. Liu, Y. Wang, L. Yoon, P. Wu, K. B. Sharpless and J. W. Kelly, Angew. Chem., Int. Ed., 2016, 55, 1835 (
Angew. Chem.
, 2016
, 128
, 1867
) CrossRef CAS PubMed;
(d) G. Ren, Q. Zheng and H. Wang, Org. Lett., 2017, 19, 1582 CrossRef CAS PubMed;
(e) T. Lim, S. Byun and B. M. Kim, Asian J. Org. Chem., 2017, 6, 1222 CrossRef CAS;
(f) L. Ravindar, S. N. A. Bukhari, K. P. Rakesh, H. M. Manukumar, H. K. Vivek, N. Mallesha, Z.-Z. Xiea and H.-L. Qin, Bioorg. Chem., 2018, 81, 107 CrossRef CAS PubMed.
-
(a) H. Wang, F. Zhou, G. Ren, Q. Zheng, H. Chen, B. Gao, L. Klivansky, Y. Liu, B. Wu, Q. Xu, J. Lu, K. B. Sharpless and P. Wu, Angew. Chem., Int. Ed., 2017, 56, 11203 CrossRef CAS PubMed;
(b) B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083 CrossRef CAS PubMed;
(c) Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680 CrossRef CAS PubMed.
-
(a) P. S. Hanley, T. P. Clark, A. L. Krasovskiy, M. S. Ober, J. P. O'Brien and T. S. Staton, ACS Catal., 2016, 6, 3515 CrossRef CAS;
(b) M. Epifanov, P. J. Foth, F. Gu, C. Barrillon, S. S. Kanani, C. S. Higman, J. E. Hein and G. M. Sammis, J. Am. Chem. Soc., 2018, 140, 16464 CrossRef CAS PubMed;
(c) T. A. Fattaha, A. Saeeda and F. Albericio, J. Fluorine Chem., 2018, 213, 87 CrossRef;
(d) Z. Liu, G. Meng, T. Guo, J. Dong and P. Wu, Curr. Protoc. Chem. Biol., 2019 DOI:10.1002/cpch.64;
(e) T. Guo, G. Meng, X. Zhan, Q. Yang, T. Ma, L. Xu, K. B. Sharpless and J. Dong, Angew. Chem., 2018, 130, 2635 (
Angew. Chem. Int. Ed.
, 2018
, 57
, 2605
) CrossRef.
-
(a) M. R. Derrick, H. D. Burgess, M. T. Baker and N. E. Binnie, J. Am. Inst. Conserv., 1990, 29, 77 CrossRef;
(b)
S. Samuels, I. Dewhurst and A. Boobis, Joint FAO/WHO Meeting on Pesticide Residues (JMPR), 2005, p. 453 Search PubMed.
-
(a) W.-T. Tsai, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2010, 28, 125 CrossRef CAS PubMed;
(b) V. C. Papadimitriou, R. W. Portmann, D. W. Fahey, J. Mühle, R. F. Weiss and J. B. Burkholder, J. Phys. Chem. A, 2008, 112, 12657 CrossRef CAS PubMed;
(c) J. Mühle, J. Huang, R. F. Weiss, R. G. Prinn, B. R. Miller, P. K. Salameh, C. M. Harth, P. J. Fraser, L. W. Porter, B. R. Greally, S. O'Doherty and P. G. Simmonds, J. Geophys. Res.: Atmos., 2009, 114, D05306 Search PubMed;
(d)
R. I. Krieger, Hayes’ Handbook of Pesticide Toxicology, Academic Press, New York, 3rd edn, 2010 Search PubMed;
(e) G. M. Calvert, C. A. Mueller, J. M. Fajen, D. W. Chrislip, J. Russo, T. Briggle, L. E. Fleming, A. J. Suruda and K. Steenland, Am. J. Public Health, 1998, 88, 1774 CrossRef CAS PubMed.
- W. Lange and E. Muller, Ber. Dtsch. Chem. Ges. B, 1930, 63, 2653 CrossRef.
- R. Cramer and D. D. Coffman, J. Org. Chem., 1961, 26, 4164 CrossRef CAS.
-
(a) W. C. Firth Jr., J. Polym. Sci., Part B: Polym. Phys., 1972, 10, 637 CrossRef;
(b)
W. C. Firth Jr., US3733304, 1973.
-
(a) M. Hedayatullah, A. Guy and L. Denivelle, Phosphorus Sulfur Relat. Elem., 1980, 8, 125 CrossRef CAS;
(b) E. R. Falardeau and D. D. DesMarteau, J. Chem. Eng. Data, 1976, 21, 386 CrossRef CAS.
-
(a) B. Yang, N. Wang, P. D. Schnier, F. Zheng, H. Zhu, N. F. Polizzi, A. Ittuveetil, V. Saikam, W. F. DeGrado, Q. Wang, P. G. Wang and L. Wang, J. Am. Chem. Soc., 2019, 141, 7698 CrossRef CAS PubMed;
(b) N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 4995 CrossRef CAS PubMed.
- S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 2903 (
Angew. Chem.
, 2017
, 129
, 2949
) CrossRef CAS PubMed.
- Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919 CrossRef CAS PubMed.
-
(a) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed;
(b) Y. Ren, M. Yan, J. Wang, Z.-C. Zhang and K. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674 CrossRef CAS PubMed;
(c) H. Sajiki, A. Mori, T. Ikawa, T. Maegawa and K. Hirota, Org. Lett., 2006, 8, 987 CrossRef CAS PubMed;
(d) N. Iranpoor and F. Panahi, Org. Lett., 2015, 17, 214 CrossRef CAS PubMed.
- X.-Y. Wang, J. Leng, S.-M. Wang, A. M. Asiri, H. M. Marwani and H.-L. Qin, Tetrahedron Lett., 2017, 58, 2340 CrossRef CAS.
- P. S. Hanley, M. S. Ober, A. L. Krasovskiy, G. T. Whiteker and W. J. Kruper, ACS Catal., 2015, 5, 5041 CrossRef CAS.
- X. Li, H. Zhang, F. Feng, Q. Hu and Z. Yuan, ChemistrySelect, 2018, 3, 12287 CrossRef CAS.
-
(a)
J. Otera and J. Nishikido, Esterification: methods, reactions, and applications, Wiley, Hoboken, NJ, 2009, p. 1 CrossRef;
(b) J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef CAS;
(c)
R. C. Larock, Comprehensive organic transformations: a guide to functional group preparations, Wiley-VCH, New York, 1999, vol. 1, pp. 1–2640 Search PubMed;
(d)
H. Maag, Prodrugs of Carboxylic Acids, in Prodrugs, ed. V. Maag, R. Borchardt, M. Hageman, R. H. Maag and J. Tilley, Springer, New York, 2007, vol. V, p. 703 Search PubMed;
(e)
S. P. Bew, Carboxylic Acids, in Comprehensive Organic Functional Group Transformations II, ed. A. R. Katritzky and R. J. K. Taylor, Elsevier, Oxford, 2005, p. 19 Search PubMed;
(f)
M. A. Ogliaruso and J. F. Wolfe, in Synthesis of Carboxylic Acids, Esters, and Their Derivatives, ed. S. Patai and Z. Rappoport, Wiley, New York, 1991 CrossRef;
(g) J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed;
(h) G. P. Roth and J. A. Thomas, Tetrahedron Lett., 1992, 33, 1959 CrossRef CAS;
(i) M. A. McGuire, E. Sorenson, F. W. Owings, T. M. Resnick, M. Fox and N. H. Baine, J. Org. Chem., 1994, 59, 6683 CrossRef CAS.
- W.-Y. Fang, J. Leng and H.-L. Qin, Chem. – Asian J., 2017, 12, 2323 CrossRef CAS PubMed.
- C. Ma, C.-Q. Zhao, X.-T. Xu, Z.-M. Li, X.-Y. Wang, K. Zhang and T.-S. Mei, Org. Lett., 2019, 21, 2464 CrossRef CAS PubMed.
-
(a)
The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg and C. M. Breneman and J. F. Liebman, Wiley-Interscience, Hoboken, 2000, pp. 1–672 Search PubMed;
(b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
-
(a) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS PubMed;
(b) J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765 CAS;
(c) T. Cupido, J. Tulla-Puche, J. Spengler and F. Albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768 CAS;
(d) A. K. Ghose, V. N. Viswanadhan and J. J. J. Wendoloski, Comb. Chem., 1999, 1, 55 CrossRef CAS;
(e) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC;
(f) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed.
- W.-Y. Fang, Y.-M. Huang, J. Leng and H.-L. Qin, Asian J. Org. Chem., 2018, 7, 751 CrossRef CAS.
-
(a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS PubMed;
(b) M. Frizler, F. Lohr, N. Furtmann, J. Kläs and M. Gutschow, J. Med. Chem., 2011, 54, 396 CrossRef CAS PubMed;
(c)
R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989, p. 819 Search PubMed;
(d)
A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substance: Synthesis, Patents, Applications, Georg Thieme, Stuttgart, 4th edn, 2001 Search PubMed.
-
(a)
A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappaport, Wiley, New York, 1983 Search PubMed;
(b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed;
(c)
M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, Hoboken, NJ, 6th edn, 2007 Search PubMed.
- C. Zhao, W.-Y. Fang, K. P. Rakesh and H.-L. Qin, Org. Chem. Front., 2018, 5, 1835 RSC.
-
(a) M. R. Kumar, K. Park and S. Lee, Adv. Synth. Catal., 2010, 352, 3255 CrossRef CAS;
(b) U. Beutler, M. Boehm, P. C. Fuenfschilling, T. Heinz, J. Mutz, U. Onken, M. Mueller and W. Zaugg, Org. Process Res. Dev., 2007, 11, 892 CrossRef CAS;
(c) S. B. Madasu, N. A. Vekariya, C. Koteswaramma, A. Islam, P. D. Sanasi and R. B. Korupolu, Org. Process Res. Dev., 2012, 16, 5025 CrossRef.
-
(a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed;
(b) J. Wu, Tetrahedron Lett., 2014, 55, 4289 CrossRef CAS.
-
(a) L. N. Markovskij, V. E. Pashinnik and A. V. Kirsanov, Synthesis, 1973, 787 CrossRef;
(b) W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS.
- S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452 CrossRef CAS PubMed.
-
(a)
J. Dong, V. Fokin, L. Krasnova, L. R. Kwisnek, J. S. Oakdale and K. B. Sharpless, US2104089078, 2014;
(b)
J. Dong and K. B. Sharpless, US2015188120, 2015.
- J. S. Oakdale, L. Kwisnek and V. V. Fokin, Macromolecules, 2016, 49, 4473 CrossRef CAS.
- E. Zhang, J. Tang, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Chem. – Eur. J., 2016, 22, 5692 CrossRef CAS PubMed.
-
(a) R. N. Brogden, Drugs, 1986, 32, 60 CrossRef PubMed;
(b) G. Varvounis, Adv. Heterocycl. Chem., 2009, 98, 143 CrossRef CAS;
(c) A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423 CrossRef CAS;
(d) S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984 CrossRef CAS PubMed;
(e) H. Fu and J. Yao, J. Am. Chem. Soc., 2001, 123, 1434 CrossRef CAS;
(f) H. Zhou, Z. Wei, J.-L. Zhang, H.-M. Yang, C.-G. Xia and G.-X. Jiang, Angew. Chem., Int. Ed., 2017, 56, 1077 CrossRef CAS PubMed;
(g)
L. Yet, in Comprehensive Heterocyclic Chemistry III, ed. C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 1–141 Search PubMed.
-
(a) A. Baranczak, Y. Liu, S. Connelly, W.-G. Han Du, E. R. Greiner, J. C. Genereux, R. L. Wiseman, Y. S. Eisele, N. C. Bradbury, J. Dong, L. Noodleman, K. B. Sharpless, I. A. Wilson, S. E. Encalada and J. W. Kelly, J. Am. Chem. Soc., 2015, 137, 7404 CrossRef CAS PubMed;
(b) W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353 CrossRef CAS PubMed;
(c) L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584 CrossRef CAS PubMed;
(d) D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200 CrossRef CAS PubMed;
(e) L. H. Jones, Angew. Chem., Int. Ed., 2018, 57, 9220 CrossRef CAS PubMed;
(f) P. Martin-Gago and C. A. Olsen, Angew. Chem., Int. Ed., 2019, 58, 957 CrossRef CAS PubMed.
- J. Leng and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 5001 RSC.
-
(a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed;
(b) K. Kacprzak, I. Skiera, M. Piasecka and Z. Paryzek, Chem. Rev., 2016, 116, 5689 CrossRef CAS PubMed.
-
(a) H. Tian, A. Fürstenberg and T. Huber, Chem. Rev., 2017, 117, 186 CrossRef CAS PubMed;
(b) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC;
(c) C. G. Parker, C. A. Kuttruff, A. Galmozzi, L. Jørgensen, C.-H. Yeh, D. J. Hermanson, Y. Wang, M. Artola, S. J. McKerrall, C. M. Josyln, B. Nørremark, G. Dünstl, J. Felding, E. Saez, P. S. Baran and B. F. Cravatt, ACS Cent. Sci., 2017, 3, 1276 CrossRef CAS PubMed;
(d) Q. Zhou, J. Gui, C.-M. Pan, E. Albone, X. Cheng, E. M. Suh, L. Grasso, Y. Ishihara and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12994 CrossRef CAS PubMed;
(e) J. S. Cisar and B. F. Cravatt, J. Am. Chem. Soc., 2012, 134, 10385 CrossRef CAS PubMed;
(f) S. E. Tully and B. F. Cravatt, J. Am. Chem. Soc., 2010, 132, 3264 CrossRef CAS PubMed.
-
(a)
P. J. Stang and F. Diederich, Modern Acetylene Chemistry, Wiley-VCH, Weinheim, 1995 CrossRef;
(b) J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906 CrossRef CAS PubMed;
(c) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed.
-
(a) F. Diederich, Nature, 1994, 369, 199 CrossRef CAS;
(b) K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC.
-
(a)
F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed;
(b)
J. Lam, H. Breteler, T. L. Arnason and L. Hansen, Chemistry and Biology of Naturally-Occurring Acetylenes and Related Compounds, Elsevier, Amsterdam, 1988 Search PubMed;
(c)
S. Patai, Chemistry of Triple-Bonded Functional Groups, Wiley-VCH, New York, 1994 Search PubMed;
(d) B. M. Trost, J. D. Sieber, W. Qian, R. Dhawan and Z. T. Ball, Angew. Chem., Int. Ed., 2009, 48, 5478 CrossRef CAS PubMed;
(e) Z.-F. Zhou, M. Menna, Y.-S. Cai and Y.-W. Guo, Chem. Rev., 2015, 115, 1543 CrossRef CAS PubMed.
- G.-F. Zha, W.-Y. Fang, Y.-G. Li, J. Leng, X. Chen and H.-L. Qin, J. Am. Chem. Soc., 2018, 140, 17666 CrossRef CAS PubMed.
-
(a) A. J. Mancuso, S.-L. Huang and D. J. Swern, Org. Chem., 1978, 43, 2480 CrossRef CAS;
(b) A. J. Mancuso and D. Swern, Synthesis, 1981, 165 CrossRef CAS;
(c) T. T. Tidwell, Synthesis, 1990, 857 CrossRef CAS.
-
(a) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642 CrossRef CAS PubMed;
(b) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC.
- S. Thorand and N. Krause, J. Org. Chem., 1998, 63, 8551 CrossRef CAS.
- A. J. Salmon, M. L. Williams, Q. K. Wu, J. Morizzi, D. Gregg, S. A. Charman, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2012, 55, 5506 CrossRef CAS PubMed.
-
(a) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed;
(b)
M. Hudlicky, Oxidation in Organic Chemistry, ACS, Washington DC, 1990 Search PubMed;
(c) T. Daisuke, T. Masayuki, M. Katsuhiko and H. Togo, Tetrahedron, 2012, 68, 6849 CrossRef;
(d) M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221 CrossRef CAS PubMed;
(e) M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227 CrossRef CAS.
-
(a) H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513 CrossRef CAS PubMed;
(b) M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751 CrossRef CAS PubMed;
(c) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed;
(d) A. Das and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 8892 (
Angew. Chem.
, 2017
, 129
, 9018
) CrossRef CAS PubMed;
(e) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406 CrossRef CAS PubMed;
(f) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 (
Angew. Chem.
, 2011
, 123
, 5138
) CrossRef CAS PubMed;
(g) D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS PubMed;
(h) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636 CrossRef CAS PubMed;
(i) S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, Adv. Synth. Catal., 2011, 353, 1005 CrossRef CAS.
-
(a) M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Organomet. Chem., 2015, 63, 91 CrossRef CAS;
(b) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC;
(c) T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem. – Asian J., 2008, 3, 196 CrossRef CAS PubMed.
- G.-F. Zha, W.-Y. Fang, J. Leng and H.-L. Qin, Adv. Synth. Catal., 2019, 361, 2262 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
-
(a) A. G. Olsson, F. McTaggart and A. Raza, Cardiovasc. Drug Rev., 2002, 20, 303 CrossRef CAS PubMed;
(b) F. McTaggart and P. Jones, Cardiovasc. Drugs Ther., 2008, 22, 321 CrossRef CAS PubMed.
-
(a) M. Watanabe, H. Koike, T. Ishiba, T. Okada, S. Seo and K. Hirai, Bioorg. Med. Chem., 1997, 5, 437 CrossRef CAS PubMed;
(b) N. Andrushko, V. Andrushko, G. König, A. Spannenberg and A. Börner, Eur. J. Org. Chem., 2008, 847 CrossRef CAS;
(c) S. Tartaggia, C. Ferrari, M. Pontini and O. De Lucchi, Eur. J. Org. Chem., 2015, 4102 CrossRef CAS;
(d) S. Tartaggia, S. Fogal, R. Motterle, C. Ferrari, M. Pontini, R. Aureli and O. De Lucchi, Eur. J. Org. Chem., 2016, 3162 CrossRef CAS.
-
(a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS;
(b) C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS;
(c) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC;
(d) D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202 CrossRef CAS PubMed;
(e) T. D. Senecal, W. Shu and S. L. Buchwald, Angew. Chem., 2013, 125, 10219 (
Angew. Chem. Int. Ed.
, 2013
, 52
, 10035
) CrossRef;
(f) P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Org. Lett., 2011, 13, 648 CrossRef CAS PubMed;
(g) T. Schareina, R. Jackstell, T. Schulz, A. Zapf, A. Cotte, M. Gotta and M. Beller, Adv. Synth. Catal., 2009, 351, 643 CrossRef CAS;
(h) S. Erhardt, V. V. Grushin, A. H. Kilpatrick, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2008, 130, 4828 CrossRef CAS PubMed;
(i) M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., 2003, 115, 1700 (
Angew. Chem. Int. Ed.
, 2003
, 42
, 1661
) CrossRef;
(j) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed;
(k) N. E. Leadbeater and R. K. Arvela, J. Org. Chem., 2003, 68, 9122 CrossRef PubMed.
- Y. Jiang, B. Sun, W.-Y. Fang and H.-L. Qin, Eur. J. Org. Chem., 2019, 3190 CrossRef CAS.
- Z.-Y. Li, P.-B. Lu, H. Ji, Q. Shao, Q.-D. You and X. Liu, Acta Pharmacol. Sin., 2009, 44, 1112 CAS.
-
(a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS;
(b) E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821 CrossRef CAS;
(c) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef;
(d)
F. Diederich and P. J. Stang, Metal-catalyzed cross-coupling reactions, John Wiley & Sons, 2008 Search PubMed;
(e)
J. F. Hartwig, Organotransition metal chemistry: from bonding to catalysis, Univ. Science Books, 2010 Search PubMed;
(f) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed;
(g) J. J. Dunsford, E. R. Clark and M. J. Ingleson, Angew. Chem., 2015, 127, 5780 (
Angew. Chem. Int. Ed.
, 2015
, 54
, 5688
) CrossRef;
(h) A. D. Benischke, I. Knoll, A. Rérat, C. Gosmini and P. Knochel, Chem. Commun., 2016, 52, 3171 RSC.
-
(a) I. Iovel, K. Mertins, J. Kischel, A. Zapf and M. Beller, Angew. Chem., 2005, 117, 3981 (
Angew. Chem. Int. Ed.
, 2005
, 44
, 3913
) CrossRef;
(b) K. Mertins, I. Iovel, J. Kischel, A. Zapf and M. Beller, Angew. Chem., 2005, 117, 242 (
Angew. Chem. Int. Ed.
, 2005
, 44
, 238
) CrossRef;
(c) V. D. Sarca and K. K. Laali, Green Chem., 2006, 8, 615 RSC;
(d) S. Ogoshi, H. Nakashima, K. Shimonaka and H. Kurosawa, J. Am. Chem. Soc., 2001, 123, 8626 CrossRef CAS PubMed;
(e) F. P. DeHaan, W. D. Covey, R. L. Ezelle, J. E. Margetan, S. A. Pace, M. J. Sollenberger and D. S. Wolf, J. Org. Chem., 1984, 49, 3954 CrossRef CAS;
(f) G. A. Olah, S. Kobayashi and M. Tashiro, J. Am. Chem. Soc., 1972, 94, 7448 CrossRef CAS.
-
(a) S. Langle, M. Abarbri and A. Duchene, Tetrahedron Lett., 2003, 44, 9255 CrossRef CAS;
(b) S. M. Nobre and A. L. Monteiro, Tetrahedron Lett., 2004, 45, 8225 CrossRef CAS;
(c) R. Singh, M. S. Viciu, N. Kramareva, O. Navarro and S. P. Nolan, Org. Lett., 2005, 7, 1829 CrossRef CAS PubMed;
(d) G. A. Molander and M. D. Elia, J. Org. Chem., 2006, 71, 9198 CrossRef CAS PubMed;
(e) M. J. Burns, I. J. S. Fairlamb, A. R. Kapdi, P. Sehnal and R. J. K. Taylor, Org. Lett., 2007, 9, 5397 CrossRef CAS PubMed;
(f) E. Alacid and C. Najera, Org. Lett., 2008, 10, 5011 CrossRef CAS PubMed;
(g) Y. Zhang, M.-T. Feng and J.-M. Lu, Org. Biomol. Chem., 2013, 11, 2266 RSC;
(h) R. Kuwano and M. Yokogi, Org. Lett., 2005, 7, 945 CrossRef CAS PubMed;
(i) J.-Y. Yu and R. Kuwano, Org. Lett., 2008, 10, 973 CrossRef CAS PubMed;
(j) Y.-Y. Sun, J. Yi, X. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2014, 50, 11060 RSC;
(k) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC;
(l) M. Kuriyama, M. Shinozawa, N. Hamaguchi, S. Matsuo and O. Onomura, J. Org. Chem., 2014, 79, 5921 CrossRef CAS PubMed;
(m) Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383 CrossRef CAS PubMed;
(n) C. Li, Y. Zhang, Q. Sun, T. Gu, H. Peng and W. Tang, J. Am. Chem. Soc., 2016, 138, 10774 CrossRef CAS PubMed;
(o) M. Ueda, D. Nakakoji, Y. Kuwahara, K. Nishimura and I. Ryu, Tetrahedron Lett., 2016, 57, 4142 CrossRef CAS;
(p) G. Wu, S. Xu, Y. Deng, C. Wu, X. Zhao, W. Ji, Y. Zhang and J. Wang, Tetrahedron, 2016, 72, 8022 CrossRef CAS;
(q) Z. He, F. Song, H. Sun and Y. Huang, J. Am. Chem. Soc., 2018, 140, 2693 CrossRef CAS PubMed;
(r) M. Ohsumi, A. Ito and N. Nishiwaki, RSC Adv., 2018, 8, 35056 RSC.
- C. Zhao, G.-F. Zha, W.-Y. Fang, K. P. Rakesh and H.-L. Qin, Eur. J. Org. Chem., 2019, 1801 CrossRef CAS.
-
(a) C. H. Basch, K. M. Cobb and M. P. Watson, Org. Lett., 2016, 18, 136 CrossRef CAS PubMed;
(b) J. Hu, H. Sun, W. Cai, X. Pu, Y. Zhang and Z. Shi, J. Org. Chem., 2016, 81, 14 CrossRef CAS PubMed.
-
(a) P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280 CrossRef CAS PubMed;
(b) T. Wang, S. Yang, S. Xu, C. Han, G. Guo and J. Zhao, RSC Adv., 2017, 7, 15805 RSC;
(c) Z.-C. Cao, D.-G. Yu, R.-Y. Zhu, J.-B. Wei and Z.-J. Shi, Chem. Commun., 2015, 51, 2683 RSC.
-
(a) C. Wanner, H. Wieland, P. Schollmeyer and W. H. Horl, Eur. J. Clin. Pharmacol., 1991, 40, S85 CrossRef PubMed;
(b) K. Mertins, I. Iovel, J. Kischel, A. Zapf and M. Beller, Adv. Synth. Catal., 2006, 348, 691 CrossRef CAS;
(c) X. Mo, J. Yakiwchuk, J. Dansereau, J. A. McCubbin and D. G. Hall, J. Am. Chem. Soc., 2015, 137, 9694 CrossRef CAS PubMed.
-
(a) J. Zhang, Z. Wang, Y. Wang, C. Wan, X. Zheng and Z. Wang, Green Chem., 2009, 11, 1973 RSC;
(b) A. Venkat Narsaiah and P. Narsimha, Med. Chem. Res., 2012, 21, 538 CrossRef CAS.
-
(a) B. Kunze, B. Bohlendorf, H. Reichenbach and G. Hofle, J. Antibiot., 2008, 61, 18 CrossRef CAS PubMed;
(b) A. M. Harned and K. A. Volp, Nat. Prod. Rep., 2011, 28, 1790 RSC;
(c) A. Vasas and J. Hohmann, Nat. Prod. Rep., 2011, 28, 824 RSC;
(d) E. Mevers, J. Sauri, Y. Liu, A. Moser, T. R. Ramadhar, M. Varlan, R. T. Williamson, G. E. Martin and J. Clardy, J. Am. Chem. Soc., 2016, 138, 12324 CrossRef CAS PubMed.
-
(a) O. Saku, H. Ishida, E. Atsumi, Y. Sugimoto, H. Kodaira, Y. Kato, S. Shirakura and Y. Nakasato, J. Med. Chem., 2012, 55, 3436 CrossRef CAS PubMed;
(b) A. Schçffmann, L. Wimmer, D. Goldmann, S. Khom, J. Hintersteiner, I. Baburin, T. Schwarz, M. Hintersteininger, P. Pakfeifer, M. Oufir, M. Hamburger, T. Erker, G. F. Ecker, M. D. Mihovilovic and S. Hering, J. Med. Chem., 2014, 57, 5602 CrossRef PubMed.
-
(a) Y.-H. Xu, W.-J. Wang, Z.-K. Wen, J. J. Hartley and T.-P. Loh, Tetrahedron Lett., 2010, 51, 3504 CrossRef CAS;
(b) Y. H. Xu, Y. K. Chok and T. P. Loh, Chem. Sci., 2011, 2, 1822 RSC;
(c) Z. K. Wen, Y. H. Xu and T. P. Loh, Chem. – Eur. J., 2012, 18, 13284 CrossRef CAS PubMed;
(d) Q.-J. Liang, C. Yang, F.-F. Meng, B. Jiang, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2017, 56, 5091 (
Angew. Chem.
, 2017
, 129
, 5173
) CrossRef CAS PubMed;
(e) M. Z. Li, L. X. Li and H. B. Ge, Adv. Synth. Catal., 2010, 352, 2445 CrossRef CAS;
(f) Y. Y. Yu, M. J. Niphakis and G. I. Georg, Org. Lett., 2011, 13, 5932 CrossRef CAS PubMed;
(g) D. Kim and S. Hong, Org. Lett., 2011, 13, 4466 CrossRef CAS PubMed;
(h) M. Min, Y. Kim and S. Hong, Chem. Commun., 2013, 49, 196 RSC;
(i) C. Yu, J. Zhang and G. Zhong, Chem. Commun., 2017, 53, 9902 RSC;
(j) T.-J. Hu, M.-Y. Li, Q. Zhao, C.-G. Feng and G.-Q. Lin, Angew. Chem., 2018, 130, 5973 CrossRef;
(k) I. T. Crouch, T. Dreier and D. E. Frantz, Angew. Chem., Int. Ed., 2011, 50, 6128 CrossRef CAS PubMed;
(l) L. Li, Y. Chu, L. Gaoa and Z. Song, Chem. Commun., 2015, 51, 15546 RSC;
(m) Q. Zhao, V. Tognetti, L. Joubert, T. Besset, X. Pannecoucke, J.-P. Bouillon and T. Poisson, Org. Lett., 2017, 19, 2106 CrossRef CAS PubMed;
(n) C. H. Yoon, K. S. Yoo, S. W. Yi, R. K. Mishra and K. W. Jung, Org. Lett., 2004, 6, 4037 CrossRef CAS PubMed;
(o) H. Tsujita, Y. Ura, S. Matsuki, K. Wada, T. Mitsudo and T. Kondo, Angew. Chem., Int. Ed., 2007, 46, 5160 CrossRef CAS PubMed;
(p) A. T. Lindhardt, M. L. H. Mantel and T. Skrydstrup, Angew. Chem., Int. Ed., 2008, 47, 2668 CrossRef CAS PubMed;
(q) N. M. Neisius and B. Plietker, Angew. Chem., Int. Ed., 2009, 48, 5752 CrossRef CAS PubMed;
(r) S. Mannathan and C.-H. Cheng, Chem. Commun., 2010, 46, 1923 RSC;
(s) H. Horie, I. Koyama, T. Kurahashi and S. Matsubara, Chem. Commun., 2011, 47, 2658 RSC;
(t) N. Saito, K. Saito, M. Shiro and Y. Sato, Org. Lett., 2011, 13, 2718 CrossRef CAS PubMed;
(u) J. Zhang, A. Ugrinov, Y. Zhang and P. Zhao, Angew. Chem., Int. Ed., 2014, 53, 8437 CrossRef CAS PubMed;
(v) S. Kiyota, S. In, N. Komine and M. Hirano, Chem. Lett., 2017, 46, 1040 CrossRef CAS;
(w) T. Li, J. Zhang, C. Yu, X. Lu, L. Xu and G. Zhong, Chem. Commun., 2017, 53, 12926 RSC;
(x) Y. Shibata, Y. Otake, M. Hirano and K. Tanaka, Org. Lett., 2009, 11, 689 CrossRef CAS PubMed;
(y) K. Meng, J. Zhang, F. Li, Z. Lin, K. Zhang and G. Zhong, Org. Lett., 2017, 19, 2498 CrossRef CAS PubMed.
- R. Lekkala, R. Lekkala, B. Moku and H.-L. Qin, Org. Chem. Front., 2019, 6, 796 RSC.
-
(a) S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252 RSC;
(b) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed;
(c) J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325 RSC.
-
(a) T. Pathak, Chem. Rev., 2002, 102, 1623 CrossRef CAS PubMed;
(b) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed;
(c) C. O. Kappe and E. V. der
Eycken, Chem. Soc. Rev., 2010, 39, 1280 RSC;
(d) G. Aromi, L. A. Barrios, O. Rubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485 CrossRef CAS.
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS;
(b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS;
(c) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed;
(d) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 2139 RSC.
- X. Zhang, K. P. Rakesh and H.-L. Qin, Chem. Commun., 2019, 55, 2845 RSC.
- S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodionov and V. V. Fokin, Org. Lett., 2010, 12, 4217 CrossRef CAS PubMed.
- D. R. Edwards and R. Wolfenden, J. Org. Chem., 2012, 77, 4450 CrossRef CAS PubMed.
- D. K. Padma, V. S. Bhat and A. R. V. Murthy, J. Fluorine Chem., 1982, 20, 425 CrossRef CAS.
-
G. Hamprecht, BASF, US3919308, 1975 Search PubMed.
- S. Li, D. Cohen-Karni, L. T. Beringer, C. Wu, E. Kallick, H. Edington, M. J. Passineau and S. Averick, Polymer, 2016, 99, 7 CrossRef CAS.
- S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087 RSC.
- J. E. Anderson, R. Davis, R. N. Fitzgerald and J. M. Haberman, Synth. Commun., 2006, 36, 2129 CrossRef.
-
(a) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed;
(b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- W.-Y. Fang, G.-F. Zha, C. Zhao and H.-L. Qin, Chem. Commun., 2019, 55, 6273 RSC.
- K. Hyodo, K. Togashi, N. Oishi, G. Hasegawa and K. Uchida, Org. Lett., 2017, 19, 3005 CrossRef CAS PubMed.
-
(a) A. Rapeyko, M. J. Climent, A. Corma, P. Concepcion and S. Iborra, ACS Catal., 2016, 6, 4564 CrossRef CAS;
(b) K. Hyodo, S. Kitagawa, M. Yamazaki and K. Uchida, Chem. – Asian J., 2016, 11, 1348 CrossRef CAS PubMed;
(c) K. Yamaguchi, H. Fujiwara, Y. Ogasawara, M. Kotani and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 922 Search PubMed;
(d) K. Ishihara, Y. Furuya and H. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 2983 CrossRef CAS;
(e) S. H. Yang and S. Chang, Org. Lett., 2001, 3, 4209 CrossRef CAS PubMed.
- Q. Wu, Y. Luo, A. Lei and J. You, J. Am. Chem. Soc., 2016, 138, 2885 CrossRef CAS PubMed.
-
(a) X.-D. An and S. Yu, Org. Lett., 2015, 17, 5064 CrossRef CAS PubMed;
(b) S. Laulhe, S. S. Gori and M. H. Nantz, J. Org. Chem., 2012, 77, 9334 CrossRef CAS PubMed;
(c) C. B. Kelly, K. M. Lambert, M. A. Mercadante, J. M. Ovian, W. F. Bailey and N. E. Leadbeater, Angew. Chem., Int. Ed., 2015, 54, 4241 CrossRef CAS PubMed;
(d) L. M. Dornan, Q. Cao, J. C. A. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030 RSC;
(e) J.-H. Noh and J. Kim, J. Org. Chem., 2015, 80, 11624 CrossRef CAS PubMed;
(f) B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364 CrossRef CAS PubMed.
-
(a) J. Gurjar, J. Bater and V. V. Fokin, Chem. – Eur. J., 2019, 25, 1906 CrossRef CAS PubMed;
(b) W.-Y. Fang and H.-L. Qin, J. Org. Chem., 2019, 84, 5803 CrossRef CAS PubMed.
- G. C. Reddy, V. Chandregowda and G. Venkateswara Rao, Heterocycles, 2007, 71, 39 CrossRef.
- P. R. Melvin, D. M. Ferguson, S. D. Schimler, D. C. Bland and M. S. Sanford, Org. Lett., 2019, 21, 1350 CrossRef CAS PubMed.
-
B. Das and K. Damodar, Epoxides and Oxetanes, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, pp. 63–90 Search PubMed.
-
(a) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed;
(b) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227 CrossRef CAS PubMed.
- R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Beilstein J. Org. Chem., 2019, 15, 976 CrossRef CAS PubMed.
- J. Kwon and B. M. Kim, Org. Lett., 2019, 21, 428 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Hua-Li
Qin
and
Hua-Li
Qin