
Stable dicationic dioxoliums and fate of their dioxolyl radicals†
Marc
Devillard
a,
Vianney
Regnier
a,
Jacques
Pecaut
b and
David
Martin
 *a
*a
aUniv. Grenoble Alpes, CNRS, DCM, 38000 Grenoble, France. E-mail: david.martin@univ-grenoble-alpes.fr
bUniv. Grenoble Alpes, CEA, CNRS, INAC-SyMMES, UMR 5819 38000 Grenoble, France
First published on 18th March 2019
Abstract
Stable dicationic dioxolium salts featuring an ancillary vinamidinium pattern were synthesized and characterized. Although highly reactive, they were found otherwise easy to handle under inert atmosphere. This offered the opportunity to generate and study the fate of unknown 1,3-dioxolyl radicals. Depending on substituents, reduction led to the formation of dimers of either dioxolyl or cyclohexadienyl radicals, stemming from a process that is related to the Surzur–Tanner rearrangement. The cyclohexadienyl radical could be characterized in the case of a tri(tert-butyl)phenyl group, which prevents dimerization processes.
Introduction
The design of stabilized C-centered reactive molecules, such as carbeniums, carbanions or radicals, through the introduction of hetero-substituents is a well-proven concept.1–4 Indeed, the diversity of main group elements provides a wide palette of electronic effects, from prototypical π-accepting B- or Al-based functions, up to strong electron-donating amino groups.5 Oxygen stands out as the most electronegative element of the periodic table (apart from fluorine),5a,b while at the same time RO-alkoxy groups are almost equivalent to amino substituents in terms of π-donation.5 Strikingly, this combination isn't synergistic and the strong, but opposite, effects often counterbalance each other. In other words, the O-substituents fail to decrease the basicity of electron-rich carbanions because of their +M donation,3 whereas at the same time the electronegativity of oxygen prevent them for taming the reactivity of electron deficient carbenium centers.1,6 Thus, simple aryl- and alkyl-oxoniums are highly reactive and their study have been essentially confined to super acidic media.2,7,8 This is in marked contrast with their nitrogen counterparts: countless bench-stable iminiums and amidiniums have been reported.9 Similarly, the introduction of alkoxy groups have little beneficial effects on the stability of C-centered radicals. Even their combination with an electron-withdrawing group, so-called capto-dative substitution,4 results in modest enhancements, especially when compared to amino groups. As a matter of fact, to date, all isolated C-radicals with a simple capto-dative substitution pattern feature N-substituents as donors.10,11In this article, we consider the case of 1,3-dioxolyl scaffolds A (Scheme 1). Radicals A˙ have never been evidenced experimentally,12 although computational studies already assessed their possible role as reactive intermediates, especially in the rearrangement of β-(acyloxy)vinyl radicals.13–15 Note that known16 parented 1,3-dioxolanyl radicals B˙ should be similarly the intermediates in the shift of β-(acyloxy)alkyl radicals, so-called Surzur–Tanner rearrangement.17 However, extensive experimental and theoretical works have demonstrated that this reaction proceeds in fact through a closely-related transition state, which lies lower in energy (see Scheme 1).17c,18 Even more, the ester-shift doesn't occur in the case of ortho-(acyloxy)aryl radicals, neither through 1,3-benzodioxolyl radicals C˙ nor through another pathway, because of the inability of the aryl radical to achieve suitable orbital overlap.19
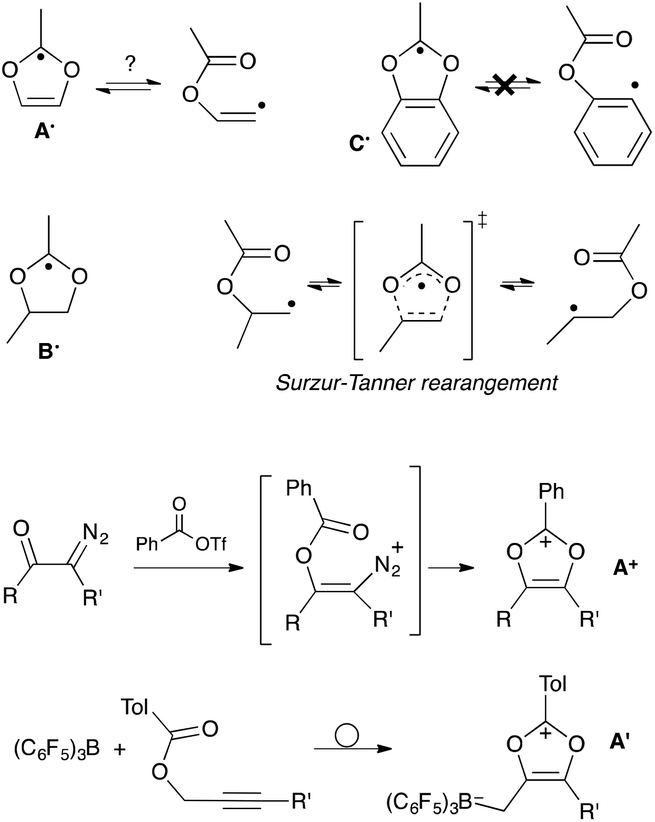 | ||
| Scheme 1 Dioxolyl, dioxolanyl and benzodioxolyl radicals A˙–C˙; their relation with “Surzur–Tanner”-type rearrangements; synthesis of dioxoliums salts A+ and A′. | ||
The absence of viable synthetic route towards the generation of A˙ certainly explains why this class of radicals still constitutes a terra incognita. In principle, they could be unambiguously produced through one-electron reduction of the corresponding dioxolium salts A+. However, these carbeniums are highly reactive themselves,20,21 apart from derivatives benefiting from further stabilization, such as a benzo-fused ring22 or an additional hetero-substituent.23 Even in this latter case, attempts to assess the fate of the corresponding dioxolyl-like radicals were inconclusive.24 To date, only neutral zwitterrionic borate-based derivatives A′, which were recently prepared by Stephan et al. from prop-2-yn-1-yl benzoates, have been structurally characterized.21b
Herein we report the synthesis of isolable dicationic dioxolium salts, featuring a vinamidinium pattern. Though reactive, they are easily handle-able under dry inert atmosphere. This allowed not only for full spectroscopic and structural characterizations, but also stepwise reactivity studies, especially the generation and fate of the corresponding cationic 1,3-dioxolyl radicals.
Results and discussion
This study was initially motivated by our interest in the reactivity of salt 1a (Scheme 2), in the context of our work on unusual vinamidinium scaffolds.25 The three-step synthesis of this novel synthon was straightforward. We first performed an iodide-catalyzed esterification of benzoic acid with N,N-(dimethyl)chloroacetamide. The resulting 2-(benzoyloxy) acetamide 2a was reacted with dichloromethylene-dimethyl iminium chloride26 to yield, after anion metathesis, 1,3-(dichloro)vinamidinium hexafluorophosphate salt 3a. Finally, the addition of one equivalent of dimethyl(trimethylsilyl)amine allowed for a clean selective mono-substitution, affording 1a in 78% yield.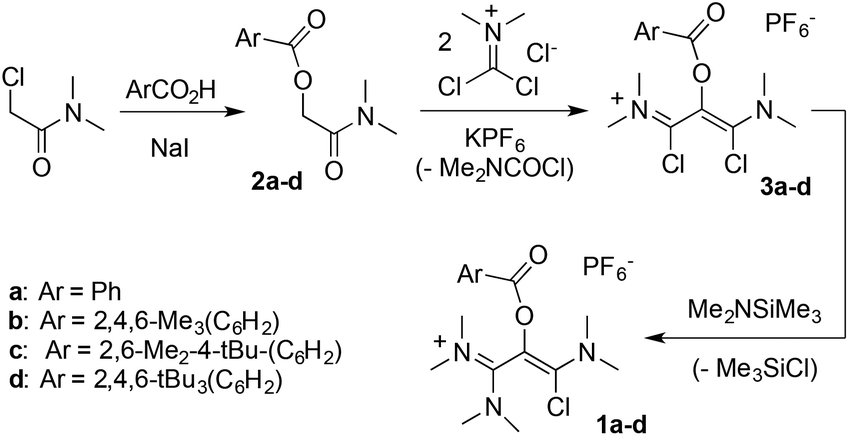 | ||
| Scheme 2 Synthesis of 1-(chloro)vinamidinium salts 1a–d. | ||
Originally, we then wished to introduce a bulky anilino group by direct nucleophilic acyl substitution of the remaining chloride. A clean and complete reaction of 1a with N-(trimethylsilyl)-2,6-di(isopropyl)aniline 4 was observed after 3 days at 60 °C in acetonitrile (Scheme 3). However, whereas HR-MS analysis was consistent with the expected substitution product, the presence of a C–H signal in 13C NMR at δ = 72.3 ppm indicated the formation of a different structural isomer. A single crystal X-ray diffraction study finally allowed for the identification of benzimidate 5 (see ESI†).27
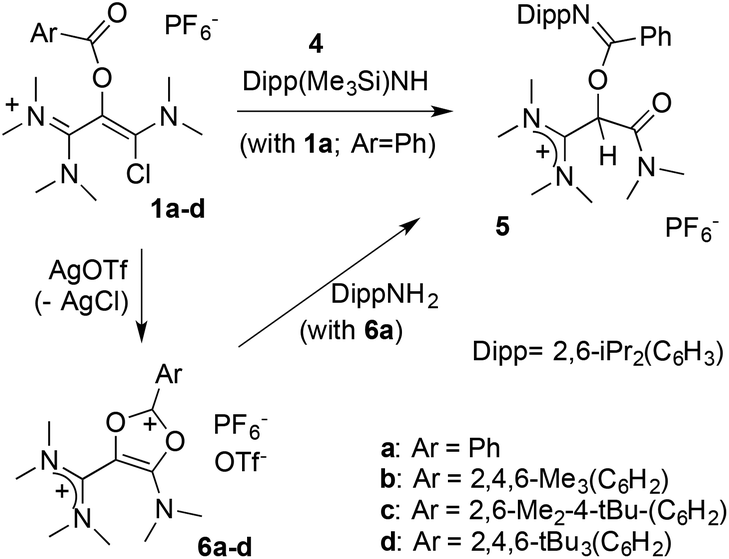 | ||
| Scheme 3 Reaction of 1a with trimethylsilylanilines; synthesis of dioxolium salts 6a–d. | ||
The outcome of this reaction suggested the transient formation of dicationic 1,3-dioxolium 6a, followed by fast aminolysis.20,21a In order to probe this hypothesis, a solution of 1a was stirred in presence of silver triflate. A bright yellow precipitate appeared, while NMR monitoring of the supernatant showed the gradual disappearance of the starting material with complete conversion after 2 days. The product was purified by crystallization and isolated in 56% yield. An X-ray diffraction analysis confirmed the formation of the dicationic dioxolium 6a (see further below).
We reacted 6a with 2,6-di(isopropyl)aniline and observed the formation of 5, as initially suggested by the reactivity of 1a. As expected, 6a is also very sensitive to moisture and solubilization in tetrahydrofuran immediately triggers the oligomerization of the solvent. However, we found it otherwise easy to handle under inert atmosphere, which encouraged us to synthesize a set of parented dicationic dioxoliums 6b–d, featuring methyl or tert-butyl groups in ortho and para positions of the aryl moieties. Starting from the corresponding benzoic acids, we synthesized 2-(aroyloxy)acetamide 2b–d. Their reaction with dichloromethylene-dimethyliminium chloride first performed very poorly, certainly due to the use of bulkier aryl substituents. Among several modifications to Viehe's original protocol,26 the use of acetonitrile as a solvent was found critical to finally isolate 1,3-di(chloro)vinamidinium salts 3b–d in 44–62% yields. Next, treatment with dimethyl(trimethylsilyl)amine yielded 1-chlorovinamidinium salts 1b–d. As frequently observed for vinamidinium cations, 1a–d consist in mixtures of interconverting E and Z isomers, which can't be separated. Moreover, the conformers have distinct or fluxional 13C and 1H NMR signals at room temperature, resulting in equivocal spectra. We confirmed further their structure by X-ray diffraction analysis. Note that, in the solid state, 1a–c adopt a Z configuration, which was therefore attributed to the most stable conformer, whereas both forms are present in the unit cell of 1d. Finally, addition of one equivalent of silver triflate afforded the desired dicationic dioxoliums in 95–98% yield.
X-ray diffraction analysis of 6a and 6c, for which suitable single crystals could be obtained, revealed similar structures for both dications (Fig. 1 and Table 1). The π interaction of oxygen atoms with the formal carbenium center is evident from the short C1–O bond lengths (6a: 1.286(2)/1.317(2) Å; 6c: 1.321(3)/1.303(3) Å, which are typical values in saturated dioxolaniums28 and oxazolium salts)29 whereas C2–O1 (6a: 1.397(2) Å; 6c: 1.390(3) Å) and C3–O2 (6a: 1.419(2) Å, 6c: 1.409(3) Å) bond lengths are significantly longer. The aryl group and the dioxolium ring are nearly co-planar and their conjugation results in a rather short C1–C11 bond (1.429(3) Å in 6a,c, compared to 1.478(4) Å in 1a, and 1.492(10) and 1.506(9) Å in 1c for the two different molecules in the unit cell, respectively). From a structural point of view, the dioxolium patterns seem to have little influence on the vinamidinium moieties. In 6a, for instance, the C6–C3 (1.439(3) Å) and C2–C3 (1.365(3) Å) bond lengths are almost identical to those in corresponding acyclic precursor 1a (C6–C3: 1.440(4) Å and C3–C2: 1.370(5) Å).
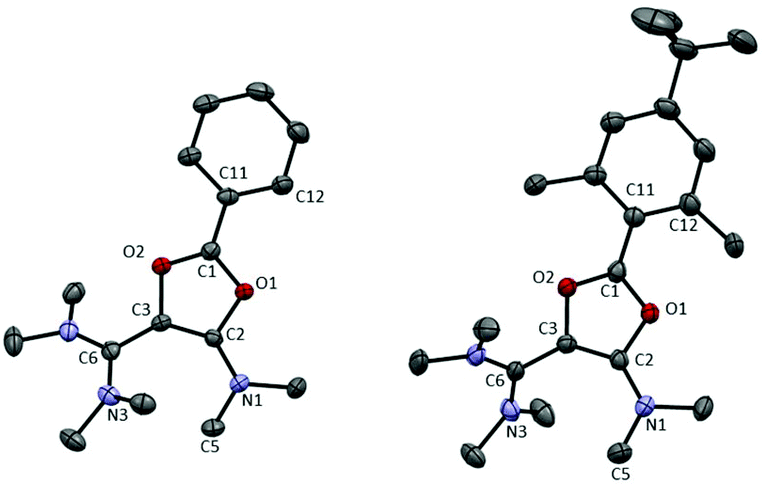 | ||
| Fig. 1 Representation of the X-ray structure of 6a and 6c with 50% probability ellipsoids. Counter-anions, hydrogen atoms and solvent are omitted for clarity. See Table 1 for key structural parameters. | ||
| 6a | 6b | 6c | 6d | |
|---|---|---|---|---|
| a From solid-state structures; values in brackets are from computed optimized structures, see ESI. b In dichloromethane. c From cyclovoltammograms of solutions in acetonitrile + (n-Bu)4NPF6 0.1 mol L−1 (carbon electrode, Φ = 3 mm; scan rate: 100 mV s−1). | ||||
| Ar: 2,6-R2-4-R′(C6H2) | ||||
| R | H | Me | Me | tBu |
| R′ | H | Me | tBu | tBu |
| Bond lengths (Å) | ||||
| C1–O1 | 1.317(2) | — | 1.321(3) | — |
| (1.3106) | (1.3196) | (1.3192) | (1.3102) | |
| C1–O2 | 1.286(2) | — | 1.303(3) | — |
| (1.2821) | (1.2909) | (1.2904) | (1.2787) | |
| O1–C2 | 1.397(2) | — | 1.390(3) | — |
| (1.3820) | (1.3768) | (1.3766) | (1.3816) | |
| O2–C3 | 1.419(2) | — | 1.409(3) | — |
| (1.4041) | (1.4021) | (1.4024) | (1.3959) | |
| C2–C3 | 1.365(3) | — | 1.365(3) | — |
| (1.3730) | (1.3705) | (1.3710) | (1.3780) | |
| C1–C11 | 1.429(3) | — | 1.429(3) | — |
| (1.4244) | (1.4189) | (1.4194) | (1.4518) | |
| C3–C6 | 1.439(3) | — | 1.444(3) | — |
| (1.4373) | (1.4349) | (1.4355) | (1.4417) | |
| N1–C2 | 1.312(2) | — | 1.319(3) | — |
| (1.3120) | (1.3135) | (1.3134) | (1.3098) | |
| Torsions (°) | ||||
| O1–C1–C11–C12 | 5.3(3) | — | 174.6(2) | — |
| (1.2) | (1.8) | (177.7) | (72.6) | |
| O2–C3–C6–N2 | 40.1(3) | — | 45.5(3) | — |
| (44.4) | (43.7) | (43.7) | (42.4) | |
| C4–N1–C2–O1 | 14.8(3) | — | 8.4(4) | — |
| (15.7) | (14.4) | (13.8) | (12.9) | |
| δ 13 C NMR (ppm) | ||||
| C1 | 171.2 | 171.0 | 170.9 | 177.8 |
| C2 | 156.3 | 156.1 | 156.1 | 155.9 |
| C3 | 114.1 | 113.1 | 113.1 | 114.5 |
| C6 | 155.5 | 155.8 | 155.8 | 154.8 |
| C11 | 117.3 | 113.2 | 113.3 | 108.3 |
| λ max UV-visb (nm) | 380.0 | 381.0 | 383.0 | 352.0 |
| E pc vs. Fc/Fc+ (V)c | −1.06 | −1.12 | −1.11 | −1.17 |
Similarly, the 13C NMR chemical shifts of C2/C6 (154.8–156.3 ppm) and C3 (113.1–114.5 ppm) in dioxoliums 6a–d parallel those in precursors 1a. They are fully consistent with polarized vinaminidinium moieties, featuring electron-rich C3 positions and electrophilic C2/C6 centers. Overall, these data suggest only a modest interaction between the π-systems of the vinamidinium and the Aryl–C1–(O1O2) moieties. This is in line with previous findings that dioxoliums are not aromatic and their five membered ring is well described as a carbenium center stabilized by two oxygen atoms along with an isolated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond.21
C double bond.21
Of note, although spectroscopic data for 6a–d are overall analogous, alkyl substituents in ortho and para positions of the aryl group results in shielding the ipso carbon atom (13C NMR chemical shift for 6a: δ = 117.3 ppm, for 6b–c: δ = 113.1 ppm). This is especially the case for 6d (δ = 108.3 ppm). In addition, 6a–c have almost identical 13C NMR chemical shifts for the carbenium center C1 (δ = 171.0–171.9 ppm), whereas the signal in 6d is deshielded by 7 ppm. We interpreted those slight, but significant, differences as the result of the twist around the C1–C11 bond in 6d, due to the bulky tert-butyl ortho-substituents. A hypsochromic shift for the main UV-vis absorption band of 6d (λ = 350 nm, whereas for 6a–c: λ ≈ 380 nm) also supported a decrease in π-conjugation.
Accordingly, the DFT-optimized structure of dication 6d features a large dihedral angle C12–C11–C1–O1 of 73°, whereas the dioxolium rings and the aryl groups are nearly coplanar in 6a–c. The twisting in 6d results in a lengthening of the C1–C11 bond (6d: 1.452 Å; 6a–c: 1.419–1.429 Å).
Next, we examined the fate of these dications upon reduction. Cyclovoltammograms of all dioxoliums 6a-d feature an irreversible reduction wave at about −1.1 V. As a two-electron reduction would lead to an unreasonable “acetal anion” equivalent, we hypothesized an initial one-electron transfer. Note that no reversibility could be evidenced even at high scan rates (up to 10 V s−1), thus indicating that the resulting radicals 7a–d must undergo a fast chemical transformation.
In order to get further insight, we performed the chemical reductions of dioxolium salts 6a–d. We first reacted 6a with half an equivalent of zinc powder. The monitoring of the reaction by 1H NMR showed the gradual disappearance of 6a and the appearance of a new set of signals, while the reaction mixture remained EPR silent. At full conversion, the crude mixture consists in closely related isomers. Fractional crystallization allowed for the isolation of two diastereomeric forms of the dimer of dioxolyl radical 7a, the bis(dioxole)s d,l-8a and meso-8a, in 53% and 7% yield respectively (Scheme 3). They were fully characterized and their structure was ascertained by X-ray analysis (Fig. 2). In the solid state, meso-8a displays a anti conformation of the two dioxolyl groups whereas both anti and gauche conformers are observed in the case of d,l-8a.
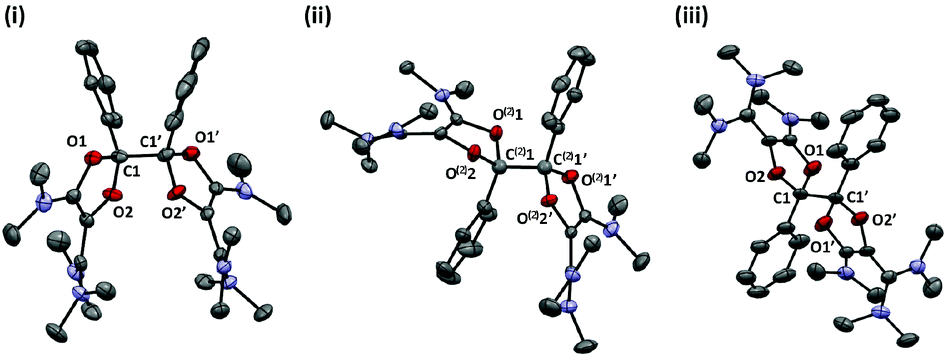 | ||
| Fig. 2 Representation of the X-ray structure of d,l-8a (i: gauche conformer; ii: anti conformer) and meso-8a (iii) with 50% probability ellipsoids. Counter-anions, hydrogen atoms and solvent are omitted for clarity. | ||
Importantly, we couldn't evidence any reversible dissociation of the dimers. Neither d,l-8a nor meso-8a afford equilibrated mixtures of diastereomers when heated in acetonitrile for several hours at 80 °C. This indicates that Gibbs free activation energies for a C–C bond breaking in dimers 8a are at least 30 kcal mol−1. Note that DFT calculations30 at the uB3LYP/6-311 g(d,p) level of theory failed to predict such a strong bond. For instance, the dimerization of 7a to afford the anti conformer of d,l-8a is predicted to be endergonic by ΔG = +12.1 kcal mol−1 and exergonic by only −24 kcal mol−1 when introducing the Polarizing Continuum Model (PCM) for acetonitrile. As dispersion forces can play a critical role in the stability of such encumbered dimers,31 we considered the long-range corrected functional ωB97XD,32 which implements a version of Grimme's D2 model for dispersion forces.33 As a matter of fact, the uωB97XD/6-311 g(d,p)/PCM level of theory predicts a more exergonic dimerization (ΔG = −47 kcal mol−1; −11 kcal mol−1 without PCM), but also affords optimized geometries that better fit experimental solid-state structures, including the length of the C1–C1′ bond that is formed upon dimerization of 7a (for the anti conformer of d,l-8a, B3LYP: 1.569 Å, ωB97XD: 1.558 Å, X-ray: 1.552(4) Å). For consistency throughout this work, all geometry optimizations were carried out at this level of theory. Note that the anti conformer of d,l-8a was found more stable than the gauche conformer and than the meso diastereomer as well, but by only few kcal mol−1, in line with the experimental observation of the three forms.
We wondered whether replacing the phenyl group of 7a with a bulkier 2,4,6-trimethylphenyl could prevent the dimerization of the corresponding dioxolyl radical 7b into the bis(dioxole) 8b. Therefore, we performed the reduction of dication 6b with 0.5 equivalent of zinc. The 1H NMR analysis of the crude mixture revealed the selective formation of a new compound 9b that could be isolated as yellow crystals. The 1H NMR spectrum of 9b features an olefinic resonance signal at 5.96 ppm suggesting the dearomatization of the aryl group and the formation of a cyclohexadiene moiety. This assumption was confirmed by a single crystal X-ray diffraction analysis. Dication 9b is a symmetrical dimer of 2-oxaspiro[4.5]deca-3,6,9-triene-1-one units, connected at their C8 position (Fig. 3). Its formation suggests that dioxolyl radical 7b undergoes a ring opening through C–O bond cleavage to generate vinyl radical 10b (Scheme 4). This latter undergoes a spiro-cyclization to afford cyclohexadienyl radical 11b, which is apparently persistent enough to build-up in solution and dimerizes.
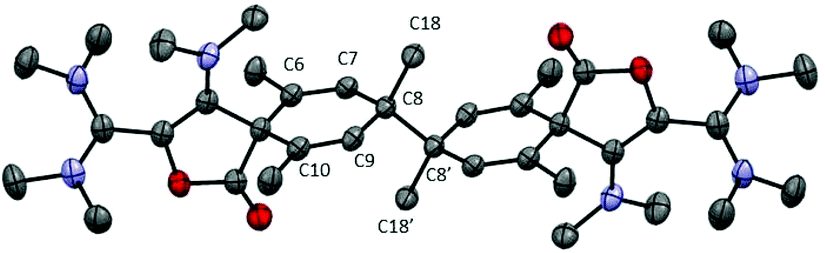 | ||
| Fig. 3 Representation of the X-ray structure of 9b with 50% probability ellipsoids. Counter-anions, hydrogen atoms and solvent are omitted for clarity. | ||
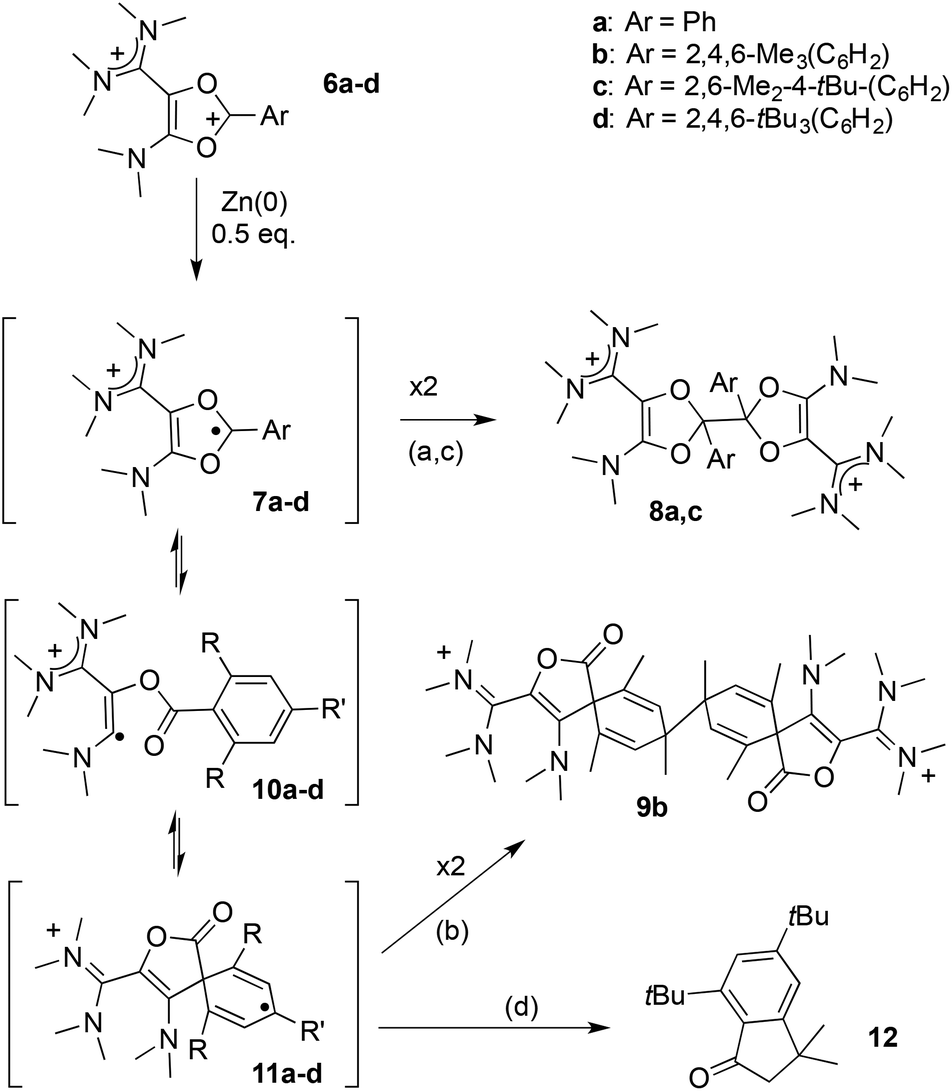 | ||
| Scheme 4 Chemical reduction of 6a–d. | ||
In the case of the reduction of 6c, which features an even more bulky 2,6-dimethyl-4-tertbutylphenyl group, the crude mixture yielded very complex NMR spectra, suggesting that the corresponding radical 7c evolved following multiple pathways. Nevertheless, a small amount of pure material could be isolated as a crystalline solid, which was attributed to dimer 8c. Indeed, although no suitable crystals for X-ray diffraction could be obtained, NMR data are clearly reminiscent of bis(dioxole) 8a, including signals for untouched aryl substituents and a peak in 13C{1H} NMR at 115.9 ppm for C1 and C1′ carbons, which are linking the two monomeric units (8a: 111.2 ppm). This attribution is also supported by HR-MS analysis, which is consistent with a dicationic dimer.
DFT calculations indicate that radicals 7a–c have almost identical electronic structures. Most of the Mulliken spin density is centered on the C1 carbon atom (51–53%, see Table 2 and Fig. 4a), the rest being spread over the aryl group (30–33%), and the two oxygen atoms (12–13%). Similarly to the corresponding dioxolium carbeniums, the π-systems of the dioxolyl and the vinamidinium moities poorly interact, the latter bearing less than 5% spin density. Dioxolyl 7a (aryl = phenyl), vinyl 10a and cyclohexadienyl radicals 11a were found very close in energy and it is likely that the three forms co-exist in solution. In the case of bulkier aryl substituents, the balance is more shifted towards the cyclohexadienyl forms 11b–c. Note that the isolated products 8a,c and 9b do not correspond to the dimer of the most stable radical form but to the overall most stable dimer, thus suggesting that dimerization is essentially under thermodynamic control. Interestingly, the introduction of methyl ortho-substituents is not detrimental to the formation of dimers 8a–c from radicals 7a–c, the dimerization being even more exergonic for 7b,c (ΔG about −70 kcal mol−1) than for 7a (ΔG = −51 kcal mol−1). Similarly, the formation of dimer 9b from cyclohexadienyl radical 11b is predicted to be more exergonic than the formation of 9a from 11a. However, the para tert-butyl group in 11c is clearly prejudicial to the formation of the corresponding dimer 9c (ΔG = −6 kcal mol−1 only). In line with this trend, radicals 7d and 11d, which stem from dioxolium 6d with a 2,4,6-tri(tert-butyl)phenyl substituent, should not dimerize. Indeed, the formation of dimer 9d from 11d was found slightly endergonic and no minimum on the hypersurface of energy could be found for the putative dimer 8d. Note that 7d is also predicted to be fundamentally different from 7a–c. The aryl group is not conjugated with the rest of the π-system anymore and does not feature significant spin density, 80% of it being now localized on the carbenium center C1 (Table 2 and Fig. 4b). The latter has no planar environment anymore (sum of bond angles around C1 in 7d: 350.6°, in 7a–c: >359.9°), indicating an increased sp3 character. This illustrates the key role of the aryl group in the stabilization of dioxolyl radicals through spin delocalization. In addition, not only is 7d poorly stabilized, but the cyclohexadienyl form is only −12 kcal mol−1 lower in energy and is likely to be highly reactive as well.
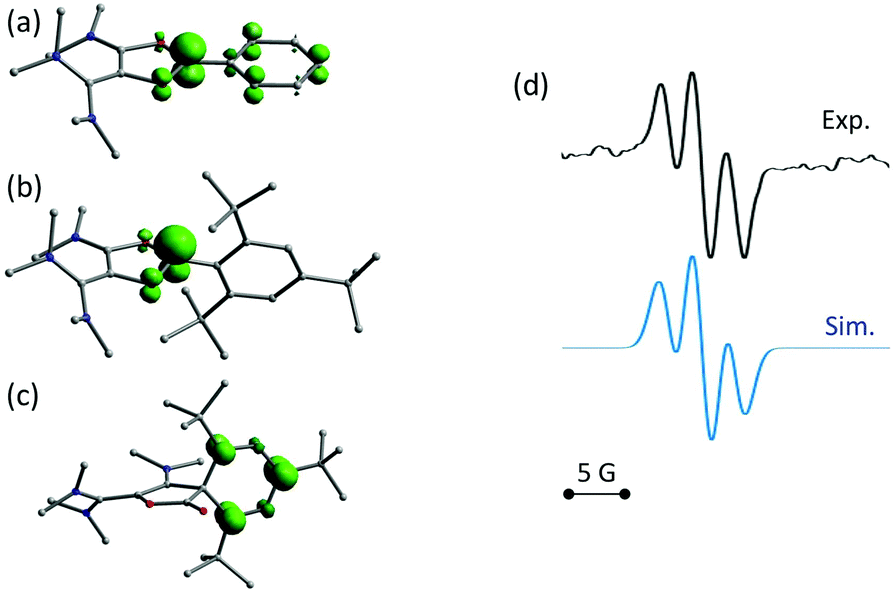 | ||
| Fig. 4 Representation of computed Mulliken spin densities for, (a) 6a, (b) 6d and (c) 11d; (d) experimental X-band isotropic EPR spectrum at room temperature in acetonitrile after reduction of 6d (top, in black), the simulated spectrum (bottom, in blue) was obtained with a Lorentzian line-broadening parameter of 0.25 and an hyperfine constant a(1H) of 8.0 MHz (2 nuclei). | ||
| Dioxolyl radical | 7a | 7b | 7c | 7d |
|---|---|---|---|---|
| a Energies are in kcal mol−1. b No minimum found for 8d. | ||||
| R | H | Me | Me | tBu |
| R′ | H | Me | tBu | tBu |
| Mulliken spin density | ||||
| C1 | 51% | 51% | 53% | 80% |
| Aryl | 32% | 33% | 30% | 1% |
| O1 and O2 | 13% | 12% | 12% | 15% |
| ΔG (relative to 7a–d ) | ||||
| 10a–d | +1.0 | −1.6 | −0.8 | −0.8 |
| 11a–d | −4.7 | −16.7 | −15.7 | −12.2 |
| [G(8a–d)–G(9a–d)]a | −11.4 | +10.2 | −32.0 | n.a.b |
| ΔG (dimer formation) | ||||
| 7a–d (×2) → 8a–d | −51.2 | −68.5 | −69.8 | n.a.b |
| 11a–d (×2) → 9a–d | −30.4 | −45.3 | −6.4 | +2.7 |
In order to experimentally assess the fate of these radicals, we examined the reduction of dioxolium 6d with half an equivalent of zinc(0) powder. EPR monitoring of the reaction showed the appearance of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 triplet, corresponding to an hyperfine coupling constant a = 8 MHz with two equivalent ½ spins34 (Fig. 4d). This value is in perfect agreement with the expected spectrum for 11d. In particular, DFT calculations predict that this organic π-radical should only feature significant isotropic hyperfine coupling constants with the two hydrogen atoms in meta position of the aryl group, with a computed value a(1H) of about 9 MHz. Though persistent at room temperature, the EPR signal evolved after few hours into a more complex unsymmetrical bandshape, indicating a mixture of radical species, and finally faded away (see ESI†). Unsurprisingly, all attempts to isolate 11d failed. Ultimately, known indanone 1235 was isolated from the crude mixture in 30% yield (Scheme 4).
:2:1 triplet, corresponding to an hyperfine coupling constant a = 8 MHz with two equivalent ½ spins34 (Fig. 4d). This value is in perfect agreement with the expected spectrum for 11d. In particular, DFT calculations predict that this organic π-radical should only feature significant isotropic hyperfine coupling constants with the two hydrogen atoms in meta position of the aryl group, with a computed value a(1H) of about 9 MHz. Though persistent at room temperature, the EPR signal evolved after few hours into a more complex unsymmetrical bandshape, indicating a mixture of radical species, and finally faded away (see ESI†). Unsurprisingly, all attempts to isolate 11d failed. Ultimately, known indanone 1235 was isolated from the crude mixture in 30% yield (Scheme 4).
Note that, at a pinch, the formation of dimers 8a,c could have been the result of an ionic mechanism. However, the formation of 9b, the observation of radical 11d, as well as DFT results as a whole, definitely support radical pathways, stemming from a one-electron reduction of the dioxoliums.
We propose a mechanism for the decay of 11d into 12, which is supported by DFT investigations (Fig. 5). The rate-determining step of the process is the ring opening of 11d, yielding vinyl radical 10d′ (ΔG‡ = + 18.7 kcal mol−1). The latter is only +4.4 kcal mol−1 higher in energy than the most stable conformer 10d. It features the relevant conformation for a hydrogen shift from an ortho tert-butyl group (ΔG‡ = +8.1 kcal mol−1). The resulting alkyl radical 13 adds intramolecularly to the carbonyl group (ΔG‡ = +8.8 kcal mol−1), yielding five-membered ring 14, which undergoes a barrier-less homolytic C–O bond breaking to afford indanone 12 and radical 15. Note that we previously showed that so-called oxyallyl radical cations, which are parented to 15, are remarkably stabilized and could even be highly air-persistent.10a,c,25a,d,e Although it appears that 15 evolve further in the reaction conditions, it is likely that it contributes to the transiently observed EPR spectrum upon decay of 11d. However, we couldn't obtain an unambiguous simulation for the EPR bandshapes by only considering a mixture of the cyclohexadienyl and oxyallyl radicals. This suggested the formation of a more complex mixture of paramagnetic species.
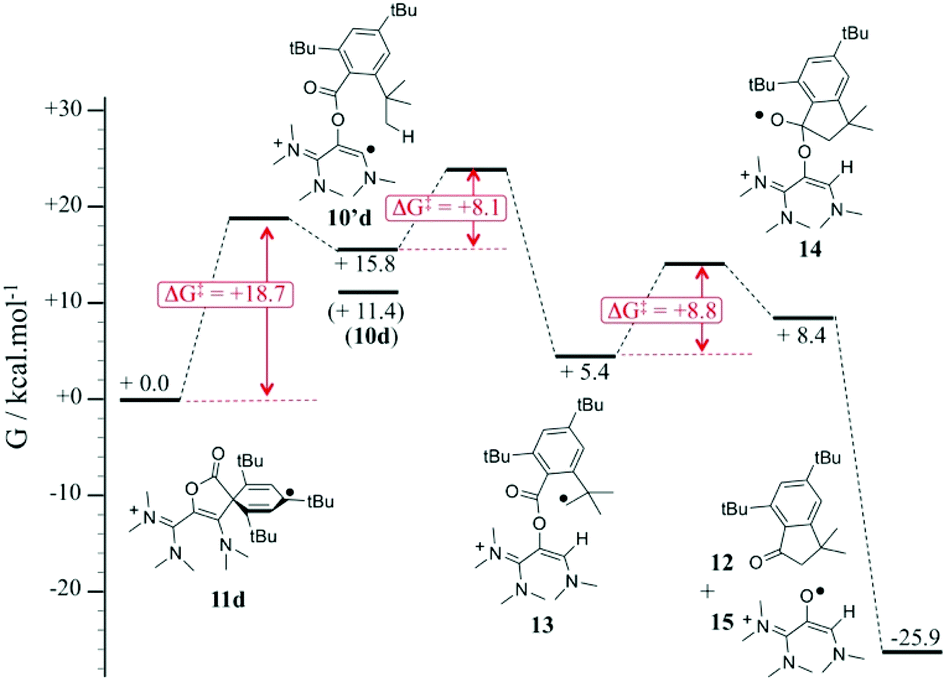 | ||
| Fig. 5 Computed mechanism for the formation of indenone 12 from 11d,b at the; uωB97XD/6-311 g(d,p)/PCM(acetonitrile) level of theory relative Gibbs energies of intermediates and Gibbs energies of activation. | ||
Conclusion
Dicationic dioxoliums 6a–d were readily synthesized from acetamides 2a–d.36–39 All spectroscopic and structural data indicate a poorly aromatic five-membered ring and a modest interaction between the π-systems of the vinamidinium and carbenium moieties. This situation is reminiscent of that of the neutral borate-based zwitterions from Stephan et al.,21b which were the only non-stabilized dioxoliums to have been previously structurally characterized by X-ray analysis.The fate of 6a–d upon reduction highly depends on the aryl substituent. All dioxolyl radicals 7a–d can undergo a ring-opening, which is reminiscent of the Surzur–Tanner rearrangement of β-(acyloxy)alkyl radicals. They are in equilibrium with the resulting vinyl radical forms 10a–d and their spiro-cyclization products, the cyclohexadienyl radicals 11a–d. In the case of a bulky tri(tert-butyl)phenyl aryl group (7d), dimerization processes are disfavored. The radical essentially exists in the cyclohexadienyl form 11d, which was observed at room temperature by EPR spectroscopy.
DFT studies show that spin-delocalization on the aryl group plays a major role in the stabilization of dioxolyl radicals, with only second-order effects of the O-substituents. As a matter of fact, dioxolyl radicals 7a–c could be depicted as well as benzyl radicals with ancillary O-substituents: more than 30% of spin density is found on the fully conjugated aryl groups, only 12–13% being spread on the two oxygen atoms. In 7d, the bulky tert-butyl ortho-substituents twist away the π-systems and prevent this delocalization. The radical is highly localized, with more than 80% of spin density on one carbon atom, which features some sp3 hybridation.
Finally, although DFT ωB97XD/6-311 g(d,p)/PCM calculations could fairly account for the experimental data and observations of this work, it is too early to fully ascertain their accuracy. We are actually considering experimental gas phase studies with a modified QhQ mass spectrometer to better calibrate DFT level of theory and further explore the uncharted territory of the reactivity of these species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Agency for Research (ANR-14-CE06-0013-01 and ANR-17-ERC2-0015). The authors are grateful to the IR-RPE CNRS FR3443 RENARD network for EPR facilities. Thanks are due to the “Centre de Calcul Intensif en Chimie de Grenoble” and the University of Grenoble-Alpes, including the Pole-Agir fund, and partial support from Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). The authors also thank the ICMG Chemistry Nanobio Platform of Grenoble, especially Dr C. Phillouze and N. Altounian for the X-ray crystallographic structure determination of compounds 5.References
- (a) R. W. Taft, R. H. Martin and F. W. Lampe, J. Am. Chem. Soc., 1965, 87, 2490 CrossRef CAS; (b) F. Bernardi, A. Bottoni and A. Venturini, J. Am. Chem. Soc., 1986, 108, 5395 CrossRef CAS; (c) J. Kapp, C. Schade, A. M. El-Nahas and P. von R. Schleyer, Angew. Chem., Int. Ed. Engl., 1996, 35, 2236 CrossRef; (d) H. Grützmacher and C. M. Marchand, Coord. Chem. Rev., 1997, 163, 287 CrossRef; (e) J. Rissler, M. Hartmann, C. M. Marchand, H. Grützmacher and G. Frenking, Chem. – Eur. J., 2001, 7, 2834 CrossRef CAS PubMed; (f) H. F. Fang, Z. Wang and X. Fu, Coord. Chem. Rev., 2017, 344, 214 CrossRef CAS.
- (a) G. A. Olah, Angew. Chem., Int. Ed. Engl., 1993, 32, 767 CrossRef; (b) G. A. Olah, Angew. Chem., Int. Ed. Engl., 1995, 34, 1393 CrossRef CAS; (c) G. A. Olah, J. Org. Chem., 2001, 66, 5943 CrossRef CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- (a) H. G. Viehe, R. Mértnyi, L. Stella and Z. Janousek, Angew. Chem., Int. Ed. Engl., 1979, 18, 917 CrossRef; (b) H. G. Viehe, Z. Janousek, R. Mértnyi and L. Stella, Acc. Chem. Res., 1985, 18, 148 CrossRef CAS; (c) H. Zipse, Top. Curr. Chem., 2006, 263, 163 CrossRef CAS; (d) Substituent Effects in Radical Chemistry, ed. V. H. Veihe, Z. Janousek and R. Mértnyi, Riedell, Boston, 1986 Search PubMed; (e) A. R. Katritzky, M. C. Zerner and M. M. Karelson, J. Am. Chem. Soc., 1986, 108, 7213 CrossRef CAS; (f) D. J. Pasto, J. Am. Chem. Soc., 1988, 110, 8164 CrossRef CAS; (g) F. G. Bordwell and T.-Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558 CrossRef CAS; (h) M. L. Coote and A. B. Dickerson, Aust. J. Chem., 2008, 61, 163 CrossRef CAS; (i) A. S. Menon, D. J. Henry, T. Bally and L. Radom, Org. Biomol. Chem., 2011, 9, 3636 RSC; (j) M. L. Coote, C. Y. Lin, A. L. Beckwith and A. A. Zavitsas, Phys. Chem. Chem. Phys., 2010, 12, 9597 RSC.
- For kinetic and thermodynamic metrics of substituent effects, see also: (a) H. O. Pritchard and H. A. Skinner, Chem. Rev., 1955, 55, 745 CrossRef CAS; (b) J. Mullay, in Electronegativity. Structure and Bonding, Vol. 66, ed. K. D. Sen and C. K. Jørgensen, Springer, Berlin, 1987 Search PubMed; (c) C. Hansch, A. Leo and W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (d) H. Mayr and A. R. Ofial, Acc. Chem. Res., 2016, 49, 952 CrossRef CAS PubMed.
- M. A. Kouskoulli, D. M. Smith and A. V. Nicolaides, J. Mol. Struct.: THEOCHEM, 2007, 811, 355 CrossRef CAS.
- For pioneering studies, see: (a) G. A. Olah, M. Calin and D. H. O'Brien, J. Am. Chem. Soc., 1967, 89, 3586 CrossRef CAS; (b) G. A. Olah and A. M. White, J. Am. Chem. Soc., 1967, 89, 3591 CrossRef CAS; (c) G. A. Olah and A. M. White, J. Am. Chem. Soc., 1968, 90, 1884 CrossRef CAS.
- Further features are usually required. Aromatic heterocyclic pyriliums, among which anthocyanidin-based natural dyes, are regarded as the prototypical stable O-substituted carbeniums. See for examples: (a) D. W. Hill, Chem. Rev., 1936, 19, 27 CrossRef CAS; (b) G. A. Iacobucci and J. G. Sweeny, Tetrahedron, 1983, 39, 3005 CrossRef CAS; (c) F. Pina, M. J. Melo, C. A. T. Laia, A. J. Parola and J. C. Lima, Chem. Soc. Rev., 2012, 41, 869 RSC.
- For preparation of simple iminiums salts, see: (a) A. Ahond, A. Cave, C. Kan-Fan, H.-P. Husson, J. de Rostolan and P. Pottier, J. Am. Chem. Soc., 1968, 90, 5622 CrossRef CAS; (b) H. Voltz and H. H. Kiltz, Tetrahedron, 1970, 26, 1917 CrossRef; (c) N. Holy, R. Fowler, E. Burnett and R. Lorenz, Tetrahedron, 1979, 35, 613 CrossRef CAS; (d) U. Jahn and W. Schroth, Tetrahedron Lett., 1993, 34, 5863 CrossRef CAS.
- (a) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014 CrossRef CAS PubMed; (b) J. K. Mahoney, D. Martin, C. Moore, A. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766 CrossRef CAS PubMed; (c) J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519 CrossRef CAS PubMed; (d) C. L. Deardorff, E. R. Sikma, C. P. Rhodes and T. W. Hudnall, Chem. Commun., 2016, 52, 9024 RSC; (e) J. K. Mahoney, R. Jazzar, G. Royal, D. Martin and G. Bertrand, Chem. – Eur. J., 2017, 23, 6206 CrossRef CAS PubMed; (f) V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109 CrossRef CAS PubMed.
- See also: (a) T. H. Koch, J. A. Olesen and J. DeNiro, J. Am. Chem. Soc., 1975, 97, 7285 CrossRef CAS; (b) R. J. Himmelsbach, A. D. Barone, D. L. Kleyer and T. H. Koch, J. Org. Chem., 1983, 48, 2989 CrossRef CAS; (c) G. Gaudiano, K. Sweeney, R. C. Haltiwanger and T. H. Koch, J. Am. Chem. Soc., 1984, 106, 7628 CrossRef CAS; (d) O. Benson, Jr., S. H. Demirdji, R. C. Haltiwanger and T. H. Koch, J. Am. Chem. Soc., 1991, 113, 8879 CrossRef; (e) O. Benson, Jr., G. Gaudiano, R. C. Haltiwanger and T. H. Koch, J. Org. Chem., 1988, 53, 3036 CrossRef; (f) D. L. Kleyer, R. C. Haltiwanger and T. H. Koch, J. Org. Chem., 1983, 48, 147 CrossRef CAS; (g) F. Welle, S. P. Verevkin, M. Keller, H.-D. Beckhaus and C. Ruchardt, Chem. Ber., 1994, 127, 697 CrossRef CAS; (h) I. Nakanishi, S. Itoh and S. Fukuzumi, Chem. – Eur. J., 1999, 5, 2810 CrossRef CAS; (i) M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579 CrossRef CAS PubMed; (j) L. Jin, M. Melaimi, L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351 RSC; (k) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441 CrossRef CAS PubMed.
- Note also that even parented oxygen equivalents of tetrathiafulvalene radical cations cannot be isolated: (a) T. Nogami, K. Tanaka and A. Kobayahi, Synth. Met., 2001, 120, 755 CrossRef CAS; (b) T. Kojima, K. Tanaka, T. Ishida and T. Nogami, J. Org. Chem., 2004, 69, 9319 CrossRef CAS PubMed.
- U. Wille, J. Org. Chem., 2006, 71, 4040 CrossRef CAS PubMed.
- U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed.
- For closely related reactions, see also: (a) U. Wille and C. Plath, Liebigs Ann./Recl., 1997, 111 CrossRef CAS; (b) U. Wille and L. Lietzau, Tetrahedron, 1999, 55, 10119 CrossRef CAS; (c) U. Wille, Org. Lett., 2000, 2, 3485 CrossRef CAS PubMed; (d) L. Lietzau and U. Wille, Heterocycles, 2001, 55, 377 CrossRef CAS; (e) U. Wille, Chem. – Eur. J., 2002, 8, 340 CrossRef CAS PubMed; (f) U. Wille, J. Am. Chem. Soc., 2002, 124, 14 CrossRef CAS PubMed; (g) C. Jargstorff and U. Wille, Eur. J. Org. Chem., 2003, 2173 Search PubMed; (h) U. Wille, J. Phys. Org. Chem., 2011, 24, 672 CrossRef CAS.
- (a) C. Brunton, K. U. Ingold, B. P. Roberts, A. L. J. Beckwith and P. J. Krusic, J. Am. Chem. Soc., 1977, 99, 3177 CrossRef; (b) C. Gaze and B. C. Gilbert, J. Chem. Soc., Perkin Trans. 2, 1978, 235 RSC; (c) C. Gaze and B. C. Gilbert, J. Chem. Soc., Perkin Trans. 2, 1979, 763 RSC; (d) V. Malatesta and K. U. Ingold, J. Am. Chem. Soc., 1981, 103, 609 CrossRef CAS; (e) A. L. J. Beckwith and C. J. Easton, J. Am. Chem. Soc., 1981, 103, 615 CrossRef CAS; (f) S. Deycard, J. Lusztyk, K. U. Ingold, F. Zerbetto, M. Z. Zgierski and W. Siebrand, J. Am. Chem. Soc., 1990, 112, 4284 CrossRef CAS; (g) A. J. Fielding, P. Franchi, B. P. Roberts and T. M. Smits, J. Chem. Soc., Perkin Trans. 2, 2002, 155 CAS.
- (a) J. M. Surzur and P. Teissier, C. R. Acad. Sci. Fr. Ser. C, 1967, 264, 1981 CAS; (b) D. D. Tanner and F. C. P. Law, J. Am. Chem. Soc., 1969, 91, 7535 CrossRef CAS; (c) A. L. J. Beckwith, D. Crich, P. J. Duggan and Q. Yao, Chem. Rev., 1997, 97, 3273 CrossRef CAS.
- (a) L. R. C. Barclay, D. Griller and K. U. Ingold, J. Am. Chem. Soc., 1982, 104, 4399 CrossRef CAS; (b) S. Saebo, A. L. J. Beckwith and L. Radom, J. Am. Chem. Soc., 1984, 106, 5119 CrossRef CAS; (c) L. R. C. Barclay, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 1793 CrossRef CAS; (d) D. Crich and D.-H. Suk, Can. J. Chem., 2004, 82, 75 CrossRef CAS; (e) H. Zipse, J. Am. Chem. Soc., 1997, 119, 1087 CrossRef CAS; (f) E. Lacôte and P. Renaud, Angew. Chem., Int. Ed., 1998, 37, 2259 CrossRef.
- (a) W. T. Evanochenko and P. Shevlin, J. Org. Chem., 1979, 44, 4426 CrossRef; (b) F. Shahidi and T. T. Tidwell, Can. J. Chem., 1982, 60, 1092 CrossRef CAS.
- (a) A. D. Allen, T. Kitamura, R. A. McClelland, P. J. Stang and T. T. Tidwell, J. Am. Chem. Soc., 1990, 112, 8873 CrossRef CAS; (b) T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868 CrossRef CAS PubMed.
- The only examples of 1,3-dioxolium compounds without charge-stabilizing atoms at C-2 are limited to: (a) W. Lorenz and G. Maas, J. Org. Chem., 1987, 52, 375 CrossRef CAS; (b) M. M. Hansmann, R. L. Melen, F. Rominger, A. S. K. Hashmi and D. W. Stephan, Chem. Commun., 2014, 50, 7243 RSC.
- (a) H. Gross and J. Rusche, Angew. Chem., Int. Ed. Engl., 1964, 3, 511 CrossRef; (b) K. Dimroth, P. Heinrich and K. Schromm, Angew. Chem., Int. Ed. Engl., 1965, 4, 873 CrossRef; (c) H. Volz and G. Zimmermann, Tetrahedron Lett., 1970, 3597 CrossRef CAS.
- (a) G. A. Olah and A. M. White, J. Am. Chem. Soc., 1968, 90, 1884 CrossRef CAS PubMed; (b) G. A. Olah and J. L. Grant, J. Org. Chem., 1977, 42, 2237 CrossRef CAS; (c) U. Timm, U. Plücken, H. Petersen and H. Meier, J. Heterocycl. Chem., 1979, 16, 1303 CrossRef CAS; (d) H. Mayr and U. von der Brüggen, Chem. Ber., 1988, 121, 339 CrossRef CAS.
- (a) P. R. Moses, J. Q. Chambers, J. O. Sutherland and D. R. Williams, J. Electrochem. Soc., 1975, 122, 608 CrossRef CAS; (b) R. D. Braun and D. C. Green, J. Electroanal. Chem., 1977, 79, 381 CrossRef CAS; (c) G. Maas and B. Singer, Chem. Ber., 1983, 116, 3639 CrossRef.
- (a) V. Regnier, F. Molton, C. Philouze and and D. Martin, Chem. Commun., 2016, 52, 11422 RSC; (b) V. Regnier, Y. Planet, C. E. Moore, J. Pecaut, C. Philouze and D. Martin, Angew. Chem., Int. Ed., 2017, 56, 1031 CrossRef CAS PubMed; (c) M. Tripathi, V. Regnier, C. Lincheneau and D. Martin, New J. Chem., 2017, 41, 15016 RSC; (d) M. Devillard, V. Regnier, M. Tripathi and D. Martin, J. Mol. Struct., 2018, 1172, 3 CrossRef CAS; (e) M. Tripathi, V. Regnier, Z. Ziani, M. Devillard, C. Philouze and D. Martin, RSC Adv., 2018, 8, 38346 RSC; (f) V. Regnier and D. Martin, Org. Chem. Front., 2015, 2, 1536 RSC.
- (a) Z. Janousek and H. G. Viehe, Angew. Chem., Int. Ed. Engl., 1971, 10, 574 CrossRef CAS; (b) H. G. Viehe, Z. Janousek, R. Gompper and D. Lach, Angew. Chem., Int. Ed. Engl., 1973, 12, 566 CrossRef; (c) A. Fürstner, M. Alcazaro and H. Krause, Org. Synth., 2009, 298 Search PubMed.
- CCDC 1898554–1898565† contain the supplementary crystallographic data for this paper.
- (a) H. Paulsen and R. Dammeyer, Chem. Ber., 1973, 106, 2324 CrossRef CAS; (b) H. Paulsen and R. Dammeyer, Chem. Ber., 1976, 109, 1837 CrossRef CAS; (c) M. R. Caira and J. F. de Wet, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 709 CrossRef; (d) R. F. Childs, R. M. Orgias, C. J. L. Lock and M. Mahendran, Can. J. Chem., 1993, 71, 836 CrossRef CAS.
- (a) A. O. Doroshenko, T. V. Sakhno and A. V. Kyrychenko, Spectrosc. Lett., 2002, 345, 171 CrossRef; (b) D. C. Graham, K. J. Cavell and B. F. Yates, Dalton Trans., 2007, 4650 RSC; (c) J. Zhang, J. Fu, X. Su, X. Qin, M. Zhao and M. Shi, Chem. Commun., 2012, 48, 9625 RSC; (d) R. L. Melen, M. M. Hansmann, A. J. Lough, A. S. K. Hasmi and D. W. Stephan, Chem. – Eur. J., 2013, 19, 11928 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, DFT calculations were performed using the program package Gaussian09: Gaussian 09, Revision 4.2.0, Gaussian Inc., Wallingford CT, 2009 Search PubMed. See ESI† for further details.
- For a seminal example and discussions, see: (a) S. Grimme and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 12639 CrossRef CAS PubMed; (b) S. Grimme, Angew. Chem., Int. Ed., 2006, 45, 4460 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- The experimental EPR spectrum was fitted with the EasySpin simulation package: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- P. J. Wagner, B.-S. Park, M. Sobczak, J. Frey and Z. Rappoport, J. Am. Chem. Soc., 1995, 117, 7619 CrossRef CAS.
- A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836 CrossRef PubMed.
- H. G. Emblem, K. Hargreaves and C. E. Oxley, J. Appl. Chem., 1968, 18, 97 CrossRef CAS.
- C. Hering-Junghans, A. Schulz, M. Thomas and A. Villinger, Dalton Trans., 2016, 45, 6053 RSC.
- J. E. Field, T. J. Hill and D. Venkataraman, J. Org. Chem., 2003, 68, 6071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, NMR spectra. CCDC 1898554–1898565. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00298g |
| This journal is © the Partner Organisations 2019 |