
Dual-directional alkyne-terminated macrocycles: Enroute to non-aggregating molecular platforms†
Ali
Husain
*a,
Asaithampi
Ganesan
a,
Basma
Ghazal
a,
Mahmut
Durmuş
b,
Xian-Fu
Zhang
 c and
Saad
Makhseed
*a
c and
Saad
Makhseed
*a
aDepartment of Chemistry, Kuwait University, P.O. Box 5969, Safat, 13060, Kuwait. E-mail: ali.husain@ku.edu.kw; saad.makhseed@ku.edu.kw
bGebze Technical University, Department of Chemistry, 41400 Gebze-Kocaeli, Turkey
cMPC Technologies, Hamilton, Ontario, Canada L8S 3H4
First published on 31st July 2019
Abstract
Derivatized phthalocyanines (Pcs) and their heteroatom analogues, azaphthalocyanines (AzaPcs), bearing a variety of highly active ligands, have many advantageous properties that make them suitable as novel macrocyclic platforms. Many peripherally/non-peripherally functionalized macrocycles have been successfully employed as molecular platforms; however, due to the lack of directional control of the outward-facing reactive species, retaining their planarity while avoiding aggregation was quite challenging. The present work demonstrates the role of dual directionality using multivalent, orthogonal propargyl moieties based on two novel Zn(II)phthalocyanine (Pc1) and azaphthalocyanine (AzaPc1) complexes. These groups prevent the macrocyclic planar cores from self-associating in solution or in the solid state, as confirmed by 1H NMR, UV-Vis and single-crystal X-ray diffraction analyses. Such activated systems are thus highly applicable as key intermediates in the development of “molecular platforms” to generate endless, applicable, non-aggregated macrocyclic materials via a variety of organic transformations, specifically, the powerful click reaction, Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC). In addition to their utility as efficient building blocks suitable for further modification, the photophysical/photochemical properties of these compounds were also investigated to determine their photocatalytic activities for valuable applications, such as in photodynamic therapy (PDT).
Introduction
Phthalocyanines (Pcs) and their heteroatom analogues, azaphthalocyanines (AzaPcs), have unique physico-chemical and thermal properties, making them an important class of compounds.1 Their remarkable architectures consisting of extensive π-electron delocalization and a central cavity that can bind different cations, as well as the relative ease of the chemical modification of their peripheral/non-peripheral positions and axial metal sites, led synthetic chemists to explore their utility in the synthesis of valuable macrocycle-based materials with a wide range of extraordinary applications in many advanced research areas, including solar cells, chemical/biological sensors, photodynamic therapy (PDT), liquid crystals, optical light-emitting diodes (OLEDs), photovoltaics, and polymer/materials science.1,2Many studies on Pcs and their analogous systems require an appropriate conjugation of suitable substituents that can provide unique structural and/or physico-chemical features that are essential for particular applications.1a,2a The most common synthetic strategy used for generating such derivatized macrocycles begins with decorating aryl ortho-dicarbonitrile substrates with appropriate conjugated species, followed by the cyclization of the resulting Pc/Pc-analogue subunits in the presence of suitable metal ions to obtain target macrocyclic complexes. Such synthetic procedures are typically feasible for expert synthetic chemists. However, the instability of the substituents under common harsh reaction conditions, the formation of undesired by-products, difficult purifications, and low yields of the desired products are major drawbacks in the synthesis of such modified materials using this classic approach.
In recent years, fabricating macrocyclic backbones with extremely reactive terminal alkyne groups has become a practical method for the construction of activated macrocycles and innovative macrocyclic platform systems.3 Many alkyne-terminated macrocycles have been described in the literature, and most of these reported structures are well established as building blocks for the attachment of different ligating sites on the ring,3 and other structures have been effectively immobilized on nanofibers,4 dendrimers,5 polymers,6 nanoparticles (NPs),7 glassy carbon electrodes (GCEs),8 and gold surfaces9 and used for the synthesis of rotaxane hybrid systems.10 All alkynyl-substituted macrocycles, except for axially metal-decorated species,3i are composed of outward-facing aliphatic/aromatic alkyne ends, and to the best of our knowledge, there have been no reports describing the utility of the dual directionality concept in the synthesis of non-aggregated, terminal alkyne-activated macrocyclic platforms.
Herein, we describe the design and synthesis of two novel alkyne-terminated phthalocyanine and azaphthalocyanine macrocycles and their applications as activated molecular platforms. Notably, the strategy presented in this work, i.e., the dual directionality of multivalent propargyl moieties based on two novel zinc(II) phthalocyanine (Pc1) and azaphthalocyanine (AzaPc1) macrocycles (Fig. 1), has many advantages that allow it to overcome the shortcomings of the traditional method noted above. The reported macrocycles, which are non-aggregated, can be synthesized in reasonable yields, and their purifications are simple. Their terminal ends are highly reactive towards various reaction conditions, and stoichiometric conversions would be expected for many organic transformations, including Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC),11 as well as Sonogashira,12 Eglinton,13 Glaser–Hay,14 Cadiot–Chodkiewicz15 carbon–carbon cross-couplings and others.16
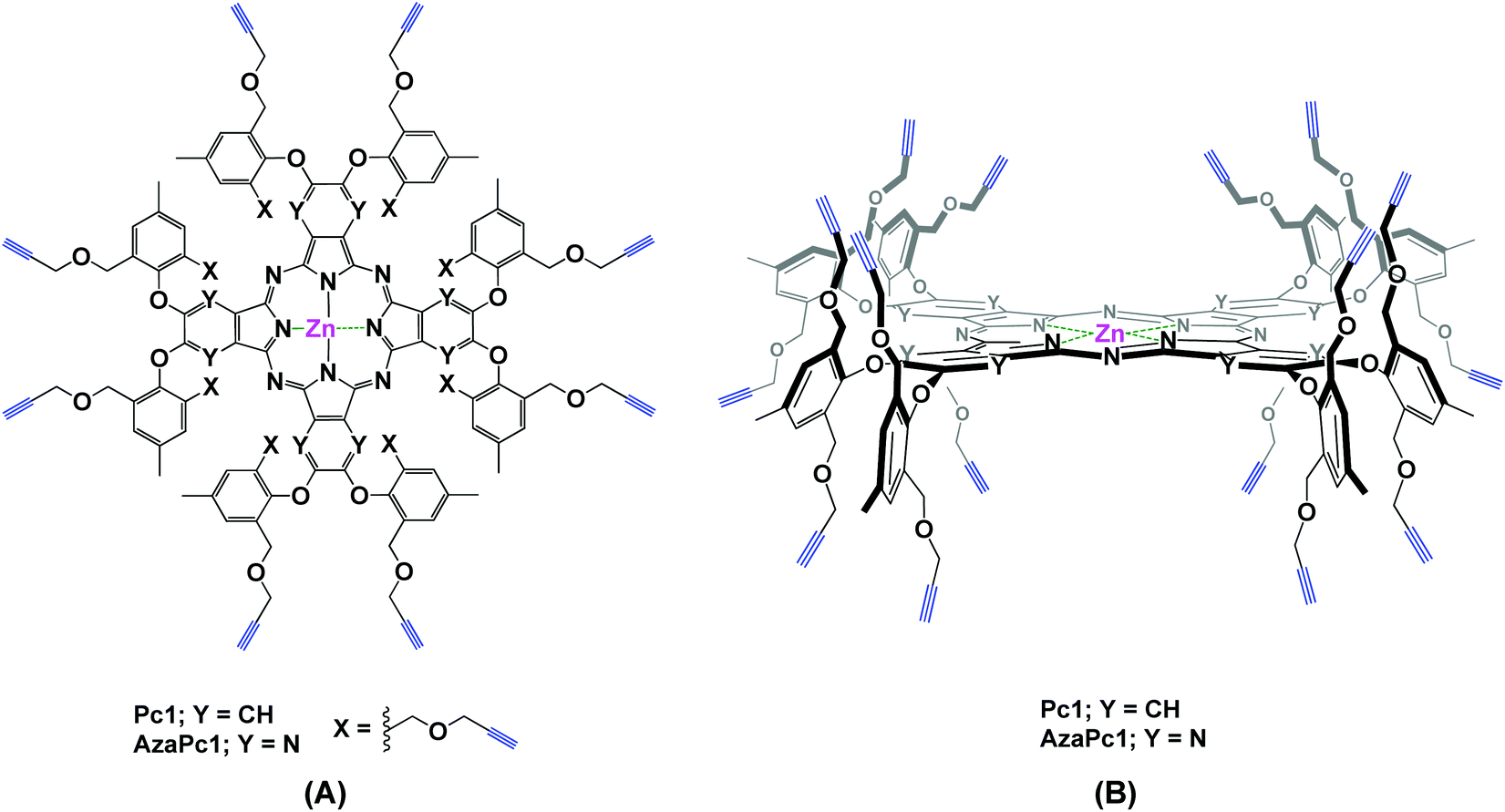 | ||
| Fig. 1 (A) Top view and (B) 3D representation of the general structures of Pc1 and AzaPc1. | ||
Results and discussion
The enforced rigid planar structures of Pc/AzaPc make them robust frameworks, and their rims can be covalently modified without compromising their planarity. As a result, they are an ideal molecular backbone for investigating the role of the dual directionality of multivalent propargyl moieties altered by orthogonal bulky phenoxyl substituents on the periphery of the macrocycle. The restricted rotation, i.e., the directional control (up/down) of the terminal alkyne units, which is a result of the bulky phenoxyl groups, remarkably prevented the macrocyclic cores from self-associating in solution or the solid state, as confirmed by their spectral data (e.g., 1H NMR, and UV-Vis), single-crystal X-ray diffraction and elemental analyses. The related spectra are provided in the ESI.†The synthesis of Pc1 and AzaPc1 was based on three major synthetic steps: (i) functionalizing p-cresol derivative 1 with propargyl groups to afford dipropargyl-p-cresol derivative 2, (ii) decorating activated phthalonitrile and pyrazine-2,3-dicarbonitrile substrates with p-cresol 2 to afford precursors 3 and 4, respectively, and (iii) cyclizing the resulting Pc/AzaPc subunits in the presence of zinc(II) to generate the target macrocycles, Pc1/AzaPc1 (Scheme 1). All novel compounds, including 2–4, Pc1 and AzaPc1, were fully characterized by NMR, FT-IR, MALDI-TOF, HRMS, and single-crystal X-ray diffraction spectral techniques and CHN elemental analysis (ESI†).
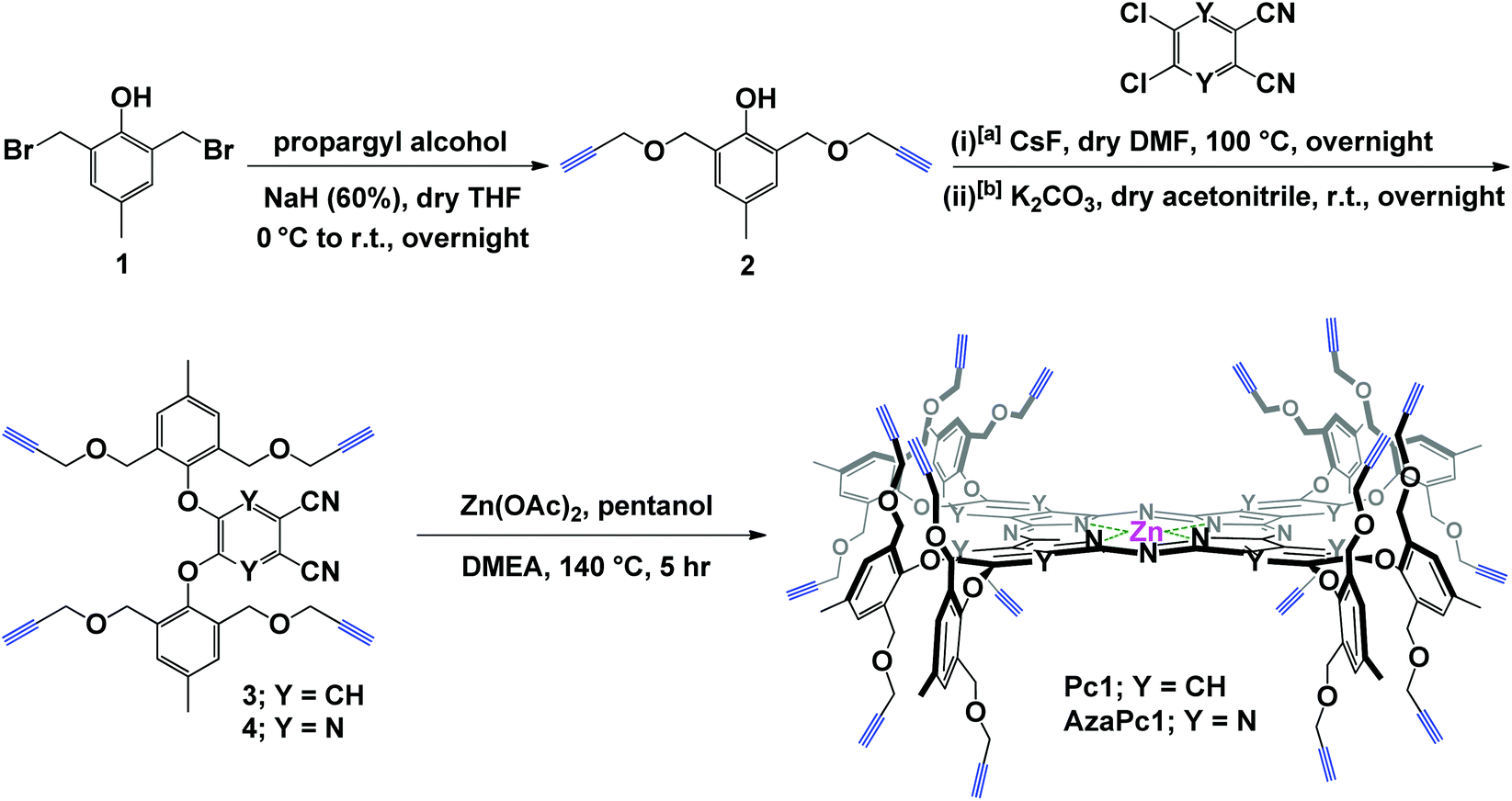 | ||
| Scheme 1 Synthesis of Pc1 and AzaPc1; reaction conditions for the synthesis of [a] Pc-precursor 3 and [b] AzaPc-precursor 4. | ||
The NMR spectra of the two platforms were well resolved, and the presence and sharpness of all the expected proton and carbon signals strongly indicate that both macrocycles are fully dissolved (Fig. 2). For example, in their 1H NMR spectra (600 MHz, DMSO-d6, 25 °C), the terminal alkyne protons (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif) , H4) were observed as triplets at 3.15 ppm (J = 2.7 Hz) for Pc1 and at 2.77 ppm (J = 2.1 Hz) for AzaPc1, while the methyleneoxy protons (–CH2OC2CCH, H3) appeared as doublets at 4.15 ppm (J = 2.4 Hz) and 4.13 ppm (J = 2.7 Hz) for Pc1 and AzaPc1, respectively. In their 13C NMR spectra (150 MHz, DMSO-d6, 25 °C), the acetylene carbons (–C
, H4) were observed as triplets at 3.15 ppm (J = 2.7 Hz) for Pc1 and at 2.77 ppm (J = 2.1 Hz) for AzaPc1, while the methyleneoxy protons (–CH2OC2CCH, H3) appeared as doublets at 4.15 ppm (J = 2.4 Hz) and 4.13 ppm (J = 2.7 Hz) for Pc1 and AzaPc1, respectively. In their 13C NMR spectra (150 MHz, DMSO-d6, 25 °C), the acetylene carbons (–C![[C with combining low line]](https://www.rsc.org/images/entities/b_i_char_0043_0332.gif) H, C4, and –CH, C5) were observed at ∼77.0 ppm and ∼80.0 ppm, respectively, whereas the methyleneoxy carbons (–CH2OH2CCH, C3) were at 57.2 ppm for Pc1 and at 52.9 ppm for AzaPc1. The MS (MALDI-TOF) of both macrocycles confirmed their chemical compositions as, m/z values; observed 2514.7826 [M]+, calculated 2514.8400 [M]+ for Pc1 and observed 2524.4539 [M]+, calculated 2523.9664 [M]+ for AzaPc1.
H, C4, and –CH, C5) were observed at ∼77.0 ppm and ∼80.0 ppm, respectively, whereas the methyleneoxy carbons (–CH2OH2CCH, C3) were at 57.2 ppm for Pc1 and at 52.9 ppm for AzaPc1. The MS (MALDI-TOF) of both macrocycles confirmed their chemical compositions as, m/z values; observed 2514.7826 [M]+, calculated 2514.8400 [M]+ for Pc1 and observed 2524.4539 [M]+, calculated 2523.9664 [M]+ for AzaPc1.
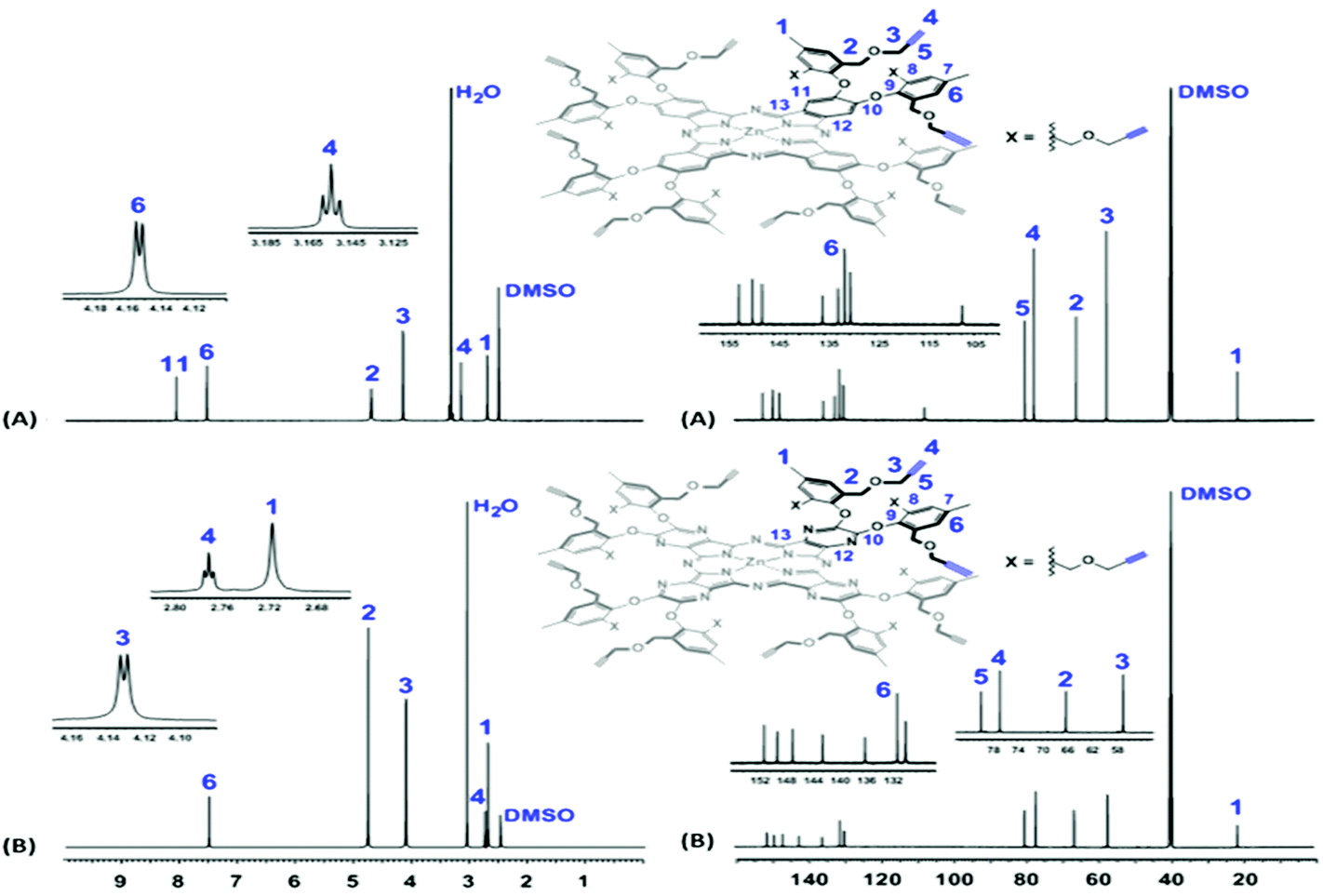 | ||
| Fig. 2 Full 1H and 13C NMR spectra of (A) Pc1 and (B) AzaPc1. | ||
The 1H NMR spectroscopic technique (600 MHz, DMSO-d6) was furthermore employed to evaluate the aggregation behavior of the studied complexes at different concentrations (ranging from 13.5 mM to 1.35 mM) (Fig. S17 & S18†) and different temperatures (25–95 °C) (Fig. S19†). With respect to different concentrations studied, well-resolved 1H NMR spectra, i.e., sharp proton signals in both aliphatic/aromatic regions, were obtained even at a high concentrated medium (13.5 mM) (Fig. S17 & S18†). In addition, the temperature-dependent 1H NMR spectra for both complexes gave similar results in terms of high-quality features with comparison to those obtained over different concentrations (Fig. S19†). From the above findings, it is evident that the resulting position of the multiple terminal alkyne ends, i.e., dual-directionality, acquired by the steric hindered bulky phenoxyl units, has a significant role in preventing the macrocyclic cores from approaching one another in solvated media.
Good quality crystals of Pc1/AzaPc1 were generated by a suitable solvent diffusion method, and the crystalline materials were analysed by X-ray diffraction (Fig. 3). The crystal structures of Pc1 (Fig. 3A and B) and AzaPc1 (Fig. 3C and D) confirmed the restricted rotation of the orthogonal phenoxyl substituents (almost 90°), and the resulting positions of the propargyl moieties clearly dismiss any possible intermolecular π–π stacking interactions between the macrocyclic planes. The asymmetric unit cell of the Pc1 crystal contains only half of the macrocycles due to the internal symmetry exhibited by the molecule, and the complete structure was obtained by symmetry expansion.
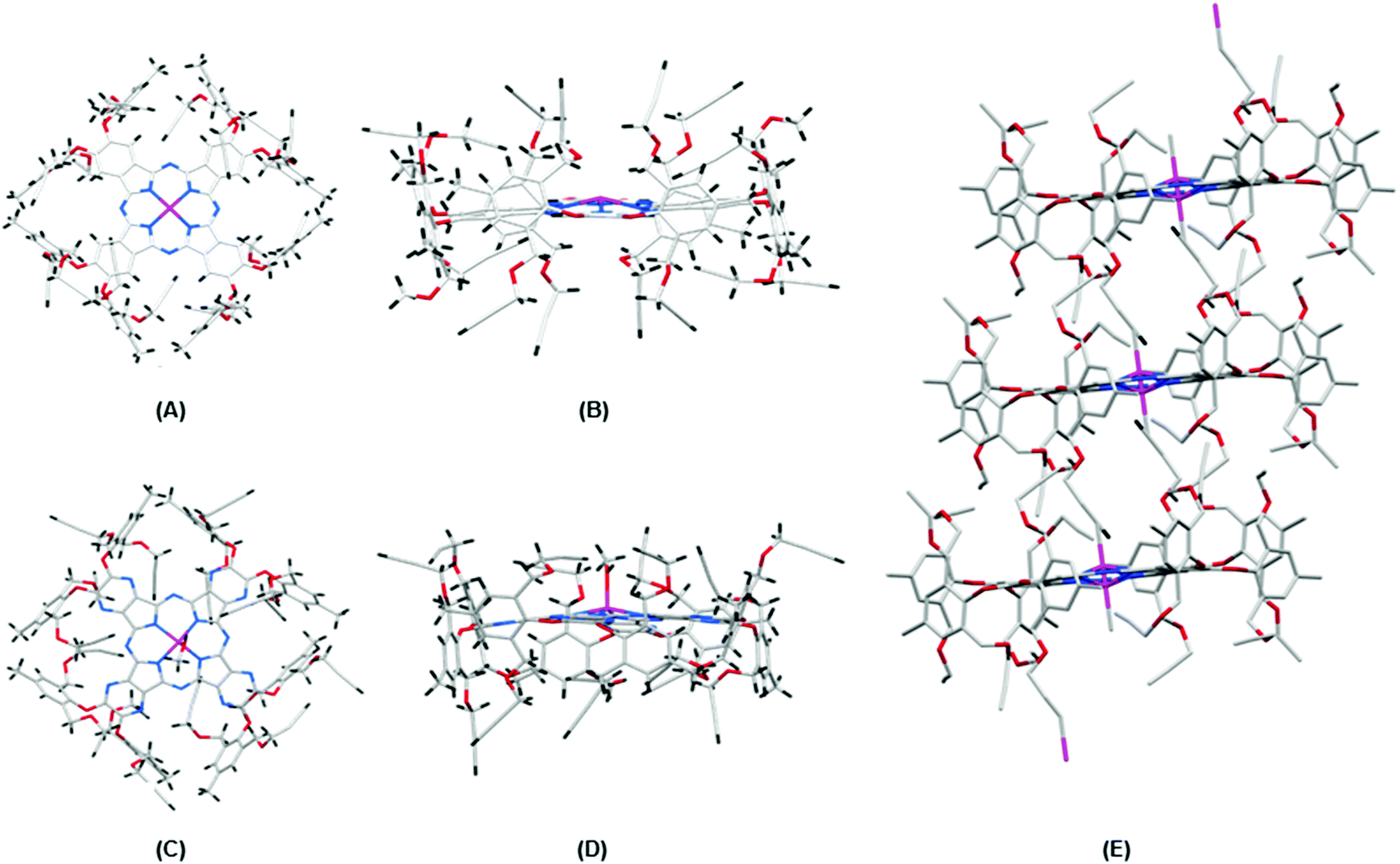 | ||
| Fig. 3 (A) Top view and (B) side view of the crystal structure of Pc1; (C) top view and (D) side view of AzaPc1; (E) Zn ← CC–H coordination observed from both sides of the Pc unit and the resulting columnar propagation of the macrocycles: colour code: blue, nitrogen; grey, carbon; red, oxygen; pink, zinc and black, hydrogen. | ||
The top and side views of the Pc1/AzaPc1 structures clearly show their exact solid-state structures. Both macrocycles have a domed geometry with Zn(II) ions at the top of the macrocyclic planes. However, due to positional disorder, the Zn(II) ion in the AzaPc1 crystal occupies both sides of the macrocyclic plane with half occupancies each. In the case of AzaPc1, the Zn(II) ion has an axial methanol ligand, whereas in that of Pc1, such axial solvent ligation is not observed. However, two terminal ethynyl groups are very close to the Zn(II) ion in Pc1, so appreciable Zn ← CC–H coordination could be possible in this crystal form (Fig. 3E). Due to positional disorder, this Zn ← CC–H coordination is observed from both sides of the macrocyclic unit and thus propagates along the crystal in a columnar manner (ESI†).
The enhanced solubility and diminished aggregation of both Pc1/AzaPc1 were further confirmed by solution-based electronic spectroscopic investigations. For example, the ground state electronic absorption spectra of Pc1/AzaPc1 were recorded in DMF. These results showed that both macrocycles are monomeric, as evidenced by their single (narrow) Q-band absorptions (ESI†).17 The Q-band for Pc1 was observed at 678 nm, while the 48 nm blue shift for the hetero-derivative, AzaPc1 (λQ = 630 nm), is caused by the presence of nitrogen atoms instead of the CH groups in the macrocyclic system. The B-bands of the novel macrocycles were observed at approximately 340 and 380 nm, respectively. In addition, the self-association of the studied complexes was evaluated in different organic media, namely, DCM, chloroform, DMF, DMSO, ethanol, methanol, THF and toluene, and no aggregation behaviour was observed in any of the solvent systems used (ESI†). The aggregation behaviours were also examined at different concentrations in DMF to establish an appropriate concentration for further photophysical and photochemical studies. The data were well fitted using the Beer–Lambert law at concentrations ranging from 1.0 × 10−5 to 1.0 × 10−6 M, and no aggregation was detected within this concentration range (ESI†). Furthermore, the fluorescence behaviours were explored in DMF, and similar fluorescence behaviours were observed, i.e., the excitation spectra were parallel to the absorption spectra, and both were mirror images of the fluorescence emission spectra of the corresponding complexes (Fig. 4A and B). The proximity of the wavelength of each component of the Q-band absorption to the Q-band maxima of the excitation spectra suggested that the nuclear configurations of the ground and excited states are similar and are not affected by excitation. The excitation maxima were 677 nm for Pc1 and 629 nm for AzaPc1, while the emission maxima were 685 nm and 642 nm for Pc1 and AzaPc1, respectively. The Stokes shifts were found between 6 nm and 10 nm, which are typical values for zinc(II)-containing phthalocyanine complexes.18 The photophysical and photochemical properties of these novel macrocyclic complexes were investigated to determine their photocatalytic applications, such as in PDT. The fluorescence quantum yields (Φf) were determined in DMF using the method reported in the literature,19 while the fluorescence lifetime (τF) values were obtained using a time-correlated single-photon counting (TCSPC) procedure (Table 1).20 Time-resolved fluorescence studies were carried out in DMF, and the fluorescence decays for both macrocycles were assessed using single-exponential curves (Fig. 4C and D). The singlet oxygen quantum yields (ΦΔ) were measured in DMF by a chemical method using 1,3-diphenylisobenzofuran (DPBF) as a singlet oxygen quencher. The disappearance of the DPBF absorbance at 414 nm was monitored using a UV-Vis spectrophotometer. The stability of the Q-band intensities suggested that neither of the studied complexes decomposed under the light irradiation used during the singlet oxygen studies. The complexes showed similar singlet oxygen generation properties and singlet oxygen quantum yield values (∼0.6). Consequently, Pc1/AzaPc1 are suggested to be suitable as photosensitizers (PSs) in PDT due to their high singlet oxygen production abilities. The photodegradation quantum yields (Φd values) were also studied in DMF, and the Φd values were on the order of 10−4 (Table S1†), making them similar to those of Pcs containing different metals and substituents (ESI†).21
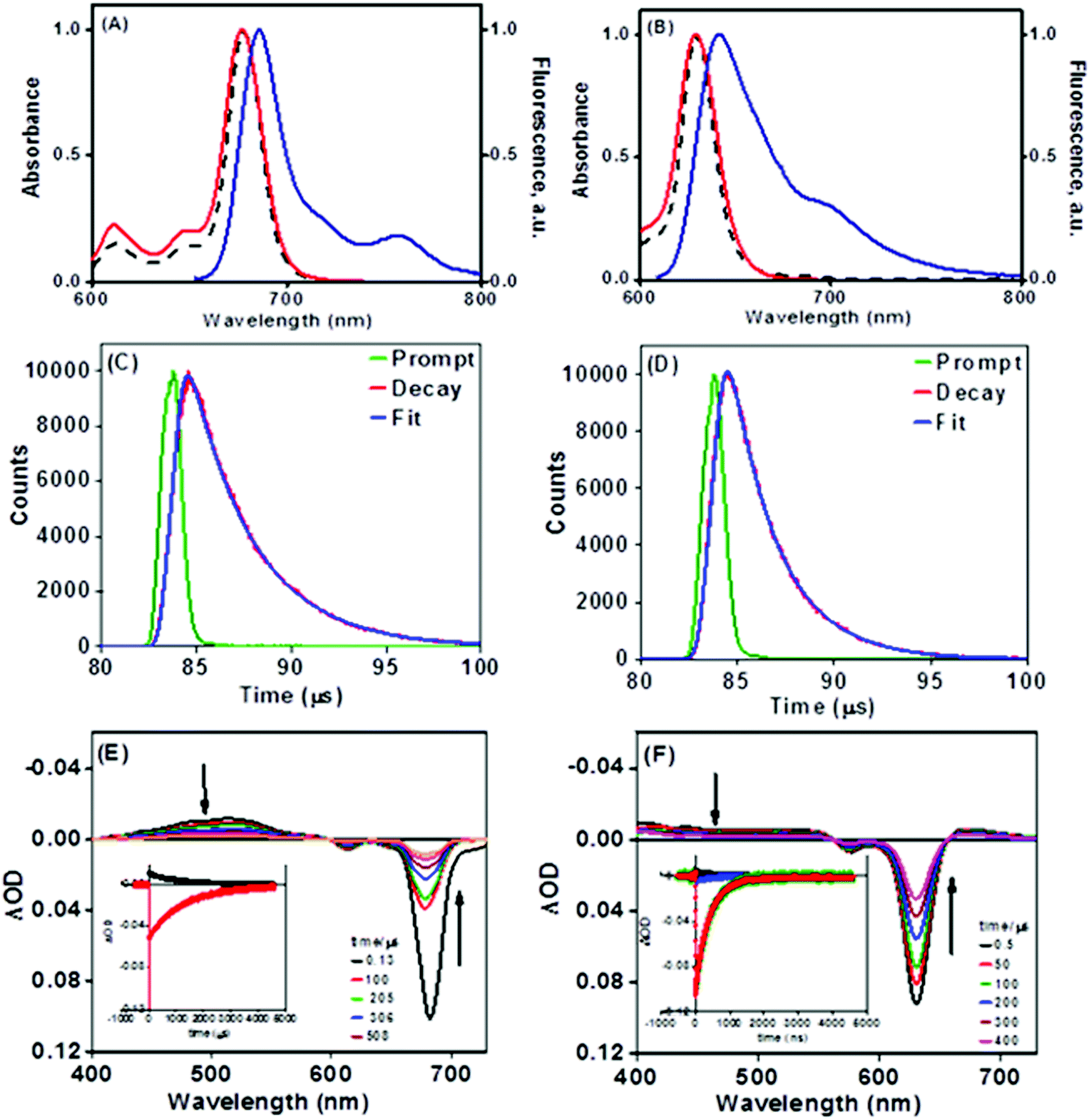 | ||
| Fig. 4 Absorption, excitation and emission spectra of (A) Pc1 and (B) AzaPc1 in DMF, time-correlated single-photon counting (TCSPC) fluorescence decay curves of (C) Pc1 and (D) AzaPc1 in DMF, transient absorption spectra in nitrogen-saturated DMF with excitation by an OPO laser at 686 nm for PC1 and 637 nm for AzaPc1. The inset shows the decay of the triplet state and the ground state recovery of (E) Pc1 and (F) AzaPc1 in nitrogen-saturated DMF. | ||
| Entry | Compd. | Φ F | τ F (ns) | Φ T | τ T (μs) | Φ Δ | Δ s |
|---|---|---|---|---|---|---|---|
| a Φ F = fluorescence quantum yield, τF = fluorescence lifetime, ΦT = triplet state quantum yield, τT = triplet lifetime, ΦΔ = singlet oxygen quantum yield and Δs = function triplet state deactivated by oxygen. b Std-ZnPc was used as a reference compound.22 | |||||||
| 1 | Pc1 | 0.16 | 3.30 | 0.62 | 1231 | 0.60 | 0.97 |
| 2 | AzaPc1 | 0.14 | 2.46 | 0.64 | 400 | 0.63 | 0.98 |
| 3 | Std-ZnPc | 0.17 | 1.03 | 0.56 | 330 | 0.56 | 1.00 |
The excited triplet states (T1) of the studied Pc1/AzaPc1 complexes were also evaluated using laser flash photolysis (LFP). Fig. 4E and F show the transient absorption spectra (TAS) in the μs scale of these macrocycles, and the spectra are typical of triplet–triplet (T1–Tn) absorptions for Pcs. The TAS in nitrogen-saturated solutions were characterized by a positive absorption maximum at 513 nm for Pc1 and at 410 nm for AzaPc1, and both bands were assigned to the T1–Tn triplet absorption. The observed blue shift in the positive absorption band exhibited by AzaPc1 relative to that of Pc1 is expected because the ground state absorption spectrum of AzaPc1 is blue-shifted by 48 nm from that of Pc1 (678 nm). The negative absorption peaks for both complexes in the Q-band region match their ground state absorptions and are separated from their positive peaks (the triplet state) by well-defined isosbestic points, indicating that only two states are involved in the excitation (T1 → S0). In addition, the similarity between the positive absorption decay and the ground state recovery behaviours is clearly shown in Fig. 4E and F along with the same increase and similar decay times. These curves can all be fitted by single-exponential functions, confirming the presence of only one transient species (τ1) in the examined solutions.
The triplet-state lifetime (τT), the quantum yield (ΦT) and the molar absorption coefficient (ΔεT–T) were calculated for both of the studied complexes (Table 1). The τT values were calculated to be 1231 μs for Pc1 and 400 μs for AzaPc1, which are longer than the lifetimes previously reported for standard ZnPc, and these values are long enough to allow the production of singlet oxygen. This very long triplet lifetime could be attributed to the obstruction of solvent collision by the multiple bulky phenoxyl units on the periphery. These large substituents in Pc1 result in higher structural rigidity and thus offer more protection, which makes τT of Pc1 larger. On the other hand, in air-saturated solutions, the measured τT values for both complexes decreased dramatically to 1.2 μs for Pc1 and to 1.9 μs for AzaPc1compared to 0.30 μs for ZnPc, and their TAS spectra did not change noticeably, i.e., the shape and spectral position remained constant, confirming the effective quenching of molecular oxygen by a physical process (T1 + O2 → S0 + 1O2), and consequently, the positive absorptions can confidently be attributed to T1–Tn triplet absorptions. The formation of the triplet species (ΦT) by each of the two complexes was evaluated, and they were found to be very close to their ΦΔ values, as shown in Table 1, suggesting an effective interaction between their triplet states and the molecular oxygen present in the system, resulting in a high quantum yield of singlet oxygen (% of ΦΔ/ΦT ≈ 96 for Pc1 and 98 for AzaPc1). However, the total yields of both florescence and the triplet state of the two macrocycles are comparable and are less than one, indicating the presence of internal conversion (ΦIC = 1 − ΦF − ΦT). LFP confirms the formation of T with a very high quantum yield and a long lifetime from the studied complexes, which make them an important class of compounds for many applications.
After successfully synthesizing and characterizing the novel alkynyl macrocyclic systems, their utility as efficient molecular platforms in the CuAAC reaction was investigated. Among all organic transformations, Cu(I)-mediated azide–alkyne couplings are one of the most attractive organic reactions, and this technique has received great attention from many researchers, as evidenced by the rapid increase in the number of publications on the topic since 2002.11 The simple setup/workup, the use of benign or easily removed solvents, fast reaction rate, high chemical/thermal stability of the resulting 1,4-disubstituted 1,2,3-triazole and high yield of the isolated product are considered to be major advantages to using this type of 1,3-dipolar cycloaddition reaction. Accordingly, the CuAAC on the studied complexes was carried out using the reaction of Pc1/AzaPc1 with benzyl azide in different organic solvents, namely, chloroform, THF, EtOAc, toluene, benzene, DMF and DMSO. All reactions were performed in the presence of CuI/DIPEA at 60 °C for 1 h, and the results are listed in Table 2. The CuAAC reactions proceeded in high yield (>90%) in refluxing chloroform with no sign of any unreacted starting material or partially benzylated side-products (Table 2, entry 1). On the other hand, the desired products were obtained in <70% when other organic solvents were used, and both products and the remaining unreacted starting materials were isolated by chromatography (Table 2, entries 2–7).
|
|
|||
|---|---|---|---|
| Entry | Product | Solvent | Yieldb (%) |
| a Reaction conditions: Pc1/AzaPc1 (0.01 mmol), benzyl azide (16.2 equiv.), CuI (10 mol%), DIPEA (6.0 equiv.), solvent (25 mL), 60 °C, 1 h. b Isolated yield after column chromatography using 5% MeOH/DCM. | |||
| 1 | Pc2/AzaPc2 | Chloroform | 93/92 |
| 2 | Pc2/AzaPc2 | THF | 65/66 |
| 3 | Pc2/AzaPc2 | EtOAc | 54/49 |
| 4 | Pc2/AzaPc2 | Toluene | 61/65 |
| 5 | Pc2/AzaPc2 | Benzene | 62/63 |
| 6 | Pc2/AzaPc2 | DMF | 59/54 |
| 7 | Pc2/AzaPc2 | DMSO | 43/49 |
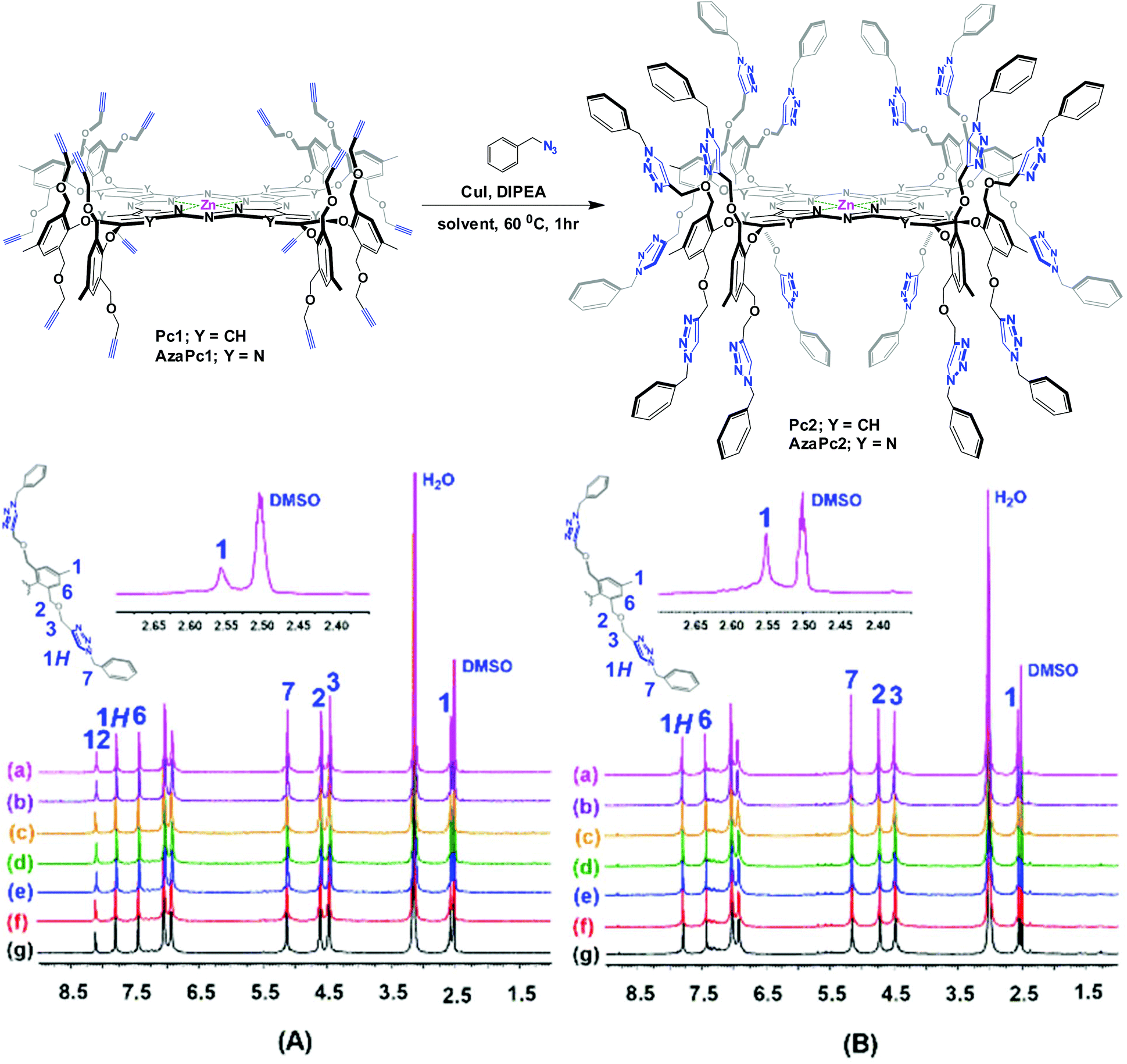
Despite these successes, some limitations associated with the formation of partially benzylated macrocyclic derivatives caused by the possible steric hindrance from the multiple propargyl arms were expected. However, these obstacles were not critical, and no partially reacted macrocycles were detected, i.e., only the desired products (in chloroform) and unreacted starting materials (when other organic solvents were used) were obtained. In fact, the 1H NMR spectra (Fig. A and B, Table 2) showed the disappearance of the terminal alkyne protons (H4) at 3.15 ppm for Pc1 and at 2.77 ppm for AzaPc1 (see Fig. 2), and new resonances at ∼8.00 ppm corresponding to the triazole protons (1H) were observed in the spectra of all isolated products, confirming that all the expected hexadeca-1,2,3-triazole linkages were fully formed. The isolation of Pc2/AzaPc2, the absence of any partially reacted macrocycles along with the fast reaction rate, suggested that all sixteen 1,2,3-triazoles were obtained in tandem in two major events (Fig. 5). The first event is based on the formation of a 1,2,3-triazole–copper complex resulting from the intermolecular CuAAC reaction between one benzyl azide substrate and one terminal alkyne unit present on the macrocyclic system (a rate-determining step). The flexibility and the position of the resulting Cu(I)-triazolide intermediate with respect to the adjacent alkyne units accelerate the subsequent azide–alkyne coupling reactions.
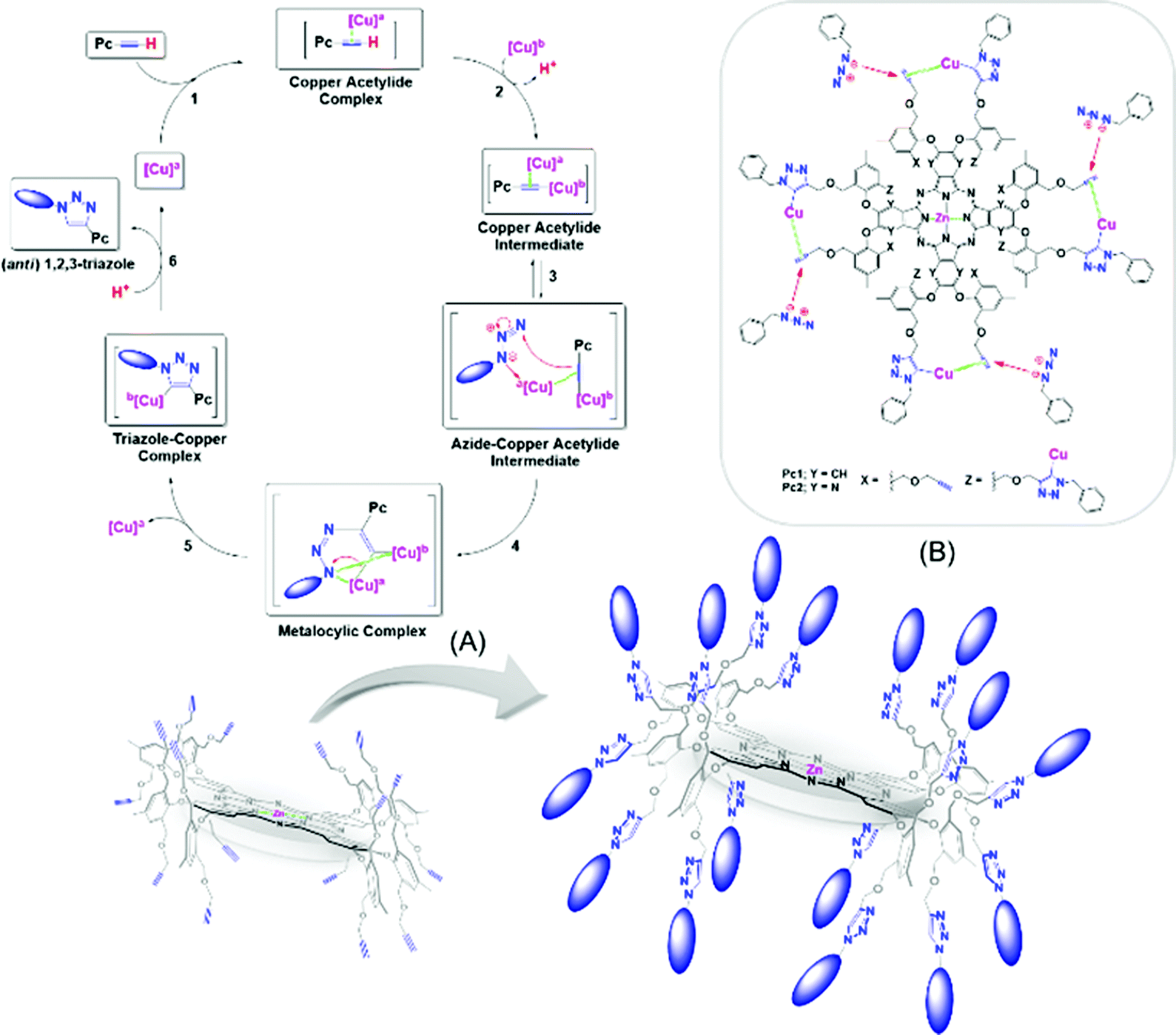 | ||
| Fig. 5 Proposed reaction mechanism; (A) CuAAC of Pc1 for the synthesis of Pc2 and (B) tandem azide–alkyne couplings mediated by the Cu(I)-triazolide intermediates based on Pc1/AzaPc1.23 | ||
After their structural identity confirmation by 1H NMR, ESI-MS and elemental analysis, the absorption spectra of the triazolide-containing complexes (Pc2 and AzaPc2) demonstrated sharp unperturbed single Q-bands, corresponding to the prevalent monomeric form. The position of their main absorption Q-bands was recorded to be almost similar to those exhibited by their platform precursors, confirming that their absorption characteristics were not influenced by the forming CuAAC reaction (ESI†). Subsequently, the essential photophysical parameters for Pc2 and AzaPc2 including fluorescence lifetimes and their quantum yields were determined primarily in DMF (Table S1†). As depicted in Fig. S33C and S33D,† the excitation and absorption spectra were perfectly overlapped along with the existence of the mirror symmetry between the excitation and emission spectra and small stock shifts as exhibited by both complexes. Such behavior can further confirm that such complexes are exclusively present in the monomeric form, and consequently, the calculated photophysical parameters were not influenced by the aggregation factor. The fluorescence lifetimes, characterized by mono-exponential functions, were not greatly affected by the CuAAC reaction and found to be approximately 2.46 nm for both complexes. Finally, their fluorescence quantum yields were calculated to be almost 0.1 and less than those of the building block precursors as shown in Tables S1 and S2.†
The unique characteristics of both novel macrocyclic systems and the remarkable CuAAC reaction combine to provide an efficient and effective synthetic procedure for attaching, orienting and directing a variety of valuable ligating substrates on the outer edges of the macrocycles for the synthesis of highly practical materials (Fig. 5).
Procedure and characterization
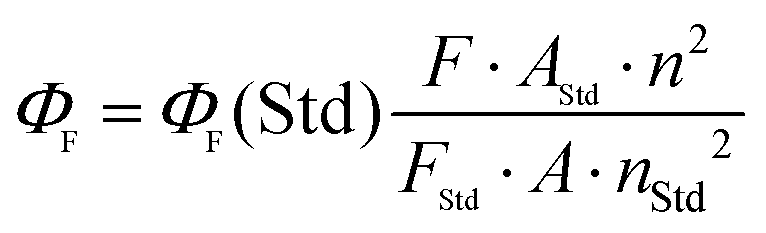 | (1) |
Time-resolved spectroscopy measurements were measured using the time-correlated single-photon-counting technique (TCSPC) integrated into the FLS-980 fluorescence spectrometer equipped with a Supercontinuum Fianium White Laser as the excitation source at 614 nm (frequency of 10 MHz). The instrument response function was measured using a diffusive reference sample (LUDOX® from Sigma-Aldrich).
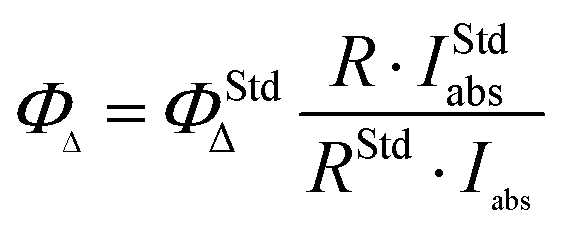 | (2) |
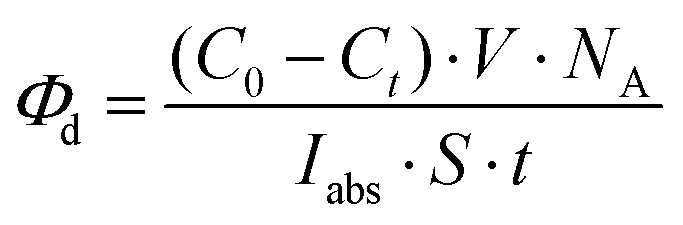 | (3) |
The triplet–triplet absorption coefficients (ΔεT) of the samples were obtained using the singlet depletion method and the following equation was used to calculate ΔεT.22d
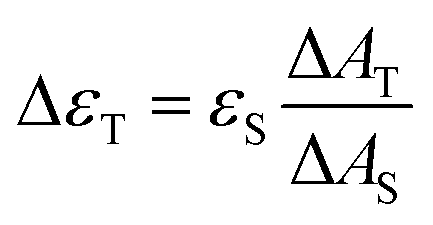 | (4) |
The triplet quantum yield ΔAT was obtained by comparing the ΔAT value of the optically matched sample solution at an excitation wavelength in a 1 cm cuvette to that of the reference using the following equation.22d
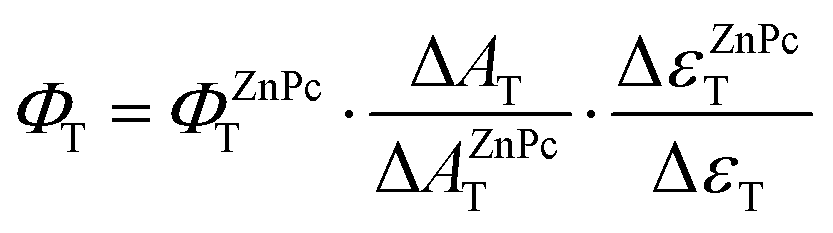 | (5) |
Experimental section
2,6-Bis(methylbromide)-p-cresol (1)29
33% HBr in glacial acetic acid (60 mL) was added to 2,6-bis(hydroxylmethyl)-p-cresol (10 g, 6.0 mmol) and the mixture was stirred at room temperature for 24 h. After completion, the reaction mixture was diluted in 100 mL cold water allowing the product to precipitate upon stirring for 30 min at room temperature. The resulting crude solid was filtered off using a Büchner funnel, which was dried and then a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 DCM/hexane solvent system to afford 2,6-bis(methyl bromide)-p-cresol 1 as white crystals in 9.0 g (51% yield); Rf = 0.29 (EtOAc/hexane 1:9); m.p. 123.6 °C; 1H NMR (600 MHz, CDCl3): δ = 7.09 (s, 2H), 5.44 (s, 1H), 4.55 (s, 4H), 2.27 (s, 3H); 13C NMR (150 MHz, CDCl3): δ = 151.20, 132.05, 130.86, 125.22, 29.76, 20.47; FT-IR, νmax/cm−1 = 3519, 3473, 3437, 1604, 1485, 1211, 966, 878, 545; HRMS (ESI): m/z calcd for C9H10Br2O: 291.9098 [M]+, found: 291.9093; elemental analysis calcd (%) for C9H10Br2O: C, 36.77; H, 3.43; Found: C, 36.70; H, 3.56.
:2 DCM/hexane solvent system to afford 2,6-bis(methyl bromide)-p-cresol 1 as white crystals in 9.0 g (51% yield); Rf = 0.29 (EtOAc/hexane 1:9); m.p. 123.6 °C; 1H NMR (600 MHz, CDCl3): δ = 7.09 (s, 2H), 5.44 (s, 1H), 4.55 (s, 4H), 2.27 (s, 3H); 13C NMR (150 MHz, CDCl3): δ = 151.20, 132.05, 130.86, 125.22, 29.76, 20.47; FT-IR, νmax/cm−1 = 3519, 3473, 3437, 1604, 1485, 1211, 966, 878, 545; HRMS (ESI): m/z calcd for C9H10Br2O: 291.9098 [M]+, found: 291.9093; elemental analysis calcd (%) for C9H10Br2O: C, 36.77; H, 3.43; Found: C, 36.70; H, 3.56.
2,6-Bis(methylpropargyl)-p-cresol (2)
NaH (60%) (4.1 g, 102 mmol) was added slowly to 100 mL ice cold dry THF. Propargyl alcohol (7.8 mL, 136 mmol) was then added dropwise to the solution, and the reaction mixture was stirred for 30 min at 0 °C. A solution of compound 1 (10 g, 34.0 mmol) in 50 mL dry THF was then added, and the reaction was allowed to stir at room temperature for 12 h. After completion, the reaction mixture was quenched using MeOH followed by concentrating the solvent mixture under reduced pressure. The crude mixture was washed with 100 mL of water and extracted using EtOAc (2 × 50 mL) and the combined organic phase was collected and dried over anhydrous Na2SO4. The resulting crude product was subjected to column chromatography using a 1:9 EtOAc/hexane solvent system as an eluent to obtain 2,6-bis(methylpropargyl)-p-cresol 2 as a white solid in 4.6 g (55% yield); Rf = 0.23 (EtOAc/hexane 1:9); 1H NMR (600 MHz, DMSO-d6): δ = 8.34 (s, 1H), 6.97 (s, 2H), 4.50 (s, 4H), 4.17 (d, J = 2.3 Hz, 4H), 3.46 (t, J = 2.6 Hz, 2H), 2.20 (s, 3H);13C NMR (150 MHz, DMSO-d6): δ = 150.13, 129.03, 127.72, 124.69, 80.35, 77.18, 66.48, 56.93, 20.14; FT-IR, νmax/cm−1 = 3414, 3289, 2914, 2858, 2740, 2117, 1761, 1484, 1073, 641; HRMS (ESI): m/z calcd for C15H16O3Na: 267.0997 [M + Na]+, found: 267.0985; elemental analysis calcd (%) for C15H16O3: C, 73.75; H, 6.60, Found: C, 73.52; H, 6.90.
4,5-Bis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy)phthalonitrile (3)
Compound 2 (3.0 g, 12.2 mmol) was dissolved in 40 mL anhydrous DMF. Anhydrous CsF (3.1 g, 20.2 mmol) was then added to the solution and the mixture was allowed to stir for 10 min at room temperature. A solution of 4,5-dichlorophthalonitrile (1.0 g, 5.1 mmol) in 10 mL anhydrous DMF was then added and the reaction mixture was heated to 100 °C for 48 hours. After completion, the formed salt was filtered off using a Büchner funnel and the crude mixture was diluted in 100 mL water and extracted with DCM (2 × 50 mL). DCM was then concentrated under reduced pressure and the crude material was purified by column chromatography using a 2:8 EtOAc/hexane solvent system as an eluent to obtain the desired product 3 as a white solid in 2.0 g (64% yield); Rf = 0.42 (EtOAc/hexane 3:7); m.p. 117.7 °C; 1H NMR (600 MHz, DMSO-d6) = 7.35 (s, 4H), 6.99 (s, 2H), 4.44 (s, 8H), 4.08 (d, J = 2.6 Hz, 8H), 3.45 (t, J = 2.6 Hz, 4H), 2.39 (s, 6H); 13C NMR (150 MHz, DMSO-d6): δ = 150.44, 146.15, 136.02, 130.69, 130.21, 119.16, 115.58, 108.82, 79.70, 77.20, 65.65, 57.07, 20.55; FT-IR, νmax/cm−1 = 3289, 3265, 2871, 2230, 2125, 1589, 1503, 1345, 878, 533; HRMS (ESI): m/z calcd for C38H32N2O6Na: 635.2158 [M + Na]+, found: 635.2292; elemental analysis calcd (%) for C38H32N2O6: C, 74.49; H, 5.26; N, 4.57; Found: C, 74.66; H, 6.01; N, 4.26.
5,6-Bis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy)pyrazine-2,3-dicarbonitrile (4)
Compound 2 (3.0 g, 12.1 mmol) was dissolved in 40 mL anhydrous acetonitrile. Anhydrous K2CO3 (2.8 g, 20.0 mmol) was then added to the solution and the mixture was allowed to stir for 10 min at room temperature. A solution of 2,3-dichloro-5,6-dicyano-1,4-pyrazine (1.0 g, 5.1 mmol) in 10 mL anhydrous acetonitrile was then added dropwise over a 5 min period of time and the reaction mixture was stirred overnight at room temperature. After completion, the formed salt was filtered off using a Büchner funnel and acetonitrile was removed under reduced pressure. The crude product was purified by column chromatography using a 1:9 EtOAc/hexane solvent system as an eluent to obtain the desired product 4 as a white solid in 3.0 g (97% yield); Rf = 0.39 (EtOAc/hexane 3:7); m.p. 168.9 °C; 1H NMR (600 MHz, DMSO-d6): δ = 7.32 (s, 4H), 4.49 (s, 8H), 4.05 (d, J = 2.3 Hz, 8H), 3.42 (t, J = 2.6 Hz, 4H), 2.39 (s, 6H); 13C NMR (150 MHz, DMSO-d6): δ = 151.09, 145.42, 136.54, 130.22, 129.77, 123.71, 113.47, 79.77, 77.23, 66.20, 56.79, 20.51; FT-IR, νmax/cm−1 = 3281, 2919, 2877, 2239, 2128, 1439, 1224, 675; HRMS (ESI): m/z calcd for C36H30N4O6Na: 637.2063 [M + Na]+, found: 637.2037; elemental analysis calcd (%) for C36H30N4O6: C, 70.35; H, 4.92; N, 9.12; Found: C, 69.61; H, 5.01; N, 8.99.
2,3,9,10,16,17,23,24-Octakis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy) phthalocyaninato zinc(II) (Pc1)
A mixture of compound 3 (2.5 g, 4.0 mmol) and Zn(OAc)2 (187.4 mg, 1.0 mmol) was dissolved in 3 mL pentanol. A catalytic amount of N,N-dimethylethanolamine was then added to the solution and the reaction mixture was heated to 140 °C for 5 hours. The reaction mixture was then precipitated from methanol and the solid material was filtered off using a Büchner funnel. The crude product was subjected to column chromatography using a 1:19 EtOAc/DCM solvent system as an eluent to afford the desired product Pc1 as a dark green solid in 1.1 g (43% yield); Rf = 0.66 (MeOH/DCM 1:9); m.p. 284.3 °C; 1H NMR (600 MHz, DMSO-d6): δ = 8.06 (s, 8H), 7.53 (s, 16H), 4.69 (s, 32H), 4.15 (d, J = 2.4 Hz, 32H), 3.15 (t, J = 2.7 Hz, 16H), 2.69 (s, 24H); 13C NMR (150 MHz, DMSO-d6): δ = 152.09, 149.32, 147.42, 135.37, 132.23, 130.92, 129.76, 107.43, 79.73, 77.28, 65.60, 57.25, 21.10; FT-IR, νmax/cm−1 = 3437, 3286, 2913, 2855, 2115, 1610, 1451, 1397, 1199, 1088, 634; MALDI-TOF: m/z calcd for C152H128N8O24Zn: 2514.8400 [M]+, found: 2514.7826; elemental analysis calcd (%) for C152H128N8O24Zn: C, 72.56; H, 5.13; N, 4.45; Found: C, 71.90; H, 5.21; N, 4.17.
2,3,9,10,16,17,23,24-Octakis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy) tetrapyrazinoporphyrazinato zinc(II) (AzaPc1)
A mixture of compound 4 (2.5 g, 4.1 mmol) and Zn(OAc)2 (186.3 mg, 1.0 mmol) was dissolved in 3 mL pentanol. A catalytic amount of N,N-dimethylethanolamine was then added to the solution and the reaction mixture was heated to 140 °C for 1 hour. The reaction mixture was then precipitated from methanol and the solid material was filtered off using a Büchner funnel. The crude product was subjected to column chromatography using a 1:19 EtOAc/DCM solvent system as an eluent to afford the desired product AzaPc1 as a dark green solid in 710 mg (28% yield); Rf = 0.54 (MeOH/DCM 1:9); m.p. 282.0 °C; 1H NMR (600 MHz, DMSO-d6): δ = 7.52 (s, 16H), 4.78 (s, 32H), 4.13 (d, J = 2.7 Hz, 32H), 2.77 (t, J = 2.1 Hz, 16H), 2.72 (s, 24H); 13C NMR (150 MHz, DMSO-d6): δ = 150.94, 148.95, 146.63, 142.10, 135.72, 130.85, 129.58, 79.87, 76.81, 66.19, 56.93, 21.14; FT-IR, νmax/cm−1 = 3286, 2919, 2858, 2115, 1625, 1542, 1396, 1248, 926, 661; MALDI-TOF: m/z calcd for C144H120N16O24Zn: 2523.9664 [M]+, found: 2524.4539; elemental analysis calcd (%) for C144H120N16O24Zn: C, 68.52; H, 4.79; N, 8.88; Found: C, 66.84; H, 4.73; N, 8.53.
2,3,9,10,16,17,23,24-Octakis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy) hexadeca-benzyl phthalocyanine (Pc2) via the CuAAC reaction
Pc1 (0.01 mmol) and benzyl azide (16.2 equiv.) were dissolved in 25 mL chloroform. CuI (10 mol%) and DIPEA (6.0 equiv.) were then added to the solution and the reaction mixture was refluxed for 1 hour. After completion, the reaction mixture was washed with ammonium hydroxide solution and the organic layer was collected and dried over anhydrous Na2SO4. Chloroform was removed over reduced pressure and the product was purified by column chromatography using a 19:1 MeOH/DCM solvent system as an eluent to afford the desired product Pc2 as a green solid in 43 mg (93% yield); Rf = 0.57 (MeOH/DCM 1:9); m.p. 300 > °C; 1H NMR (600 MHz, DMSO-d6): δ = 8.11 (s, 8H), 7.73 (s, 16H), 7.43 (s, 16H), 7.01–7.03 (m, 48H), 6.92–6.94 (m, 32H), 5.11 (s, 32H), 4.61 (s, 32H), 4.47 (s, 32H), 2.56 (s, 24H); 13C NMR (150 MHz, DMSO-d6): δ = 152.16, 149.56, 147.28, 144.01, 135.41, 135.12, 130.99, 128.32, 127.43, 123.69, 66.16, 63.00, 52.42, 20.86; FT-IR, νmax/cm−1 = 3134, 3063, 3031, 2918, 2859, 1605, 1451, 1396, 1198, 1088, 885, 720; deconvoluted HRMS (ESI): m/z calcd for C264H240N56O24Zn: 4641.8572 [M]+, found: 4642.0000; elemental analysis calcd (%) for C264H240N56O24Zn: C, 68.24; H, 5.21; N, 16.88; Found: C, 67.89; H, 4.81; N, 16.47.
2,3,9,10,16,17,23,24-Octakis(4-methyl-2,6-bis((prop-2-yn-1-yloxy)methyl)phenoxy) hexadeca-benzyl azaphthalocyanine (AzaPc2) via the CuAAC reaction
AzaPc1 (0.01 mmol) and benzyl azide (16.2 equiv.) were dissolved in 25 mL chloroform. CuI (10 mol%) and DIPEA (6.0 equiv.) were then added to the solution and the reaction mixture was refluxed for 1 hour. After completion, the reaction mixture was washed with ammonium hydroxide solution and the organic layer was collected and dried over anhydrous Na2SO4. Chloroform was removed over reduced pressure and the product was purified by column chromatography using a 19:1 MeOH/CHCl3 solvent system as an eluent to afford the desired product AzaPc2 as a green solid in 43 mg (92% yield); Rf = 0.43 (MeOH/DCM 1:9); m.p. 300 > °C; 1H NMR (600 MHz, DMSO-d6): δ = 7.79 (s, 16H), 7.43 (s, 16H), 7.01–7.03 (m, 48H), 6.91–6.94 (m, 32H), 5.15 (s, 32H), 4.72 (s, 32H), 4.48 (s, 32H), 2.55 (s, 24H); 13C NMR (150 MHz, DMSO-d6): δ = 150.78, 148.88, 146.08, 141.78, 135.33, 128.50, 128.18, 127.26, 123.95, 66.65, 62.73, 52.31, 20.73; FT-IR, νmax/cm−1 = 3133, 3064, 3030, 2920, 2859, 1680, 1456, 1398, 923, 721; deconvoluted HRMS (ESI): m/z calcd for C256H232N64O24Zn: 4649.8192 [M]+, found: 4650.0000. Elemental analysis calcd (%) for C256H232N64O24Zn + CHCl3: C, 64.66; H, 4.92; N, 18.78; Found: C, 64.50; H, 4.92; N, 18.59.
Conclusion
In conclusion, the rational design and synthesis of two novel Zn(II)-containing macrocycles, Pc1 and AzaPc1, bearing multivalent, dual-directional propargyl functionalities are described. The presence and orientation of the terminal ethynyl group on macrocyclic backbones is of great interest because the resulting structures do not aggregate in solution or in their crystalline forms as confirmed by various spectral techniques (1H NMR, UV-Vis and single-crystal X-ray diffraction). Their photocatalytic activities towards photodynamic therapy (PDT) for cancer treatment, along with their utility as activated molecular platforms via CuAAC, have been investigated. Their CuAAC reactions with benzyl azide were successfully carried out within a short period (1 h), resulting in triazolide-containing macrocycles, Pc2/AzaPc2, in excellent yields (>90%). The fast reaction rate, the isolation of fully reacted macrocycles and the absence of any partially 1,2,3-triazolide-substituted by-products suggested that the CuAAC reactions were carried out in tandem and that the alkyne activation favours subsequent modifications, making this system suitable for the synthesis of numerous applicable macrocyclic derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Kuwait Foundation for the Advancement of Science (Grant Number P115-14-SC05) and the RSP unit general facilities of the Faculty of Science GFS (GS 01/01, GS 03/01, GS 01/03, GS 01/05, and GS 02/13). We thank the Chemistry Department at Kuwait University for the use of their MALDI-TOF mass spectrometer.References
- (a) H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed; (b) B. Ghazal, M. Machacek, M. A. Shalaby, V. Novakova, P. Zimcik and S. Makhseed, J. Med. Chem., 2017, 60, 6060–6076 CrossRef CAS PubMed.
- (a) C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed; (b) B. M. Luby, C. D. Walsh and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 2558–2569 CrossRef CAS PubMed; (c) X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS PubMed; (d) C. Duan, G. Zango, M. G. Iglesias, F. J. M. Colberts, M. M. Wienk, M. V. Martínez-Díaz, R. A. J. Janssen and T. Torres, Angew. Chem., Int. Ed., 2017, 56, 148–152 CrossRef CAS PubMed; (e) E. Fazio, J. Jaramillo-García, M. Medel, M. Urbani, M. Grätzel, M. K. Nazeerudin, G. de la Torre and T. Torres, ChemistryOpen, 2017, 6, 121–127 CrossRef CAS PubMed; (f) N. B. McKeown, Phthalocyanine materials: synthesis, structure, and function, Cambridge University Press, Cambridge, U.K., New York, 1998 Search PubMed; (g) N. B. McKeown, S. Makhseed, K. J. Msayib, L.-L. Ooi, M. Helliwell and J. E. Warren, Angew. Chem., Int. Ed., 2005, 44, 7485–7485 CrossRef; (h) M. Kimura, K. Wada, K. Ohta, K. Hanabusa, H. Shirai and N. Kobayashi, Macromolecules, 2001, 34, 4706–4711 CrossRef CAS.
- (a) X. Chen, C.-W. Lu, Y. Huang and D. V. McGrath, Tetrahedron, 2015, 71, 9154–9160 CrossRef CAS; (b) M. Li, E. Khoshdel and D. M. Haddleton, Polym. Chem., 2013, 4, 4405–4411 RSC; (c) M. Mayukh, C.-W. Lu, E. Hernandez and D. V. McGrath, Chem. – Eur. J., 2011, 17, 8472–8478 CrossRef CAS PubMed; (d) Y. A. Sütcüler, A. M. Sevim, T. Ç. Çanak, İ. E. Serhatlı and A. Gül, Dyes Pigm., 2017, 144, 58–68 CrossRef; (e) H. Dinçer, H. Mert, E. Çalışkan, G. Y. Atmaca and A. Erdoğmuş, J. Mol. Struct., 2015, 1102, 190–196 CrossRef; (f) B. N. Şen, H. Mert, H. Dinçer and A. Koca, Dyes Pigm., 2014, 100, 1–10 CrossRef; (g) J. Aimi, M. Komura, T. Iyoda, A. Saeki, S. Seki, M. Takeuchi and T. Nakanishi, J. Mater. Chem. C, 2015, 3, 2484–2490 RSC; (h) H. Dinçer, H. Mert, B. N. Şen, A. Dağ and S. Bayraktar, Dyes Pigm., 2013, 98, 246–254 CrossRef; (i) Y. Bandera, M. K. Burdette, J. A. Shetzline, R. Jenkins, S. E. Creager and S. H. Foulger, Dyes Pigm., 2016, 125, 72–79 CrossRef CAS.
- C. Zhenjun, F. Ying, Z. Qinghua and C. Dajun, Chem. Lett., 2009, 38, 1144–1145 CrossRef.
- (a) S. Y. S. Chow, R. C. H. Wong, S. Zhao, P.-C. Lo and D. K. P. Ng, Chem. – Eur. J., 2018, 24, 5779–5789 CrossRef CAS PubMed; (b) S. Y. S. Chow, P.-C. Lo and D. K. P. Ng, Dalton Trans., 2016, 45, 13021–13024 RSC.
- (a) D. Akyüz and A. Koca, J. Electrochem. Soc., 2018, 165, B508–B514 CrossRef; (b) G. Tillet, P. De Leonardis, A. Alaaeddine, M. Umeda, S. Mori, N. Shibata, S. M. Aly, D. Fortin, P. D. Harvey and B. Ameduri, Macromol. Chem. Phys., 2012, 213, 1559–1568 CrossRef CAS.
- (a) O. M. Bankole and T. Nyokong, J. Mol. Struct., 2017, 1136, 309–320 CrossRef CAS; (b) A. Fashina, E. Amuhaya and T. Nyokong, Spectrochim. Acta, Part A, 2015, 140, 256–264 CrossRef CAS PubMed; (c) A. Wichmann, G. Schnurpfeil, J. Backenköhler, L. Kolke, V. A. Azov, D. Wöhrle, M. Bäumer and A. Wittstock, Tetrahedron, 2014, 70, 6127–6133 CrossRef CAS.
- (a) D. Akyüz, B. Keskin, U. Şahintürk and A. Koca, Appl. Catal., B, 2016, 188, 217–226 CrossRef; (b) C. S. J. N. O'Donoghue, G. Fomo and T. Nyokong, Electroanalysis, 2016, 28, 3019–3027 CrossRef; (c) C. S. J. N. O'Donoghue, M. Shumba and T. Nyokong, Electroanalysis, 2017, 29, 1731–1740 CrossRef; (d) S. R. Nxele, P. Mashazi and T. Nyokong, Electroanalysis, 2015, 27, 2468–2478 CrossRef CAS.
- D. Mwanza, S. Mvango, S. Khene, T. Nyokong and P. Mashazi, Electrochim. Acta, 2017, 254, 89–100 CrossRef CAS.
- S. V. Kirner, C. Henkel, D. M. Guldi, J. D. Megiatto Jr. and D. I. Schuster, Chem. Sci., 2015, 6, 7293–7304 RSC.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- (a) W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763–2772 CrossRef CAS; (b) P. J. Low and M. I. Bruce, in Advances in Organometallic Chemistry, ed. R. West and A. F. Hill, Academic Press, 2001, vol. 48, pp. 71–288 Search PubMed.
- L. Su, J. Dong, L. Liu, M. Sun, R. Qiu, Y. Zhou and S.-F. Yin, J. Am. Chem. Soc., 2016, 138, 12348–12351 CrossRef CAS PubMed.
- K. S. Sindhu, A. P. Thankachan, P. S. Sajitha and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 6891–6905 RSC.
- (a) R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed; (b) J. Blanco-Urgoiti, L. Añorbe, L. Pérez-Serrano, G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2004, 33, 32–42 RSC; (c) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- J. Mack and M. J. Stillman, J. Am. Chem. Soc., 1994, 116, 1292–1304 CrossRef CAS.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS.
- M. Göksel, M. Durmuş and D. Atilla, J. Porphyrins Phthalocyanines, 2012, 16, 895–906 CrossRef.
- (a) B. Ghazal, A. Husain, A. Ganesan, M. Durmuş, X.-F. Zhang and S. Makhseed, Dyes Pigm., 2019, 164, 296–304 CrossRef CAS; (b) B. Ghazal, E. N. Kaya, A. Husain, A. Ganesan, M. Durmuş and S. Makhseed, J. Porphyrins Phthalocyanines, 2019, 23, 46–55 CrossRef CAS.
- (a) F. P. Crisóstomo, T. Martín and R. Carrillo, Angew. Chem., Int. Ed., 2014, 53, 2181–2185 CrossRef PubMed; (b) M.-j. Bu, G.-p. Lu and C. Cai, Synlett, 2015, 26, 1841–1846 CrossRef CAS.
- (a) Y. Zorlu, F. Dumoulin, M. Durmuş and V. Ahsen, Tetrahedron, 2010, 66, 3248–3258 CrossRef CAS; (b) M. Durmuş, V. Ahsen and T. Nyokong, J. Photochem. Photobiol., A, 2007, 186, 323–329 CrossRef; (c) A. Ogunsipe, J.-Y. Chen and T. Nyokong, New J. Chem., 2004, 28, 822–827 RSC; (d) I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- W. L. F. Armarego and D. D. Perrin, Purification of laboratory chemicals, 1997, http://books.google.com/books?id=e0xqAAAAMAAJ Search PubMed.
- (a) S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260 CrossRef CAS; (b) M. D. Maree, T. Nyokong, K. Suhling and D. Phillips, J. Porphyrins Phthalocyanines, 2002, 06, 373–376 CrossRef CAS.
- M. Durmuş, Photochemical and Photophysical Characterization, Springer Dordrecht Heidelberg, London New York, 2012 Search PubMed.
- W. Spiller, H. Kliesch, D. Wöhrle, S. Hackbarth, B. Röder and G. Schnurpfeil, J. Porphyrins Phthalocyanines, 1998, 2, 145–158 CrossRef CAS.
- S. M. Bishop, A. Beeby, A. W. Parker, M. S. C. Foley and D. Phillips, J. Photochem. Photobiol., A, 1995, 90, 39–44 CrossRef CAS.
- B. M. Trost, V. S. C. Yeh, H. Ito and N. Bremeyer, Org. Lett., 2002, 4, 2621–2623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1915920–1915923 and 1941881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00695h |
| This journal is © the Partner Organisations 2019 |