
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen solvent assisted synthesis of cesium bismuth halide perovskite nanocrystals and the influences of slow and fast anion exchange rates†
Rana Faryad
Ali
 ,
Irene
Andreu
and
Byron D.
Gates
*
,
Irene
Andreu
and
Byron D.
Gates
*
Department of Chemistry and 4D LABS, Simon Fraser University, 8888 University Drive Burnaby, BC V5A 1S6, Canada. E-mail: bgates@sfu.ca
First published on 8th October 2019
Abstract
Replacing lead in halide perovskites to address the concerns of their toxicity and stability has driven a recent surge in research toward alternative lead-free perovskite materials. Lead-free all inorganic cesium bismuth halide (Cs3Bi2X9) perovskite nanocrystals have attracted attention in recent years due to the air-stability and non-toxic nature of bismuth. Herein, we demonstrate a facile sonication-assisted approach for the preparation of all-inorganic cesium bismuth iodide (Cs3Bi2I9) perovskite nanocrystals (NCs) using propylene carbonate as a green, alternative solvent. The photoluminescence (PL) spectra of the Cs3Bi2X9 NCs have a peak emission that can be tuned from 410 to 550 nm by controlling the composition of the NCs through an anion exchange reaction using tetraalkylammonium halides as a source of halide ions. The rate of this anion exchange reaction is demonstrated to have a significant influence on the dimensions of the NCs obtained from the parent Cs3Bi2I9 NCs. The PL emission of these nanocrystals is predominately due to exciton recombination processes. The NCs also exhibit air-stability for at least 150 days.
Introduction
Metal halide perovskite nanomaterials with a general formula ABX3 [A = CH3NH3 (MA), Cs; B = Pb, Bi, Sn, Ge, etc.; and X = Cl, Br, I] have drawn extensive attention due to their unique optical and electronic properties.1–3 This progress has been spurred by their excellent optical properties (i.e., high photoluminescence quantum efficiency and sharp absorption edge), and the prospect for low-cost synthetic processes.4,5 Additionally, metal halide perovskites offer considerable tunability of their optical bandgaps through either the direct incorporation or exchange of different halides into the crystal structure, or through reducing the crystallite dimensions to the nanometer scale.6–9 This change in the bandgaps of the halide perovskites relies on the different ionization potentials of the halide ions, which enable tuning of the halide perovskite emission throughout the entire visible spectrum.10 Among these materials, lead halide perovskite based nanomaterials have been extensively explored for photonic applications, such as solar cells, light emitting diodes, and photodetectors.11,12 The intrinsic toxicity of lead-based materials limits their utility due to their hazards to human health and the environment.13,14 The substitution of lead with relatively non-toxic metals in the perovskite crystal structure is highly desirable for the commercialization and widespread utilization of these materials.13,14In recent years, lead-free perovskite systems have been a primary focus of research into perovskite based materials and sought after for their use in optoelectronic applications.15–17 Several less toxic metals such as Sn, Ge, Bi, and Sb have been explored to prepare lead-free perovskite nanomaterials.18–20 Among lead-free perovskites, Sn and Ge containing perovskites are unstable due to the oxidative transformation of Sn2+ and Ge2+ to Sn4+ and Ge4+, respectively.21 For decades, Bi has been used as a nontoxic replacement for Pb in quantum dots and piezoelectric materials.21 Bismuth is also a promising candidate for use in perovskite based materials due to the isoelectronic structure of Bi3+ with Pb2+ and its improved chemical stability over that of Sn2+and Ge2+.20,22–26 Furthermore, Bi3+ and Pb2+ are adjacent in the periodic table and have very similar ionic radii, which may ease the incorporation of Bi3+ into the perovskite lattice.13 The interest in bismuth-based perovskites quickly led to the synthesis of nanocrystals (NCs), first in the form of organic/inorganic hybrid perovskites, and later also as purely inorganic cesium-based perovskites.27,28 Among these bismuth halide based perovskites, the all inorganic based cesium bismuth halide perovskite nanomaterials have exhibited a better chemical stability and have a lower density of defects than the organic based methylammonium (MA) perovskite nanomaterials.20,22
To the best of our knowledge, there are relatively few reports to prepare all-inorganic Cs3Bi2X9 nanocrystals through solution-phase methods.20,22–24 Many of these methods can provide good control over the size, shape, and purity of Cs3Bi2X9 nanomaterials. These methods used to prepare nanomaterials of Cs3Bi2X9 include hot-injection approaches and ligand-assisted re-precipitation (LARP) methods. The hot-injection approach uses an inert atmosphere for processing the reagents and temperatures above 100 °C to initiate the decomposition of the precursors to form the perovskite nanocrystals.24 The ligand-assisted re-precipitation process is a low cost and facile approach to synthesize perovskite NCs.20,29 In LARP, the precursors are dissolved in a ‘‘good’’ polar solvent (e.g., dimethyl sulfoxide or DMSO; dimethylformamide or DMF) to form a precursor solution. This dissolution step is followed by the injection of the precursor solution into a ‘‘poor’’ solvent, (e.g., isopropanol, toluene, or octane) to form the perovskite NCs.29,30 Similar to the hot-injection methods, the nucleation and growth stages progress rapidly and can both occur simultaneously for the LARP approaches.31 The LARP processes can result in the formation of aggregated and poorly crystallized nanostructures, and can result in relatively low reaction yields. Furthermore, some mixtures of polar and nonpolar solvents could degrade or otherwise dissolve the perovskite NCs.29 Additionally, a common polar solvent used in LARP is DMF, which is relatively toxic.31 As an alternative, ultrasonication-based techniques provide a direct, single-step approach to preparing relatively large-scale quantities of lead halide perovskites with the ability to also tune the dimensions and shapes of the products.32–35 These ultrasonication-based approaches to prepare lead halide perovskites provide access to relatively low temperature processes, and produce a relatively less aggregated product. These approaches also enable the ability to incorporate a wide range of reagents.32,35 The sonication-based approaches have not yet been extended to the synthesis of lead-free bismuth halide perovskites.
Herein, we demonstrate a facile, green solution-phase approach for the synthesis of cesium bismuth iodide (Cs3Bi2I9) perovskite NCs along with a characterization of their optical properties. Colloidal NCs of Cs3Bi2I9 were prepared by a simple sonication-assisted approach carried out in propylene carbonate (PC), a green solvent. This solution-phase approach to prepare Cs3Bi2I9 NCs has a number of advantages including a relatively low reaction temperature (e.g., ∼55 °C), and the manipulation of the reagents and products under an ambient atmosphere. This process prepared colloidal Cs3Bi2I9 NCs with an average diameter of 9 nm. These NCs also had a photoluminescence (PL) emission at 550 nm with a full-width-half-maximum (FWHM) of 65 nm, and exhibited photostability when stored as a suspension in PC and exposed to air over a period of five months. In addition, the peak position of the PL emission of the NCs was tuned through an anion exchange reaction using either tetraoctylammonium bromide (TOA-Br) or tetrabutylammonium chloride (TBA-Cl) to adjust the halide composition of the NCs.
Experimental section
All of the chemicals were of analytical grade and were used as received without further purification. Cesium bismuth iodide (Cs3Bi2I9) NCs were prepared in a single step process through a sonication-assisted solution-phase approach. In brief, 60 mM of bismuth acetate [Bi(OOCCH3)3, >99%, Sigma Aldrich] and 90 mM of cesium iodide (CsI, >99%, Sigma Aldrich) were prepared in 5 mL of propylene carbonate (PC, >99%, Sigma Aldrich). This process was followed by the addition of 0.25 mL oleic acid (OA, 99%, Alfa Aesar) as a capping agent. The mixture was subjected to tip-assisted sonication for 15 min at 22% of the maximum ultrasound amplitude (Fisher Scientific Sonic Dismembrator, Model No. 500, maximum output of 500 W) with intervals of 1 s on and 1 s off. During the course of the reaction, the colour of the reaction mixture changed from colorless to dark orange, indicating the formation of the Cs3Bi2I9 NCs. After completion of the reaction, any larger particles were removed from the suspension via a process of centrifugation (Model No. AccuSpin 400, Fisher Scientific) at 5000 rpm for 5 min to obtain a transparent orange colored solution that contained a suspension of the NCs.The solution containing the Cs3Bi2I9 NCs was used for the subsequent halide substitution reactions, and for studying the optical properties of these materials. Both Cs3Bi2Br9 and Cs3Bi2Cl9 NCs were prepared by halide substitution of the Cs3Bi2I9 NCs at ambient temperatures. In brief, a 0.03 M (for a slower anion exchange reaction) and 0.08 M (for a faster anion exchange reaction) solution of either tetraoctylammonium bromide (TOA-Br, 99%, Fluka) or tetrabutylammonium chloride (TBA-Cl, 99%, Fluka) were prepared in propylene carbonate as the source of halide to be used in the anion exchange processes. A solution of the appropriate halide species was added either relatively slowly (e.g., 1 μL per 5 s) or relatively fast (e.g., 10 μL per 5 s) to the as-prepared colloidal solution of Cs3Bi2I9 NCs. A series of ultraviolet (UV)-visible absorbance spectra were obtained as a function of reaction time to monitor the anion exchange processes.
For analysis of the products by powder X-ray diffraction (XRD) and Raman spectroscopy, the Cs3Bi2X9 NCs were precipitated from solution by adding chloroform to the suspension of NCs in propylene carbonate, which was followed by centrifugation of the solution at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. The supernatant was decanted and the obtained precipitates were dried before performing these analyses.
000 rpm for 15 min. The supernatant was decanted and the obtained precipitates were dried before performing these analyses.
Characterization of the cesium bismuth halide (Cs3Bi2X9) nanocrystals
The morphology, dimensions, crystallinity, and lattice parameters of the Cs3Bi2X9 NCs were characterized using an FEI Osiris X-FEG 8 transmission electron microscope (TEM) operated at an accelerating voltage of 200 kV. Analyses by energy dispersive X-ray spectroscopy (EDX) were performed using the FEI Osiris scanning/TEM, which was equipped with a Super-X EDX system with ChemiSTEM Technology integrating the signal from four spectrometers. Samples were prepared for TEM analysis by dispersing each of the purified products in hexanes followed by drop casting 5 μL of each suspension onto separate TEM grids (200 mesh copper grids coated with Formvar/carbon) purchased from Cedarlane Laboratories. Each TEM grid was dried under vacuum (∼230 torr) for at least 8 h prior to analysis.Phase and crystallinity of each sample was also assessed by XRD analyses performed using a Rigaku R-Axis Rapid diffractometer equipped with a 3 kW sealed tube copper source (Kα radiation, λ = 0.15418 nm) collimated to 0.5 mm. Powder samples were packed into cylindrical recesses drilled into glass microscope slides (Leica 1 mm Surgipath Snowcoat X-tra Micro Slides) for acquiring their diffraction patterns.
Purity and phase of the products were further assessed using Raman spectroscopy techniques. Raman spectra were collected using a Renishaw inVia Raman microscope with a 50× long working distance (LWD) objective lens (Leica, 0.5 NA) and a 785 nm argon ion laser (Modu-Laser, Model No. Stellar-Pro 514/50) set to 10% laser power with an exposure time of 30 s. The Raman spectrometer was calibrated by collecting the Raman spectrum of a polished silicon (Si) standard with a distinct peak centered at 520 cm−1. Raman spectra were collected for each of the powdered samples, which were supported on glass slide (Leica 1 mm Surgipath Snowcoat X-tra Micro Slides). The spectra were acquired from 50 to 300 cm−1 using a grating with 1800 lines per mm.
The optical absorption spectra of the Cs3Bi2X9 NCs were measured using an Agilent Technologies UV-visible spectrophotometer (Agilent 8453, Model No. G1103). For these measurements, the NCs suspended in PC were held in 1 cm path length poly(methyl methacrylate) cuvettes (VWR™, Catalog No. 634-8537). For the stability experiments, the NCs were stored as a suspension in PC over a period of 2 months. A series of UV-Visible spectra were recorded throughout this period of time (e.g., 4, 8, 12, 30, 60 and 150 days). Photoluminescence measurements of the Cs3Bi2X9 NCs were carried out using a Photon Technology International (PTI) Quantamaster spectrofluorometer using excitation wavelengths that matched the extinction maxima for each composition of NCs. The absolute photoluminescence quantum efficiency (PLQE) of the Cs3Bi2I9 NCs was measured using an FS5 spectrofluorometer (Edinburgh instruments) equipped with an integrating sphere (FS5 SC-30). To minimize the self-quenching effects, the absorbance of the sample was set below 0.2 at the excitation wavelength with a 10 mm quartz cuvette. The blank emission (e.g., from a solution of PC) was collected with the same cuvette containing only the solvent as a blank in the same spectral range used for the sample emission measurement. The absolute PLQE values were calculated using eqn S1 in ESI.† The time-resolved PL spectra were collected on a Horiba PTI QuantaMaster™ 400 with a DeltaFlex time-correlated single photon counting (TCSPC) lifetime measurement system equipped with a 370 nm excitation laser pulse. The instrument response function was measured using a non-fluorescent LUDOX®-50 scatterer (50 wt% suspension of silica beads in water, Sigma Aldrich).
Results and discussion
The Cs3Bi2I9 NCs were prepared in one-step by a solution-phase sonication process. We used PC, a green solvent,31 for the synthesis of the halide perovskite. Propylene carbonate has a relatively high boiling point (i.e., 240 °C), and is considered to be a nonhazardous solvent. The polar nature of the PC enables the ability to readily dissolve a variety of ionic species, such as the precursors to the Cs3Bi2I9 NCs. In this method, tip-assisted sonication was used to induce the formation of Cs3Bi2I9 NCs through a one-pot process directly from its molecular constituents (i.e., bismuth acetate and cesium iodide) in the presence of a surfactant, oleic acid. The formation of the Cs3Bi2I9 NCs during the reaction was noted by a change in the appearance of the solution, changing from colorless to a dark orange color. Transmission electron microscopy (TEM) analysis of the as-synthesized Cs3Bi2I9 NCs indicated the formation of uniform and well-dispersed NCs with quasi-spherical shapes (Fig. 1a). The average diameter of these NCs was 9 ± 2 nm (Fig. 1b). The atomic-scale crystallinity of the Cs3Bi2I9 nanoparticles was analyzed by high-resolution TEM (HRTEM) (Fig. 1c). A uniform lattice structure was observed throughout each of the nanocrystals. These results suggested that each nanoparticle was formed as a single crystal. The periodic fringe patterns observed by HRTEM for some of these nanocrystals had a spacing of 2.2 Å. This spacing matched the inter-planar spacing for the (315) planes of Cs3Bi2I9. Composition of the Cs3Bi2I9 NCs was further confirmed by energy dispersive X-ray spectroscopy (EDX), which indicated the presence of Cs, Bi and I in the NCs (Fig. S1 in ESI†).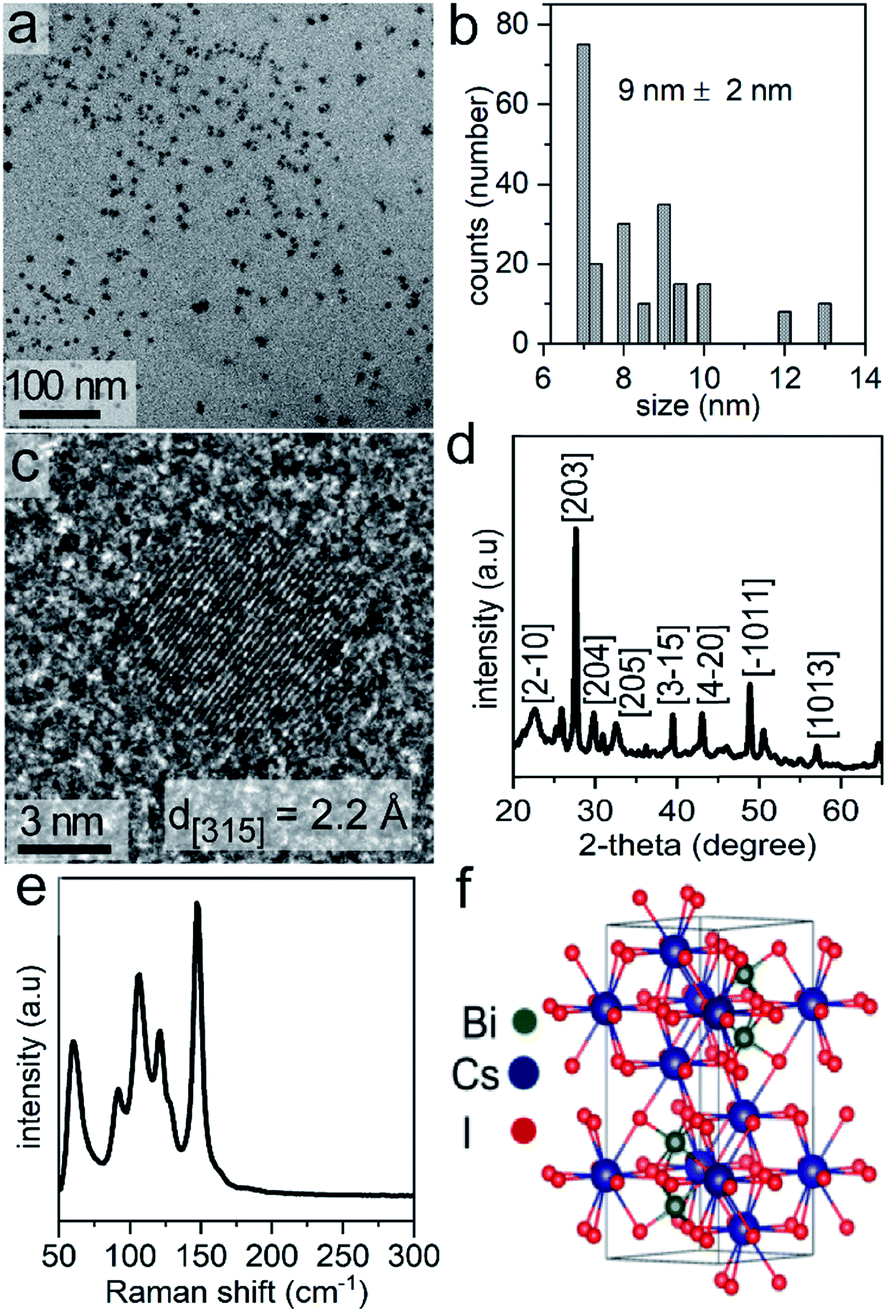 | ||
| Fig. 1 Cesium bismuth iodide (Cs3Bi2I9) nanocrystals (NCs) as characterized by: (a) transmission electron microscopy (TEM) imaging; (b) a histogram of their size distribution obtained from TEM analyses; (c) a high-resolution (HR) TEM analysis; (d) a powder X-ray diffraction analysis; (e) Raman spectroscopy techniques; and (f) a model of the hexagonal crystal structure for Cs3Bi2I9. | ||
Bismuth based perovskite NCs containing other halides (e.g., Cs3Bi2Br9, Cs3Bi2Cl9, and mixed halide species) were prepared through a simple process of anion exchange. Post-synthetic chemical transformations through ion-exchange reactions are a simple and versatile approach to creating new materials that are not readily accessible by other techniques, and can provide a fine control over the final composition of a material.36,37 Recently, anion exchange reactions of halide perovskites have rapidly emerged as an approach to tuning their composition and to prepare mixed halide phases.29,38 Due to the high mobility of the halide ions and the rigid nature of the cationic sub-lattice of halide perovskites, anion-exchange reactions are a versatile approach to tune the optical properties of a variety of perovskites.3,29 There is, however, little known with regards to the effects of the rate of the anion-exchange reactions on the properties of the product, such as the optical properties and the dimensions of the resulting perovskites.38 The anion exchange process is relatively fast (e.g., several seconds) and can be achieved at ambient or lower temperatures to deliberately achieve either with a partial or complete exchange of the anions. The anion exchange reactions reported herein were conducted by suspending the NCs in propylene carbonate and adding tetraalkylammonium halides (TAA-X; X = Cl− or Br−) as a source of the desired anion for the exchange. We selected tetraalkylammonium halides to facilitate the exchange of I− with either Br− or Cl− by hindering the reverse reactions. This unidirectional ion exchange process is attributed to the interactions of hard and soft acids and bases. The tetraalkylammonium cation (TAA+) with its four alkyl chains is a soft acid that prefers to bind to softer halide ions.38 Therefore, the TAA+ cation prefers to associate with I− over Br− or Cl−. When reacting the Cs3Bi2I9 NCs with tetraoctylammonium bromide (TOA-Br) the Br− readily exchanged with I− from the NCs lattice. In other words, the transformation from TOA-Br to TOA-I is favorable. The exchange for I− in Cs3Bi2I9 NCs with Cl− available from tetrabutylammonium chloride (TBA-Cl) is also feasible as the transformation from TBA-Cl to TBA-I is favorable (e.g., the I− ions are softer than Cl−).
The dimensions of the NCs were significantly affected by the rate of the anion exchange reaction. A relatively slow anion exchange reaction (Fig. 2) used to prepare the Cs3Bi2Br9 and Cs3Bi2Cl9 NCs resulted the formation of particles with comparable dimensions to those of Cs3Bi2I9 (Fig. 3, and S2 in ESI†). The TEM analyses of the Cs3Bi2Br9 NCs acquired after completing a relatively slow halide exchange process indicated that these NCs were also crystalline and maintained a quasi-spherical shape. The average diameter of these Cs3Bi2Br9 NCs was 7 ± 2 nm (Fig. S3 in ESI†). The TEM analyses of the Cs3Bi2Cl9 NCs, also prepared by the relatively slow anion exchange process, revealed that these NCs were also crystalline, but that these products exhibited more irregular shapes and an increase in the polydispersity of the particles. The average diameter of these Cs3Bi2Cl9 NCs was 11 ± 5 nm (Fig. S3 in ESI†). These variations in the shape and size of the Cs3Bi2Cl9 NCs from the as-synthesized, parent Cs3Bi2I9 NCs could be attributed to the larger difference in radii of the Cl− and I− ions. The replacement of the I− by Cl− within the crystalline lattice likely disrupts the structure of these materials.39,40 This ion exchange process could lead to a partial dissolution, reshaping, and aggregation of the NCs. Increasing the relative rate of the anion exchange reactions (Fig. S4 and S5 in ESI†) resulted in an increase in the average diameter and polydispersity of the Cs3Bi2Br9 and Cs3Bi2Cl9 NCs. These Cs3Bi2Br9 and Cs3Bi2Cl9 NCs had average dimensions of 12 ± 4 nm and 22 ± 6 nm, respectively (Fig. 4). A number of smaller particles with average dimensions of ∼4 nm were also observed along with these larger Cs3Bi2Cl9 NCs, which may result from a partial dissolution of the NCs during the anion exchange process. It is possible that the overall increase in size of the NCs observed during this fast ion exchange process could be attributed to an initial dissolution of the Cs3Bi2I9 NCs, forming smaller nuclei. Subsequent nucleation and aggregative growth of these nuclei would result in the formation of the larger Cs3Bi2Cl9 NCs.41,42 During aggregative growth, the deposition of the nuclei on the higher energy facets also lead to a reshaping and an increase in the dimensions of the NCs.43 The ion-exchange process begins from the edges of the particles and proceeds toward their core.44,45 These ion exchange reactions have a small activation energy as they are thought to proceed through a kick-out mechanism, and that the rate-determining step is not the rate of diffusion but instead the rate of the ion exchange.44–46 These faster rates for the ion exchange reaction result in a significant mass transport between the inner core and exterior of the particles, which could result in a dissolution or partial deterioration of their structure.44
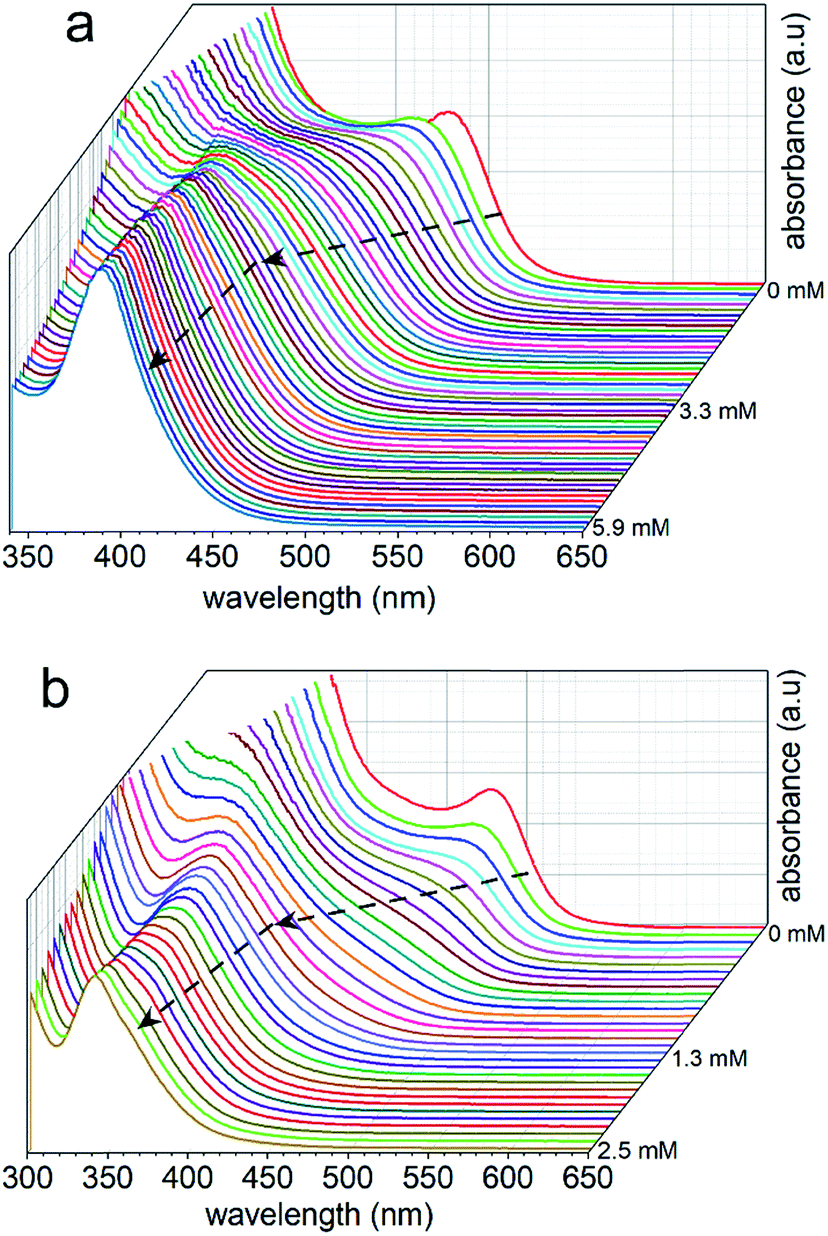 | ||
| Fig. 2 Ultraviolet (UV)-visible absorption spectra acquired over the course of the reaction to monitor a relatively slow anion exchange process for the parent Cs3Bi2I9 NCs through the formation of mixed (a) I/Br and (b) I/Cl nanocrystals by the substitution of I− with Br− and Cl−, respectively. The addition of tetraalkylammonium halides in an increasing concentration (as noted next to the spectra) were used to initiate the anion exchange reaction, which resulted in a blue-shift in the absorption spectra. The absorption spectra quickly stabilized, exhibiting no further shifts with subsequent additions of either Br− or Cl− anions to the solution. This stabilization of the spectra indicated a completion of the anion exchange process. | ||
 | ||
| Fig. 3 Transmission electron microscopy (TEM) analyses of the nanocrystals (NCs) after a relatively slow anion exchange reaction: (a and b) cesium bismuth bromide (Cs3Bi2Br9) nanocrystals; and (c and d) cesium bismuth chloride (Cs3Bi2Cl9) nanocrystals. The larger, dark regions observed in (c) are associated with assemblies of smaller nanoparticles as shown in (d). | ||
 | ||
| Fig. 4 Transmission electron microscopy (TEM) analyses of the nanocrystals (NCs) after a relatively fast ion exchange reaction: (a and b) cesium bismuth bromide (Cs3Bi2Br9) nanocrystals; and (c and d) cesium bismuth chloride (Cs3Bi2Cl9) nanocrystals. The average dimensions of the Cs3Bi2Br9 and Cs3Bi2Cl9 NCs increased to ∼12 nm and ∼22 nm, respectively. | ||
Powder X-ray diffraction (XRD) patterns were obtained for the as-synthesized Cs3Bi2I9 NCs (Fig. 1d, and S6 in ESI†), as well as for the Cs3Bi2Cl9 and Cs3Bi2Br9 NCs resulting from the relatively slow halide exchange reactions (Fig. S7 in ESI†). The major peaks in the XRD patterns of the Cs3Bi2I9 NCs matched the hexagonal crystal structure expected for Cs3Bi2I9 (space group P63/mmc, ICSD No. 410726). The Cs3Bi2Cl9 NCs and Cs3Bi2Br9 NCs were also crystalline, and had major diffraction peaks similar to the XRD profiles to those previously reported for bulk crystals of Cs3Bi2Cl9 and Cs3Bi2Br9. In comparison to these bulk single-crystals, the diffraction peaks for the samples of NCs were significantly broader, which can be attributed to the nanoscale dimensions of the colloidal NCs.47,48 Differences in the peak positions for some of the peaks observed in the XRD plots for the NCs relative to those for the ICSD standards, as seen in Fig. S6 and S7 in ESI,† are attributed to the intrinsic structural properties of the perovskite NCs. These intrinsic properties include unit cell distortions and other structural strains that result from the small dimensions of the nanocrystals, and the preferred directions of crystal growth for the nanocrystals relative to the bulk crystals used as reference materials.49 Further distortions could result from the processes of precipitating the NCs from solution and drying them in preparation for the XRD analyses. These induced transformations of the perovskite NCs may also lead to the formation of polycrystalline and/or aggregated products.50 Similar interferences and environmental influences on the reflections observed in the XRD plots have been observed in many previous reports for perovskite-based nanomaterials.20,21,23,24 The relative intensities of some reflections are also distinct from those observed in the bulk crystals, which is due to changes in the relative growth directions of the lattices in the form of the NCs.
Phase, crystallinity, and purity of the products were further analyzed by Raman spectroscopy techniques. Raman spectra were recorded from 50 to 300 cm−1. No peaks were observed above 300 cm−1. Raman spectra of Cs3Bi2I9 contain two major peaks above 110 cm−1, along with three strong modes between 50 and 110 cm−1 (Fig. 1e). The Raman bands associated with the Cs3Bi2I9 NCs further indicated the formation of a crystalline product.51 The Raman spectra of the Cs3Bi2Cl9 and Cs3Bi2Br9 NCs also indicated the formation of crystalline products (Fig. S8 in ESI†).52 The observed vibrational modes of the Cs3Bi2I9 lattice (space group P63/mmc) are derived mainly from the vibrations of the [Bi2I9]3− anion. The crystal structure consists of molecular [Bi2I9]3− anions bound together by three Cs+ cations per unit cell (Fig. 1f). The dominant vibrations in the crystalline lattice of Cs3Bi2I9 arise from the strong bonds of the [Bi2I9]3− sub-unit and the weaker modes arise from the ionic interactions of that sub-unit with the bridging Cs+ cations (Table S1 in ESI†).51,53,54 The peak positions within the Raman spectra of the Cs3Bi2Br9 and Cs3Bi2Cl9 NCs were also in agreement with the data previously reported for larger Cs3Bi2Br9 and Cs3Bi2Cl9 crystals.52
The optical properties of the Cs3Bi2X9 NCs were investigated as colloidal suspensions under ambient conditions to evaluate their excitation and PL response. The Cs3Bi2I9 NCs had an excitation peak maximum centered at ∼485 nm and an associated emission peak at 550 nm (2.25 eV) with an FWHM of 65 nm. The PLQE of the Cs3Bi2I9 NCs was measured using a fluorescence spectrometer equipped with an integrating sphere, which yielded a PLQE value of ∼0.3%. The absolute PLQE value of these Cs3Bi2I9 NCs is higher than those previously reported in the literature for Cs3Bi2I9 NCs prepared by other methods.22,55 The PL spectra of the Cs3Bi2X9 NCs were finely tuned from 550 to 410 nm by varying their anion composition (X = Br, Cl, and I). The source of halide species for these anion exchange reactions were the tetraalkylammonium halides, which were highly soluble in PC. The extent of the halide exchange reaction was assessed by monitoring changes to the optical absorption spectra of the NCs (e.g., Fig. 2). After the anion exchange with Br− or Cl− ions, the excitation peak of the NCs exhibited a blue-shift (Fig. 5 and Table S2 in ESI†). This blue-shift in the absorption spectra was associated with an increase in the bandgap energy of the NCs after the anion exchange. The Cs3Bi2Cl9 NCs and Cs3Bi2Br9 NCs exhibited absorption peaks at 337 nm and 385 nm, and their corresponding PL emission peaks were centered at 410 nm (3.02 eV) and 460 nm (2.70 eV), respectively (Fig. 5). The bandgaps of the Cs3Bi2I9, Cs3Bi2Cl9, and Cs3Bi2Br9 NCs were each assessed to determine if they contained either direct or indirect transitions through the use of Tauc plots.56 These plots were obtained by replotting the absorption spectra as (αhν)1/nversus hν. The parameter n denotes the type of electronic transition, with a value of 1/2 used for direct transitions.55 The bandgaps associated with direct electronic transitions, as determined from these Tauc plots, were 2.33 eV, 2.81 eV and 3.37 eV for the Cs3Bi2I9 NCs, Cs3Bi2Br9 NCs, and Cs3Bi2Cl9 NCs, respectively (Fig. S9 in ESI†). Peak PL emissions for the Cs3Bi2I9 NCs and Cs3Bi2Br9 NCs were in close agreement to the bandgaps predicted for direct electronic transitions through the Tauc plots. These results do not rule out contributions from an indirect electronic transitions.55 The peak PL emission for the Cs3Bi2Cl9 NCs were, however, not as closely matched with the bandgaps predicted for a direct electronic transition. This mismatch could be attributed to defects resulting from the partial dissolution and aggregation of these NCs during the anion exchange of I− with Cl−, as well as the polydispersity of this product (Fig. S3 in ESI†).
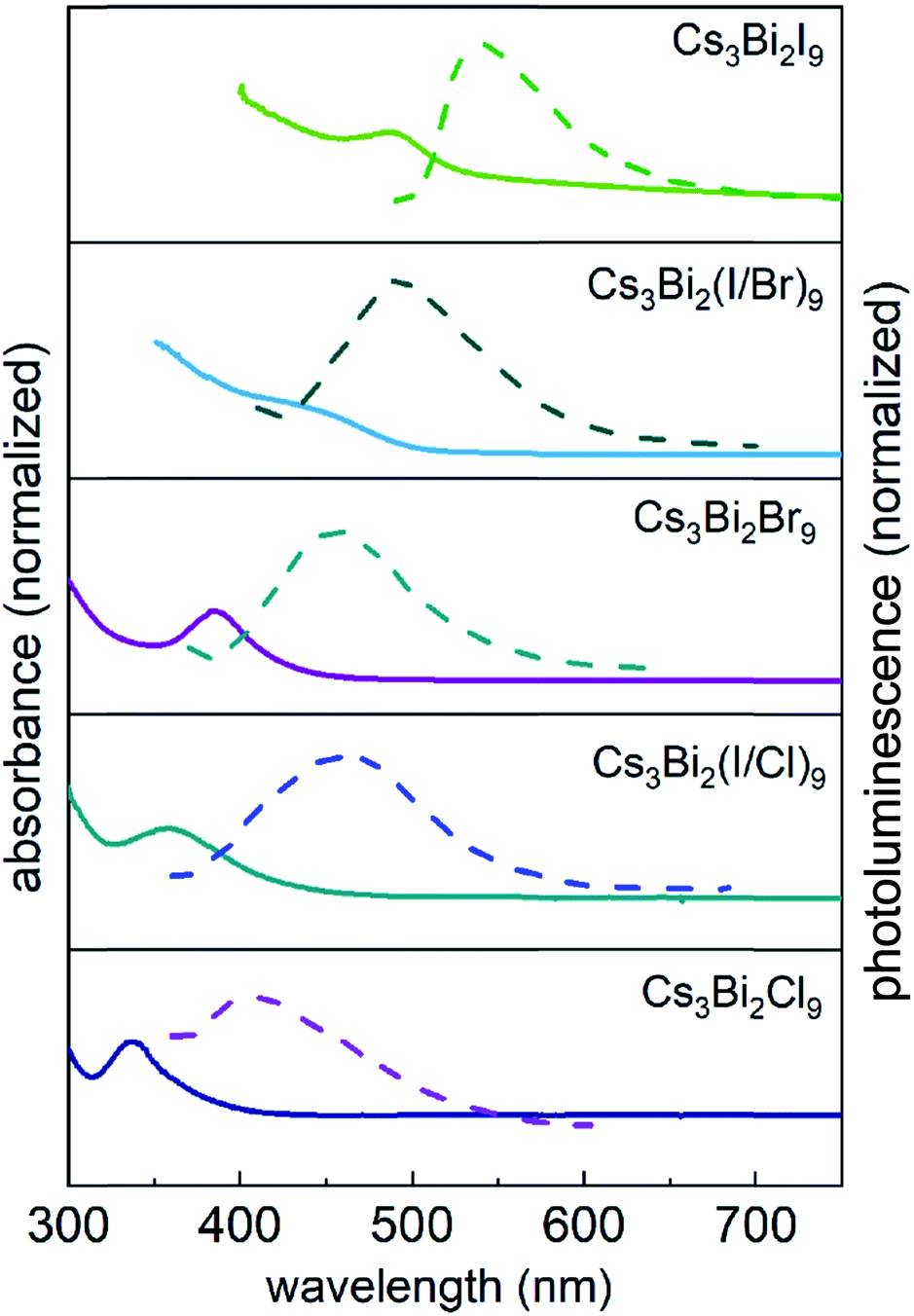 | ||
| Fig. 5 Absorption (solid) and photoluminescence (dashed) spectra of the perovskite Cs3Bi2I9 NCs, as well as NCs containing Br− and Cl− prepared by the substitution of I− with Br− (2nd and 3rd panels from the top) and Cl− (4th and 5th panels from the top). | ||
Time-resolved PL spectra were acquired using time-correlated single photon counting (TCSPC) to investigate the dynamics of exciton recombination associated with the NCs (Fig. 6, and S10 in ESI†). The PL decay of the Cs3Bi2I9 NCs was fit with a biexponential decay function composed of a short-lived PL lifetime (τ1) of 0.92 ns and a long-lived PL lifetime (τ2) of 5.3 ns (Fig. S11 in ESI†). Similar lifetimes were observed for the Cs3Bi2Br9 and Cs3Bi2Cl9 NCs. Each of the NCs exhibited a larger contribution from the short-lived PL decay processes over the long-lived PL decay processes (Table S3 in ESI†). Previous studies suggest that the processes associated with the short-lived lifetime can be attributed to direct electronic transitions within the NCs. In contrast, the long-lived component can be attributed to indirect electronic transitions, such as through recombination events at surface defects.55,57 The fluorescence decay associated with the Cs3Bi2X9 NCs is likely governed predominantly by exciton recombination within the NCs.22,58 The Cs3Bi2I9 NCs were also studied over a period of 150 days while stored under ambient conditions to assess the stability of their optical properties. A small blue-shift (∼8 nm) and slight peak broadening (∼3 nm) were observed in the UV-Visible absorbance spectra of the nanocrystals over the course of the five months of storage (Fig. S14 in ESI†). This indicated that the Cs3Bi2I9 NCs exhibited stable optical properties when stored in air as no significant changes were observed to their absorption spectra over this period of time. A slight blue-shift (∼25 nm) was observed in their PL spectra along with a similar FWHM following the 150 days of exposure to air, which indicated the relative stability of the optical properties of these NCs (Fig. S15 in ESI†).
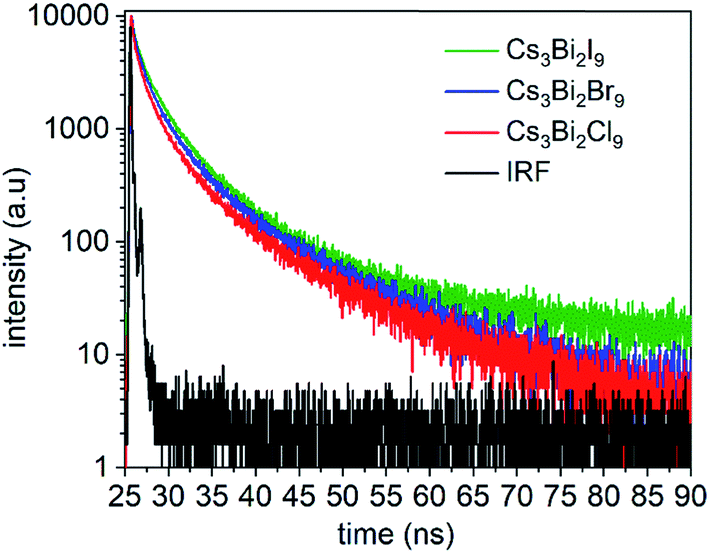 | ||
| Fig. 6 Time-resolved PL spectra for Cs3Bi2I9 NCs (green), Cs3Bi2Br9 NCs (blue), and Cs3Bi2Cl9 NCs (red) as measured by time-correlated single photon counting (TCSPC) using an excitation wavelength of 370 nm. The instrument response function (IRF) of the TCSPC set-up was measured using a non-fluorescent scatterer, LUDOX®-50. | ||
Conclusions
In summary, we have demonstrated a solution-phase route to prepare single-crystalline Cs3Bi2I9 NCs via a facile sonication-assisted method. This single-step process uses propylene carbonate as a green solvent for the synthesis. The PL spectra of the perovskite Cs3Bi2X9 NCs were tuned from 410 to 550 nm by controlling their composition through an anion exchange process carried out at room temperature using tetraalkylammonium halides (TAA-X) as a source of halide ions (X = Br− or Cl−). Controlling the rates of the anion exchange process had a significant influence on the quality of the products. For example, following a relatively slow anion exchange reaction the dimensions of the Cs3Bi2Br9 NCs and Cs3Bi2Cl9 NCs were comparable to those of the parent Cs3Bi2I9 NCs. A faster anion exchange reaction yielded NCs of Cs3Bi2Br9 and Cs3Bi2Cl9 with an increase in the relative dimensions of these products and an increase in their polydispersity. Time-resolved PL measurements indicated that the fluorescence decay associated with the NCs proceeds predominately through an exciton recombination within the NCs. The Cs3Bi2I9 NCs were also air stable for over 150 days with a slight blue-shift of their absorbance and PL properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grant No. 1077758), and through the Collaborative Health Research Projects (CHRP) Partnership Program supported in part by the Canadian Institutes of Health Research (Grant No. 134742) and the Natural Science Engineering Research Council of Canada (Grant No. CHRP 462260), the Canada Research Chairs Program (B. D. Gates, Grant No. 950-215846), and CMC Microsystems (MNT Grants No. 6345 and No. 5954). This work made use of 4D LABS (http://www.4dlabs.com) and the Center for Soft Materials shared facilities supported by the Canada Foundation for Innovation (CFI), British Columbia Knowledge Development Fund (BCKDF), Western Economic Diversification Canada, and Simon Fraser University. We also thank Dr Saeid Kamal (4D LABS), Dr Ryan Roberts, and Prof. Daniel Leznoff for discussions and assistance with acquiring the fluorescence lifetimes and PLQE data for the samples described herein.References
- S.-T. Ha, R. Su, J. Xing, Q. Zhang and Q. Xiong, Chem. Sci., 2017, 8, 2522–2536 RSC
.
- W. Zhang, G. E. Eperon and H. J. Snaith, Nat. Energy, 2016, 1, 16048 CrossRef CAS
- Q. A. Akkerman, G. Rainò, M. V Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS PubMed
- A. R. Srimath Kandada and A. Petrozza, Acc. Chem. Res., 2016, 49, 536–544 CrossRef CAS PubMed
- C. C. Stoumpos and M. G. Kanatzidis, Acc. Chem. Res., 2015, 48, 2791–2802 CrossRef CAS PubMed
- Y. Wang and H. Sun, Small Methods, 2018, 2, 1700252 CrossRef
- H. Huang, L. Polavarapu, J. A. Sichert, A. S. Susha, A. S. Urban and A. L. Rogach, NPG Asia Mater., 2016, 8, e328 CrossRef CAS
- A. Walsh, J. Phys. Chem. C, 2015, 119, 5755–5760 CrossRef CAS PubMed
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 6, 668–679 CrossRef PubMed
- M. V Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef PubMed
- Z. Shi, J. Guo, Y. Chen, Q. Li, Y. Pan, H. Zhang, Y. Xia and W. Huang, Adv. Mater., 2017, 29, 1605005 CrossRef PubMed
- L. Liang and P. Gao, Adv. Sci., 2018, 5, 1700331 CrossRef PubMed
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 1803792 CrossRef PubMed
- X. Wang, T. Zhang, Y. Lou and Y. Zhao, Mater. Chem. Front., 2019, 3, 365–375 RSC
- W. Ning and F. Gao, Adv. Mater., 2019, 1900326 CrossRef PubMed
- D. S. Dolzhnikov, C. Wang, Y. Xu, M. G. Kanatzidis and E. A. Weiss, Chem. Mater., 2017, 29, 7901–7907 CrossRef CAS
- J. Zhang, Y. Yang, H. Deng, U. Farooq, X. Yang, J. Khan, J. Tang and H. Song, ACS Nano, 2017, 11, 9294–9302 CrossRef CAS PubMed
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., 2017, 129, 12645–12649 CrossRef
- M. Leng, Y. Yang, K. Zeng, Z. Chen, Z. Tan, S. Li, J. Li, B. Xu, D. Li and M. P. Hautzinger, Adv. Funct. Mater., 2018, 28, 1704446 CrossRef
- M. Leng, Z. Chen, Y. Yang, Z. Li, K. Zeng, K. Li, G. Niu, Y. He, Q. Zhou and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15012–15016 CrossRef CAS PubMed
- Y. Lou, M. Fang, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 3779–3782 RSC
- R. D. Nelson, K. Santra, Y. Wang, A. Hadi, J. W. Petrich and M. G. Panthani, Chem. Commun., 2018, 54, 3640–3643 RSC
- B. Park, B. Philippe, X. Zhang, H. Rensmo, G. Boschloo and E. M. J. Johansson, Adv. Mater., 2015, 27, 6806–6813 CrossRef CAS PubMed
- J. Pal, A. Bhunia, S. Chakraborty, S. Manna, S. Das, A. Dewan, S. Datta and A. Nag, J. Phys. Chem. C, 2018, 122, 10643–10649 CrossRef CAS
- L. Zhang, K. Wang and B. Zou, ChemSusChem, 2019, 12, 1612–1630 CrossRef CAS PubMed
- V. K. Ravi, N. Singhal and A. Nag, J. Mater. Chem. A, 2018, 6, 21666–21675 RSC
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119(5), 3296–3348 CrossRef CAS PubMed
- K. Hills-Kimball, Y. Nagaoka, C. Cao, E. Chaykovsky and O. Chen, J. Mater. Chem. C, 2017, 5, 5680–5684 RSC
- K. L. Gardner, J. G. Tait, T. Merckx, W. Qiu, U. W. Paetzold, L. Kootstra, M. Jaysankar, R. Gehlhaar, D. Cheyns and P. Heremans, Adv. Energy Mater., 2016, 6, 1600386 CrossRef
- Y. Tong, E. Bladt, M. F. Aygüler, A. Manzi, K. Milowska, V. A. Hintermayr, P. Docampo, S. Bals, A. Urban, P. Lakshminarayana and J. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 13887–13892 CrossRef CAS PubMed
- D. M. Jang, D. H. Kim, K. Park, J. Park, J. W. Lee and J. K. Song, J. Mater. Chem. C, 2016, 4, 10625–10629 RSC
- Y. Tong, B. J. Bohn, E. Bladt, K. Wang, P. Müller-Buschbaum, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2017, 56, 13887–13892 CrossRef CAS PubMed
- H. Huang, Q. Xue, B. Chen, Y. Xiong, J. Schneider, C. Zhi, H. Zhong and A. L. Rogach, Angew. Chem., 2017, 129, 9699–9704 CrossRef
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Nano Lett., 2011, 11, 4964–4970 CrossRef CAS PubMed
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed
- R. Agarwal, N. M. Krook, M.-L. Ren, L. Z. Tan, W. Liu, A. M. Rappe and R. Agarwal, Nano Lett., 2018, 18, 1620–1627 CrossRef CAS PubMed
- S. Gupta, S. V Kershaw and A. L. Rogach, Adv. Mater., 2013, 25, 6923–6944 CrossRef CAS PubMed
- B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 19977–19980 CrossRef CAS PubMed
- O. Ramirez, P. Ramasamy, Y. Chan Choi and J.-S. Lee, Chem. Mater., 2018, 31, 268–276 CrossRef
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2013, 26, 5–21 CrossRef
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed
- Z. Fan, L.-C. Lin, W. Buijs, T. J. H. Vlugt and M. A. Van Huis, Nat. Commun., 2016, 7, 11503 CrossRef CAS PubMed
- V. Lesnyak, R. Brescia, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2015, 137, 9315–9323 CrossRef CAS PubMed
- P. Scardi, M. Leoni and K. R. Beyerlein, Z. Kristallogr. - Cryst. Mater., 2011, 226, 924–933 CrossRef CAS
- A. V Vorontsov and S. V Tsybulya, Ind. Eng. Chem. Res., 2018, 57, 2526–2536 CrossRef
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed
- E. Fanizza, F. Cascella, D. Altamura, C. Giannini, A. Panniello, L. Triggiani, F. Panzarea, N. Depalo, R. Grisorio and G. P. Suranna, Nano Res., 2019, 12, 1155–1166 CrossRef CAS
- K. M. McCall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 29, 4129–4145 CrossRef CAS
- G. Bator, J. Baran, R. Jakubas and M. Karbowiak, Vib. Spectrosc., 1998, 16, 11–20 CrossRef CAS
-
O. V Vakulenko, V. O. Gubanov, S. V Kun, F. V Motsnyi, E. Y. Peresh and V. A. Terekhov, in Optical Diagnostics of Materials and Devices for Opto-, Micro-, and Quantum Electronics, 1997, vol. 3359, pp. 351–355 Search PubMed
- A. Nila, M. Baibarac, A. Matea, R. Mitran and I. Baltog, Phys. Status Solidi, 2017, 254, 1552805 CrossRef
- Y. Zhang, J. Yin, M. R. Parida, G. H. Ahmed, J. Pan, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 3173–3177 CrossRef CAS PubMed
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef CAS
- V. Sarritzu, N. Sestu, D. Marongiu, X. Chang, Q. Wang, S. Masi, S. Colella, A. Rizzo, A. Gocalinska and E. Pelucchi, Adv. Opt. Mater., 2018, 1701254 CrossRef
- G. D. Scholes and G. Rumbles, Nat. Mater., 2006, 5, 683–696 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Energy dispersed X-ray spectroscopy data; UV-visible spectroscopy data; histograms of particle size; transmission electron microscopy data; powder X-ray diffraction analyses; Raman spectroscopy data; Tauc plots; fluorescence lifetime data; and optical images. See DOI: 10.1039/c9na00586b |
| This journal is © The Royal Society of Chemistry 2019 |