
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective activation of organocatalysts by specific signals†
Chandan
Maity‡
 ,
Fanny
Trausel‡
and
Rienk
Eelkema
*
,
Fanny
Trausel‡
and
Rienk
Eelkema
*
Department of Chemical Engineering, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: r.eelkema@tudelft.nl
First published on 20th June 2018
Abstract
Reminiscent of signal transduction in biological systems, artificial catalysts whose activity can be controlled by physical or chemical signals would be of high interest in the design of chemical systems that can respond to their environment. Self-immolative chemistry offers a generic method for the development of catalysts that can be activated by different signals. To demonstrate the versatility of that concept, we synthesized organocatalysts that can be activated by three different signals and that can be used to control two different reactions. In this way the organocatalyst proline is designed as a pro-catalyst that is activated either by the chemical signal H2O2, by light or by the enzyme penicillin acylase. The pro-catalysts were used to exert temporal control over the rate of an aldol reaction and a Michael reaction.
Introduction
In nature, cells communicate and respond to signals from their environment by changing the activity of enzymes.1,2 Artificial catalysts that can be activated by different signals are scarce and would be of high interest to design systems that can respond to the environment. Some catalysts that can be activated by signals have been reported.3 Many of these catalysts use light as a signal.4 Only a small number of catalysts that can be activated by chemical signals are found in the literature.3,5–7 Many of these catalysts suffer from complex syntheses and a lack of generic design. A versatile tool to design catalysts that can respond to different signals is self-immolative chemistry.8–10 A self-immolative molecule contains a signal-labile functional group. When this group reacts with the signal, the molecule fragments and releases a molecule of interest, in our case a catalyst. Recently we reported an organocatalyst that can be activated by a chemical signal and that controls the formation of a polymer or a self-assembled material.11Here we report a general strategy to design organocatalysts that can be selectively activated in response to a specific signal. To demonstrate the versatility of the design we report how to render an organocatalyst responsive to three different signals and that it can be used to control the rates of two different reactions. Specifically, we synthesized three different protected proline catalysts that can be activated by three different signals (Fig. 1): PP-1 is activated by the chemical signal H2O2, PP-2 is activated by light and PP-3 is activated via catalytic hydrolysis by the enzyme Penicillin Acylase (PA). When the pro-prolines are activated by the relevant signal, the catalyst proline P-4 is released. In addition, we used the pro-prolines to catalyse an aldol reaction between a ketone and an aldehyde and a Michael reaction between an nitro-olefin and an aliphatic aldehyde. The strategy constitutes a versatile method for the development a wide range of pro-catalysts to control a large variety of reactions.
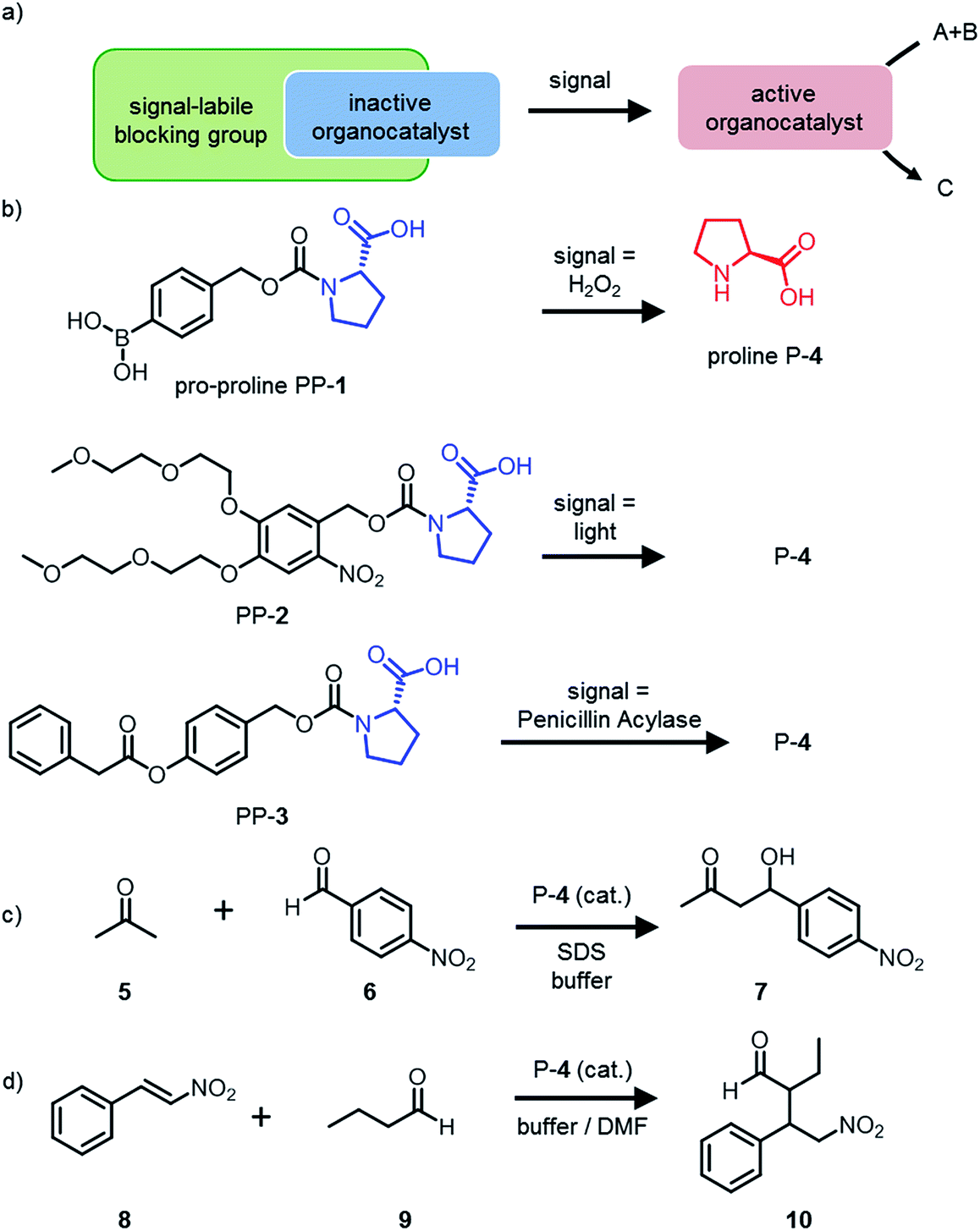 | ||
| Fig. 1 A generic design for a pro-catalyst that can be activated by a signal to catalyse a reaction. (a) Schematic representation of the protected organocatalyst that is activated by the signal and then catalyses a reaction. (b) The pro-proline PP-1 is activated by the chemical signal H2O2 and releases the organocatalyst proline P-4. PP-2 is activated by light and releases P-4. PP-3 is activated by the enzyme Penicillin Acylase (PA) and releases P-4. (c) The aldol reaction between acetone 5 and 4-nitrobenzaldehyde 6 is catalysed by P-4. (d) The Michael reaction between trans-β-nitrostyrene 8 and butanal 9 is catalysed by P-4. | ||
Results and discussion
Catalyst design
The general strategy to design an organocatalyst that can be activated by a signal to control a catalytic target reaction, is based on the following considerations (Fig. 1a):(1) The catalytic centre of the organocatalyst should allow protection with a self-immolative moiety: organocatalysts which have a primary or secondary amine or an alcohol as a catalytic centre are promising candidates.
(2) It should be possible to activate the catalyst in less than 30 minutes (depending on the timescale of the reaction) under the same conditions as are required for the catalytic target reaction. To enable the facile release of the catalyst it may be important to include a carbamate or carbonate group.8
(3) All reagents, the catalyst and the pro-catalyst should be soluble in the same solvent and the solvent should be compatible with the signal and the self-immolative reaction.
(4) The signal that activates the catalyst should not lead to a different outcome of the catalytic target reaction.
(5) The pro-catalyst should be stable under the conditions used in the catalytic target reaction and should not release the catalyst without being triggered by a specific signal.
(6) The catalytic target reaction should be susceptible to catalysis (show at least a 2-fold increase in reaction rate upon addition or activation of the catalyst) under the operating conditions.
Based on these guidelines we recently designed an organocatalyst that can be activated by a chemical signal and that controls the formation of a polymer or supramolecular network material.11 There, hydrazone bond formation12 was catalysed by a signal-responsive aniline procatalyst.
In the current work, we want to demonstrate the versatility of the design. We chose proline as an organocatalyst for two reasons. Firstly, proline is a simple organocatalyst with a catalytic centre that can easily be modified. Secondly, proline is a nucleophilic catalyst used for many different reactions.13 We decided to protect proline with three different self-immolative groups that react to three different signals (Fig. 1b). PP-1 is protected with a boronic acid self-immolative group that is oxidatively cleaved by hydrogen peroxide. Hydrogen peroxide is a relevant disease related biomarker as it is generated during oxidative stress by the oxidation of enzyme substrates such as glucose.14–16 PP-2 contains a light-sensitive group that is cleaved upon irradiation. To extend the scope of our design to biological systems we designed PP-3: the hydrolysis of the ester group in PP-3 to induce the self-immolative reaction is catalysed by the enzyme Penicillin Acylase (PA).
Synthesis of the catalysts
The pro-prolines were synthesized via chloroformate intermediates in two steps (Fig. 2). The total yield over all steps for PP-1 is 74%, for PP-2 it is 45% and PP-3 it is 48% (see ESI† for more information). By incorporating a carbamate group in the design we took into account design guideline 2. The synthetic route is generic: it enables the protection of a large range of organocatalysts and the use of a variety of self-immolative protecting groups to allow response to different signals. | ||
Fig. 2 Synthetic route to form pro-prolines PP-1, PP-2 and PP-3. (a) Synthesis of PP-1, reaction conditions: (i) Na2CO3, triphosgene, toluene, 0 °C – room temperature, 6 h. (ii) NaHCO3, proline P-4, water, 0 °C – room temperature, overnight, (iii) NaIO4, NH4OAc, water/acetone (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). (b) Synthesis of PP-2, reaction conditions: (i) HNO3 (70% in water), 0–20 °C, 2 h, (ii) NaBH4, methanol, 1 h, room temperature, (iii) K2CO3, triphosgene, toluene, 0 °C – room temperature, 6 h, (iv) NaHCO3, P-4, water, 0 °C – room temperature, overnight. (c) Synthesis of PP-3, reaction conditions: (i) phenylacetyl chloride, triethylamine, THF, 0 °C – room temperature, overnight, (ii) NaBH4, 2-propanol, 1 h, room temperature, (iii) K2CO3, triphosgene, toluene, 0 °C – room temperature, 6 h, (iv) trimethylsilylchloride (TMSCl), diisopropylamine, DCM, 0 °C – room temperature, 4 h. :1 v/v). (b) Synthesis of PP-2, reaction conditions: (i) HNO3 (70% in water), 0–20 °C, 2 h, (ii) NaBH4, methanol, 1 h, room temperature, (iii) K2CO3, triphosgene, toluene, 0 °C – room temperature, 6 h, (iv) NaHCO3, P-4, water, 0 °C – room temperature, overnight. (c) Synthesis of PP-3, reaction conditions: (i) phenylacetyl chloride, triethylamine, THF, 0 °C – room temperature, overnight, (ii) NaBH4, 2-propanol, 1 h, room temperature, (iii) K2CO3, triphosgene, toluene, 0 °C – room temperature, 6 h, (iv) trimethylsilylchloride (TMSCl), diisopropylamine, DCM, 0 °C – room temperature, 4 h. | ||
Activation of the catalysts
We tested whether the pro-prolines would release proline when the relevant signal was applied and whether this conversion was complete (see ESI† for more information). To PP-1 (50 mg) in methanol (2 mL) we added H2O2 (1 mL, 30%). After 10 min, thin layer chromatography showed complete conversion of PP-1. The reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate and washed with sodium sulfite (sat., aq). The obtained solution was concentrated under reduced pressure and analysed by 1H NMR spectroscopy and mass spectrometry to confirm complete conversion of PP-1 and the formation of proline P-4. When we followed the activation of PP-1 (10 mM in 20% dimethylformamide-d7 in 10 mM sodium phosphate buffer of pH 8.0) in NMR, we found that 10 equivalents of H2O2 are sufficient to convert PP-1 completely into P-4 in within 10 minutes. We noticed that although the boronic acid group is efficiently removed in up to 80% organic solvent, for the release of proline, 70–80% buffer is necessary to decrease the activation time (from 2 hours in 50% buffer to 10 minutes in 80% buffer). To investigate the activation of PP-2 we irradiated PP-2 (50 mg) in methanol (2 mL) in a 4 mL vial with light for 30 minutes using a Nikon Intensilight C-HGFI lamp (130 W mercury lamp with 100% light intensity, 320–600 nm). The activation of PP-2 was analysed by UV/vis spectroscopy, 1H-NMR spectroscopy and mass spectrometry and confirmed complete conversion and release of P-4. To activate PP-3 we added a solution of PA (5.5 mg, 98 U) in sodium phosphate buffer (100 mM, pH 7.4) to a solution of PP-3 (0.5 mg) in acetone (10 μL). After stirring for 10 minutes we concentrated the reaction mixture, added chloroform and filtered. The solution was concentrated under reduced pressure and analysed by 1H NMR and mass spectroscopy and confirmed complete conversion of PP-3 and release of P-4. In short, all pro-prolines were converted completely into proline within 30 minutes, thereby keeping with guideline 2. The design of the pro-catalysts thus enables efficient signal triggered release of the catalyst.Using the pro-catalysts to control reaction kinetics
We investigated whether we could use the pro-prolines to control reaction kinetics. We focussed on two reactions that obey guidelines 3–6 reasonably well: an aldol reaction between a ketone and an aldehyde and a Michael reaction between an nitro-olefin and an aldehyde.Control over the aldol reaction
We chose the proline-catalysed aldol reaction between acetone 5 and 4-nitrobenzaldehyde 6. For the activation of both PP-1 and PP-3 an aqueous environment is necessary. As reported in the literature, proline catalysis of aldol reactions is typically severely hindered by water. Still, in buffered media with the help of the surfactant sodium dodecyl sulfate (SDS), the reaction is accelerated by proline P-4.17 Proline acts as a nucleophilic catalyst by forming an enamine intermediate with acetone.18 The aldol reaction was followed using 1H-NMR spectroscopy, reaction conditions: 4-nitrobenzaldehyde 6 (10 mM), PP-1 (2 mM) in 20% acetone 5 in sodium phosphate buffer (100 mM, pH 7.4) with 10% D2O and SDS (1 mM) as an additive17 (Fig. 3 and Table 1). Without catalyst the aldol product 7 is still formed, it reaches 65% conversion in 48 h. The native catalyst P-4 (2 mM) induces a 4.2-fold increase in reaction rate and reaches 99% conversion in 48 h: this indicates that the aldol reaction is indeed catalysed by P-4. The reactions in the presence of unactivated PP-1, PP-2 and PP-3 (2 mM) show the same reaction rate as the uncatalysed reaction and reach a 65% conversion as well after 48 h. Without activation the pro-catalysts thus do not show any catalytic activity. Addition of H2O2 (2 mM, 1 equivalent to PP-1) to the reaction with PP-1 (2 mM) induces a 2.6-fold increase in reaction rate and the reaction reaches 93% conversion in 48 h. This indicates an efficient activation of the catalyst. H2O2 alone does not have any influence on the reaction rate. It is important not to use an excess of H2O2 with respect to PP-1 to prevent oxidation of nitro-benzaldehyde 6 (see ESI†). Irradiating the reaction mixture containing PP-2 (2 mM) leads to a 2.3-fold increase of reaction rate and a conversion of 93% after 48 h, another demonstration of efficient catalyst activation. Light-irradiation in the absence of PP-2 does not have any influence on the reaction rate. Addition of PA (5.5 mg, 98 U) to a reaction mixture containing PP-3 leads to a 2.7-fold increase in reaction rate and a conversion of 97% after 48 h. These results indicate efficient activation of pro-catalysts when the relevant signal is applied. Additionally, we applied ‘wrong’ signals to check whether each pro-catalyst is selectively activated by a specific signal. Irradiation of the reaction mixture with PP-1 or PP-3 did not result in any increase in reaction rate. To the reaction mixture with PP-2 we added H2O2 as a ‘wrong’ signal, and again, we did not observe an increase in reaction rate. These results demonstrate that the pro-catalysts are specific in their response, keeping with guideline 5. Altogether, the pro-catalyst-aldol system adheres to the guidelines reasonably well, however, because of the relatively large background reaction rate for aldol reactions,19 response to catalyst activation is only modest.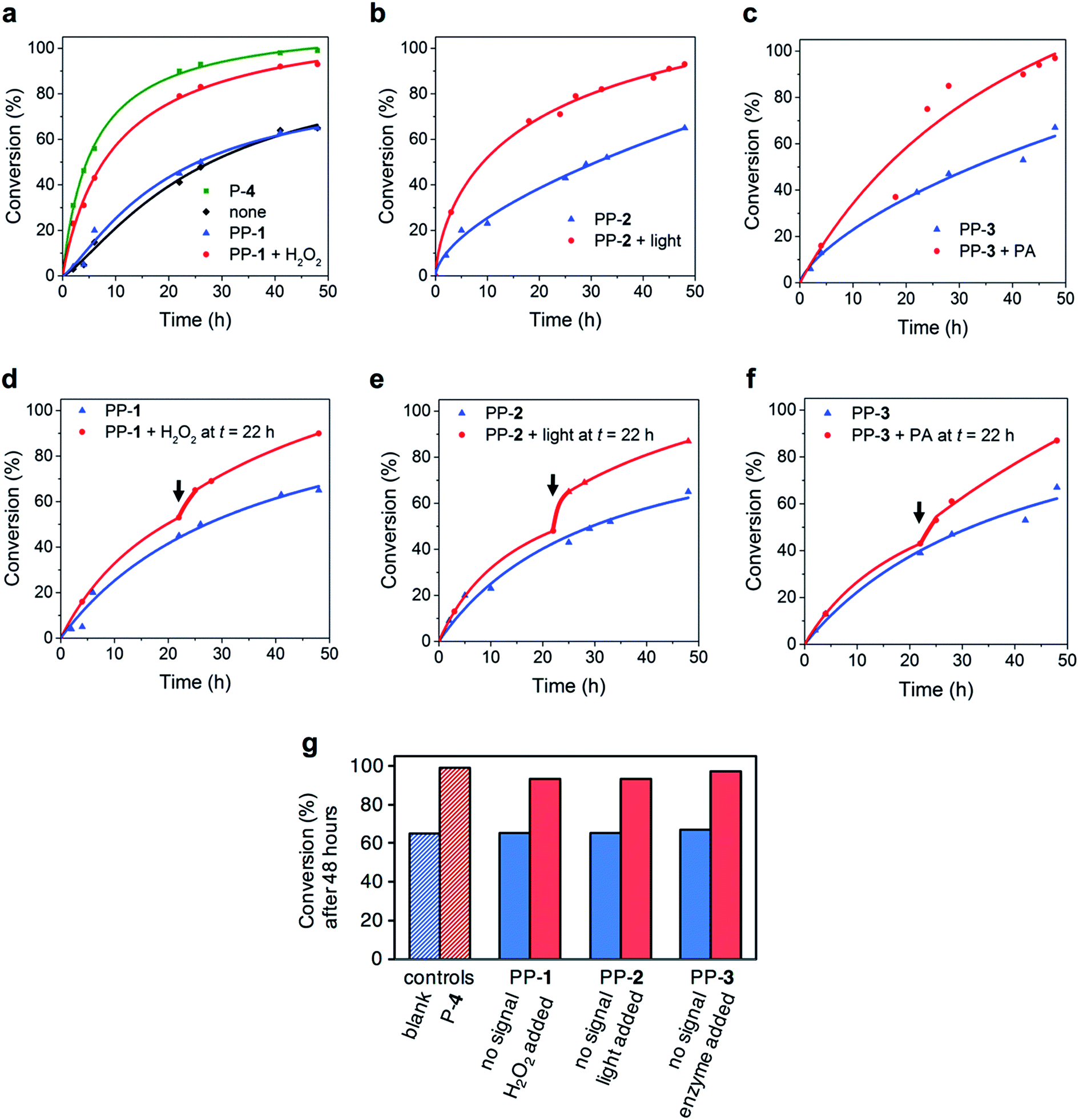 | ||
| Fig. 3 Kinetic analysis of the aldol reaction between 4-nitrobenzaldehyde 6 and acetone 5. Conditions: 10 mM 4-nitrobenzaldehyde 6, 20 volume% acetone 5, 2 mM (20 mol%) pro-proline, and a signal, in sodium phosphate buffer (100 mM, pH 7.4) with 10 v% D2O and 1 mM sodium dodecyl sulfate (SDS). (a) Conversion of the aldol product follow with NMR spectroscopy in the presence of P-4 (green), without catalyst (black), PP-1 without activation (blue), PP-1 and H2O2 (red). (b) Conversion of the aldol reaction in the presence of PP-2 without activation (blue) and in the presence of PP-2 after irradiation with light (red). (c) Conversion of the aldol reaction in the presence of PP-3 without activation (blue) and in the presence of PP-3 after addition of penicillin acylase (red). (d) Conversion of the aldol reaction in the presence of unactivated PP-1 (blue) or when the signal H2O2 was added after 22 h (red). (e) Conversion of the aldol reaction in the presence of PP-2 (blue), after 22 h, the reaction mixture was irradiated with light (red). (f) Aldol reaction in the presence of PP-3 (blue), after 22 h, penicillin acylase was added (red). In (d–f) the arrow indicates the moment of signal addition. (g) Conversion to the aldol product after 48 hours, in the absence or presence of the appropriate signal. | ||
| Catalyst system | k 2 (M−1 s−1) | k rel |
|---|---|---|
| None | 2.4 × 10−6 | 1.0 |
| P-4 | 1.0 × 10−5 | 4.2 |
| PP-1 + H2O2 | 6.2 × 10−6 | 2.6 |
| PP-1 | 2.4 × 10−6 | 1.0 |
| PP-1 + light | 2.4 × 10−6 | 1.0 |
| PP-2 + light | 5.5 × 10−6 | 2.3 |
| PP-2 | 2.3 × 10−6 | 1.0 |
| PP-2 + H2O2 | 2.4 × 10−6 | 1.0 |
| PP-3 + PA | 6.6 × 10−6 | 2.7 |
| PP-3 | 2.2 × 10−6 | 0.9 |
| PP-3 + light | 2.3 × 10−6 | 0.9 |
Importantly, addition of the signal at any given time during the reaction should lead to a change in reaction rate at that particular moment. To investigate this, we followed the reaction with PP-1 (2 mM) and added H2O2 (2 mM) after 22 hours (Fig. 3). The reaction rate increased 1.6-fold. This result demonstrates an autonomous response to a chemical change in the environment. The increase in reaction rate is lower than when the signal was added at the beginning of the reaction: this can be explained because a large part of the 4-nitrobenzaldehyde 6 has already been consumed and lower concentrations of reagents that are part of the rate equation lead to lower reaction rates. Similarly, when we followed the reaction with PP-2 (2 mM) and irradiated with light after 22 h we found a 1.5-fold increase in reaction rate. Furthermore, addition of PA (5.5 mg, 98 U) after 22 h to the reaction containing PP-3 led to a 1.4-fold increase in reaction rate. The pro-catalysts thus allow temporal response to signals from the environment.
Control over the Michael reaction
To demonstrate the versatility of the responsive catalyst system, we also used PP-1 to control the rate of a Michael reaction between trans-β-nitrostyrene 8 and butanal 9 (Fig. 1 and 4). Proline acts as a nucleophilic catalyst in the Michael reaction by forming an enamine intermediate with the aldehyde.20 Even though the reaction is commonly performed in organic solvents, the organocatalyzed Michael reaction is known to proceed in water.21 Reaction conditions: 10 mM trans-β-nitrostyrene 8, 100 mM butanal 9, 10 mM P-4 or PP-1, 100 mM (10 eq.) H2O2 in phosphate buffer (10 mM, pH 8.0) + dimethyl formamide-d7 (DMF-d7). DMF-d7 (20% v/v) was used to ensure solubility of the product. An advantage of the Michael reaction is the complete lack of background reactivity: without catalyst the reaction does not show any detectable conversion. Addition of the native catalyst P-4 (10 mM) increases the reaction rate constant to 7.7 × 10−3 M−1 s−1. This indicates that the Michael reaction is catalysed by P-4, in fact for the reaction to proceed on a reasonable timescale, it requires a catalyst. The reaction in the presence of PP-1 (10 mM) without the chemical signal does not show any conversion. The reaction with PP-1 (10 mM) and H2O2 (10 equivalents, 100 mM) proceeds with a rate constant of 2.0 × 10−4 M−1 s−1. These results indicate that the catalyst is efficiently activated (Table 2). The rate for the reaction with PP-1 and H2O2 is considerable lower than the rate for the reaction with P-4. This decrease in reaction rate can partially be explained by the use of H2O2: the Michael reaction in the presence of P-4 and H2O2 has a lower rate constant (9.3 × 10−4 M−1 s−1) than the reaction with only P-4 (7.7 × 10−3 M−1 s−1). Analysis of the reaction mixture by NMR spectroscopy reveals that the trans-β-nitrostyrene 8 degrades slowly in the presence of H2O2. This negative influence of the signal H2O2 on the reaction is in conflict with guideline 3. The rate constant for the Michael reaction in the presence of PP-1 and H2O2 is even lower: because we confirmed with NMR measurements that PP-1 is completely converted during the reaction, a reason for the decrease in reaction rate might be that more trans-β-nitrostyrene 8 is degraded in the time (10 min) that it takes to activate PP-1. Importantly, because of the complete lack of background reaction we should be able to use our responsive catalyst to switch the system from ‘off’ (no conversion) to ‘on’ (the reaction proceeds). Indeed, the reaction (or lack thereof) was followed for 1 hour, after which H2O2 (100 mM) was added: this resulted in an immediate response and again a reaction rate constant of 2.0 × 10−4 M−1 s−1. All in all, the pro-catalyst can be used to design a system that can autonomously switch on when a specific chemical signal is detected.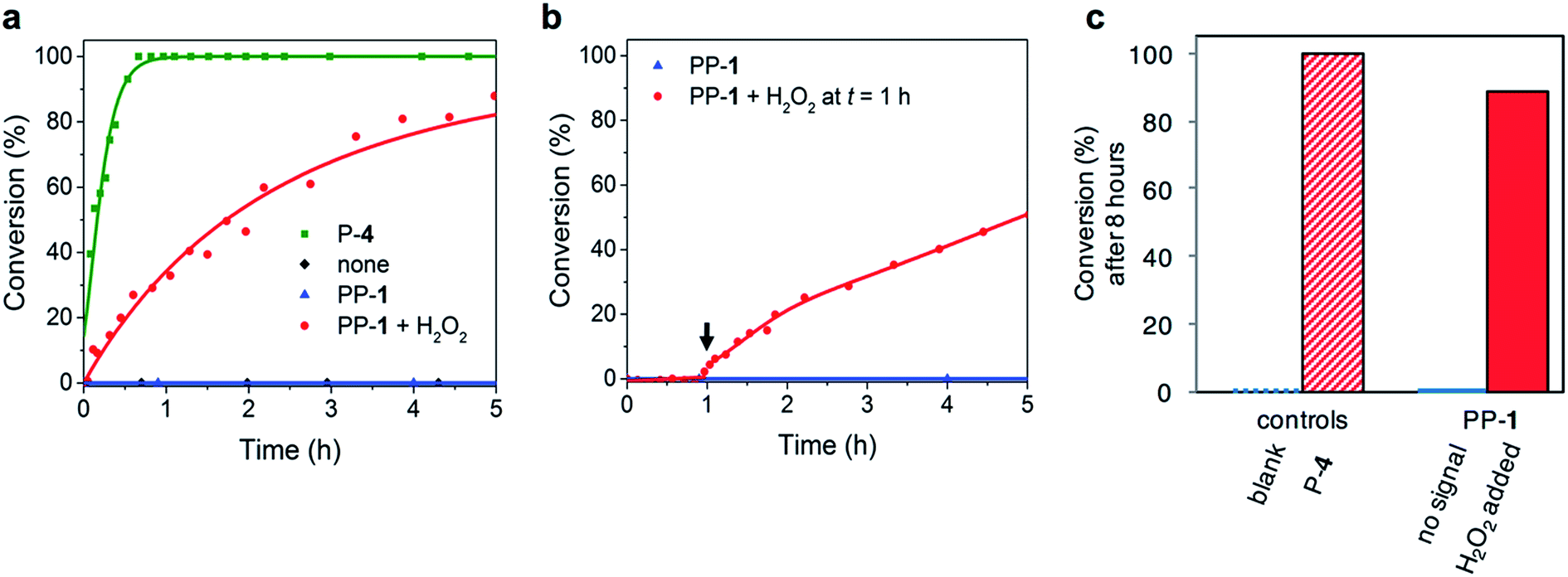 | ||
| Fig. 4 Kinetic analysis of the Michael reaction between trans-β-nitrostyrene 8 and butanal 9. Reaction conditions: 10 mM trans-β-nitrostyrene 8, 100 mM butanal 9, 10 mM PP-1, 100 mM (10 eq.) H2O2 in phosphate buffer (10 mM, pH 8.0) + dimethyl formamide-d7 (DMF-d7). (a) Conversion to the Michael product 10 followed with NMR spectroscopy, reaction with PP-1 (10 mM, blue), without catalyst (black, overlapping with blue), with P-4 (10 mM, green) and with PP-1 (10 mM) and H2O2 (100 mM, red). (b) Conversion to the Michael product 10 followed with 1H-NMR spectroscopy, reaction with PP-1 (10 mM, blue) and the reaction with PP-1 (10 mM) where H2O2 (100 mM) was added after 1 hour (red). The arrow indicates the moment of signal addition. (c) Conversion (%) after 8 hours of reaction time, without signal (blue), with signal (red). With P-4 the reaction reaches >99% conversion in 8 h and with PP-1 and the H2O2 the reaction reaches 89% conversion. Without catalyst or signal, there is no conversion. | ||
| Catalyst system | k 2 (M−1 s−1) |
|---|---|
| None | 0 |
| PP-1 | 0 |
| H2O2 | 0 |
| P-4 | 7.7 × 10−3 |
| PP-1 + H2O2 | 2.0 × 10−4 |
| P-4 + H2O2 | 9.3 × 10−4 |
Conclusions
In summary, we demonstrate a versatile design for the development of responsive catalysts that can be selectively activated by various signals. The generic design allows for a straightforward synthesis for the blocking of catalysts with a variety of self-immolative groups. The self-immolative design enables efficient activation of the pro-catalysts with a reasonable amount of signal. We demonstrated an application of our catalyst design by synthesizing three examples of blocked proline catalysts, of which one can be activated by H2O2 (PP-1), the second by light (PP-2) and the third by the enzyme penicillin acylase (PP-3). PP-1, PP-2 and PP-3 were used to control the rate of an aldol reaction: activation of the pro-catalyst showed the same increase in rate as when the native catalyst was added. The pro-catalysts could also be activated efficiently during the reaction, allowing temporal control over the reaction rate. The versatility of the pro-catalyst system was made more apparent as we additionally used PP-1 to control a Michael reaction. This reaction does not show any conversion without active catalyst. Activation of PP-1 enables the reaction to proceed, even though the reaction rate is lower than when the native proline catalyst is used. The reaction can be initialized at any moment in a mixture of the starting materials for the Michael reaction with PP-1 and leads to an immediate response in reaction rate. The pro-catalysts can thus autonomously respond to signals from the environment. A next step in responsive catalysis would be autonomous or controlled deactivation of the activated catalyst, to allow for transient signal amplification. We are currently working on a system to gain reversible control over catalyst activity. We envision that our design for organocatalysts that can be selectively activated by specific signals may be applied to create systems and materials that can respond to their environment, as the next step towards communication between artificial chemical systems.Experimental
Instrumentation and characterization
NMR spectra were recorded on an Agilent-400 MR DD2 (400 MHz for 1H and 100.5 MHz for 13C) at 298 K using residual protonated solvent signals as internal standard.Data availability
Data relevant to the findings of this study are available from the corresponding author on request.Author contributions
C. M., F. T. and R. E. conceived the research. R. E. directed the research. C. M. and F. T. carried out the experiments. R. E. revised the manuscript; C. M. and F. T. wrote the manuscript, all authors commented on the work and the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research through a VIDI grant and by the European Research Council (ERC consolidator grant 726381). We thank Dr Stephen Eustace for help with NMR measurements.References
- E. Whitehead, Prog. Biophys. Mol. Biol., 1970, 21, 321 CrossRef PubMed.
- T. Traut, Allosteric regulatory enzymes, Springer, Boston, 2008 Search PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC.
- (a) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef PubMed; (b) S. Neri, S. Garcia Martin, C. Pezzato and L. J. Prins, J. Am. Chem. Soc., 2017, 139, 1794 CrossRef PubMed; (c) H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kučanda, D. Manna, P. K. Kundu, J.-W. Lee, P. Král and R. Klajn, Nat. Nanotechnol., 2016, 11, 82 CrossRef PubMed; (d) C. Maity, W. E. Hendriksen, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998 CrossRef PubMed.
- N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508 CrossRef PubMed.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161 CrossRef PubMed.
- J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340 CrossRef PubMed.
- A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492 CrossRef PubMed.
- G. I. Peterson, M. B. Larsen and A. J. Boydston, Macromolecules, 2012, 45, 7317 CrossRef.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309 CrossRef PubMed.
- F. Trausel, C. Maity, J. M. Poolman, D. S. J. Kouwenberg, F. Versluis, J. H. van Esch and R. Eelkema, Nat. Commun., 2017, 8, 879 CrossRef PubMed.
- (a) D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358 CrossRef PubMed; (b) F. Trausel, B. Fan, S. A. P. van Rossum, J. H. van Esch and R. Eelkema, Adv. Synth. Catal., 2018 DOI:10.1002/adsc.201800342; (c) F. Trausel, F. Versluis, C. Maity, J. M. Poolman, M. Lovrak, J. H. van Esch and R. Eelkema, Acc. Chem. Res., 2016, 49, 1440 CrossRef PubMed.
- B. List, Tetrahedron, 2002, 58, 5573 CrossRef.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511 CrossRef PubMed.
- K. G. M. M. Alberti and P. Z. Zimmet, Diabetic Med., 1998, 15, 539 CrossRef PubMed.
- A. Sreekumar, L. M. Poisson, T. M. Rajendiran, A. P. Khan, Q. Cao, J. Yu, B. Laxman, R. Mehra, R. J. Lonigro, Y. Li, M. K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X. Cao, J. Byun, G. S. Omenn, D. Ghosh, S. Pennathur, D. C. Alexander, A. Berger, J. R. Shuster, J. T. Wei, S. Varambally, C. Beecher and A. M. Chinnaiyan, Nature, 2009, 457, 910 CrossRef PubMed.
- A. Cordova, W. Notz and C. F. Barbas III, Chem. Commun., 2002, 3024 RSC.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef.
- X. Zhang and K. N. Houk, J. Org. Chem., 2005, 70, 9712 CrossRef PubMed.
- (a) B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423 CrossRef PubMed; (b) H. Yang and M. W. Wong, Org. Biomol. Chem., 2012, 10, 3229 RSC.
- Z. Zheng, B. L. Perkins and B. Ni, J. Am. Chem. Soc., 2010, 132, 50 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General methods, synthesis of the pro-prolines, release of the catalysts by the signals, aldol reactions, Michael reactions, reaction kinetics, NMR spectra. See DOI: 10.1039/c8sc02019a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |