
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriple action Pt(IV) derivatives of cisplatin: a new class of potent anticancer agents that overcome resistance†
Emanuele
Petruzzella
 a,
Roman
Sirota
a,
Irene
Solazzo
b,
Valentina
Gandin
*b and
Dan
Gibson
*a
a,
Roman
Sirota
a,
Irene
Solazzo
b,
Valentina
Gandin
*b and
Dan
Gibson
*a
aInstitute for Drug Research, School of Pharmacy, The Hebrew University, Jerusalem, 91120, Israel. E-mail: dang@ekmd.huji.ac.il
bDipartimento di Scienze del Farmaco, Università di Padova, Via Marzolo 5, 35131, Padova, Italy
First published on 24th April 2018
Abstract
A series of triple action Pt(IV) prodrugs was designed to test the hypothesis that multi-action compounds, where each bioactive moiety intervenes in several cellular processes, might be more effective than a single agent at killing cancer cells. In particular, “triple action” Pt(IV) derivatives of cisplatin, where the axial ligands are inhibitors of cyclooxygenase (COXi), histone deacetylase (HDACi) or pyruvate dehydrogenase kinase (PDKi) were developed. All compounds, ctc-[Pt(NH3)2(COXi)(PDKi)Cl2], ctc-[Pt(NH3)2(COXi)(HDACi)Cl2] and ctc-[Pt(NH3)2(HDACi)(PDKi)Cl2], where COXi = aspirin or ibuprofen, PDKi = dichloroacetate and HDACi = valproate or phenylbutyrate, were significantly more cytotoxic than cisplatin against all cell lines of an in-house panel of human cancer cells. They were particularly effective against thyroid and pancreatic cancer cells in monolayer cytotoxicity tests. Remarkably, in 3D spheroid cancer cell cultures, some triple action compounds showed an antitumor potency up to 50-fold higher than cisplatin against a KRAS mutated pancreatic cancer cell line (PSN-1 cells). Standard biochemical assays classically employed to explore structure activity relationships of platinum drugs, such as cellular uptake and binding to potential biological targets (DNA, HDAC, mitochondria, and COX), do not provide linear correlations with the overall cytotoxicity data. We observed a preferential induction of ROS production and of an anti-mitochondrial effect in cancer cells compared to rapidly dividing non-cancerous cells. Thus, we propose that these new triple action Pt(IV) derivatives of cisplatin are a novel and interesting class of potent and selective cytotoxic agents.
Introduction
Cisplatin, carboplatin and oxaliplatin (Fig. 1) are square planar Pt(II) anti-cancer drugs that are in widespread clinical use.1 The first two compounds are particularly effective against testicular and ovarian cancers, but are also widely used for many other indications in combination with other drugs, while oxaliplatin is used to treat colorectal cancers. Platinum drugs are used in 50% of all clinical regimens.2 The platinum drugs are believed to trigger cancer cell death by losing their non-am(m)ine ligands and covalently binding to two adjacent guanines on the same strand of the nuclear DNA. Binding to the DNA creates a significant distortion of the double helical structure, and the responses of the cancer cells to the distortion determine the fate of the cancer cells.3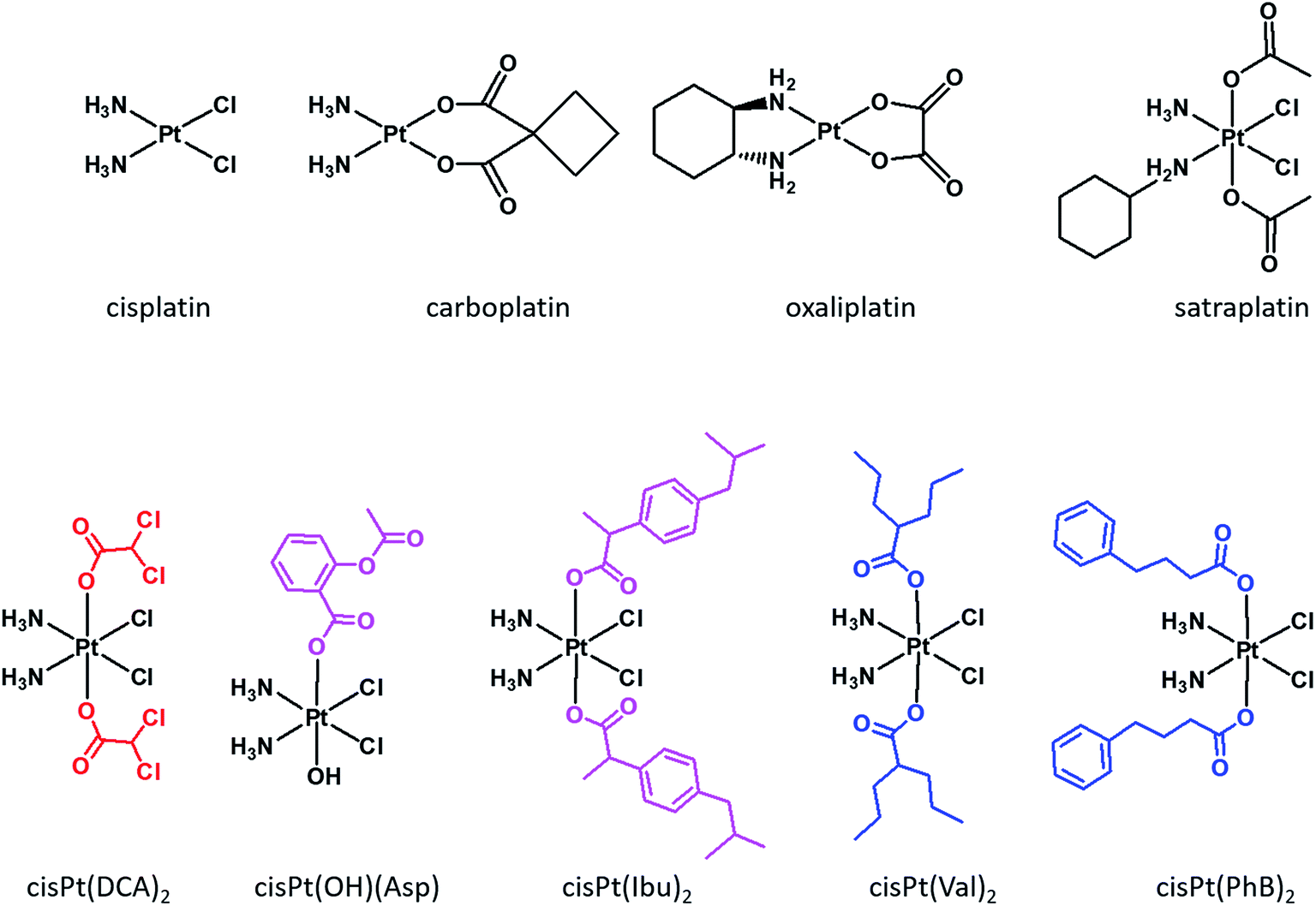 | ||
| Fig. 1 Structures of the three Pt(II) drugs approved by FDA as well as satraplatin (top) and examples of dual action Pt(IV) prodrugs with DPKi (DCA), COXis (ASP and Ibu) and HDACis (Val and PhB) (bottom). | ||
Despite its efficacy and success, severe side effects and resistance limit the usage of cisplatin.4 In order to overcome resistance, clinicians administer the platinum drugs in combination with other drugs.5 The main advantage of combination chemotherapy is that several anti-proliferative agents that have different mechanisms of action and different cellular targets, attack the tumors thereby increasing the chances of killing the cancer cells and of overcoming resistance to a single drug. The problem with co-administration of several drugs is that each drug has a different pharmacokinetic profile, thus complicating the prediction of the overall therapeutic outcome.
Although currently all the Pt drugs used in the clinic are Pt(II) complexes, Pt(IV) complexes have recently attracted a lot of attention since their chemical properties allow great flexibility in the design of novel drugs, including multi-target drugs.6 Pt(IV) complexes, such as satraplatin (Fig. 1), are low spin d6 octahedral complexes that are kinetically more inert than their Pt(II) precursors and may be administered orally, a feature that can improve the quality of life of patients and reduce hospitalization costs.7 Pt(IV) complexes are believed to act as prodrugs that are activated inside the cancer cells by reductive elimination resulting in the concurrent release the two axial ligands as well as the square planar Pt(II) drug (Scheme 1).8 “Dual action” Pt(IV) prodrugs that are Pt(IV) derivatives of cisplatin, carboplatin or oxaliplatin with bioactive axial ligands have gained popularity, unfolding interesting horizons for improving Pt-based chemotherapy.9
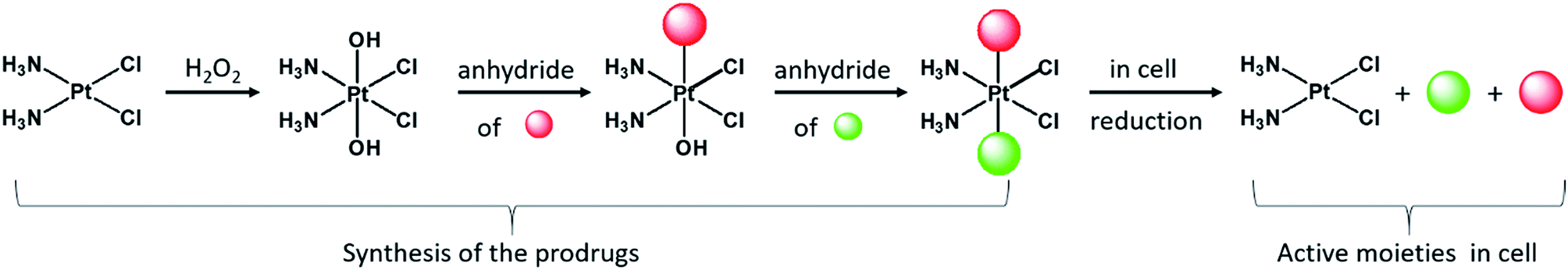 | ||
| Scheme 1 The Pt(IV) prodrugs were prepared from cisplatin that was oxidized to oxoplatin with H2O2. Two successive carboxylations of oxoplatin with the different anhydrides yield “triple action” prodrugs that are reduced in the cell releasing cisplatin as well as the two bioactive axial ligands. | ||
Mitaplatin is the Pt(IV) derivative of cisplatin that has two dichloroacetate (DCA) moieties in the axial positions (Fig. 1).10 DCA is an orphan drug that inhibits the pyruvate dehydrogenase kinase (PDK), an enzyme that phosphorylates the pyruvate dehydrogenase complex (PDHC), a key enzyme in the cellular respiration process. The inhibition of PDHC by PDK shifts the cellular metabolism from glucose oxidation to glycolysis (Warburg effect).11 The inhibition of PDK reverses the Warburg effect, compromising the survival of the tumour cells. Mitaplatin combines the action of cisplatin that binds to the DNA with that of DCA that acts at mitochondrial level.
Pt(IV) complexes with axial histone deacetylase (HDAC) inhibitors are another example of dual action compounds. Histones are proteins that control the structure of chromatin and nucleosomes. Their deacetylation results in a more open chromatin structure, exposing DNA to potential platination. HDAC inhibitors can reactivate dormant tumor-suppressor genes.12 HDAC inhibitors, such as vorinostat or belinostat, are best characterized as anticancer agents. Some HDACis were approved by the FDA for treatment of lymphomas, and others are in clinical trials.13 Tethering the HDACis phenylbutyrate (PhB) or valproate (Val) to the axial positions of the Pt(IV) derivative of cisplatin yielded very potent cytotoxic agents being up to 100-fold more potent than cisplatin.14–16 PhB is also an inhibitor of PDK, and its combination with DCA led to an increase of PDHC activity that is greater than the sum of the effects of the single drugs alone.17
Another class of dual action prodrugs are Pt(IV) complexes with cyclooxygenase (COX) inhibitors in the axial positions. The rationale for the design of such complexes stems from the observation that cancer is often associated with chronic inflammation and that COX-2 is overexpressed in many tumors and is involved in their initiation and progression.18 Also, there are reports that combining cisplatin with COXis enhances the potency of cisplatin and/or reduces the side effects.19,20 The nonsteroidal anti-inflammatory drugs (NSAIDs) that were conjugated to the axial positions of Pt(IV) complexes include, ibuprofen and indomethacin. The Pt(IV) derivative of cisplatin with one axial aspirin and one hydroxido ligand is a potent cytotoxic agent and also exhibits anti-inflammatory activity.21,22
The Pt(IV) derivative of cisplatin with two ibuprofens in the axial positions, cis-Pt(Ibu)2 (Fig. 1) is a very active compound, with IC50 values in the nM range.23 Moreover, combining HDAC and COX-2 inhibitors increases the efficacy by a factor of 10 compared with the single drugs.24
The hypotheses underlying the preparation of triple action compounds were also reinforced by the high effectiveness of a quadruple action Pt(IV) prodrug that simultaneously releases four different bioactive moieties inside the cancer cell. Its cytotoxicity against cancer cells was up to 450-fold more potent than cisplatin against KRAS mutated cancer cells.25 Surprisingly, we are not aware of any reports on systematic studies on triple action Pt(IV) based prodrugs.
Herein we report on the design, synthesis and biological studies on eight triple action Pt(IV) compounds, that upon reduction release three different bioactive moieties in the cancer cell. There are a lot of possible rationales and options for choosing the two different bioactive axial ligands. We elected to begin by choosing the above mentioned bioactive ligands because of the demonstrated ability of each one, by a different mechanism, to act synergistically with cisplatin to enhance cytotoxicity. The eight compounds reported here represent some possible combinations of these ligands. The compounds are Pt(IV) derivatives of cisplatin that can be divided to three classes; (a) those with one PDKi (DCA) and one COXi (Asp or Ibu), (b) those with one PDKi and one HDACi (PhB or Val) and (c) those with one HDACi and one COXi.
Results and discussion
Synthesis
The complexes presented in this work are Pt(IV) derivatives of cisplatin with two different bioactive ligands in the axial positions making them “triple action” Pt(IV) anticancer prodrug candidates. They were obtained from oxoplatin by two successive carboxylations with the anhydrides of the axial ligands (Fig. 2). The first carboxylation was performed in DMSO using a low concentration of oxoplatin (∼10 mg mL−1)26 and the second carboxylation was performed in acetonitrile at higher concentrations.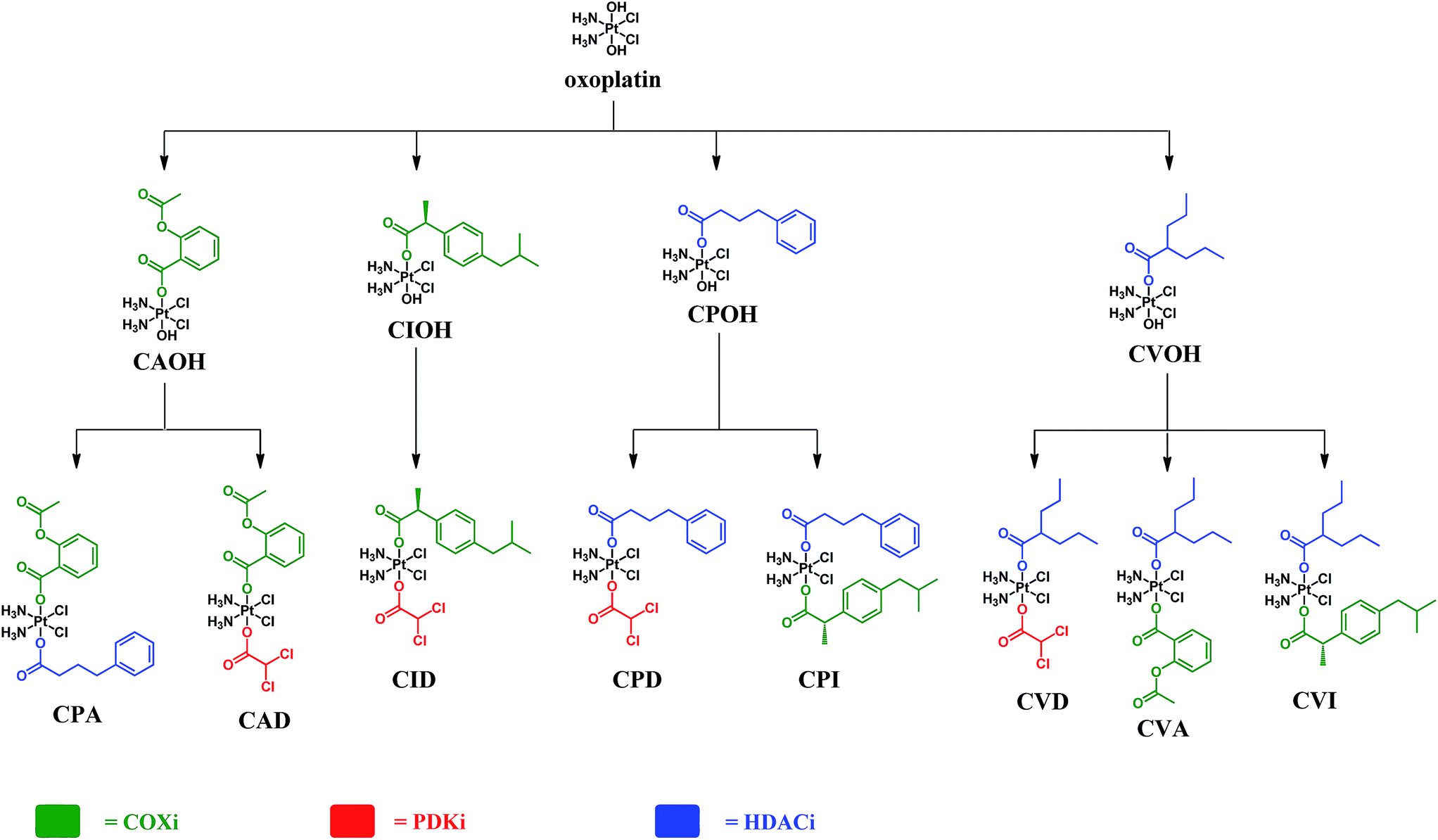 | ||
| Fig. 2 Schemes of the synthetic procedures of the triple action Pt(IV) derivatives of cisplatin. | ||
The efficiency of monocarboxylations depends of the solubility of the Pt(IV) dihydroxido complex and on the reactivity of the anhydride. Oxoplatin is poorly soluble in common solvents making it difficult to isolate the monocarboxylated complexes, especially with highly reactive anhydrides. Initially, the reaction mixtures were pale yellow suspensions and upon completion of the first carboxylation the reaction mixtures turned into pale yellow solutions. The 195Pt NMR resonances at ∼1050 ppm, are consistent with monocarboxylato derivatives of oxoplatin. The monocarboxylation derivatives with aspirin (CAOH), ibuprofen (CIOH), phenylbutyrate (CPOH), and valproate (CVOH) were used, without further purification, as intermediates for the following step.
Because of the high reactivity of the DCA anhydride we were not able to selectively obtain the monocarboxylato derivative ctc-[Pt(NH3)2(DCA)(OH)Cl2]. Aspirin anhydride reacted with oxoplatin very slowly, so the concentration of oxoplatin was raised to ∼15 mg mL−1, to increase the speed of reaction. The second carboxylation reactions were performed in acetonitrile and monitored by RP-HPLC. All the biscarboxylato derivatives were characterized by 1H, 195Pt NMR and HPLC and their purity was ascertained by elemental analysis (Fig. S1–S8 and Table S1†). The compounds did not fly in the ESIMS.
These compounds can be divided into three classes based on the combination of the biological activity of their axial ligands: the first has one COXi and one PDKi (CAD and CID); the second has one HDACi and one COXi (CPA, CPI, CVA and CVI) and the third has one HDACi and one PDKi (CPD and CVD) – Fig. 2.
Stability studies
The stability of the eight triple action compounds in phosphate buffer solution at 37 °C was monitored by HPLC. CID, CPD, CVD and CVI were stable under these conditions. After one day, CPA, CVA and CPI underwent slow ∼10, 20 and 25% degradation, respectively. Degradation continued in the following three days. CAD was the least stable compound, with >50% degradation after the first day, and complete degradation in the following days. The nature of these degradation processes is currently under investigation in order to elucidate their mechanism. Interestingly, compounds with an aspirin ligand were less stable, leading us to suspect that this ligand may play a central role in the degradation process.Cytotoxicity
The cytotoxicity of the eight triple action compounds was assessed in a panel of human cancer cell lines comprised of colorectal (HCT-15), thyroid (BCPAP), pancreatic (PSN-1), and colon (LoVo) cancer cell lines by the MTT test, using cisplatin and oxaliplatin as references. IC50 values were calculated from the dose–survival curves obtained after 72 h of drug treatment (Table 1). In addition, the cytotoxic properties of the complexes were evaluated in a pair of tumor cell lines selected for their sensitivity and resistance to cisplatin (ovarian adenocarcinoma cells 2008/C13*). We calculated cross-resistance profiles by means of the resistance factor (RF), defined as the ratio between IC50 values calculated for the resistant cells and those obtained with the sensitive ones. Interestingly, regardless of the composition of the triple action compounds, all were more potent than cisplatin in all the cell lines tested and they were more effective than oxaliplatin against thyroid (BCPAP), pancreatic (PSN-1), colon (LoVo) and ovarian (2008) cancer cells. While the average IC50 of cisplatin over the six cell lines was 12.46 μM, those for the triple action compounds range from 0.37–1.46 μM. Three cell lines are more sensitive than the others to the triple action compounds. The average IC50 values of all eight compounds against BCPAP (0.14 μM), PSN-1 (0.26 μM) and C13* (1.13 μM) are 51, 71 and 20 times lower than those of cisplatin while against HCT-15, 2008 and LoVo they are about 6, 3 and 24 fold better.| Class | Cmpd | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT-15 colon | BCPAP thyroid | PSN-1 pancreas | LoVo colon | 2008 ovary | C13* ovary | RF | Average | ||
| COXi, PDKi | CAD | 1.03 ± 0.25 | 0.06 ± 0.005 | 0.06 ± 0.008 | 0.755 ± 0.06 | 0.61 ± 0.19 | 1.66 ± 0.09 | 2.7 | 0.70 |
| CID | 0.65 ± 0.17 | 0.14 ± 0.04 | 0.08 ± 0.01 | 0.285 ± 0.02 | 0.32 ± 0.09 | 0.97 ± 0.22 | 3.0 | 0.41 | |
| HDACi, COXi | CPA | 1.86 ± 0.41 | 0.06 ± 0.004 | 0.09 ± 0.02 | 0.055 ± 0.01 | 0.29 ± 0.11 | 0.43 ± 0.12 | 1.5 | 0.46 |
| CPI | 4.98 ± 1.25 | 0.08 ± 0.01 | 0.92 ± 0.2 | 0.211 ± 0.08 | 0.89 ± 0.19 | 1.65 ± 0.11 | 1.9 | 1.46 | |
| CVA | 4.51 ± 0.85 | 0.01 ± 0.003 | 0.13 ± 0.04 | 0.97 ± 0.08 | 0.69 ± 0.08 | 0.77 ± 0.04 | 1.1 | 1.18 | |
| CVI | 3.98 ± 0.89 | 0.68 ± 0.08 | 0.07 ± 0.01 | 0.034 ± 0.03 | 1.35 ± 0.29 | 1.61 ± 0.42 | 1.2 | 1.29 | |
| HDACi, PDKi | CPD | 1.06 ± 0.13 | 0.11 ± 0.03 | 0.10 ± 0.02 | 0.019 ± 0.01 | 0.33 ± 0.04 | 0.58 ± 0.06 | 1.8 | 0.37 |
| CVD | 3.11 ± 1.02 | 0.01 ± 0.004 | 0.61 ± 0.1 | 0.757 ± 0.23 | 1.12 ± 0.20 | 1.39 ± 0.37 | 1.2 | 1.17 | |
| Average IC 50 | 2.65 | 0.14 | 0.26 | 0.39 | 1.13 | 0.71 | |||
| Ref. | CDDP | 15.28 ± 2.63 | 7.38 ± 1.53 | 18.25 ± 3.11 | 9.15 ± 2.07 | 2.22 ± 1.02 | 22.52 ± 3.15 | 10.1 | 12.47 |
| OXP | 1.15 ± 0.43 | 4.37 ± 1.07 | 8.25 ± 3.42 | 1.01 ± 0.34 | 1.53 ± 0.88 | 3.06 ± 1.00 | 2.0 | 3.23 | |
The triple action compounds comprise three groups, based on the nature of their bioactive axial moieties. The first group combines COX and PDK inhibitors and consists of CAD and CID. These two compounds have average IC50 values of 0.70 and 0.41 μM, respectively, about 18 and 31 times lower than cisplatin (average IC50 = 12.47 μM) and roughly 5 and 8 times lower than oxaliplatin (average IC50 = 3.23 μM). Both compounds were extremely effective against the thyroid (BCPAP, IC50 = 0.06 and 0.14 μM, respectively) and pancreatic cancer (PSN-1, IC50 = 0.06 and 0.08 μM, respectively) cell lines. CAD and CID were somewhat less active against the resistant ovarian cancer C13* but still much better than cisplatin. They were slightly cross-resistant with cisplatin (RF = 2.7 and 3.0, respectively).
The second group of triple action compounds combines HDAC and COX inhibitors, and consists of four compounds: CPA, CPI, CVA, and CVI. The average IC50 against all tested cell lines of CPA (0.46 μM) is 27 fold lower than that of cisplatin, while those for CPI, CVA, and CVI are ca. 10 times more potent than cisplatin. CPA is 7 times more effective than oxaliplatin and CPI, CPV, and CVI are about 3 times more potent than oxaliplatin. These compounds are less potent against colorectal cancer HCT-15 cells (low μM IC50s) but extremely potent against thyroid and pancreatic cancers, with some IC50s under 100 nM. Against the colorectal HCT-15 cancer cells, the compounds were more potent than cisplatin. In the ovarian cancer cell pair (2008 and C13* cells), they showed no cross resistance with cisplatin, eliciting RF values lower than 2.
The last class of compounds consists of CPD and CVD that combine HDAC and PDK inhibitors. The average IC50 values for CPD (0.37 μM) are 34 and 9-fold more potent than cisplatin and oxaliplatin, respectively, and 3 times more potent than CVD. Excluding HCT-15 cells, CPD has sub-micromolar IC50 values against all tested cell lines. Replacing PhB with Val (as the HDACi) resulted in reduced potency of CVD compared to CPD in all cell lines, with the exception of thyroid tumor cells, against which CVD has a low nM IC50. Both compounds retained their activity against cisplatin-resistant ovarian cancer cells with RF values lower than 2.
In order to assess whether triple action compounds are more active than their dual action analogues, we synthesized and screened five dual action compounds that have one bioactive ligand in an axial position and an inert acetato group in the other. We used HDACis (PhB and Val), PDKi (DCA) or COXis (Asp and Ibu) as the bioactive ligands. Complexes CPAc, CVAc, CDAc, CAAc, and CIAc, are depicted in Fig. 3. Although they are not the exact analogues of the triple action compounds (differ in lipophilicity, solubility etc.), the dual action compounds are all capable of delivering the cisplatin and one bioactive ligand into the cancer cells. Thus, a comparison with the triple action compounds could be helpful for assessing the relevance of adding the second bioactive ligand.
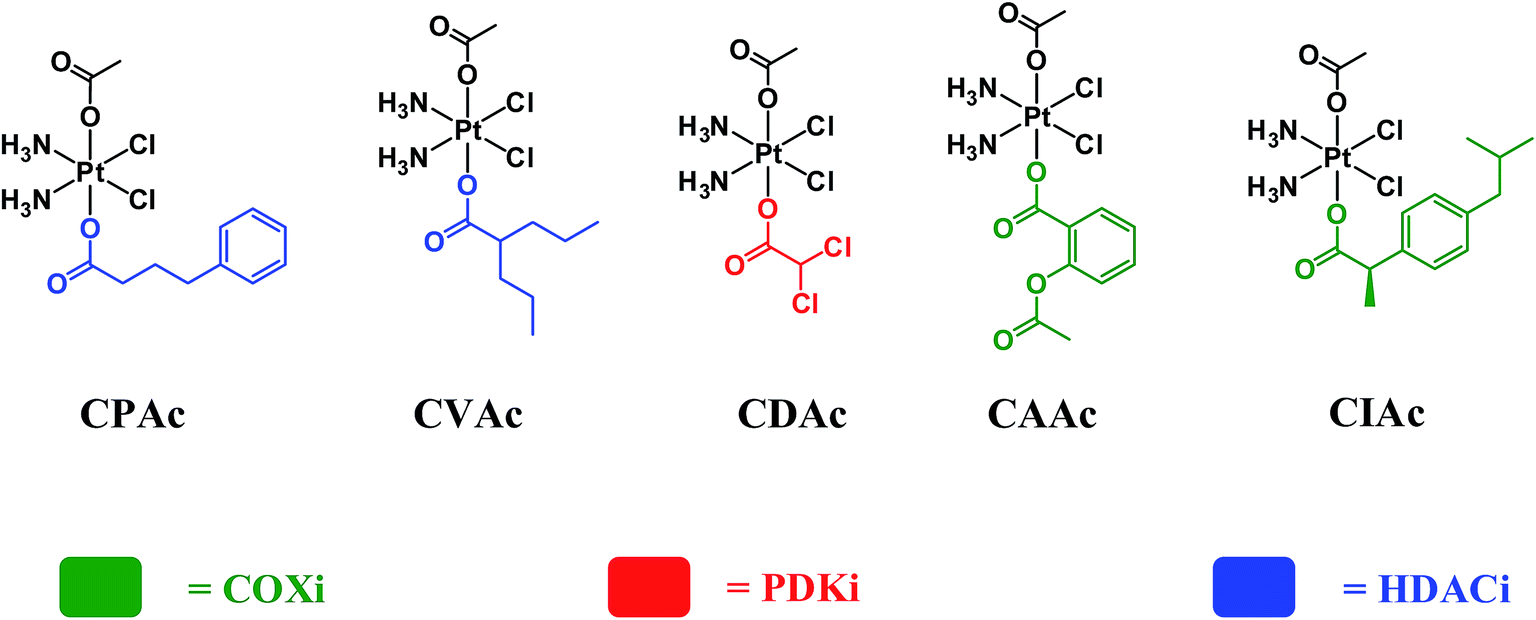 | ||
| Fig. 3 Chemical structures of acetato dual action compounds CPAc, CVAc, CDAc, CAAc and CIAc. | ||
Comparing the average IC50 values of the five dual action compounds to the eight triple action compounds and to cisplatin (Table S2†), we see that in general the triple action compounds are more potent than the dual action compounds that in turn are more effective than cisplatin and oxaliplatin. Only in some cases, dual action compounds were slightly more effective than their corresponding triple action complexes (Table S2†).
All the triple action compounds are potent cytotoxic agents and hence it is difficult to select lead compounds from these data. Although the average IC50 values of the compounds are very impressive, we are interested primarily in identifying specific compounds that are effective against specific cancers. Judging from their IC50 values, CID, CPD, CPA and CAD seem like the most potent compounds.
To assess whether combining the three bioactive components in a single triple action prodrug is superior to exposing the cells to a mixture of its components, we evaluated the cytotoxicity of a combination of cisplatin, PhB and DCA at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio against five cancer cell lines. The results reported in Table S4† clearly confirm that co-treatment of cancer cells with cisplatin, PhB and DCA, yielded IC50 values similar to those of cisplatin itself (significantly higher than CPD) and did not result in an synergistic or additive cytotoxic effect.
:1:1 ratio against five cancer cell lines. The results reported in Table S4† clearly confirm that co-treatment of cancer cells with cisplatin, PhB and DCA, yielded IC50 values similar to those of cisplatin itself (significantly higher than CPD) and did not result in an synergistic or additive cytotoxic effect.
To assess further the potential of these compounds, we screened them against 3D spheroids of PSN-1 pancreatic cancer cells. Conventional 2D cell cultures cannot mimic the complexity of in vivo tumors and hence they are not always good predictors of in vivo activity. On the other hand, 3D cell cultures possess several features such as cell–cell interactions, hypoxia, drug penetration, response and resistance, and production/deposition of the extracellular matrix that more closely mimic in vivo tumors, being potentially more predictive for in vivo results than conventional 2D cell cultures.27
We tested the triple action Pt(IV) cisplatin derivatives on spheroids of PSN-1 pancreatic tumor cells, using cisplatin as reference (Table 2). We chose to conduct the 3D spheroids screen on the pancreatic cancer cell line (rather than the thyroid cancer cell line) since pancreatic cancer survival rates are dramatically lower than those for thyroid cancer, and there is interest in agents that are effective against pancreatic cancer. All the newly synthesized triple action compounds were significantly more active than cisplatin. Three compounds, CVA, CVI and CPD, have sub-μM IC50 values, being more than 50 times more active than cisplatin. The other five compound have IC50s ranging from ca. 3 to 8 μM, being 15 to 8 fold more potent than cisplatin.
| Class | IC50 (μM) | |
|---|---|---|
| Compound | PSN-1 3D | |
| COXi, PDKi | CAD | 3.73 ± 0.35 |
| CID | 5.25 ± 0.71 | |
| HDACi, COXi | CPA | 7.75 ± 0.01 |
| CPI | 3.63 ± 0.53 | |
| CVA | 0.79 ± 0.06 | |
| CVI | 0.93 ± 0.46 | |
| HDACi, PDKi | CPD | 0.95 ± 1.12 |
| CVD | 3.38 ± 0.88 | |
| Reference | CDDP | 52.56 ± 3.78 |
Since all the triple action compounds are not or just slightly cross-resistant with cisplatin we decided to check whether they can also circumvent oxaliplatin resistance. We screened them against LoVo colon cancer cells and the oxaliplatin resistant form LoVo OXP (Table S3†). Four compounds circumvented resistance to oxaliplatin having RF values < 2: the two compounds combining HDAC and PDK inhibitors (CPD and CVD) and two combining the HDACis (PhB and Val) with the COXi aspirin. Interestingly, all compounds containing ibuprofen had very high RF values. CPD and CPA exhibit remarkably low IC50 values, and are the most active among the all eight triple action compounds.
One of the major drawbacks of chemotherapeutics, including platinum drugs, are the side effects originating in part from toxic effects to non-cancerous cells. We measured the cytotoxicity of the compounds against non-cancerous cells (HEK293) and calculated the selectivity index (SI) defined as the ratio of the IC50s of the non-cancerous cells to those of the cancer cells that are more sensitive to triple action complexes, namely PSN-1, BCPAP and LoVo cells. The cytotoxicity data for HEK293 cells and the selectivity indices appear in Table 3.
| Class | Compound | IC50 (μM) HEK293 | Selectivity index (SI) | ||
|---|---|---|---|---|---|
| PSN-1 | BCPAP | LoVo | |||
| COXi, PDKi | CAD | 0.09 ± 0.02 | 1.5 | 1.5 | 0.1 |
| CID | 0.07 ± 0.03 | 0.9 | 0.5 | 0.2 | |
| HDACi, COXi | CPA | 0.11 ± 0.01 | 1.2 | 1.8 | 2.0 |
| CPI | 0.13 ± 0.03 | 0.1 | 1.6 | 0.6 | |
| CVA | 0.59 ± 0.21 | 4.5 | 59.0 | 0.6 | |
| CVI | 0.47 ± 0.08 | 6.7 | 0.7 | 13.8 | |
| HDACi, PDKi | CPD | 1.56 ± 0.22 | 15.6 | 14.2 | 82.1 |
| CVD | 1.98 ± 0.26 | 3.2 | 198.0 | 2.6 | |
| Reference | CDDP | 19.62 ± 2.33 | 1.1 | 2.6 | 2.1 |
CPD has high selectivity indices against PSN-1 (15.6), BCPAP (14.2) and LoVo (82.1) cancer cells. CVA and CVD have high SI against BCPAP cancer cells (59 and 198 respectively) and showed a moderate selectivity against PSN-1 cells (SI 4.5 and 3.2 respectively). CVI has SI of 6.7 and 13.8 against PSN-1 and LoVo respectively. The remaining complexes have no selectivity towards those cancer cells.
Among the eight compounds studied, CPD is the most promising compound since it is one of the most active compounds in 2D and 3D cell cultures, is active against cisplatin- and oxaliplatin-resistant cell lines, and has high selectivity towards BCPAP and the KRAS mutated PSN-1 and LoVo cancer cell lines.
Biochemical assays
When trying to kill a cancer cell with a single agent such as cisplatin it is intuitively appealing to assume that increased cellular accumulation should correlate with enhanced platination of nuclear DNA resulting in higher potency. Many reports describe attempts to increase the cellular accumulation of cisplatin, including using Pt(IV) prodrugs with lipophilic ligands.28 The presence of lipophilic ligands in the axial positions increases the lipophilicity of the whole Pt(IV) complex, improving the cellular internalization of both cisplatin and of the anionic axial ligands (“synergistic accumulation”).14 This in turn is expected to increase the efficiency of DNA plantation by cisplatin.We measured the cellular uptake and the DNA platination levels of the triple action compounds and cisplatin in PSN-1 cells (Fig. 4A and B). Clearly, there is no correlation between the cellular accumulation and the potency. The most potent compound, CAD, has the lowest accumulation level among the triple action compounds, significantly lower than CID and CVI whose IC50 values are similar to that of CAD. This suggests that CAD might be the most effective compound at killing the PSN-1 cancer cells. Although we might expect that higher cellular accumulation would result in higher platination levels of nuclear DNA, this is not the case here (Fig. 4B). Interestingly, there is no correlation between platination levels of the nuclear DNA and the cytotoxicity. CPI is the least potent triple action compound yet has the highest levels of DNA platination. One of the most potent compounds, CAD, has the lowest platination level of all compounds, 5.6 fold lower than cisplatin although it is 304 fold more cytotoxic than cisplatin. DNA platination by the triple action compounds suggests that the prodrugs were reduced in the cell releasing cisplatin that modified the DNA. In summary, with the triple action compounds we find no correlation between the cytotoxicity and cellular accumulation or DNA platination.
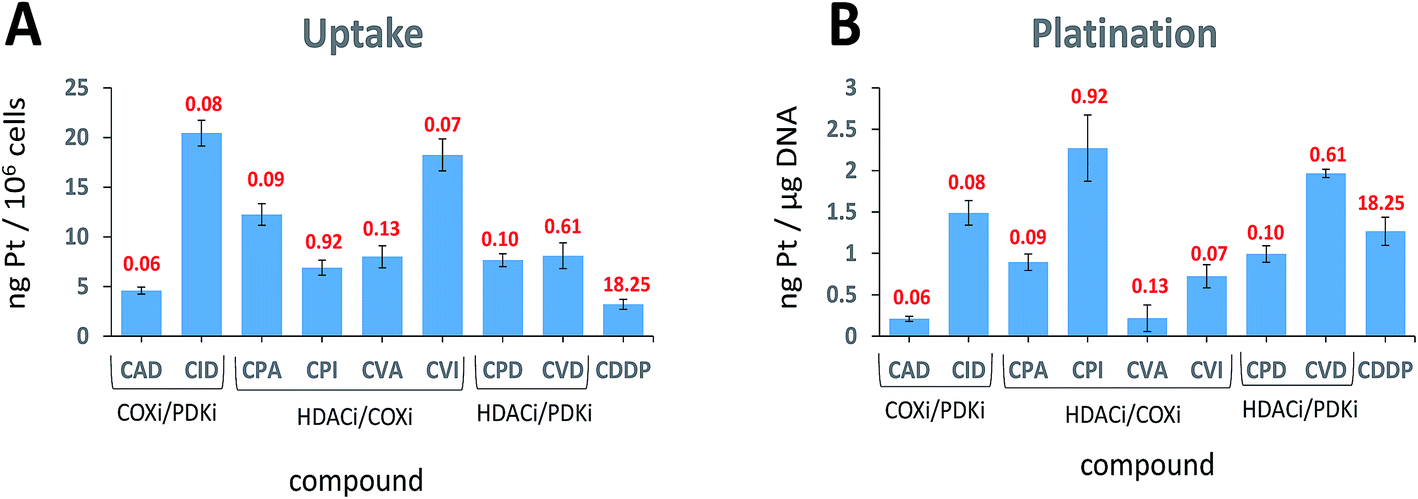 | ||
| Fig. 4 Cellular uptake (A) and DNA platination (B). (A) PSN-1 cells were incubated for 24 h with 1 μM of tested complexes. (B) Platination levels of nuclear DNA extracts. PSN-1 cells were treated for 24 h with 1 μM of tested complexes. DNA was extracted, quantified and the amount of Pt bound to DNA was estimated by GF–AAS. The IC50 values of the compounds against the PSN-1 cells are depicted in red. | ||
Although we realize that each bioactive component can interact with several cellular targets, it is interesting to see whether they actually inhibit in the cancer cells the enzymes they were designed to inhibit.
We investigated their ability to inhibit HDAC activity (Fig. 5A). As expected, only the compounds with a PhB or Val ligands inhibited HDAC activity. PhB and Val in the axial positions were equally effective at inhibiting HDAC activity. Again, there is no correlation between inhibition of HDAC activity and cytotoxicity.
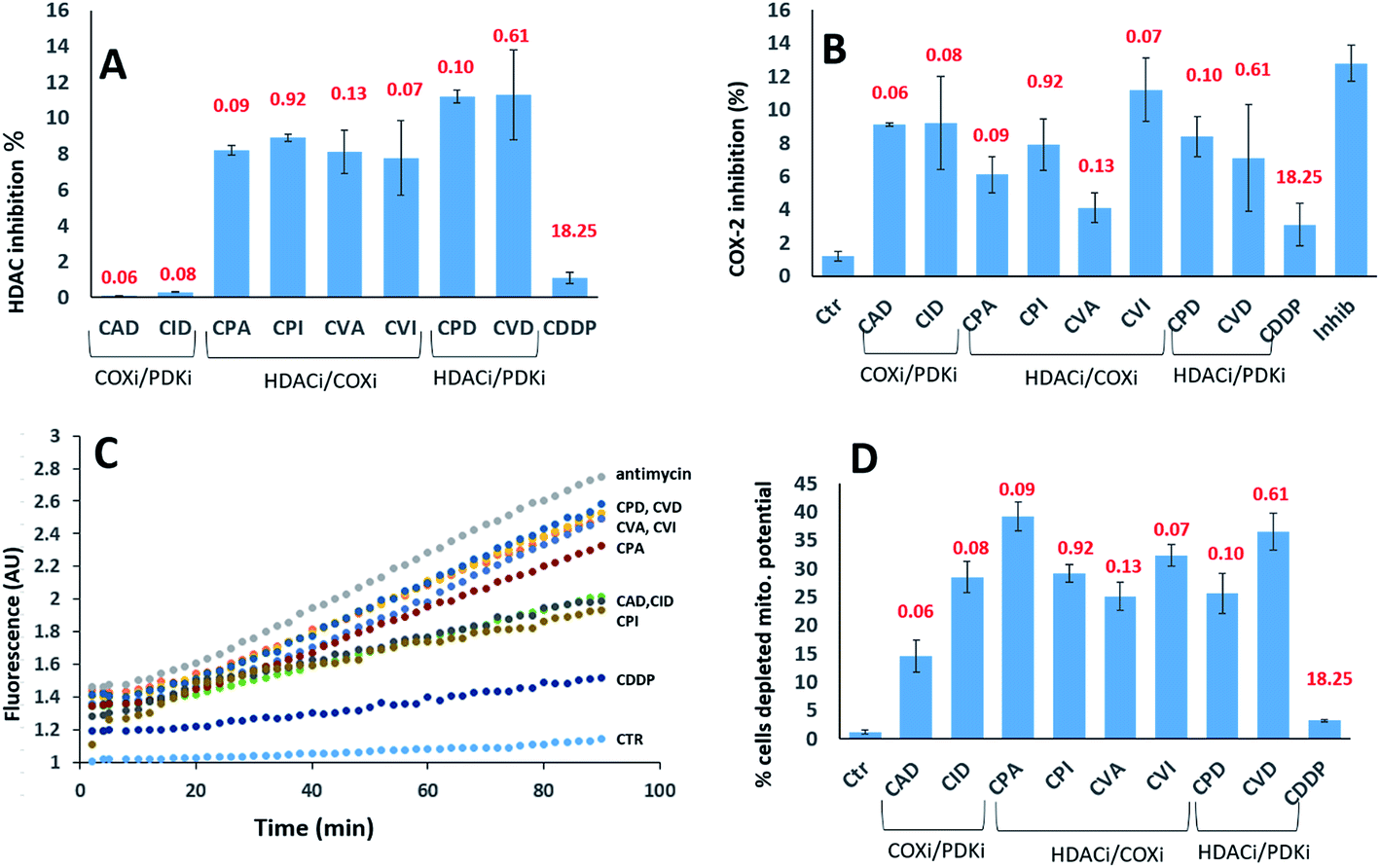 | ||
| Fig. 5 (A) HDAC inhibition. PSN-1 cells were incubated for 24 h with 1 μM of tested complexes. The HDAC activity was determined in cells by FLUOR DE LYS® HDAC fluorometric activity assay kit (Enzo Life Sciences) following the manufacturers' instructions. In all cases, data are the means of at least three independent experiments. (B) COX-2 expression. PSN-1 cells were incubated for 24 h with 1 μM of tested complexes. The inhibition of COX-2 was measured by using COX activity assay kit (Cayman Chemical, Ann Arbor, USA) following the manufacturers' instructions. In all cases, data are the means of at least three independent experiments. Error bars indicate SD. (C) ROS production. PSN-1 cells were preincubated in PBS/10 mM glucose medium for 20 min at 37 °C in the presence of 10 μM CM–DCFDA and then treated with equimolar doses (10 μM) of Pt(IV) derivatives. The fluorescence of DCF was measured. In all cases, data are the means of three independent experiments. (D) Effects on cellular mitochondrial membrane potential. PSN-1 cells were treated for 24 h with 1 μM of tested compounds. The percentage of cells with hypopolarized mitochondrial membrane potential was determined by Mito-ID® Membrane Potential Kit. The IC50 values of the compounds against the PSN-1 cells are depicted in red. | ||
We assayed their ability to inhibit the expression of COX-2 in PSN-1 treated cells (Fig. 5B). Previous reports indicated that aspirin–Pt(IV) derivatives maintained their COX inhibition effect,21 while ibuprofen–Pt(IV) derivatives did not.22 In the case of triple action Pt(IV) compounds, CVI was the most effective COXi (11.2% inhibition), while its aspirin analogue CVA was the least effective (4.1% inhibition). A similar trend was observed for CPI and CPA, causing 7.9 and 6.1% inhibition respectively. COXi/PDKi functionalized compounds CAD and CID caused ∼9% COX-2 inhibition, with the aspirin derivative slightly more potent than the ibuprofen analogue. Surprisingly, CPD and CDV complexes that do not have any COXi moiety caused 8.4 and 7.1% COX-2 inhibition, respectively. These results suggest that also complexes not containing any COXi moiety can somehow inhibit the activity of COX-2 in cancer cells.
It is well known that DCA treatment causes an imbalance in the redox homeostasis, leading to increased production of reactive species, de-structuration of mitochondrial pathophysiology and loss of mitochondrial membrane potential.27 Therefore, we assayed the effects induced by tested complexes on mitochondria by measuring the cellular ROS production (Fig. 5C) and mitochondrial membrane potential (Fig. 5D). All tested complexes were able to induce a time-dependent increase in the cellular basal ROS production. The ROS production induced by triple action complexes was significantly superior than that induced by cisplatin but it was lower than that provoked by a classical inhibitor of the mitochondrial respiratory chain at the level of complex III, namely antimycin A. The most effective complexes were CVD and CPD, being able to induce an increase in cellular basal ROS production that is only slightly inferior to that elicited by antimycin A. It is well known that a substantial increase of ROS production at mitochondrial level leads to the hypopolarization of the mitochondrial membrane. Consistently, all the triple action compounds are significantly more active than their cisplatin precursor in inducing mitochondrial membrane depolarization in cancer cells. Complexes CPA, CVD and CVI induced hypopolarization in 30–40% of the cancer cell population. CID, CPD, CPI and CVA induced hypopolarization in 25–30%, while CAD in ∼15%. These results clearly showed that all complexes increase the cancer cell population with depolarized mitochondria. Interestingly, a mitochondrial effect was observed also for complexes lacking of DCA and PhB moieties that are known to act on mitochondria. This behaviour can be explained by taking into account that COX-2 modulators can affect mitochondrial homeostasis by raising the cellular basal ROS production, resulting in mitochondrial membrane potential loss. It is important to note that in rapidly dividing non-cancerous HEK293 cells, all triple action Pt(IV) complexes, and in particular CPD and CVD, were barely effective in inducing cellular ROS production and in affecting the mitochondrial membrane potential (Fig. S13,† panel A and B). This is in sharp contrast to the results obtained with the pancreatic cancer cells (PSN-1), demonstrating their preferential activity toward cancer cells compared with non-tumor cells.
Conclusions
In this work we studied three classes of triple action Pt(IV) derivatives of cisplatin having; (1) COXi/PDKi (CAD and CID); (2) HDACi/COXi (CPA, CPI, CVA and CVI); (3) HDACi/PDKi (CPD and CVD). All compounds were more cytotoxic than cisplatin against all cell lines tested and were particularly effective against thyroid and pancreatic cancer cells.As 2D cytotoxicity studies indicate that all eight compounds were potent cytotoxic agents, further experiments, such as 3D cytotoxicity and selectivity studies were performed to help select lead compounds. Among the eight compounds studied, CPD is the most promising compound, being active against cisplatin- and oxaliplatin-resistant cell lines, and having high selectivity towards PSN-1, BCPAP and LoVo cancer cell lines.
The triple action Pt(IV) prodrugs were designed to test the hypothesis that multi-action compounds, where each bioactive moiety intervenes in several cellular processes, might be more effective than a single agent as several mechanisms may be involved in killing the cells.
Thus, it is not surprising that the standard biochemical essays employed for platinum drugs such as investigating the cellular uptake and the possible biological targets (DNA, HDAC, and COX) do not provide correlations with the overall cytotoxicity data. As all compounds are active, it is unlikely that there is a single mechanism of action common to all compounds. The potency of the triple action compounds probably depends primarily on the specific cellular interactions of each of the components and on possible synergistic effects to which the cell responds, and not necessarily just on the amount of the drug that accumulates in the cell.
Interestingly, the most promising triple action compound, CPD, shares the bioactive axial ligands (DCA and PhB) with the potent and selective dinuclear quadruple action Pt(IV) complex we recently described.25
In light of the cellular studies reported above, we believe that triple action Pt(IV) derivatives of cisplatin represent a novel and interesting class of potent and selective cytotoxic agents.
Conflicts of interest
There are no conflicts to declare.List of abbreviations
| Asp | Aspirin |
| CDDP | Cisplatin |
| DCA | Dichloroacetic acid/dichloroacetate |
| Ibu | Ibuprofen |
| OAc | Acetate |
| PhB | Phenylbutyric acid/phenylbutyrate |
| Val | Valproic acid/valproate |
Acknowledgements
DG acknowledges the support of the Israel Science Foundation. V. G. acknowledges the University of Padova for financial support (grant 60A04-4015/15, DOR2016 and DOR2017). The authors wish to acknowledge Maisalon Ishan on her help with the synthesis of the compounds.References
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- A. M. Florea and D. Busselberg, Cancers, 2011, 3, 1351–1371 CrossRef CAS PubMed.
- (a) T. Boulikas and M. Vougiouka, Oncol. Rep., 2004, 11, 559–595 CAS; (b) L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- E. Gabano, M. Ravera and D. Osella, Dalton Trans., 2014, 43, 9813–9820 RSC.
- H. Choy, Expert Rev. Anticancer Ther., 2006, 6, 973–982 CrossRef CAS PubMed.
- (a) M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS; (b) E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- (a) W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, J. Am. Chem. Soc., 2005, 127, 1382–1383 CrossRef CAS PubMed; (b) L. L. Ma, R. Ma, Y. P. Wang, X. Y. Zhu, J. L. Zhang, H. C. Chan, X. F. Chen, W. J. Zhang, S. K. Chiu and G. Y. Zhu, Chem. Commun., 2015, 51, 6301–6304 RSC; (c) K. R. Barnes, A. Kutikov and S. J. Lippard, Chem. Biol., 2004, 11, 557–564 CrossRef CAS PubMed; (d) V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, J. Inorg. Biochem., 2014, 140, 72–79 CrossRef CAS PubMed; (e) A. Curci, N. Denora, R. M. Iacobazzi, N. Ditaranto, J. D. Hoeschele, N. Margiotta and G. Natile, Inorg. Chim. Acta, 2018, 472, 221–228 CrossRef CAS; (f) A. R. Z. Almotairy, V. Gandin, L. Morrison, C. Marzano, D. Montagner and A. Erxleben, J. Inorg. Biochem., 2017, 177, 1–7 CrossRef CAS PubMed.
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS.
- R. Ferriero and N. Brunetti-Pierri, Oncotarget, 2013, 4, 804–805 CrossRef PubMed.
- A. Villar-Garea and M. Esteller, Int. J. Cancer, 2004, 112, 171–178 CrossRef CAS PubMed.
- (a) M. R. Acharya, A. Sparreboom, J. Venitz and W. D. Figg, Mol. Pharmacol., 2005, 68, 917–932 CrossRef CAS PubMed; (b) T. Beckers, C. Burkhardt, H. Wieland, P. Gimmnich, T. Ciossek, T. Maier and K. Sanders, Int. J. Cancer, 2007, 121, 1138–1148 CrossRef CAS PubMed; (c) A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- R. Raveendran, J. P. Braude, E. Wexselblatt, V. Novohradsky, O. Stuchlikova, V. Brabec, V. Gandin and D. Gibson, Chem. Sci., 2016, 7, 2381–2391 RSC.
- M. Alessio, I. Zanellato, I. Bonarrigo, E. Gabano, M. Ravera and D. Osella, J. Inorg. Biochem., 2013, 129, 52–57 CrossRef CAS PubMed.
- J. Yang, X. Sun, W. Mao, M. Sui, J. Tang and Y. Shen, Mol. Pharm., 2012, 9, 2793–2800 CrossRef CAS PubMed.
- R. Ferriero, C. Iannuzzi, G. Manco and N. Brunetti-Pierri, J. Inherited Metab. Dis., 2015, 38, 895–904 CrossRef CAS PubMed.
- N. Ghosh, R. Chaki, V. Mandal and S. C. Mandal, Pharmacol. Rep., 2010, 62, 233–244 CrossRef CAS PubMed.
- (a) D. W. Knapp, N. W. Glickman, W. R. Widmer, D. B. DeNicola, L. G. Adams, T. Kuczek, P. L. Bonney, A. E. DeGortari, C. Han and L. T. Glickman, Cancer Chemother. Pharmacol., 2000, 46, 221–226 CrossRef CAS PubMed; (b) P. A. Boria, D. J. Murry, P. F. Bennett, N. W. Glickman, P. W. Snyder, B. L. Merkel, D. L. Schlittler, A. J. Mutsaers, R. M. Thomas and D. W. Knapp, J. Am. Vet. Med. Assoc., 2004, 224, 388–394 CrossRef CAS PubMed; (c) K. Hattori, R. Matsushita, K. Kimura, Y. Abe and E. Nakashima, Biol. Pharm. Bull., 2001, 24(10), 1214–1217 CrossRef CAS PubMed; (d) H. Endo, M. Yano, Y. Okumura and H. Kido, Cell Death Dis., 2015, 5, e1027 CrossRef PubMed.
- M. N. Bijman, C. A. Hermelink, M. P. van Berkel, A. C. Laan, M. L. Janmaat, G. J. Peters and E. Boven, Biochem. Pharmacol., 2008, 75, 427 CrossRef CAS PubMed.
- R. K. Pathak, S. Marrache, J. H. Choi, T. B. Berding and S. Dhar, Angew. Chem., Int. Ed., 2014, 53, 1963–1967 CrossRef CAS PubMed.
- Q. Cheng, H. Shi, H. Wang, Y. Min, J. Wang and Y. Liu, Chem. Commun., 2014, 50, 7427–7430 RSC.
- W. Neumann, B. C. Crews, M. B. Sarosi, C. M. Daniel, K. Ghebreselasie, M. S. Scholz, L. J. Marnett and E. Hey-Hawkins, ChemMedChem, 2015, 10, 183–192 CrossRef CAS PubMed.
- O. Peulen, A. Gonzalez, P. Peixoto, A. Turtoi, D. Mottet, P. Delvenne and V. Castronovo, PLoS One, 2013, 8, e75102 CAS.
- E. Petruzzella, J. P. Braude, J. R. Aldrich-Wright, V. Gandin and D. Gibson, Angew. Chem., Int. Ed., 2017, 56, 11539–11544 CrossRef CAS PubMed.
- (a) C. F. Chin, Q. Tian, M. I. Setyawati, W. Fang, E. S. Tan, D. T. Leong and W. H. Ang, J. Med. Chem., 2012, 55, 7571 CrossRef CAS PubMed; (b) S. Q. Yap, C. F. Chin, A. H. Hong Thng, Y. Y. Pang, H. K. Ho and W. H. Ang, ChemMedChem, 2017, 12, 300 CrossRef CAS PubMed.
- S. Kankotia and P. W. Stacpoole, Biochim. Biophys. Acta, 2014, 1846, 617–629 CAS.
- I. Zanellato, I. Bonarrigo, D. Colangelo, E. Gabano, M. Ravera, M. Alessio and D. Osella, J. Inorg. Biochem., 2014, 140, 219–227 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00428e |
| This journal is © The Royal Society of Chemistry 2018 |