
Tri- and di-fluoroethylation of alkenes by visible light photoredox catalysis†
Xiao-Yang
Pan
ab,
Yue
Zhao
ab,
Hong-An
Qu
c,
Jin-Hong
Lin
 *b,
Xiao-Chun
Hang
*a and
Ji-Chang
Xiao
*b
*b,
Xiao-Chun
Hang
*a and
Ji-Chang
Xiao
*b
aKey Laboratory of Flexible Electronics (KLOFE) and Institute of Advanced Materials (IAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211800, China. E-mail: iamxchhang@njtech.edu.cn; Tel: (+86)25-8358-7982
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: jlin@sioc.ac.cn; jchxiao@sioc.ac.cn; Tel: (+86)21-5492-5541 Tel: +86-21-54925340
cShandong NHU Amino Acid Co., LTD, China
First published on 6th March 2018
Abstract
The tri- and di-fluoroethylation of alkenes with sulfonium salts, (Ph2S+CH2RF TfO−) (RF = CF3 or HCF2), by visible light photoredox catalysis to give tri-/di-fluoroethyl alkenes or methoxytri-/di-fluoroethylation products are described. It was found that varying the reaction solvent led to changes in the reaction path.
Since both trifluoromethyl (CF3) and difluoromethyl (HCF2) substituents have emerged as valuable functionalities for modulating the physicochemical properties of pharmaceuticals and agrochemicals,1 significant efforts have been directed towards the development of efficient methods for the incorporation of these two groups into organic molecules. Although both tri-/di-fluoromethylation2 and tri-/di-fluoroethylation are efficient approaches for CF3 or HCF2 incorporation, tri-/di-fluoroethylation (only 2,2,2-trifluoroethylation and 2,2-difluoroethylation, respectively, are under discussion here) has been far less explored compared with tri-/di-fluoromethylation. In particular, only limited examples have been disclosed for difluoroethylation.3 Various trifluoroethylation reagents including CF3CH2I,4 CF3CH2OTs,5 CF3CHN2,6 CF3CHCl2,7 (CF3CH2SO2)2Zn,8 CF3CO2H9 and (CF3CH2I+Ar TfO−)10 have been developed, but most of them are volatile, explosive (CF3CHN2) or water sensitive (CF3CH2I+Ar TfO−).11 The only difluoroethylation reagent so far is HCF2CH2I, which is a volatile liquid (bp: 87 °C) and thus could lead to practical inconvenience. Apparently, the development of operationally convenient tri- and di-fluoroethylation reagents is highly desirable.
Tri- and di-fluoroethylation of alkenes are straightforward approaches for CF3 and HCF2 incorporation. In 2013, Carreira and coworkers described the photocatalytic trifluoroethylation of styrenes to give trifluoroethyl alkenes (Scheme 1, eqn (1)).12 Shortly afterwards, the group of Guo found that oxytrifluoroethylation occurred under photocatalytic conditions in the presence of an oxygen source (eqn (1)).13 Xiang et al. disclosed a copper/silver-cocatalyzed oxidative coupling to give β-CF3/HCF2-substituted ketones (eqn (2)).3b Decarboxylation of cinnamic acids catalyzed by copper could also afford trifluoroethyl alkenes using the silver complex as an oxidizing reagent (eqn (3)).14 Wang and coworkers found that microwave conditions could accelerate this process and the varied positions of the CO2H substituent would result in different products (eqn (4)).3c All of these reactions are efficient and attractive, but the use of a volatile reagent (CF3CH2I or HCF2CH2I) is required.
 | ||
| Scheme 1 Tri- and di-fluoroethylation of alkenes. | ||
We have previously shown that tri- and difluoroethyl sulfonium salts, (Ph2S+CH2RF TfO−) (RF = CF3 or HCF2), could act as valuable sulfonium ylide reagents and fluorinated carbene precursors.15 As visible light photoredox catalysis has proven to be a valuable synthetic tool for the generation of radical species from electrophilic reagents,16 we speculated that reactive fluorinated radicals (CF3CH2˙ or HCF2CH2˙) may be produced from these sulfonium salts by visible light photoredox catalysis. In continuation of our research interest in the chemistry of fluorinated organic salts,15,17 we have now investigated the use of these sulfonium salts as reagents for visible light photoredox catalyzed tri- and -di-fluoroethylation of alkenes. Like other fluorinated sulfonium salts,18 these sulfonium salts show sufficient oxidizing power and therefore could enable tri- and di-fluoroethylation. Interestingly, we found that varying reaction solvents led to changes in the reaction process (Scheme 1, eqn (5)). Tri- and di-fluoroethylation occurred to give alkenes in DMAc, while difunctionalization was observed in MeOH.
Our initial attempts at the trifluoroethylation of alkene 1a with trifluoroethylsulfonium salt, [Ph2S+CH2CF3 TfO−] (reagent I), were successful to afford the desired product 2a albeit in a low yield (Table 1, entry 1). The examination of the reaction solvent (entries 1–4) indicated that dimethylacetamide (DMAc) was a suitable solvent (entry 4). A brief survey of the photocatalyst revealed that only Ir(ppy)3 was capable of catalyzing this reaction (entry 4 vs. entries 5–7) probably due to a high reduction potential of Ir(ppy)3 in the excited state (EIV/*III1/2 = −1.73 V vs. SCE).16a The concentration had slight effect on the reaction, and increasing the concentration led to an increase in the yield to 62% (entry 8 vs. entries 4 and 9). Increasing the loading of reagent I (entry 10) or prolonging the reaction time (entry 11) did not increase the yield. This reaction should be accompanied by a deprotonation process, and thus the presence of a base may be favorable. Various organic and inorganic bases were investigated (entries 12–17) and it was found that the use of CuO gave the product in 75% yield (entry 16). The photocatalyst was essential for this transformation, and no product was observed without using the photocatalyst (entry 18).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Base | Yieldb |
| a Reaction conditions: Substrate 1a (0.2 mmol), (Ph2S+CH2CF3 TfO−) (3 equiv.), Ir(ppy)3 (3 mol%) and base (2 equiv.) in solvent irradiated with blue LEDs at room temperature for 12 h. b The yields were determined by 19F NMR spectroscopy. c [Ir(ppy)2(dtbbpy)]PF6 was used as the photocatalyst instead of Ir(ppy)3. d [Ru(bpy)3](PF6)2 was used as the photocatalyst instead of Ir(ppy)3. e [Ru(phen)3](PF6)2 was used as the photocatalyst instead of Ir(ppy)3. f 4 equivalents of reagent I were used. g The reaction time was 18 h. h No photocatalyst was used. | |||
| 1 | MeCN (2 mL) | — | 7 |
| 2 | DMF (2 mL) | — | 44 |
| 3 | DMSO (2 mL) | — | 35 |
| 4 | DMAc (2 mL) | — | 52 |
| 5c | DMAc (2 mL) | — | 0 |
| 6d | DMAc (2 mL) | — | 0 |
| 7e | DMAc (2 mL) | — | 0 |
| 8 | DMAc (1.5 mL) | — | 62 |
| 9 | DMAc (2.5 mL) | — | 49 |
| 10f | DMAc (1.5 mL) | — | 62 |
| 11g | DMAc (1.5 mL) | — | 60 |
| 12 | DMAc (1.5 mL) | iPr2NEt | 14 |
| 13 | DMAc (1.5 mL) | Et3N | 15 |
| 14 | DMAc (1.5 mL) | NaHCO3 | 30 |
| 15 | DMAc (1.5 mL) | KHCO3 | 25 |
| 16 | DMAc (1.5 mL) | CuO | 75 |
| 17 | DMAc (1.5 mL) | ZnO | 48 |
| 18h | DMAc (1.5 mL) | — | 0 |
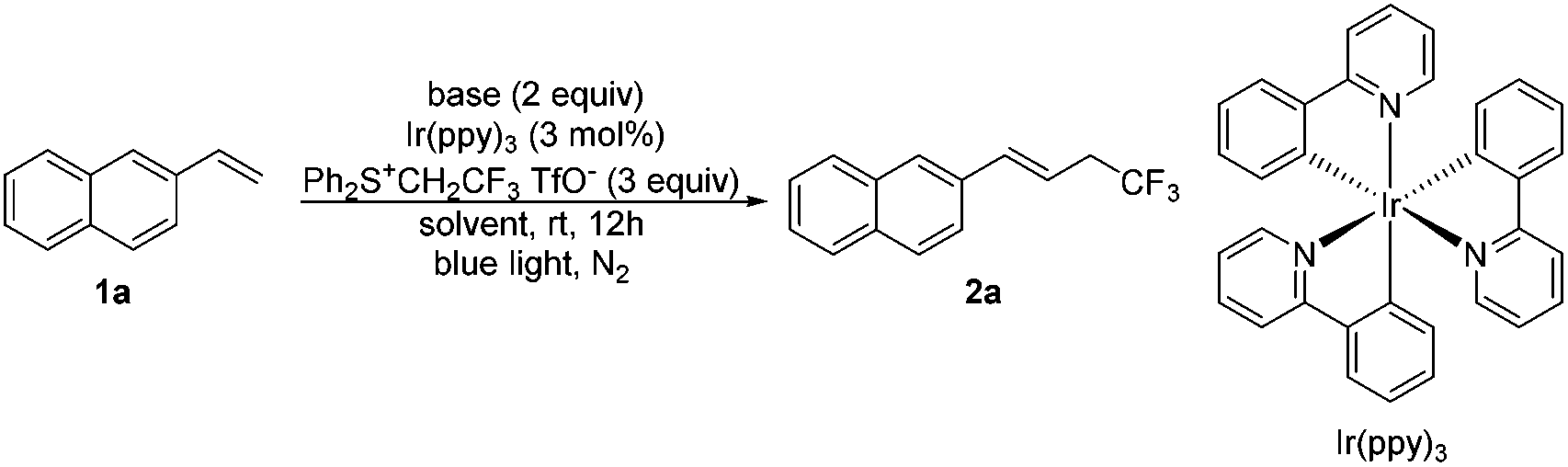
With the optimal conditions (Table 1, entry 16) in hand, we explored the substrate scope of tri- and di-fluoroethylation of alkenes to give CF3CH2- and HCF2CH2-substituted alkenes (Scheme 2). Various aryl alkenes were converted smoothly into the desired trifluoroethylation products (2a–2g), and a good reactivity was observed even for sterically hindered 1,1-disubstituted alkene (2g). The reaction was apparently affected by electronic effects of substituents. A strong electron donating group (2f) or an electron withdrawing group (2h) would suppress the desired conversion. A Cl or CN substituent present in the phenyl group in styrene also resulted in low yields (<30%). The internal alkene was inert towards trifluoroethylation under these conditions, probably due to strong steric effects (2i). No desired product was observed for the transformation of α,β-unsaturated alkene (2j). In the case of alkyl alkenes such as 4-phenyl-1-butene, complex mixtures were obtained partially because deprotonation can occur at two different positions to give regioisomers. Compared with trifluoroethyl sulfonium salt I, difluoroethyl sulfonium salt II shows lower reactivity and therefore a longer reaction time (48 h) was required (3a–3g). Moderate yields were obtained for trifluoroethylation. All the products obtained above (except 2g and 3g) were E-isomers, as indicated by the 1H NMR coupling constant of around 16.0 Hz for the two H atoms in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and by comparing the NMR data with the reported literature values (see the ESI†).
C bond and by comparing the NMR data with the reported literature values (see the ESI†).
 | ||
Scheme 2 Tri- and di-fluoroethylation to give alkenes. Isolated yields. Reaction conditions: Substrate 1a (0.5 mmol), reagent I or II (3 equiv.), Ir(ppy)3 (3 mol%) and CuO (2 equiv.) in DMAc (3 mL) irradiated with blue LEDs at room temperature for 24 h or 48 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The yield was determined by 19F NMR spectroscopy. The yield was determined by 19F NMR spectroscopy. | ||
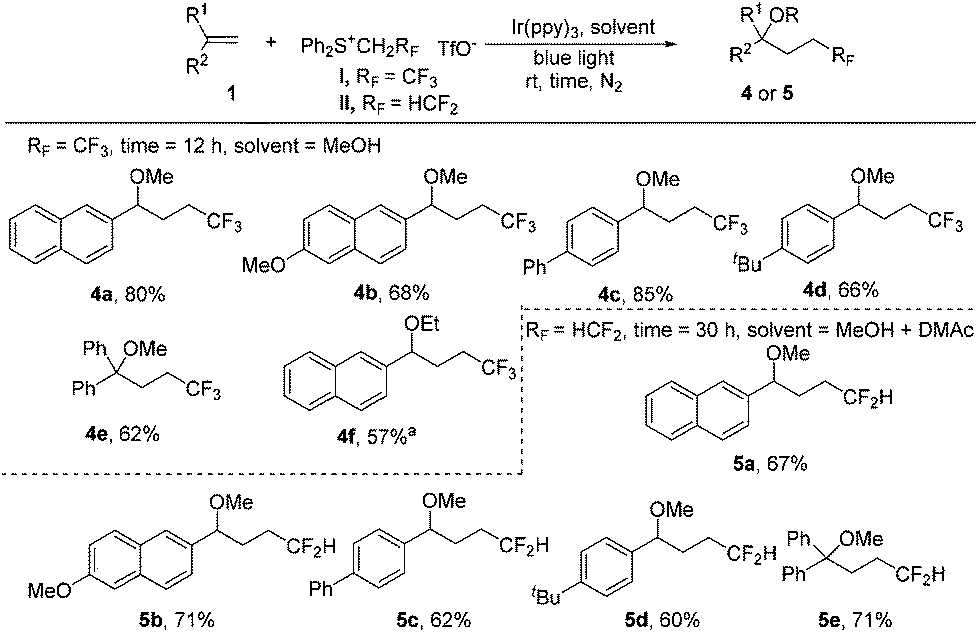
Interestingly, we found that subtle changes in reaction solvents resulted in a different reaction process. The use of methanol as the solvent for trifluoroethylation of the substrate 1a gave the methoxytrifluoroethylation product 4a in 77% 19F NMR yield. Low solubility of sulfonium salt I (completely soluble in DMAc) in MeOH could lead to a decrease in the efficiency of light absorption, meaning that the yield may be increased by reducing the loading of salt I. Indeed, the 19F NMR yield was slightly increased to 82% without the presence of CuO even by reducing the loading of salt I to 2 equiv. The substrate scope of methoxytrifluoroethylation of alkenes was investigated by using 2 equiv. of sulfonium reagents (Scheme 3). Various aryl alkenes were reactive towards methoxytrifluoroethylation and moderate yields were obtained (4a–4e). The use of ethanol instead of methanol as the reaction solvent gave the ethoxytrifluoroethylation product in a low yield. Gratifyingly, 57% yield could be obtained (4f) in the presence of a cosolvent, DMAc. Methoxydifluoroethylation was found to be quite sluggish using methanol as the single solvent. To our delight, the MeOH/DMAc cosolvent could afford the desired products in moderate yields by prolonging the reaction time (5a–5e).
 | ||
| Scheme 3 Methoxytri- and methoxydi-fluoroethylation of alkenes. Isolated yields. Reaction conditions: Substrate 1a (0.5 mmol), reagent I or II (2 equiv.) and Ir(ppy)3 (2 mol%) in DMAc (3 mL) or DMAc/MeOH (v/v = 2 mL/1 mL) irradiated with blue LEDs at room temperature for 12 h or 30 h. aEtOH/DMAc (v/v = 1 mL/2 mL) was used as the solvent instead of MeOH to obtain product 4f. | ||
The redox potentials of trifluoroethylsulfonium salt and difluoroethylsulfonium salt measured by cyclic voltammetry were −1.517 V vs. SCE and −1.237 V vs. SCE, respectively (see the ESI†) (for comparison, the reduction potential of CF3CH2I is −1.70 V),19 indicating that these two sulfonium salts may be reduced by reducing the intermediate, photoexcited complex [Ir(ppy)3*] (EIV/*III1/2 = −1.73 V vs. SCE),16a to generate radical species (CF3CH2˙ and HCF2CH2˙), thus allowing for the above tri- and di-fluoroethylation reactions. Indeed, we found that trifluoroethylation of alkene 1a was dramatically suppressed in the presence of a radical scavenger, TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) (Scheme 4). Moreover, TEMPO-CH2CF3 was obtained in 53% yield, indicating the generation of CF3CH2˙ radicals in this process.
 | ||
| Scheme 4 Evidence for the radical process. aThe yield was determined by 19F NMR spectroscopy; bthe yield was calculated based on TEMPO as the limiting reagent. | ||
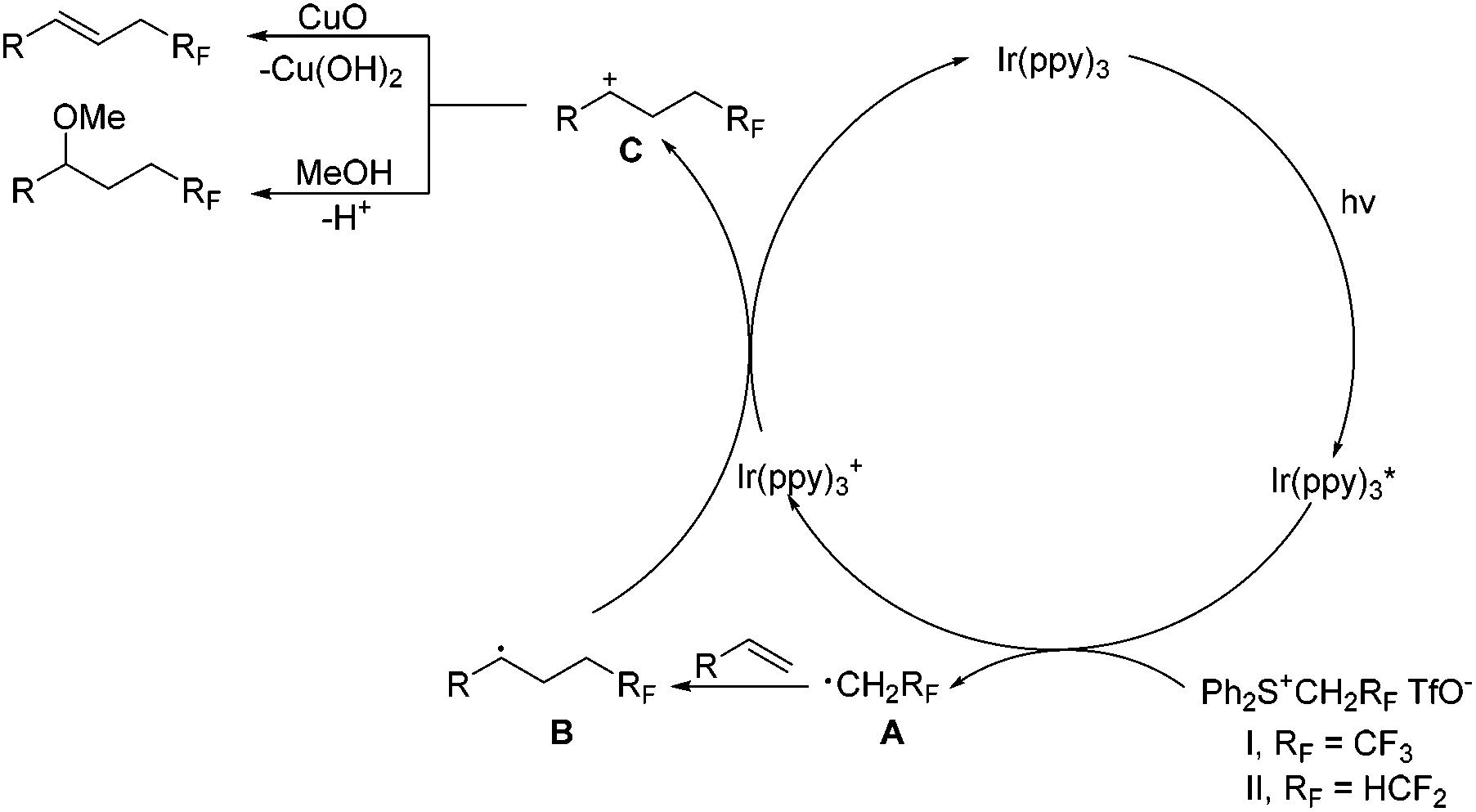
On the basis of the above results, the reaction mechanism is proposed as shown in Scheme 5. Upon irradiation with visible light, Ir(ppy)3 undergoes photoexcitation to give an excited species Ir(ppy)3*, which is a strong reductant (EIV/*III1/2 = −1.73 V vs. SCE)16a and capable of donating an electron to sulfonium salts to generate an oxidized catalyst Ir(ppy)3+ and radical species A, RFCH2˙ (RF = CF3 or HCF2). The radical species A is readily trapped by alkenes to produce the radical intermediate B, oxidation of which with Ir(ppy)3+ releases the photocatalyst and affords the cation intermediate C. Deprotonation of the intermediate C furnishes alkenes, and the nucleophilic attack of methanol gives methoxytri-/di-fluoroethylation products.
 | ||
| Scheme 5 The proposed reaction mechanism. | ||
Conclusions
In summary, we have described tri- and di-fluoroethylation of alkenes with sulfonium salts, (Ph2S+CH2RF TfO−) (RF = CF3 or HCF2), by visible light photoredox catalysis to give tri-/di-fluoroethyl alkenes or methoxytri-/di-fluoroethylation products. It is interesting that varying the reaction solvent led to changes in the reaction process, and difunctionalization of alkenes was observed in the presence of methanol. This work represents the first protocol for the use of convenient sulfonium reagents in the solvent-dependent tri-/di-fluoroethylation reactions. Sulfonium salts, (Ph2S+CH2RF TfO−) (RF = CF3 or HCF2), may become attractive tri-/di-fluoromethylation reagents because of their stability, facile accessibility, and ease of handling.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the National Basic Research Program of China (2015CB931903), the National Natural Science Foundation (21421002, 21472222, 21502214, 21672242), the Chinese Academy of Sciences (XDA02020105, XDA02020106), and the Key Research Program of Frontier Sciences (CAS) (QYZDJ-SSW-SLH049) for financial support.Notes and references
- (a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing, Chichester, 2009 Search PubMed; (c) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2nd edn, 2013 Search PubMed; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (e) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed; (c) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS PubMed; (d) C. Alonso, E. Martinez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed; (e) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed; (f) C.-P. Zhang, Q.-Y. Chen, Y. Guo, J.-C. Xiao and Y.-C. Gu, Coord. Chem. Rev., 2014, 261, 28–72 CrossRef CAS; (g) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139–152 CrossRef CAS; (h) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- (a) X. Zhang and C. Yang, Adv. Synth. Catal., 2015, 357, 2721–2727 CrossRef CAS; (b) N. Yi, H. Zhang, C. Xu, W. Deng, R. Wang, D. Peng, Z. Zeng and J. Xiang, Org. Lett., 2016, 18, 1780–1783 CrossRef CAS PubMed; (c) Y. Zhu, J. Gong and Y. Wang, J. Org. Chem., 2017, 82, 7428–7436 CrossRef CAS PubMed.
- (a) M. Zhu, X. Han, W. Fu, Z. Wang, B. Ji, X.-Q. Hao, M.-P. Song and C. Xu, J. Org. Chem., 2016, 81, 7282–7287 CrossRef CAS PubMed; (b) X. Yu and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 12320–12323 CrossRef CAS PubMed; (c) S.-Y. Yan, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2017, 53, 10287–10290 RSC; (d) Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 1033–1036 CrossRef CAS PubMed; (e) Y.-S. Feng, C.-Q. Xie, W.-L. Qiao and H.-J. Xu, Org. Lett., 2013, 15, 936–939 CrossRef CAS PubMed.
- F. Leng, Y. Wang, H. Li, J. Li, D. Zou, Y. Wu and Y. Wu, Chem. Commun., 2013, 49, 10697–10699 RSC.
- H. Luo, G. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14503–14507 CrossRef CAS PubMed.
- E.-J. Han, Y. Sun, Q. Shen, Q.-Y. Chen, Y. Guo and Y.-G. Huang, Org. Chem. Front., 2015, 2, 1379–1387 RSC.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed.
- (a) B. L. Tóth, S. Kovács, G. Sályi and Z. Novák, Angew. Chem., Int. Ed., 2016, 55, 1988–1992 CrossRef PubMed; (b) J. Yang, Q.-Y. Han, C.-L. Zhao, T. Dong, Z.-Y. Hou, H.-L. Qin and C.-P. Zhang, Org. Biomol. Chem., 2016, 14, 7654–7658 RSC; (c) S. Kovács, B. L. Tóth, G. Borsik, T. Bihari, N. V. May, A. Stirling and Z. Novák, Adv. Synth. Catal., 2017, 359, 527–532 CrossRef; (d) M. Maraswami, S. Pankajakshan, G. Chen and T.-P. Loh, Org. Lett., 2017, 19, 4223–4226 CrossRef CAS PubMed.
- D. D. DesMarteau and V. Montanari, Chem. Commun., 1998, 2241–2242 RSC.
- L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634–1637 CrossRef CAS PubMed.
- L. Li, M. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen, Y. Guo and Z.-G. Zhao, Org. Lett., 2015, 17, 4714–4717 CrossRef CAS PubMed.
- Y. Zhang, H. Du, M. Zhu, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron Lett., 2017, 58, 880–883 CrossRef CAS.
- (a) Y. Duan, B. Zhou, J.-H. Lin and J.-C. Xiao, Chem. Commun., 2015, 51, 13127–13130 RSC; (b) Y. Duan, J.-H. Lin, J.-C. Xiao and Y. C. Gu, Org. Lett., 2016, 18, 2471–2474 CrossRef CAS PubMed; (c) Y. Duan, J.-H. Lin, J.-C. Xiao and Y.-C. Gu, Org. Chem. Front., 2017, 4, 1917–1920 RSC; (d) Y. Duan, J.-H. Lin, J.-C. Xiao and Y. C. Gu, Chem. Commun., 2017, 53, 3870–3873 RSC; (e) Q.-x. Huang, Q.-T. Zheng, Y. Duan, J.-H. Lin, J.-C. Xiao and X. Zheng, J. Org. Chem., 2017, 82, 8273–8281 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- (a) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC; (b) X.-Y. Deng, J.-H. Lin, J. Zheng and J.-C. Xiao, Chem. Commun., 2015, 51, 8805–8808 RSC; (c) J. Zheng, J.-H. Lin, L.-Y. Yu, Y. Wei, X. Zheng and J.-C. Xiao, Org. Lett., 2015, 17, 6150–6153 CrossRef CAS PubMed; (d) J. Zheng, L. Wang, J.-H. Lin, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2015, 54, 13236–13240 CrossRef CAS PubMed; (e) X.-Y. Deng, J.-H. Lin and J.-C. Xiao, Org. Lett., 2016, 18, 4384–4387 CrossRef CAS PubMed; (f) Z. Deng, J.-H. Lin, J. Cai and J.-C. Xiao, Org. Lett., 2016, 18, 3206–3209 CrossRef CAS PubMed; (g) J. Zheng, R. Cheng, J.-H. Lin, D. H. Yu, L. Ma, L. Jia, L. Zhang, L. Wang, J.-C. Xiao and S. H. Liang, Angew. Chem., Int. Ed., 2017, 56, 3196–3200 CrossRef CAS PubMed; (h) J. Yu, J.-H. Lin and J.-C. Xiao, Angew. Chem., Int. Ed., 2017, 56, 16669–16673 CrossRef CAS PubMed.
- (a) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed; (b) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Medebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed; (c) Q. H. Deng, J. R. Chen, Q. Wei, Q. Q. Zhao, L. Q. Lu and W. J. Xiao, Chem. Commun., 2015, 51, 3537–3540 RSC; (d) R. Tomita, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2015, 54, 12923–12927 CrossRef CAS PubMed; (e) H. Xiang, Q. Zhao, Z. Tang, J. Xiao, P. Xia, C. Wang, C. Yang, X. Chen and H. Yang, Org. Lett., 2017, 19, 146–149 CrossRef CAS PubMed.
- S. I. Scherbinina, O. V. Fedorov, V. V. Levin, V. A. Kokorekin, M. I. Struchkova and A. D. Dilman, J. Org. Chem., 2017, 82, 12967–12974 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00082d |
| This journal is © the Partner Organisations 2018 |