
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral and cationic tungsten(VI) fluoride complexes with tertiary phosphine and arsine coordination†
William
Levason
 ,
Francesco M.
Monzittu
,
Gillian
Reid
* and
Wenjian
Zhang
,
Francesco M.
Monzittu
,
Gillian
Reid
* and
Wenjian
Zhang
School of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 3rd September 2018
Abstract
Reaction of WF6 with AsR3 (R = Me or Et) in anhydrous CH2Cl2 at low temperature forms the neutral seven-coordinate, [WF6(AsR3)] (R = Me, Et), the first arsine complexes of WF6, whilst o-C6H4(EMe2)2 (E = P, As) produces [WF4{o-C6H4(EMe2)2}2][WF7]2. Crystal structures show the latter contain dodecahedral cations, and present the highest oxidation state metal fluoride complexes known (and the highest possible for tungsten) with soft neutral phosphine and arsine coordination.
Fluoride ligands bind very strongly to metal ions and often confer properties that are significantly different to those for analogous complexes bearing heavier halides, also giving rise to quite different chemistries. For example, metal fluoride complexes can exhibit different catalytic behaviour,1,2 can behave as specific fluorinating agents,2,3 while the strong affinity of Lewis acidic centres for fluoride is the basis of new F− sensors4 and the development of metal chelate scaffolds for new classes of 18F carriers for medical imaging (PET).5,6 On the other hand, the soft, neutral group 15 pnictines (ER3; E = P, As, R = alkyl, aryl) have found wide utility as ligands towards many Lewis acids, most typically those from the middle and late d-block in medium or low oxidation states. The capacity to tune the electronic and steric properties of the pnictine strongly influences the resulting chemistry. Through judicious choice of the metal source and the specific pnictine, recent work has established the existence of homoleptic phosphine complexes with Group 1 cations, including the distorted octahedral [M{o-C6H4(PMe2)2}3]+ cations (M = Li, Na)7 and a series of seven- and eight-coordinate complexes with the hard, oxophilic Sc(III) and Y(III) ions.8
Tungsten hexafluoride, which contains tungsten in its highest possible oxidation state, is known to form complexes with a variety of hard Lewis bases, mostly neutral N-donor ligands.9–17 Well-documented examples including [WF6(2-F-py)] (capped trigonal prism),13 [WF6(py)2] (bicapped trigonal prism),14 [WF6(2,2′-bipy)] (structure unknown)16 and the [WF4(2,2′-bipy)2]2+ cation (distorted dodecahedron).15,16 On the other hand, reaction of WF6 with sulfite esters, (RO)2SO (R = alkyl), or phosphites, (RO)3P, result in OR/F exchange to give, for example, [WF5(OR)],12,18 while reaction of WF6 with OMe2 causes O/F exchange, forming [WOF4(OMe2)].12 Similar chemistry has been further developed and exploited to provide a useful entry into WOF4 chemistry, by reaction of WF6 with (Me3Si)2O in MeCN solution to form [WOF4(MeCN)], from which the MeCN is easily displaced by other neutral ligands.19 In contrast, complexes of high oxidation state metal fluorides with soft donor ligands are extremely rare.10 The first phosphine example, [WF6(PMe3)], was briefly described in 196811 and later work17 showed this compound to have a capped trigonal prismatic geometry while [WF6(PMe2Ph)] is a capped octahedron. We recently reported20 the six-coordinate oxide-fluoride species, [WOF4(PMe3)] and seven-coordinate (pentagonal bipyramidal) [WOF4(diphosphine)] (diphosphine = Me2P(CH2)2PMe2 or o-C6H4(PMe2)2); notably, no analogues with arsenic ligands could be formed.19,20 Here we describe the synthesis and properties of arsine complexes of WF6 for the first time, together with the first cationic complexes derived from WF6 with neutral bidentate diphosphine and diarsine ligands, whose structures are confirmed crystallographically.
For the AsR3 (R = Me, Et) complexes the synthesis route involved condensation of WF6in vacuo onto a frozen solution of the appropriate ligand in anhydrous CH2Cl2 at 77 K, then allowing the mixture to warm slowly to room temperature (Scheme 1).
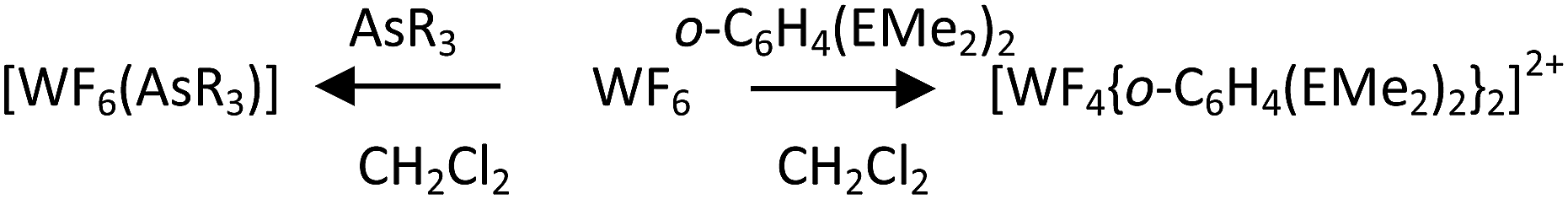 | ||
| Scheme 1 Preparative method. | ||
Upon melting (176 K) the reaction mixture containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of WF6 and AsMe3 turned deep orange-red, and deposited an orange-red powder upon removal of the volatiles in vacuo at room temperature. The reaction solution and products are extremely moisture sensitive, turn dark blue upon trace hydrolysis, and showing varying amounts of [W2O2F9]− and [WOF5]−21 in the 19F{1H} NMR spectra of such solutions. The orange-red solid, identified as [WF6(AsMe3)] by microanalysis, decomposes in a few days in the glove box at ambient temperature, but can be kept in a sealed tube in a freezer (−18 °C) for several weeks; the complex is decomposed by MeCN. The corresponding AsEt3 complex is a viscous orange-red oil and even more reactive, decomposing at room temperature over a few hours and reacting more readily with trace moisture. Neither AsPh3 nor the heavier SbEt3 yielded identifiable products under similar reaction conditions.
:1 molar ratio of WF6 and AsMe3 turned deep orange-red, and deposited an orange-red powder upon removal of the volatiles in vacuo at room temperature. The reaction solution and products are extremely moisture sensitive, turn dark blue upon trace hydrolysis, and showing varying amounts of [W2O2F9]− and [WOF5]−21 in the 19F{1H} NMR spectra of such solutions. The orange-red solid, identified as [WF6(AsMe3)] by microanalysis, decomposes in a few days in the glove box at ambient temperature, but can be kept in a sealed tube in a freezer (−18 °C) for several weeks; the complex is decomposed by MeCN. The corresponding AsEt3 complex is a viscous orange-red oil and even more reactive, decomposing at room temperature over a few hours and reacting more readily with trace moisture. Neither AsPh3 nor the heavier SbEt3 yielded identifiable products under similar reaction conditions.
The 19F{1H} NMR spectra of [WF6(AsR3)] show singlet resonances at +130.8 (R = Me) and +134.4 ppm (R = Et). They did not exhibit 183W satellites, but the chemical shifts may be compared with those in [WF6(PMe3)], δ = +133.617 and WF6 itself, δ = +167.0.16 The 19F{1H} spectra are little changed on cooling the sample to 180 K, indicating fluxionality down to low temperatures. Fluxionality is also evident in the 19F{1H} NMR spectra of the pyridine complexes [WF6(R-py)] (R = H or F) at ambient temperatures, but on cooling the solutions the separate resonances of the inequivalent fluorines of capped trigonal prismatic geometries are resolved.13,14 The IR spectra have very broad strong features at 610 (R = Me) and 622 cm−1 (R = Et), assigned to overlapping W–F stretches. The UV/visible spectrum of [WF6(AsMe3)] shows a very broad absorption at ∼22700 cm−1, which accounts for the orange-red colour and may be assigned as a ligand to metal charge transfer, As(σ) → W(d), since the F(π) → W(d) transitions are expected to lie in the far-UV region.22
In an attempt to increase the phosphine/arsine coordination and develop further the reaction chemistry with WF6, the rigid o-phenylene ligands, o-C6H4(EMe2)2 (E = P or As) were employed. These are amongst the strongest σ-donor neutral pnictines and are pre-organised for chelation. The reaction of WF6 with o-C6H4(AsMe2)2 or o-C6H4(PMe2)2 in frozen anhydrous CH2Cl2 solution in either a 1:1 or 2:1 molar ratio gave, on melting, orange-red or orange-yellow solutions, respectively, from which similarly coloured powders precipitated on concentration of the solutions in vacuo. Microanalytical data and the IR spectra obtained lead to a formulation of [WF4{o-C6H4(EMe2)2}2][WF7]2 (E = P, As). In particular, the IR spectra show very strong, broad features at ∼615 cm−1 and a sharper, medium intensity band at ∼330 cm−1, characteristic of [WF7]−.23 Weaker bands at ∼650(sh) and ∼575 cm−1 are tentatively assigned at ν(WF) in the cations. The UV-visible spectrum of solid [WF4{o-C6H4(AsMe2)2}2][WF7]2 shows broad features at 20800 and 31500 cm−1, the former assigned as As(σ) → W(d) and the latter to the π → π* transition of the aryl ring.24 In [WF4{o-C6H4(PMe2)2}2][WF7]2 the corresponding transitions lie at 23100, 31650 cm−1, the higher energy of the former compared to that in the diarsine analogue is consistent with higher electronegativity of the P(σ) orbital.25
Obtaining solution spectroscopic data and growing crystals for X-ray analysis was very challenging as a result of the extreme moisture sensitivity of the samples. The complexes were poorly soluble in CH2Cl2 and the dilute solutions rapidly decompose with loss of the colour. However, they are more soluble in MeCN and decomposition is slower in this medium. Several batches of crystals were grown by evaporation of MeCN solutions in the glove box. X-ray crystal structure solution revealed that all contained well defined [WF4{o-C6H4(EMe2)2}2]2+ cations, but contain various anions, sometimes disordered. Disordered MeCN was also present in some crystals. These results are reminiscent of the [WF4(2,2′-bipy)2]2+ systems described above.15,16 The structures of the cations are shown in Fig. 1 and 2. The cation in [WF4{o-C6H4(PMe2)2}2][WOF5]2 is a distorted dodecahedron with (cis) F-W-F angles ∼94° and <P–W–P ∼ 73°, the latter reflecting the constrained bite angle of the chelating ligand. The d(W–F) of 1.91(3)–1.94(3) Å are longer than those in either WF6 (1.826(2) Å)26 or in the six-coordinate [WOF4(OPPh3)] 1.857(3)–1.871(3) Å,19 but similar to those in the seven-coordinate [WOF4(Me2PCH2CH2PMe2)], 1.923(9)–1.960(4) Å.20 The d(W–P) = 2.5584(18)–2.5714(17) Å, are also similar to those in the latter complex (2.572(17)–2.592(18) Å). The cation geometry is very similar to that in the isoelectronic [TaF4{o-C6H4(PMe2)2}2]+.24 The [WOF5]− anions were disordered.
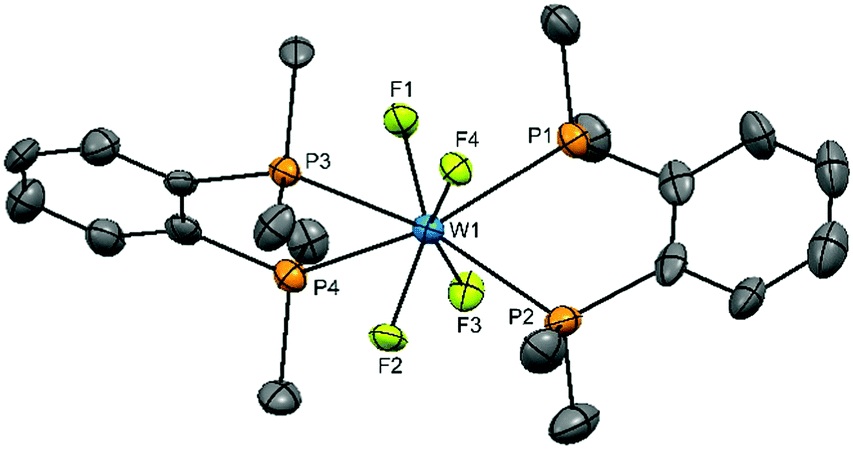 | ||
| Fig. 1 The cation present in [WF4{o-C6H4(PMe2)2}2][WOF5]2·MeCN showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): W(1)–F(3) = 1.91(4), W(1)–F(2) = 1.92(4), W(1)–F(1) = 1.92(4), W(1)–F(4) = 1.93(4), W(1)–P(3) = 2.572(17), W(1)–P(1) = 2.579(18), W(1)–P(2) = 2.582(18), W(1)–P(4) = 2.592(18), P(3)–W(1)–P(4) = 72.6(6), P(1)–W(1)–P(2) = 72.8(6). | ||
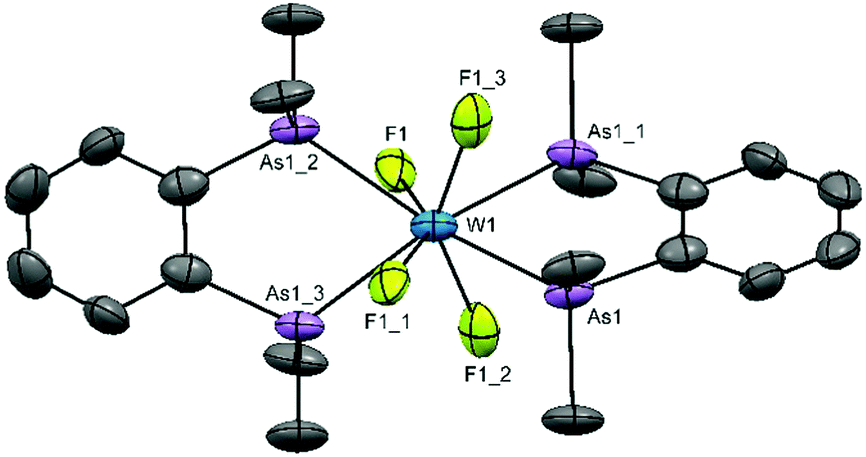 | ||
| Fig. 2 The cation present in [WF4{o-C6H4(AsMe2)2}2][WF8] showing the atom numbering scheme and with ellipsoids drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): W(1)–F(1) = 2.114(6), W(1)–As(1) = 2.6279(10), As(1)–W(1)–As(1) #1 = 75.74(4). Symmetry operators: #1 = −x + 5/4, −y + 5/4, z; #2 = x, −y + 5/4, −z + 1/4; #3 = −x + 5/4, y, −z + 1/4. | ||
Multinuclear NMR spectra were obtained from freshly prepared solutions in anhydrous MeCN. These showed that the cations were relatively stable and their resonances were only slowly lost over several days, but that the 19F{1H} resonance of the [WF7]− ion diminished rapidly over time, with new resonances attributed to [WOF5]−, [W2O2F9]− and possibly [WOF4(MeCN)]19,27,28 appearing. The new resonances must result from trace hydrolysis and/or attack on the glass, and are consistent with the identification of these species in the X-ray structure analyses.
The 1H NMR spectra of the [WF4{o-C6H4(EMe2)2}2]2+ salts showed resonances significantly to high frequency of the values in the parent ligands29 and are consistent with a single environment of the coordinated ligand. The 19F{1H} NMR spectra showed a singlet at δ = +142.8 attributed to [WF7]−.16,28 The 19F{1H} NMR resonance of the cation [WF4{o-C6H4(AsMe2)2}2]2+ was highly shielded, with δ = −25.9 and with 183W satellites (1JWF = 88 Hz). For the corresponding [WF4{o-C6H4(PMe2)2}2]2+ cation, the 19F{1H} resonance was a binomial quintet at δ = −17.5 (2JPF = 67 Hz) (Fig. 3(b)). In this case the 183W satellites were not clearly resolved. The significant shielding of the fluorine resonances in [WF4{o-C6H4(EMe2)2}2]2+cf. [WF4(2,2′-bipy)2]2+ (δ = +153)16 is characteristic of the presence of the soft donor P and As groups. Similar trends were seen in complexes of niobium and tantalum, [MF4(2,2′-bipy)2][MF6]: δ(19F{1H}) = +139.7 (Nb) or +68.1 (Ta), compared to [MF4{o-C6H4(AsMe2)2}2][MF6]: δ(19F{1H}) = +27.1 (Nb) or = −28.0 (Ta) and [MF4{o-C6H4(PMe2)2}2][MF6]: δ(19F{1H}) = −7.8 (Nb) or −39.8 (Ta).24,30
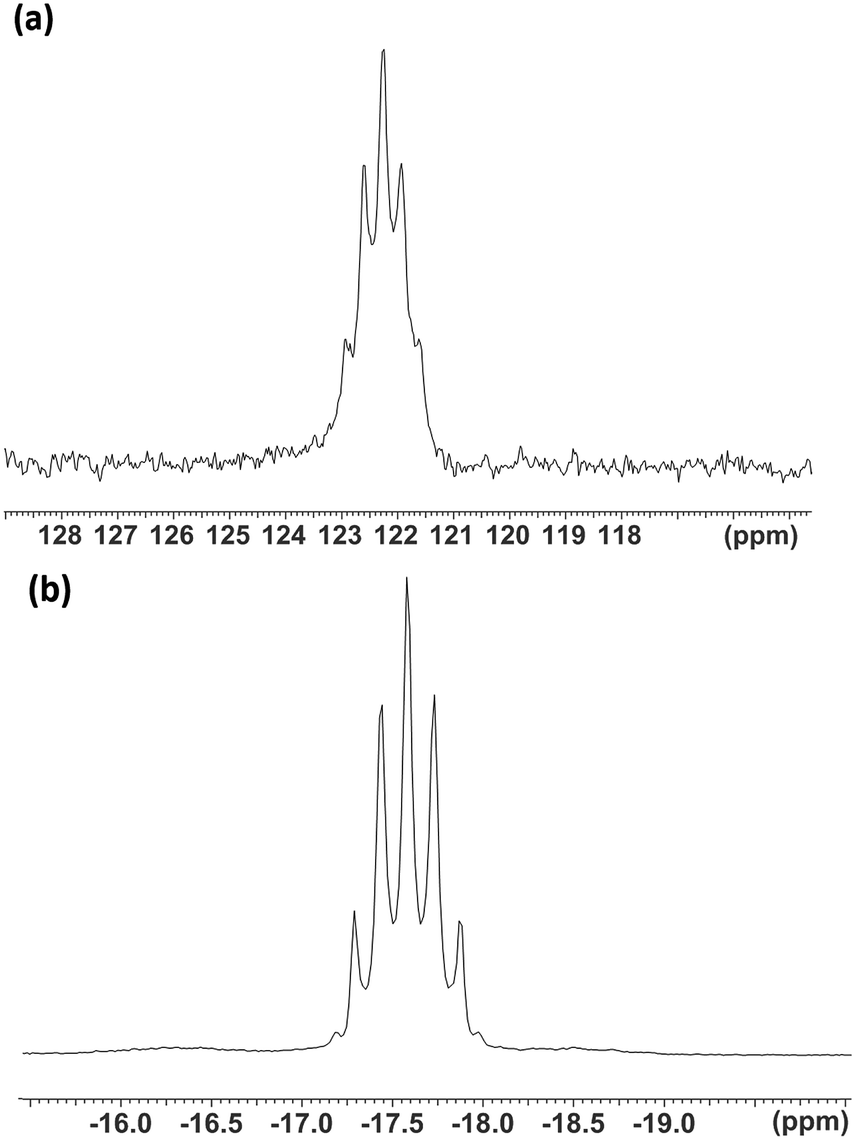 | ||
| Fig. 3 The NMR spectra of [WF4{o-C6H4(PMe2)2}2]2+: (a) 31P{1H} and (b) 19F{1H} in MeCN solution. | ||
The diphosphine complex also exhibited a quintet 31P{1H} NMR resonance (CD3CN) at δ = +122.3 (2JPF = 67 Hz). This constitutes a remarkably large coordination shift of +177 (Fig. 3(a)) for the five-membered chelate ring and may be compared with the coordination shift of +131 observed in [WOF4{o-C6H4(PMe2)2}].20
A similar reaction of WF6 with the more flexible Me2PCH2CH2PMe2 afforded a pale orange powder in low yield. Multinuclear NMR (1H, 19F{1H}, 31P{1H}) studies showed this was an inseparable mixture of two species, one identified as [WF4(Me2PCH2CH2PMe2)2][WF7]2, the second tentatively assigned as the diphosphine-bridged [F6W(μ-Me2PCH2CH2PMe2)WF6].
Reaction of WF6 with RS(CH2)2SR (R = Me, iPr) in rigorously dried CH2Cl2 gave orange brown solutions at 180 K, but the colour was lost on warming, and removal of the volatiles in vacuo resulted in recovery of the dithioether (ESI†).
In summary, this work has identified the first examples of eight-coordinate tetrafluorotungsten(VI) cations with chelating soft, neutral diphosphine and -diarsine co-ligands, whose structures are confirmed by X-ray crystallographic and spectroscopic analyses. Neutral, seven-coordinate W(VI) complexes with trialkylarsines have also been established, whereas triarylarsines and trialkylstibines yield intractable materials. While WF6 is less oxidising than the other metal hexafluorides,9 successful incorporation of the soft group 15 donor ligands by taking advantage of the pre-organised o-phenylene backbone may suggest that under suitable reaction conditions coordination chemistry with neutral ligands may also exist for other members of the little studied family of very hard and more highly oxidising transition metal hexafluorides.
We thank the EPSRC for support through EP/L505651/1, EP/N035437/1 and EP/R513325/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. F. Murphy, R. Murugavel and H. W. Roesky, Chem. Rev., 1997, 97, 3425 CrossRef PubMed.
- B. L. Pagenkopf and E. M. Carreira, Chem. – Eur. J., 1999, 5, 3437 CrossRef.
- H. C. S. Clark and J. H. Holloway, in Advanced Inorganic Fluorides, ed. T. Nakajima, B. Žemva and A. Tressaud, Elsevier, Oxford, 2000, ch. 3 Search PubMed.
- I.-S. Ke, M. Myahkostupov, F. N. Castellano and F. P. Gabbai, J. Am. Chem. Soc., 2012, 134, 15309 CrossRef PubMed.
- D. Shetty, S. Y. Choi, J. M. Jeong, J. Y. Lee, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J.-K. Chung, M. C. Lee and Y. K. Chung, Chem. Commun., 2011, 47, 9732 RSC; W. J. McBride, R. M. Sharkey, H. Karacay, C. A. D’Sousa, E. A. Rossi, P. Laverman, C.-H. Chang, O. C. Boerman and D. M. Goldenberg, J. Nucl. Med., 2009, 50, 991 CrossRef PubMed.
- R. Bhalla, C. Derby, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson, W. Zhang and G. Reid, Chem. Sci., 2014, 5, 381 RSC; W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu and G. Reid, Dalton Trans., 2017, 46, 14519 RSC; F. M. Monzittu, I. Khan, W. Levason, S. K. Luthra, G. McRobbie and G. Reid, Angew. Chem., Int. Ed., 2018, 57, 6658 CrossRef PubMed.
- M. Carravetta, M. Concistre, W. Levason, G. Reid and W. Zhang, Chem. Commun., 2015, 51, 9555 RSC.
- M. Carravetta, M. Concistre, W. Levason, G. Reid and W. Zhang, Inorg. Chem., 2016, 55, 12890 CrossRef PubMed.
- M. J. Molski and K. Seppelt, Dalton Trans., 2009, 3379 RSC.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1450 RSC.
- F. N. Tebbe and E. L. Muetterties, Inorg. Chem., 1968, 7, 172 CrossRef.
- A. M. Noble and J. M. Winfield, J. Chem. Soc. A, 1970, 2574 RSC.
- L. Arnaudet, R. Bougon, B. Buu, M. Lance, M. Nierlich and J. Vigner, Inorg. Chem., 1993, 32, 1142 CrossRef.
- L. Arnaudet, R. Bougon, B. Buu, M. Lance, P. Theury, M. Nierlich and J. Vigner, J. Fluorine Chem., 1995, 71, 123 CrossRef.
- L. Arnaudet, R. Bougon, B. Buu, M. Lance, A. Navaza, M. Nierlich and J. Vigner, J. Fluorine Chem., 1992, 59, 141 CrossRef.
- L. Arnaudet, R. Bougon, B. Buu, M. Lance, A. Navaza, M. Nierlich and J. Vigner, J. Fluorine Chem., 1994, 67, 17 CrossRef.
- S. El-Kurdi, A.-A. Al-Terkawi, B. M. Schmidt, A. Dimitrov and K. Seppelt, Chem. – Eur. J., 2010, 16, 595 CrossRef PubMed.
- A. M. Noble and J. M. Winfield, J. Chem. Soc. A, 1970, 501 RSC.
- W. Levason, G. Reid and W. Zhang, J. Fluorine Chem., 2016, 194, 50 CrossRef.
- J. W. Emsley, W. Levason, G. Reid, W. Zhang and G. De Luca, J. Fluorine Chem., 2017, 197, 74 CrossRef.
- L. Arnaudet, R. Bougon, B. Buu and M. Lance, J. Fluorine Chem., 1991, 53, 171 CrossRef.
- J. H. Holloway, G. Stanger, E. G. Hope, W. Levason and J. S. Ogden, J. Chem. Soc., Dalton Trans., 1988, 1341 RSC.
- A. Beuter, W. Kuhlmann and A. Sawodny, J. Fluorine Chem., 1975, 6, 367 CrossRef.
- W. Levason, M. E. Light, G. Reid and W. Zhang, Dalton Trans., 2014, 43, 9557 RSC.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd edn, 1984 Search PubMed.
- T. Drews, J. Supel, A. Hagenbach and K. Seppelt, Inorg. Chem., 2006, 45, 3782 CrossRef PubMed.
- R. Bougon, T. Bui Huy and P. Charpin, Inorg. Chem., 1973, 14, 1822 CrossRef.
- S. G. Sakharov, Y. V. Kokunov, M. P. Gustyakova and Y. A. Buslaev, Dokl. Akad. Nauk SSSR, 1984, 276, 148 Search PubMed.
- W. Levason, K. G. Smith, C. A. McAuliffe, F. P. McCullough, R. D. Sedgwick and S. G. Murray, J. Chem. Soc., Dalton Trans., 1979, 1718 RSC.
- W. Levason, G. Reid, J. Trayer and W. Zhang, Dalton Trans., 2014, 43, 3649 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterisation data, original IR and NMR spectra for the compounds reported. CCDC 1854218 and 1854219. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc05598j |
| This journal is © The Royal Society of Chemistry 2018 |