
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H functionalisation of aldehydes using light generated, non-stabilised diazo compounds in flow†
Paul
Dingwall
 a,
Andreas
Greb
a,
Lorène N. S.
Crespin
a,
Ricardo
Labes
a,
Biagia
Musio
a,
Jian-Siang
Poh
a,
Patrick
Pasau
b,
David C.
Blakemore
c and
Steven V.
Ley
*a
a,
Andreas
Greb
a,
Lorène N. S.
Crespin
a,
Ricardo
Labes
a,
Biagia
Musio
a,
Jian-Siang
Poh
a,
Patrick
Pasau
b,
David C.
Blakemore
c and
Steven V.
Ley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: svl1000@cam.ac.uk; Web: http://www.leygroup.ch.cam.ac.uk
bUCB Biopharma SPRL, Chemical Research R5, Chemin du Foriest 1420, Braine-L’Alleud, Belgium
cMedicine Design, Pfizer Inc., Eastern Point Road, Groton, Connecticut 06340, USA
First published on 11th September 2018
Abstract
The difficulty in accessing and safely utilising non-stabilised diazo species has in the past limited the application of this class of compounds. Here we explore further the use of oxadiazolines, non-stabilised diazo precursors which are bench stable, in direct, non-catalytic, aldehyde C–H functionalisation reactions under UV photolysis in flow and free from additives. Commercially available aldehydes are coupled to afford unsymmetrical aryl–alkyl and alkyl–alkyl ketones while mild conditions and lack of transition metal catalysts allow for exceptional functional group tolerance. Examples are given on small scale and in a larger scale continuous production.
Diazo compounds have a long-standing history as extremely versatile tools for the creation of carbon–carbon and carbon–heteroatom bonds.1–4 As a class of compounds, however, diazo derivatives are notorious for their toxic and hazardous nature, and associated difficulty in preparation and handling. These difficulties are compounded when the diazo moiety is not stabilised by either a proximal electron withdrawing group or π-system, meaning that the chemistry of non-stabilised diazo compounds is still a relatively underexplored area. The use of flow techniques as an enabling technology5–8 can mitigate these classical issues to a large extent via the in situ generation and consumption of diazo compounds, so avoiding accumulation, allowing safer access under reproducible process windows.9–19 Accordingly, our group has recently published a mild method for the generation of non-stabilised diazo compounds from oxadiazolines using UV light and their aryl–alkyl cross-coupling with boronic acids.20
Oxadiazolines are prepared in a one-pot, two step procedure by the condensation of an alkyl ketone21 with acetic hydrazide followed by cyclisation promoted by oxidants such as lead tetraacetate22 or (diacetoxyiodo)benzene23 or by electrochemical methods24 (Scheme 1). Oxadiazolines are bench stable at room temperature but, when exposed to UV irradiation, decompose to form the relevant non-stabilised diazo compound and methyl acetate.25,26 These compounds are particularly attractive for use in flow chemistry due to the certain safety issues outlined above, as well as the additional associated benefits that accrue under continuous processing conditions.
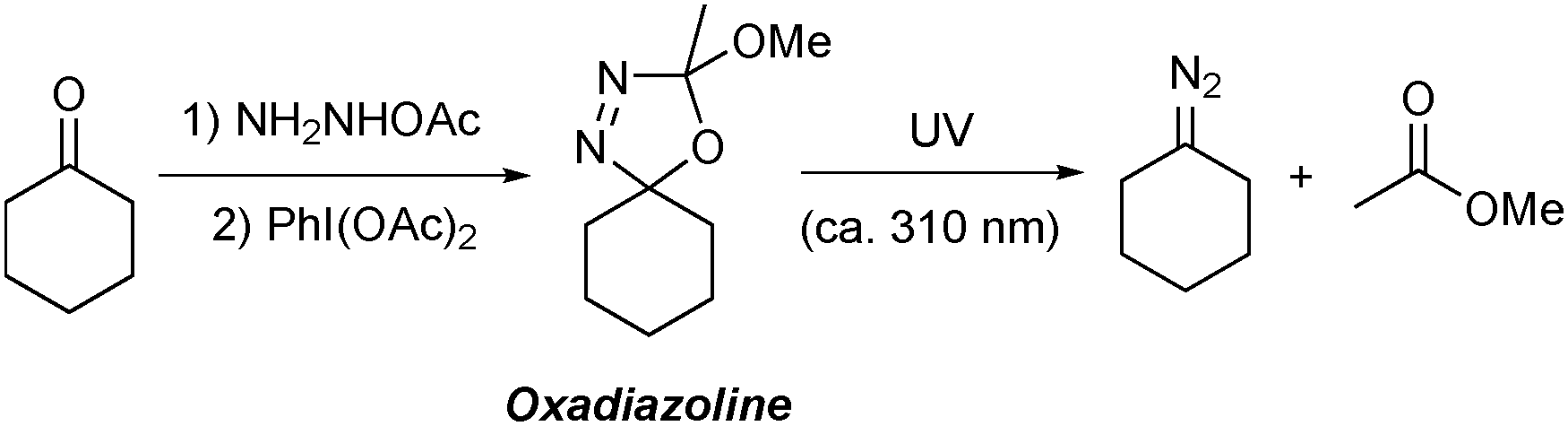 | ||
| Scheme 1 Synthesis and UV photolysis of oxadiazolines. | ||
The addition of a diazo compound to an aldehyde with subsequent 1,2-hydride shift affords the corresponding ketone product (Scheme 2).27 We have previously reported the thermally activated insertion of diazo compounds into a formyl C–H bond to generate unsymmetrical ketones using tosylhydrazones as non-stabilised diazo precursors in 2014.28
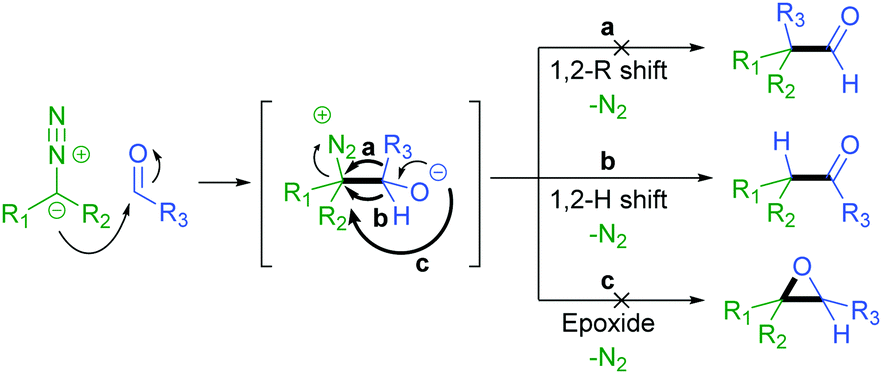 | ||
| Scheme 2 Mechanistic pathways available for diazo addition to an aldehyde. | ||
Although a ubiquitous functional group, the synthesis of unsymmetrical ketones can still prove a synthetic challenge, making this an active area of research. In the recent literature, a number of methods coupling activated carbonyl substrates, such as acetyltrimethylsilanes,29,30 acyl chlorides,31,32 amides,33–35 anhydrides,36,37 phthalimides,38 or carboxylic acids,39 with various partners have been reported. Formation of unsymmetrical ketones directly from aldehydes is more desirable than from activated substrates but more challenging still. Direct formyl C–H insertion to create a new sp2–sp3 or sp2–sp2 carbon–carbon bond is achievable via Palladium,40–43 Rhodium,44,45 or Nickel46–49 catalysed processes as well as through NHC mediated organocatalysis50,51 or indeed diazo chemistry27,28 (Scheme 3).
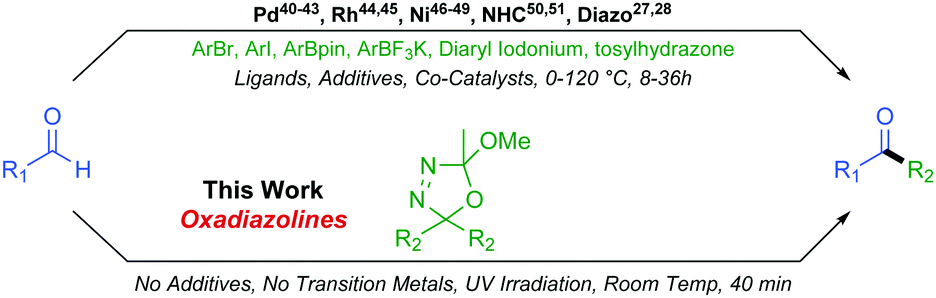 | ||
| Scheme 3 Methods for direct aldehyde coupling reactions to form unsymmetrical ketones. | ||
These existing methods afford valuable reactivity and useful chemistry. However, some drawbacks commonly encountered include long reaction times, occasionally harsh conditions, necessity for a plethora of additives, the presence of up to three catalysts, and relatively poor functional group compatibility due to the reactivities of associated transition metal catalysts. In this work, we extend our original oxadiazoline methodology and we report their very mild and rapid coupling with aldehydes to form unsymmetrical alkyl–alkyl and aryl–alkyl ketones with excellent functional group tolerance and resulting structural diversity.
Employing similar conditions to those of our previous work resulted in an excellent yield of the product ketone (Table 1, entry 1).20 Removal of the DIPEA base resulted in no change to the reaction yield (Table 1, entry 2).52 Reducing the residence time to 40 minutes resulted in a decrease in yield, however this could be increased again by raising the temperature of the reaction to 20 °C (Table 1, entries 3 and 4). Increasing the temperature to 30 °C resulted in a precipitous drop of the yield, presumably due to decomposition of the diazo intermediate (Table 1, entry 5). Reducing the equivalents of the oxadiazolines relative to the aldehyde from 2 to 1.1 resulted in a decrease in yield and no difference was observed on change of solvent (Table 1, entries 6, 7, and 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Oxadiaz] (M) | [Aldehyde] (M) | t R (min) | T (°C) | Solvent | Yielda (%) |
| a GC yield unless stated otherwise. b 0.1 M DIPEA. c Isolated yield. | ||||||
| 1b | 0.1 | 0.05 | 80 | 10 | CH2Cl2 | 94c |
| 2 | 0.1 | 0.05 | 80 | 10 | CH2Cl2 | 93 |
| 3 | 0.1 | 0.05 | 40 | 10 | CH2Cl2 | 78 |
| 4 | 0.1 | 0.05 | 40 | 20 | CH 2 Cl 2 | 91 |
| 5 | 0.1 | 0.05 | 40 | 30 | CH2Cl2 | 28 |
| 6 | 0.06 | 0.05 | 40 | 20 | CH2Cl2 | 72 |
| 7 | 0.06 | 0.05 | 40 | 20 | MeTHF | 70 |
| 8 | 0.06 | 0.05 | 40 | 20 | Dioxane | 70 |
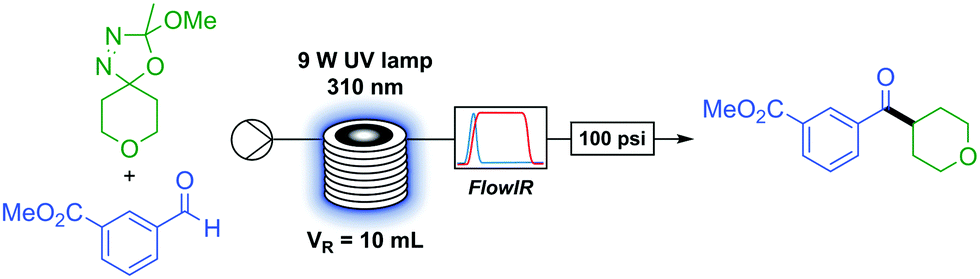
With optimised conditions in hand we began to investigate the scope of the transformation. The in situ generation and full consumption of diazo compounds in flow is an ideal case and one which, when screening new reactions, may not always hold. Inline IR53,54 is a particularly advantageous analytical technique in diazo chemistry as diazo species have a unique IR stretch (ca. 2040 cm−1) which, under our experimental setup, can be detected in a simple manner. As the IR is present upstream from the back-pressure regulator and system outlet (Table 1) if at any time the user observes a non-trivial concentration of diazo in the output of the reactor this can be simply dealt with through addition of an appropriate quenching agent (i.e. acetic acid) to the collection vessel prior to any diazo material exiting the system. An additional benefit of inline IR in this methodology is the detection of methyl acetate, which is produced in the breakdown of the oxadiazoline (Scheme 1) and can be used to observe the progress of the reaction.
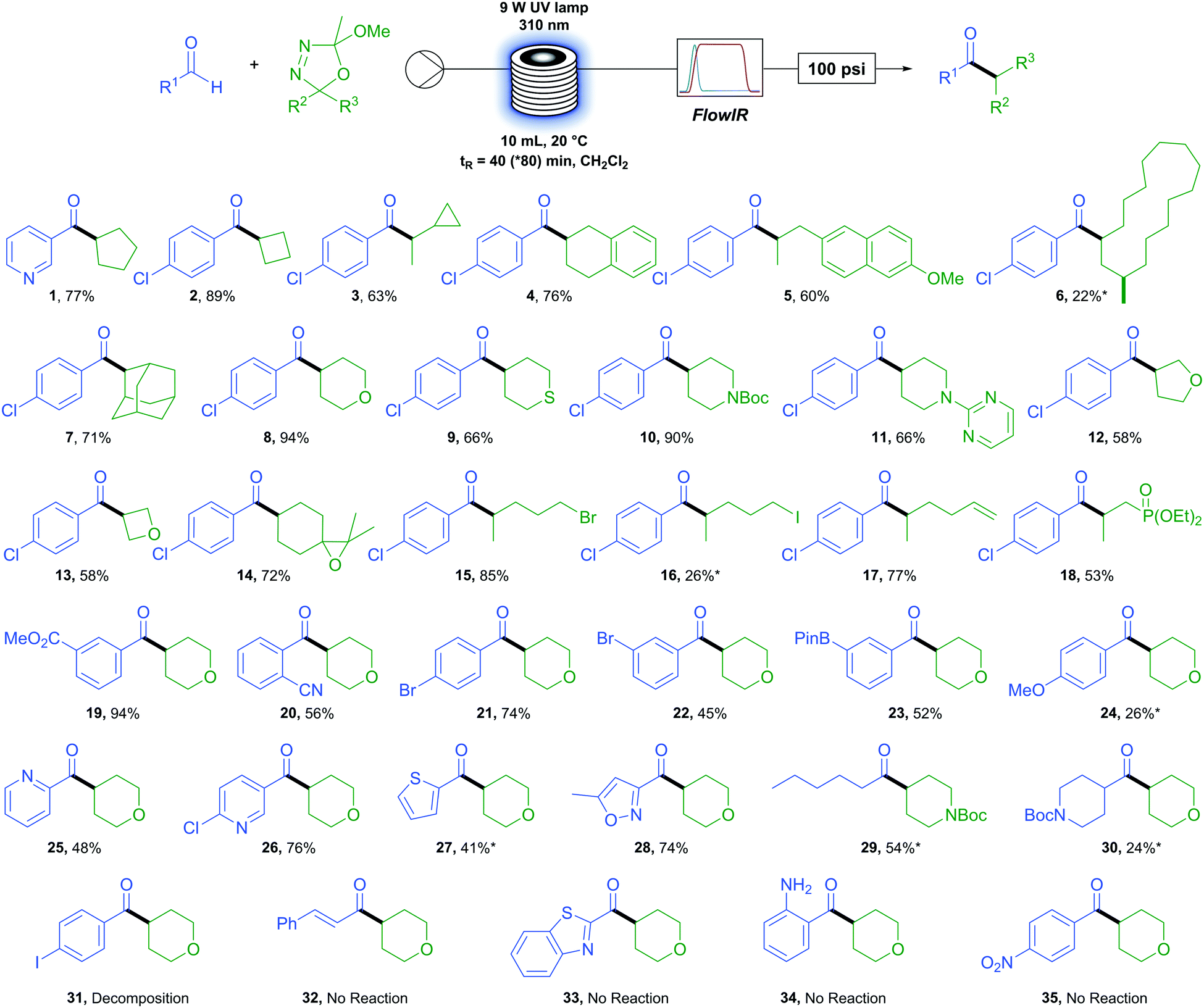
We first sought to determine the generality of the oxadiazoline component (Table 2). A variety of carbon rings were tolerated, from cyclopentane (1) and cyclobutane (2) to an oxadiazoline incorporating a cyclopropane moiety (3). No ring-opened product was observed, serving as evidence of a polar rather than a radical process. Tetralin (4) and methoxy naphthyl (5) were viable substrates. An oxadiazoline derived from the macrocyclic natural product Muscone (6) also proved viable, albeit in low yield. A bulky adamantly group (7) reacted in good yield. Six membered saturated heterocycles tetrahydropyran (8), tetrahydrathiopyran (9), N-boc and N-pyrimidyl (10 and 11) piperidine reacted in moderate to excellent yields. Five- and four-membered saturated oxygen containing heterocycles tetrahydrofuran (12) and oxetane (13) reacted in more moderate yields. Functional group tolerance proved to be excellent, with examples of an epoxide (14), primary alkyl bromide and iodide (15 and 16), terminal alkene (17), and phosphonate ester (18) exemplifying the extremely mild conditions of this reaction and accessing products which would be otherwise difficult or impossible to access via metal-catalysed methods.
|
We next turned our attention to the aldehyde scope. Methyl ester (19) resulted in excellent yields while an ortho-nitrile group (20) displayed tolerance to bulk beside the reacting position in an aromatic system. Functional group tolerance is again excellent with para- (21) and meta-bromo (22) benzaldehydes as well as the synthetically useful boron-pinacol ester (23) which would otherwise be challenging to incorporate under transition metal catalysis.55 However, the electron rich 4-methoxy benzaldehyde (24) resulted in a low yield. A variety of heterocyclic aldehydes were also successfully coupled. For example, several pyridyl containing aldehydes (1, 25 and 26) as well as thiophene (27) and isoxazole (28). We were pleased to find that aliphatic aldehydes proceed although they appear more challenging than aromatic aldehydes, with hexanal (29) resulting in a moderate yield but cyclic N-boc piperidone carboxaldehyde (30) only a low yield.
When employing 4-iodobenzaldehyde (31) as the aldehyde coupling partner only decomposition to benzaldehyde was observed and the use of cinnamaldehyde (32), benzothiazole (33), and amino (34) or nitro (35) functional groups resulted in no reaction. In each case above, little or no conversion of the oxadiazoline was observed due to the absorbance of UV irradiation by the aldehyde. With this knowledge in hand, we found that a simple test can be carried out prior to performing the reaction which allows the user to determine the feasibility and potentially adjust conditions accordingly to maximise the yield. If the λmax of the desired aldehyde coupling partner is at or above 310 nm (the wavelength of UV irradiation employed) then the reaction is unlikely to proceed (see Fig. S2, ESI†). We also found that, to some extent, this limitation can be overcome by lowering the concentration of the reactants and increasing the residence time of the reaction. This is demonstrated in the case of compound (27) (starting material aldehyde having a λmax at 311 nm) where, under our standard operating conditions the yield was 16% but was increased to 41% by simply halving the reaction concentration and doubling the residence time.
As a flow process, the methodology is eminently scalable by simply running the reaction for longer. Without accumulation of any diazo intermediate, a four hour run under steady state at standard conditions provided 580 mg of ketone 19, corresponding to a theoretical productivity of 3.48 g d−1, with similar yield to the smaller scale run (91 to 94%) with this particular reactor set-up.
In conclusion, this work expands the scope and application of oxadiazolines as highly effective precursors to non-stabilised diazo compounds. Mild reaction conditions, short reaction times, and ease of continuous operation means this methodology offers a complementary alternative to existing literature procedures. In particular, the lack of transition metals or commonly used additives such as oxidants or bases allows for the incorporation of sensitive functional groups into the ketone products, laying groundwork for their immediate further functionalisation.
The authors kindly acknowledge funding by the H2020-FETOPEN-2016-2017 programme of the European commission (P. D., S. V. L., 737266-ONE FLOW), postdoctoral fellowships from Pfizer (A. G. and L. C.), EPSRC Critical Mass Grant (EP/K009494/1) (B. M.), Cambridge Home and EU Scholarship Scheme (J. S. P.), and EPSRC (S. V. L., grant no. EP/K009494/1, EP/K039520/1 and EP/M004120/1) for financial support. The authors are also grateful to Duncan Guthrie at Vapourtec for the generous loan of a UV-150 photoreactor.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef PubMed.
- M. P. Doyle, M. A. McKervey and T. Ye, Modern catalytic methods for organic synthesis with diazo compounds, Wiley, 1998 Search PubMed.
- H. Zollinger, Diazo Chemistry I and II, Wiley-VCH, 1995 Search PubMed.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892 RSC.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131 CrossRef PubMed.
- G. Bernhard, C. David and K. C. Oliver, Angew. Chem., Int. Ed., 2015, 54, 6688 CrossRef PubMed.
- K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2018, 24, 10571 CrossRef PubMed.
- P. Rulliere, G. Benoit, E. M. D. Allouche and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 5777 CrossRef PubMed.
- B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298 CrossRef PubMed.
- S. T. R. Müller and W. Thomas, ChemSusChem, 2015, 8, 245 CrossRef PubMed.
- E. Rossi, P. Woehl and M. Maggini, Org. Process Res. Dev., 2012, 16, 1146 CrossRef.
- F. Mastronardi, B. Gutmann and C. O. Kappe, Org. Lett., 2013, 15, 5590 CrossRef PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef PubMed.
- N. Kockmann, P. Thenee, C. Fleischer-Trebes, G. Laudadio and T. Noel, React. Chem. Eng., 2017, 2, 258 RSC.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef PubMed.
- A. Clément, G. M. O. Javier and L. Hélène, Angew. Chem., Int. Ed., 2017, 56, 6294 CrossRef PubMed.
- D. Rackl, C.-J. Yoo, C. W. Jones and H. M. L. Davies, Org. Lett., 2017, 19, 3055 CrossRef PubMed.
- É. Lévesque, S. T. Laporte and A. B. Charette, Angew. Chem., Int. Ed., 2017, 56, 837 CrossRef PubMed.
- A. Greb, J. S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602 CrossRef PubMed.
- Acetophenones and benzophenes react sluggishly in the cyclisation step and form unstable oxadiazolines while aldehydes eliminate to the oxadiazole during cyclisation. For further information see: M. Békhazi, P. J. Smith and J. Warkentin, Can. J. Chem., 1984, 62, 1646–1652 CrossRef.
- J. Warkentin, J. Chem. Soc., Perkin Trans. 1, 2000, 2161 RSC.
- R. Y. Yang and L. X. Dai, J. Org. Chem., 1993, 58, 3381 CrossRef.
- T. Chiba and M. Okimoto, J. Org. Chem., 1992, 57, 1375 CrossRef.
- M. W. Majchrzak, M. Bekhazi, I. Tsesheepy and J. Warkentin, J. Org. Chem., 1989, 54, 1842 CrossRef.
- J. P. Pezacki, B. D. Wagner, C. S. Q. Lew, J. Warkentin and J. Lusztyk, J. Am. Chem. Soc., 1997, 119, 1789 CrossRef.
- N. Guttenberger and R. Breinbauer, Tetrahedron, 2017, 73, 6815 CrossRef.
- D. M. Allwood, D. C. Blakemore and S. V. Ley, Org. Lett., 2014, 16, 3064 CrossRef PubMed.
- S. D. Ramgren and N. K. Garg, Org. Lett., 2014, 16, 824 CrossRef PubMed.
- J. Schmink and S. Krska, J. Am. Chem. Soc., 2011, 133, 19574 CrossRef PubMed.
- Z. Sun, N. Kumagai and M. Shibasaki, Org. Lett., 2017, 19, 3727 CrossRef PubMed.
- J. Amani and G. A. Molander, J. Org. Chem., 2017, 82, 1856 CrossRef PubMed.
- J. Amani, R. Alam, S. Badir and G. A. Molander, Org. Lett., 2017, 19, 2426 CrossRef PubMed.
- N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2015, 8, 75 CrossRef PubMed.
- T. B. Boit, N. A. Weires, J. Kim and N. K. Garg, ACS Catal., 2018, 8, 1003 CrossRef PubMed.
- C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040 CrossRef PubMed.
- C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 11938 CrossRef PubMed.
- A. Tlahuext-Aca, R. A. Garza-Sanchez, M. Schafer and F. Glorius, Org. Lett., 2018, 20, 1546 CrossRef PubMed.
- J. Amani and G. A. Molander, Org. Lett., 2017, 19, 3612 CrossRef PubMed.
- T. Wakaki, T. Togo, D. Yoshidome, Y. Kuninobu and M. Kanai, ACS Catal., 2018, 8, 3123 CrossRef.
- J. Ruan, O. Saidi, J. A. Iggo and J. Xiao, J. Am. Chem. Soc., 2008, 130, 10510 CrossRef PubMed.
- B. Suchand and G. Satyanarayana, J. Org. Chem., 2016, 81, 6409 CrossRef PubMed.
- S. Ko, B. Kang and S. Chang, Angew. Chem., Int. Ed., 2005, 44, 455 CrossRef PubMed.
- M. Pucheault, S. Darses and J.-P. Genet, J. Am. Chem. Soc., 2004, 126, 15356 CrossRef PubMed.
- M. L. N. Rao and B. S. Ramakrishna, Eur. J. Org. Chem., 2017, 5080 CrossRef.
- J. K. Vandavasi, X. Hua, H. B. Halima and S. G. Newman, Angew. Chem., Int. Ed., 2017, 56, 15441 CrossRef PubMed.
- Y.-C. Huang, K. K. Majumdar and C.-H. Cheng, J. Org. Chem., 2002, 67, 1682 CrossRef PubMed.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353 CrossRef PubMed.
- L. J. Gu, C. Jin and H. T. Zhang, Chem. – Eur. J., 2015, 21, 8741 CrossRef PubMed.
- Q. Y. Toh, A. McNally, S. Vera, N. Erdmann and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 3772 CrossRef PubMed.
- V. P. Mehta, A. k. Sharma, S. G. Modha, S. Sharma, T. Meganathan, V. S. Parmar and E. Van der Eycken, J. Org. Chem., 2011, 76, 2920 CrossRef PubMed.
- Probing the role of the DIPEA in our previously published boronic acid coupling, we found the base to be essential in only a physical requirement to dissolve the boronic acid in dichloromethane. Further optimisation found that the amount of DIPEA could be reduced to 16 mol% in this solvent or removed entirely through use of a coordinating solvent such at THF. In the coupling of non-stabilised diazo compounds with boronic acids or aldehydes we find no evidence that the DIPEA stabilises the diazo intermediate as some have suggested.
- C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393 CrossRef.
- C. F. Carter, I. R. Baxendale, M. O'Brien, J. B. J. Pavey and S. V. Ley, Org. Biomol. Chem., 2009, 7, 4594 RSC.
- Boron-pinacol esters are significantly less reactive towards diazo compounds than their boronic acid or boroxine counterparts and we found only 3% yield each of addition to the Bpin of the starting material aldehyde or product.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc06202a |
| This journal is © The Royal Society of Chemistry 2018 |