
The rise of lithium–selenium batteries
Ali
Eftekhari
 *ab
*ab
aThe Engineering Research Institute, Ulster University, Newtownabbey BT37 OQB, UK. E-mail: eftekhari@elchem.org
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, UK
First published on 13th January 2017
Abstract
The lithium–selenium (Li–Se) battery is an alternative to its sulfur counterpart with some noticeable advantages, such as the significantly higher electrical conductivity of Se and better electrochemical performance. Although the idea of a Li–Se battery dates to the 1960s, over the past four years it has been revisited as a practical potential candidate for electrochemical energy storage. Despite similar problems of Li–Se and lithium–sulfur (Li–S) batteries, such as the so-called shuttle effect, the research strategies go in different directions because of new opportunities offered by the Li–Se batteries. For instance, Li–Se batteries can perform better in the conventional carbonate-based electrolytes, in which polysulfides are chemically unstable. The present manuscript comprehensively reviews the current status of the Li–Se battery based on the achievements during its short history. Furthermore, a promising possibility is the lithium–(sulfur/selenium) battery, utilizing a sulfur/selenium cathode that can gain the advantages of both batteries. Other batteries in this family such as sodium–selenium and magnesium–selenium are also considered.
 Ali Eftekhari | Ali Eftekhari is a professor of chemistry temporarily with the University of Ulster and Queen's University, Belfast. He has worked in different capacities in various universities across 4 continents and founded two internationally recognized academic schools from scratch, which were considered as exceptional success stories by major media such as the British newspaper, The Guardian. His research interest is focused on the material design of electrochemical systems, and he has worked on energy storage systems for 20 years. He is the principal author of over 100 papers published in leading scholarly journals and has supervised over 110 postdoctoral researchers and graduate students. He is the President of the American Nano Society. |
Introduction
The history of lithium–selenium (Li–Se) batteries is as long as that of the famous lithium–sulfur (Li–S) batteries. In the 1960s when Li–S batteries were considered as a potential primary electrochemical power source, Li–Se was also considered as a potential alternative. In 1970, a news article mentioned the forthcoming prospect of Li–Se batteries for medical and vehicle applications following the pioneering research conducted on lithium–sulfur, –selenium, and –tellurium batteries under the supervision of Elton J. Cairns at Argonne National Laboratory.1 However, none of these batteries was successful for practical development and the research attention faded in subsequent decades until the last few years when all of the old and new possibilities for energy storage were revisited due to the massively growing demand. The turning point was the new generation of rechargeable Li–S batteries introduced in the 1990s.2 In 2012, Amine and his co-workers extended this concept to a Se cathode and pioneeringly fabricated a prototype for the modern Li–Se batteries, and this possibility for electrochemical energy storage was reborn.3Of the possible electrochemical energy storage systems introduced during the 1960s and 1970s, the lithium-ion battery (LIB) is definitely the most successful. After the first commercial product was introduced to the market by Sony in 1991, the LIBs have become an essential requirement of everyday life because of their importance in all portable electronic devices. Although LIB can be scaled up for electric vehicles or household energy storage, other alternatives seem more practical; hence, considerable attention has been paid to similar cells such as Li–S and lithium-air batteries. These cells can have a similar reaction at the anode side, compared to the LIBs, but the cathode undergoes a reactive or catalytic reaction. Therefore, the cell design might be different because of the harmful cathodic products, which should be separated from the anode half cell.
While a lithium-air battery is based on an open system, a Li–S battery is more similar to the LIB. To enhance the specific capacity of graphite anodes in LIBs, a wide range of anode materials has been examined. The most promising anode materials, having a high specific capacity and low redox potential, are the group IVB and VB elements such as Si, Ge, Sn, Sb, etc. In this case, the anode process is a direct reaction (based on a conversion mechanism) transforming the elemental anode into a lithium compound instead of intercalation. This is what happens in the cathode of lithium–sulfur, –selenium, or –tellurium batteries. In fact, a Li–S battery is based on the same principle and mechanism as the LIBs, but both the anode and cathode are based on conversion-based materials, rather than intercalation compounds (such as graphite in the anode and LiCoO2 in the cathode).
The first advantage of Li–S batteries is the high specific capacity and low-cost of sulfur. However, sulfur has some disadvantages, such as high chemical reactivity, which causes the formation of harmful by-products. This is also accompanied by the dissolution of polysulfides in the electrolyte causing the infamous shuttle phenomenon, which is still one of the main obstacles of Li–S batteries. Selenium has recently attracted enormous attention in the family of chalcogenides. In comparison to sulfur, selenium is less reactive and more controllable in an electrochemical cell. One may consider that a disadvantage of a Li–Se battery is a lower specific capacity (678 mA h g−1) because of the selenium mass compared to that of a Li–S battery (1672 mA h g−1). However, owing to the significantly higher density of selenium compared to sulfur, a Li–Se battery can have almost the same volumetric capacity (3253 mA h cm−3 for Li–Se, and 3467 mA h cm−3 for Li–S batteries), and thus, similar energy and power densities (i.e., in W h and W per unit of volume, where specific energy and power are W h and W per unit of mass). Owing to the miniaturization of the portable devices, the importance of energy density is greater than that of specific energy.4
On the other hand, the electrical conductivity of selenium (1 × 10−3 S m−1) is much higher than that of sulfur (5 × 10−28 S m−1), which is practically an insulator. As a result, Li–Se batteries have attracted considerable attention during the past few years. Of course, this is just the inception of this possibility, and all research works are at very early stages. However, both experimental and theoretical considerations reasonably suggest that the Li–Se batteries have huge potential in the future of energy storage. The present manuscript attempts to review the research works that have been done so far on this recently revisited battery, to highlight the promising potentials (Table 1). It should be emphasized that selenium is the immediate neighbor of sulfur, which makes them very similar in many aspects. Thus, the common problems of Li–S batteries also exist for Li–Se batteries, but the selenium advantages can make this class of rechargeable batteries more practical, although the main focus is still on the Li–S batteries.
| Reference | Material | Synthesis | Electrolyte | Capacity | C rate | Potential range | Cyclability (low rate) | Note |
|---|---|---|---|---|---|---|---|---|
| 13 | Se/C nanocomposite | Derived from pomelo peel | Carbonate | 710 | 0.22 | 1.8–1.6 | 8% (30)@0.22C | |
| 42 | Se/micro–mesoporous carbon sphere nanocomposite | Carbonate | 705 | 0.1 | 2.0–1.7 | 70% (500) 0.25C | ||
| 10 | Se–C | Carbonate | 700 | 0.1 | 2.0–1.9 | |||
| 30 | Se confined in metal complex-derived porous carbon | Salt-baked approach | Carbonate | 685 | 0.1C | 2.1–1.6 | 15% (150)@0.1C | |
| 25 | One-dimensional organic Se-containing fiber | Carbonized polyacrylonitrile/selenium | Carbonate | 670 | 0.1 | 1.9–0.9 | 120% (500) 0.1C | |
| 53 | Se/heteroatom-doped microporous carbon | Carbonate | 670 | 0.5 | 2.0–1.7 | 80% (150)@1C | ||
| 22 | Se/interconnected porous hollow carbon bubbles composites | Carbonate | 670 | 0.1 | 1.9–1.6 | 85% (120)@0.1C | ||
| 47 | Graphene-encapsulated selenium/polyaniline core–shell nanowires | Carbonate | 660 | 0.2 | 1.9–1.6 | 8% (200)@0.2C | ||
| 3 | Se–C | Carbonate | 660 | 50 mA g−1 | 2.0–0.8 | 45% (100)@50 mA g−1 | ||
| 78 | Se confined in MWCNT encapsulated by highly porous carbon (tube-in-tube carbon) | From hard template assisted synthesis | Carbonate | 625 | 0.2 | 1.9–1.7 | 55% (800)@2C | |
| 50 | Flexible one-dimensional Se/C composite nanofibers | From electrospun polyacrylonitrile | Carbonate | 625 | 0.05 A g−1 | 1.9–1.6 | 105% (80)@0.05 A g−1 | |
| 29 | Se confined in macro-/micro-porous biochar | Derived from the inner spongy layer of pomelo pericarp | Carbonate | 600 | 0.2 | 1.9–1.6 | 15% (100)@0.2C | |
| 14 | Se confined in N-doped, hierarchically porous carbon sponges | Derived from MOF | Carbonate | 600 | 0.5 | 1.9–1.6 | 80% (200)@0.5 | |
| 24 | Carbon bonded and encapsulated selenium composites | In situ carbonization of a mixture of perylene-3,4,9,10-tetracarboxylic dianhydride and selenium | Carbonate | 590 | 40 mA h g−1 | 2.0–0.8 | 18% (250)@0.1 A g−1 | |
| 38 | Se/nitrogen-doped microporous carbon spheres | Carbonate | 580 | 0.5 | 1.9–1.6 | 100% (1600)@2C | ||
| 8 | Protective solid electrolyte layer | Electrochemical pretreatment | Carbonate | 570 | 0.2 | 2.0–1.5 | 90% (100)@0.2C | |
| 17 | Se/porous carbon composite | Derived from potassium tartrate | Carbonate | 560 | 0.24 | 1.9–1.6 | 15% (80)@0.24 | |
| 9 | Se@mesoporous carbon composite | Carbonate | 500 | 0.1 | 1.7–0.9 | 100% (1000)@0.1C | ||
| 5 | Se-confined microporous carbon cathode | Carbonate | 510 | 0.1C | 1.9–1.6 | 90% (3000)@1C | ||
| 7 | Se confined in micro- and mesoporous carbon | Carbide-derived | Carbonate | 490 | 0.1 | 2.1–1.7 | 120% (150)@0.2C | 5 M LiTFSI |
| 28 | Selenium-confined porous carbon | From silk cocoons | Carbonate | 460 | 0.5 | 1.9–1.6 | First cycle | |
| 45 | Selenized polyacrylonitrile | Carbonate | 415 | 0.5 mA cm−2 | 2.1–0.8 | 100% (1000)@0.5 mA cm−2 | ||
| 48 | Nano-fibrous Se/polypyrrole/graphene composite | Carbonate | 350 | 0.17 | 1.0–0.9 | |||
| 76 | Nanoporous selenium | Simple mechanical method adopting nano-CaCO3 as a template | Carbonate | 270 | 0.1 A g−1 | 3.0–0.8 | 70% (20)@0.1 A g−1 | First cycle |
| 20 | Se confined within porous carbon nanospheres | Ether | 660 | 0.2 | 2.0–1.6 | 70% (1200)@1C | ||
| 31 | Se on Ni foam | Coated by graphene | Ether | 670 | 0.1 | 2.4–1.0 | 40% (100)@0.1C | First cycle |
| 62 | Free-standing carbon interlayer | Ether | 660 | 0.1 | 2.1–1.8 | 80% (20)@0.1C | First cycle | |
| 19 | Se/3d mesoporous carbon/graphene | Ether | 655 | 0.1C | 2.1–1.0 | First cycle | ||
| 77 | Graphene–Se hybrid microballs | Ether | 642 | 0.1 | 2.0–1.6 | 85% (100)@0.1C | ||
| 21 | Se confined in nitrogen-containing hierarchical porous carbon | Ether | 635 | 0.5 | 2.2–2.0 | 60% (60)@1C | ||
| 32 | Graphene-coated polymer separator | Ether | 630 | 0.5 | 2.2–2.0 | First cycles | ||
| 52 | Se/nitrogen doped carbon tubes | Carbonization of polyaniline tubes | Ether | 610 | 0.2 | 1.9–1.6 | 55% (100)@0.2C | |
| 27 | Se/multi-walled carbon nanotube | Solution-based synthesis | Ether | 605 | 0.5 | 2.2–1.0 | 75% (100)@2C | |
| 16 | Se/porous carbon nanofibers | Derived from polymer carbonization | 564 | 0.2 | 75% (100)@0.2C | |||
| 44 | Selenium@carbon spheres | Microwave synthesis | Ether | 560 | 0.1 | 2.1–1.6 | 30% (100)@0.1C | |
| 26 | Se/carbon-rich core shell | One-step hydrothermal | Ether | 555 | 0.3 A g−1 | 2.2–1.0 | 50% (80)@0.3 A g−1 | |
| 51 | Se/bimodal porous carbon | Hydrothermal route | Ether | 550 | 1 | 2.2–1.2 | 50% (80)@1C | |
| 33 | Se encapsulated in porous hollow carbon spheres | Ether | 540 | 0.1 | 2.1–1.6 | |||
| 43 | Reduced graphene oxide encapsulated selenium nanoparticles | Self-assembly | Ether | 533 | 0.2 | 2.2–1.7 | 65% (200)@0.2C | |
| 23 | Se/mesoporous carbon microsphere | Ether | 505 | 0.5 | 2.1–2.0 | 65% (100)@0.5C | First cycle | |
| 18 | Se encapsulated into 3D interconnected hierarchical porous carbon aerogels | Ether | 505 | 0.5 | 2.2–1.9 | |||
| 49 | Li2Se nanoparticles | CVD carbon coating | Ether | 440 | 0.1C | 2.1–1.9 | 10% (100)@0.5C | |
| 60 | Flexible self-standing graphene–Se@CNT composite film | Ether | 350 | 0.1 | 4.0–1.0 | 80% (100) 0.1C |
Fig. 1 compares similar Li–S and Li–Se batteries. The Li–Se battery shows two key advantages in performance: significantly better cyclability and well-defined electrochemical behavior. The difference between the charge and discharge voltages, which is characteristic of the anode and cathode overpotentials is much smaller than that of the Li–S battery.
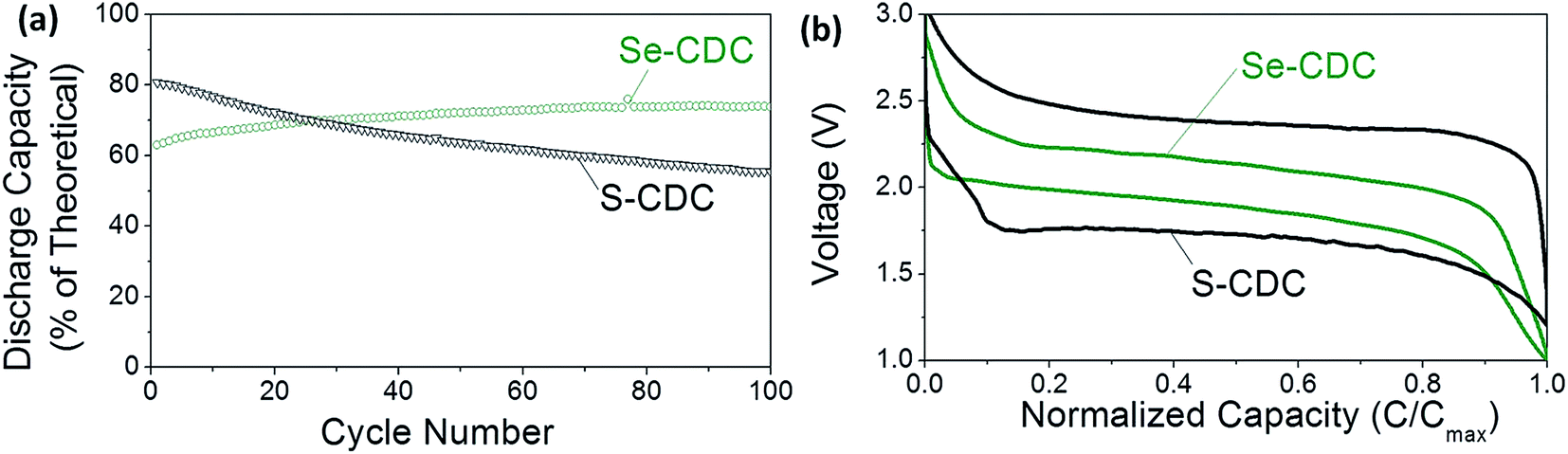 | ||
| Fig. 1 Comparison of S and Se performance: (a) cycle stability and (b) charge–discharge profiles of S-CDC and Se-CDC cathodes in identical electrolytes (5 M LiTFSI). S and Se loading in S-CDC and Se-CDC are also almost the same (≈60%). CDC stands for carbide-derived carbon. Reproduced with permission.7 Copyright 2015, Wiley-VCH. | ||
Selenium cathode
Like all conversion-based electrode materials (such as the LIB anodes), the Se cathode is subject to volume expansion upon reaction with lithium because the Se lattice structure does not have enough space to accommodate the insertion of Li atoms. To overcome this issue, the common methods that have been widely utilized to suppress the volume expansion of the LIB anodes can be employed in the Li–Se batteries. Note that it is technically possible to have a pure selenium cathode, but it is almost impossible to have a pure sulfur cathode because of its poor electrical conductivity.Instead of having a concrete selenium electrode, it is more practical to spread the electroactive material throughout a porous matrix in which there is enough space for the cathode material expansion. A practical architecture for this purpose is microporous carbon.5–7 The composite is typically prepared by the impregnation of a micro-/meso-porous carbon (or a similar nanostructured architecture) with Se at a moderate temperature of around 600 °C, owing to the low melting point of Se (221 °C).7–10 The host matrix porosity provides space for the Se volume expansion. This space can also be provided by hollow objects as the reaction in the lateral shell is faster, and the expansion can be handled internally.11
Most of the research in this area is focused on utilizing carbon with different structures derived from various sources such as biomass,12,13 metal–organic frameworks,14,15 polymers,16etc. Zhao et al. prepared a porous carbon by the simple thermal carbonation of potassium tartrate and subsequent impregnation by selenium.17 Although the overall weight of the selenium incorporated into the mesopores was 50 wt%, the SEM images show no morphological change upon Se impregnation (Fig. 2). This indicates that Se is confined within the carbon matrix.
 | ||
| Fig. 2 SEM images of as-prepared (a and b) carbon and (c and d) Se/carbon composite. Reproduced with permission.17 Copyright 2016, Royal Society of Chemistry. | ||
There are typically two approaches to conducting the so-called melt infusing process: in a tube furnace under flowing inert gas,7,18,19 or a sealed vessel with an inert atmosphere10,20–23 or vacuum.5,9 It should be taken into account that the Se content is always less than the initial ratio in the precursors due to the considerable vapor pressure of Se at high temperatures, which causes a waste of Se in the reactor.
There are also possibilities for the chemical reaction of a carbon precursor with Se24,25 or Se compounds.26 However, the Se content is normally below 50%, which is not in favor of a high energy density. Wang et al. introduced a solution-based method utilizing ethylenediamine as the solvent for the impregnation of carbon nanotubes with Se nanoparticles.27 In comparison to the conventional melt-infusion method widely employed for the impregnation of microporous carbons, they concluded that the solution-based method results in a better battery performance, accompanied by a higher discharge capacity. Nevertheless, this method cannot be employed for the impregnation of micro- or even meso-porous carbons because the Se nanoparticles are much larger than the pores. Instead, this method can be used for the surface decoration of carbon nanomaterials. Therefore, the melt-infusion method remains the dominant approach for incorporating Se into porous carbons. In this case, the melted Se utterly fills the micropores, as can be judged by the significantly reduced specific surface area of the carbon.28,29
Li et al. devised a baked-in-salt approach in which the Se infiltration occurs in a sealed reactor between two NaCl layers.30 Since the precursors are not exposed to the reactive atmosphere, no flow of inert gas is required, and this makes the process simpler. The reversible discharge capacity was over 600 mA h g−1 with well-defined battery performance displaying a flat plateau between 2.1 and 1.6 V vs. Li/Li+.
Zhang et al. synthesized the Se@C core–shell by a hydrothermal method, starting with sodium selenate as the Se precursor.26 The resulting material was carbon microtubes filled with Se (Fig. 3). The cathode could reach a discharge capacity of 558 mA h g−1, but the rate capability was poorer than the pristine Se cathode, suggesting that the carbon coating was not electrochemically permeable enough at high discharge rates.
 | ||
| Fig. 3 SEM micrograph (a and b), TEM micrograph (c and d) and elemental mapping images (e) of the Se/carbon-rich core–shell composites in the border area enclosed by the square in the TEM micrograph (d). Reproduced with permission.26 Copyright 2015, Elsevier. | ||
Fig. 4 shows the typical electrochemical performance of a Li–Se battery. The galvanostatic profiles are in good agreement with the cyclic voltammograms, suggesting one-step conversion of Se into Li2Se. This is a noticeable advantage of Li–Se batteries over Li–S batteries in which the reaction of Li and S occurs at different steps, resulting in several plateaus in the battery performance. In fact, Li–Se batteries have greater promise for delivering a constant voltage.
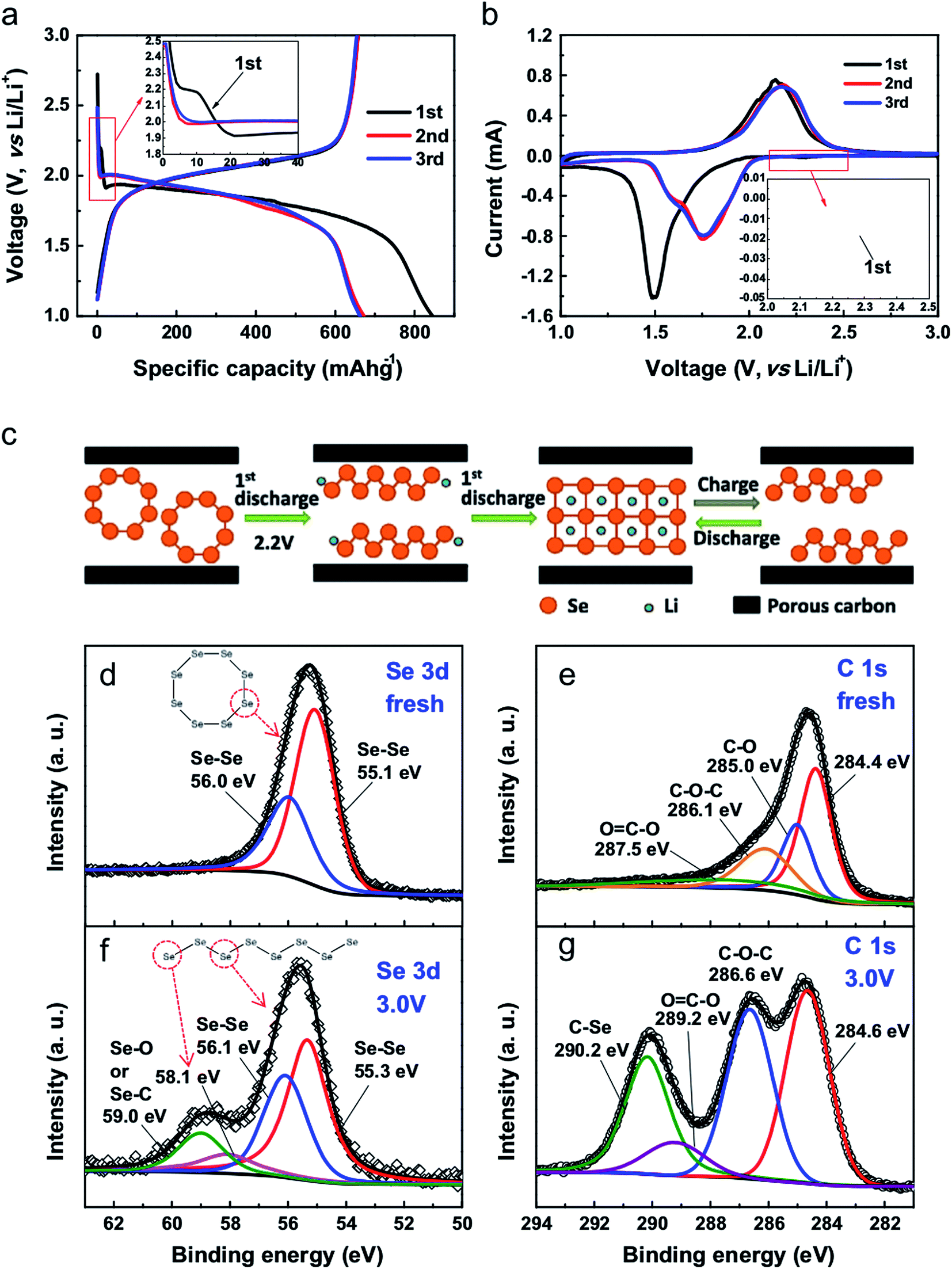 | ||
| Fig. 4 (a) Galvanostatic discharge–charge voltage profiles of the initial 3 cycles at 0.2C. (b) CV curves for Se/PCNs at a scan rate of 0.2 mV s−1 in the voltage range of 3.0–1.0 V vs. Li/Li+. (c) Proposed (de)lithiation processes of Se/PCNs. XPS spectra of Se 3d and C 1s for (d and e) fresh electrode film, and (f and g) the electrode film after the 1st discharge–charge cycle at 3.0 V. Reproduced with permission.20 Copyright 2014, Elsevier. | ||
However, this is not directly because of the difference between the conversion mechanism of Se and S, but due to the different reaction pathways, depending on the electrolyte solution. Fig. 5 compares the charge/discharge profiles of the Li–S and Li–Se batteries in different electrolytes. The multi-step conversion mechanism is the same for both systems in ether-based electrolytes. Due to the instability of polysulfides, carbonate-based electrolytes are not suitable for the Li–S batteries. It should be emphasized that the conversion mechanism in carbonate-based electrolytes is not exactly one-step (as it seems), but in the absence of middle steps, the galvanostatic profile shows a gradual transition.
 | ||
| Fig. 5 Schematic models illustrating the battery performance of Li–S and Li–Se cells in different electrolytes. | ||
As illustrated in Fig. 4c, the essential difference during the first cycles is the different lithiation process of the Se8 molecules; this transformation has been detected by XPS. Owing to the electrochemical activity of the chain-like Sen molecules, the delithiation of the Li2Se formed during the first cycle results in the formation of chain-like Sen molecules instead of the original Se8 rings.10 This is indeed general behavior, which has been reported for all Se cathode materials. However, depending on the electrode material and the electrolyte, the impact of the first cycle difference can be noticeably small,19,23,26,28,31–33 or the first cycle may follow a different pattern.25
The shape of the discharge profile is imperative to having a constant cell voltage in the battery performance. As shown in Table 1, the potential window in which the capacity is mainly delivered strongly depends on the material structure and the electrolyte. In addition to the possibility of having multiple plateaus due to multi-step lithiation, the voltage drop after the flat plateau can be gradual, delivering a significant capacity after the main lithiation process. This latter region mostly has a pseudocapacitive nature, since it is known that the electrochemical behavior of the LIB electrode materials may transform from battery performance to pseudocapacitive behavior.34,35 This pseudocapacitive region can expand to merge with the flat plateau, delivering the capacity over a broad potential window (Table 1).
Finding the optimum size of the micropores is a challenging task, which strongly depends on the overall architecture. If the micropores are too small, it is difficult to accommodate the electroactive material therein,36 and the Li diffusion is not fast and facile.37 From a basic calculation, one can conclude that the size of the micropores should be larger than 1 nm to accommodate S8 or Se8 molecules. On the other hand, larger micropores facilitate Se dissolution during the cycling performance and consequently capacity fading, but larger micropores provide better electrochemical performance.19,22,23,29,38
Mesoporous carbon also provides a suitable matrix for accommodating the Se electroactive material, since ordered mesoporous carbon (OMC) can be easily synthesized by utilizing a silica template.39 Nazar and her coworkers were the first to employ OMC for confining sulfur for the Li–S battery application.40 With the same approach, Amine and his coworkers impregnated OMC with Se to build a stable Se cathode.3 Although the Se content is less than what is expected for the practical performance, this Se/OMC architecture paved the path for the development of Li–Se batteries.
Obviously, the pore size and structure have an enormous impact on the battery performance. Liu et al. employed similar OMCs with different pore size and distribution.23 Although the initial capacity was almost independent of the pore size, the cyclability had a high dependency. As expected, the cyclability was improved by reducing the pore size, since smaller pores could better trap the electroactive material; however, quite interestingly, the rate capability was also improved. Since the rate capability is directly dependent on the Li diffusion, it is expected to have a more facile diffusion through larger channels, but it seems that the Se matrix formed in smaller pores provides a better diffusion pathway.
In addition to the carbon morphological structure, its chemical functionality also plays a substantial role in the battery performance of a Se cathode. Graphitization of the porous carbon, as well as the alteration of the sp2 hybrid, is beneficial to improving the electrochemical activity of carbon.41 The regular sp2 carbon atoms are chemically inert and do not interact with the Se atoms placed within the pores. The presence of a small amount of oxygen-based functional groups on the carbon can build strong interactions with the Se atoms, improving the Se/C composite structure and the electrode hydrophilicity.42 It is believed that the strong C–O and Se–O bonds in the composite components facilitate chemical interactions, resulting in a higher mechanical stability and better electrochemical performance.30
Peng et al. placed Se nanoparticles between the layers of reduced graphene oxide, and the cyclability was substantially improved by a capacity fading of less than 30%, while the pristine Se cathode lost over 70% of its capacity during 100 cycles at 0.2C.43 Another effect of the reduced graphene oxide was to reduce the overpotentials of charging/discharging. In addition to the protective role of the reduced graphene oxide, the chemical interactions between the composite components assisted the charge transfer across the composite matrix, as could be judged by a significantly (at least 50%) lower charge transfer resistance recorded by electrochemical impedance spectroscopy.
Doping can distribute the charge distribution across the sp2 basal plane and thus strengthen the chemical interaction between the carbon substrate and the polyselenide ions (Sen2− where 2 ≤ n ≤ 8) to avoid the possible dissolution. Several works utilized nitrogen-doped carbon for better interaction with the Se electroactive material.12,14,21,23,38 Nitrogen-doping of the carbon component improves the electrochemical performance of a Se cathode in favor of a flatter plateau and lower overpotentials (in both lithiation and delithiation processes).23
Another approach for eliminating the shuttle effect is to trap the Se electroactive material in a core–shell architecture. The carbon coating acts as a permeable membrane for the Li diffusion while preventing the dissolution of low-chain polyselenides. This can significantly improve the cyclability as there is only 30% capacity fading in the first 60 cycles (from the second cycle), while the capacity loss is negligible after that.44
Luo et al. synthesized C/Se nanocomposites by in situ encapsulation of Se during the carbon formation via a thermal carbonization of perylene-3,4,9,10-tetracarboxylicdianhydride.24 Because of this protective architecture, the cathode cyclability was maintained at 85% of its initial capacity after 250 cycles. The cell also had a good rate capability with only 50% capacity loss when increasing the charge/discharge rate from 40 mA h g−1 to 1.2 A g−1.
Shen and coworkers employed biochar as the basis and encapsulated selenium into a macro/micro porous architecture.29 They prepared a conventional coin-type cell by mixing the electroactive material with 10% acetylene black and 10% sodium alginate binder on aluminum foil as the current collector and utilized a commercial LiPF6 electrolyte. The discharge profile was almost a flat plateau delivering the cell capacity in the voltage range of 1.9 to 1.6 V. The corresponding well-defined redox system could also be observed by cyclic voltammetry. A reversible discharge capacity of 597 mA h g−1 was achieved in the second cycle at 0.2C, and less than 20% capacity fading was observed after 100 cycles. Since the micropores were generated by KOH treatment, the size distribution and structure of the micropores were dependent on the KOH concentration. For various samples with different microporosities, the specific capacity and cyclability were somehow inversely proportional.
Incorporating Se during the carbonization process is a practical approach to the preparation of Se/C composites with appropriate confinement. A Se/C cathode prepared by the carbonization of polyacrylonitrile showed excellent cyclability since the reversible discharge capacity could be maintained during 200 cycles.45 The interesting feature was that the capacity increased upon cycling at low rates. Since the rate capability was relatively good, the reversible capacity (e.g., after the 30th cycle) showed less than 30% capacity loss when increasing the charge/discharge rate by two orders of magnitude. On the other hand, the discharge voltage slightly increased with cycling. The cell exhibited minor capacity fading after one month of storage in the fully charged or discharged states.
In addition to the carbon matrix, the binder also plays a substantial role in Li diffusion within the electrode, and this effect is more important for electroactive materials such as Se, which undergo chemical changes and volume expansion during electrochemical performance. Sun et al. examined the battery performance of Se/C cathodes prepared by common binders.13 Comparison of three cases of sodium alginate (SA), carboxymethylcellulose (CMC), and polyvinylidene fluoride (PVDF) showed that the cell voltage could be changed by over 0.2 V while having about 15% change in the discharge capacity. Among these binders, SA showed the best cyclability and specific capacity, while the cyclability of PVdF was noticeably poorer.
A binder-free design of the Se cathode can provide a better opportunity for fast charging/discharging as the binder can be a blocking obstacle for the diffusion of Li atoms. Note that the binder is not as immobile as in the LIB cathodes where there is no volume change. He et al. designed a three-dimensional hierarchical architecture utilizing CNT and graphene. The Se cathode could deliver over 600 mA h g−1 at low charge/discharge rates while retaining over 200 mA h g−1 at 10C,46 which is still a higher capacity and better rate performance compared to the available LIB cathodes.
Han et al. impregnated the mesoporous carbon nanoparticles (MCNs) with Se and placed the nanoparticles between the graphene oxide layers.19 The prepared graphene oxide paper could serve as a binder-free electrode when the Se content was 62%. Although the Se-impregnated MCNs could deliver the same capacity as that of the Se/MnC-rGO, the rate capabilities of these electrodes were significantly different. While the Se/MNC cathode delivered a negligible capacity at 2C, the Se/MNC-rGO retained 60% of its capacity when increasing the discharge rate by one order of magnitude. This difference could be well understood by the significant difference in the charge transfer resistance.
Shen and coworkers synthesized a binder-free Se cathode by electroless deposition of Se over a nickel foam.31 The cathode could achieve 82% of its theoretical capacity, but the cyclability was still poor, as the specific capacity dropped from 554 mA h g−1 to 137 mA h g−1 after 100 cycles at 0.1C. To reduce the Se shuttle effect, they coated the Se surface with graphene oxide, and this significantly improved both the specific capacity and cyclability. The initial capacity was increased to 665 mA h g−1 and the cathode maintained 226 mA h g−1 after 100 cycles with the same rate.
Carbon is not the only compositing component that can confine the Se electroactive material, and other bonding matrices can also be employed to hold the Se electroactive material while providing enough space for the volume expansion during discharge. Zhang et al. reported that a nanocomposite of Se/polyaniline can enhance the discharge capacity significantly (almost double).47 However, the real difference appeared in the cyclability where the Se nanowires could not deliver a considerable capacity after 30 cycles, while the Se/polyaniline cathode delivered over 450 mA h g−1 after 200 cycles. Adding graphene to this nanocomposite further improved both the capacity and cyclability. The impedance spectra suggested that the interfacial resistance was significantly reduced by adding polyaniline and graphene. Polypyrrole has also been employed for this purpose.48 The electrochemical redox systems of the host conductive polymer may also contribute to the overall capacity.
Li2Se cathode
Owing to the extremely poor electrical conductivity of S for the cathode in Li–S batteries, there is a growing interest in utilizing Li2S as the starting cathode material because of its electrical conductivity (1 × 10−13 S m−1). Furthermore, the volume change is smaller for the Li2S cathode, resulting in 44% shrinkage, compared to the 80% volume expansion of a S cathode. Although the electrical conductivity is not an issue for the Se cathode in Li–Se batteries, utilizing Li2Se as a cathode material can be beneficial for reducing the essential volume expansion occurring in the course of charging/discharging.49 However, the utilization of Li2Se is not exactly like that of Li2S, since Li2Se is not still available commercially and has to be synthesized. On the other hand, the high melting temperature of Li2Se makes the conventional carbon impregnation difficult. This too has an advantage since the thermal stability of Li2Se can be gained in a high-temperature carbonization process for the preparation of Li2Se/C composites.Wu et al. examined Li2Se as a potential cathode material for Li–Se batteries.49 They synthesized the Li2Se nanoparticles via a chemical reaction of Se powder and a Li reducing agent plus Li2Se/C nanocomposite and Li2Se@C core/shell. All the materials delivered a specific capacity of about 500 mA h g−1 (with respect to the Li2Se mass) at 0.1C. The battery performance of the pure Li2Se nanoparticles was much better, delivering the majority of the capacity via a flat plateau; however, the cyclability was poor. In a comparison of the Li2Se nanocomposite and Li2Se@C core–shell, the former produced a higher specific capacity (even higher than that of Li2Se nanoparticles), but the cyclability of the latter was slightly better. The poor cyclability of the pure Li2Se nanoparticles was ascribed to the Li dendrites formed in the course of cycling.
Electrolytes and separators
Experimental studies showed that the performance of a Li–Se battery is better in carbonate-based electrolytes, compared to ether-based electrolytes, because of the one-step formation of the Li2Se product without the interference of unstable intermediates.13,24,30,47,50 The Li–Se battery performance in ether-based electrolytes was accompanied by multiple plateaus, suggesting the formation of Li2Se via the intermediates of Li2Se8 and Li2Se4.31,44,51 Nevertheless, Babu and Ramesha discussed that this was due to a confinement effect and the direct conversion of Se to Li2Se could also occur even in ether-based electrolytes.52 Yi et al. reported that the cathode process might be a one-step process in an ether-based electrolyte, but the battery performance was definitely less well-defined in comparison to a carbonate-based electrolyte.53 Another major difference was a noticeably higher initial capacity and lower reversible capacity in an ether-based electrolyte.In addition to the one-step cathode reaction in carbonate-based electrolytes, a stable solid electrolyte interphase (SEI) can be formed on the Se cathode, which significantly improves the cycling performance.54,55 This behavior has also been reported for the Li–S batteries.56 In general, this is a significant advantage for the Li–Se batteries to perform in the carbonate-based electrolyte, as the practical performance of Li–S batteries usually needs expensive ether-based electrolytes. Carbonate solvents chemically react with polysulfides in Li–S batteries57 and thus, the performance of Li–S batteries in the carbonate-based electrolyte is not satisfactory.58,59 Although there has been no direct investigation of the possible chemical interaction of polyselenides with carbonate-based electrolytes, it seems that polyselenides are considerably stable in the conventional carbonate electrolytes.
It should be emphasized that the battery performance is not merely dependent on the electrolyte, since a multi-step cathode reaction has also been reported in a conventional carbonate-based electrolyte.60 However, the majority of reports show one-step, well-defined electrochemical performance in the conventional carbonate-based electrolytes, an advantage that Li–S batteries lack.
The electrolyte concentration has a huge impact on the electrochemical performance of the Se cathodes, not only because of the Li transport, but also lower solubility of the polyselenides at higher concentrations, due to a common ion effect. For a higher concentration of the lithium salt, the rate capability is significantly improved.7
Ionic liquids (ILs) are novel electrolyte candidates for lithium batteries, but utilizing pure ILs as the electrolyte is neither economical nor efficient; instead, they can be used as additives to facilitate ion transport in the electrolyte.61 The influence of an IL salt was investigated in an ether-based electrolyte.18 Although the cyclability was improved during the first cycle, it was not advantageous in long-term cycling. However, the coulombic efficiency was substantially improved by the IL presence.
In addition to the capacity fading due to the shuttle effect, in which the electroactive material is dissolved in the electrolyte, the dissolved polyselenides can interfere with the anode process. A common approach in Li–S batteries is to utilize a separator to divide the anode and cathode processes. Therefore, the dissolved species cannot interact with the anode. In this case, not only is there less room for the dissolution, but the dissolved species might somehow return to the cathode matrix. In a sense, this is similar to carbon coating, but the coating layer is over the entire electrode rather than the electroactive particles. This approach has also been applied to the Li–Se batteries.32,62
Fang et al. fabricated a Li–Se battery with pure Se as the cathode, separated from the Li anode by a graphene-coated polymer.32 The cathode displayed excellent cyclability with no capacity fading over 1000 cycles at 0.5C (except a slight increase during the first cycle). They showed that the graphene coating plays a crucial role in this capacity retention, as the cell made of the polymer separator only lost 30% of its capacity during the first 200 cycles; however, the capacity was maintained afterward, probably due to the formation of a protective membrane over the polymer separator. The role of graphene was not limited to the mass transfer protection, but also included reducing the interfacial resistance according to the significantly different charge transfer resistance detected by impedance spectroscopy.
The formation of a stable but permeable SEI is an essential requirement for long-term stability. Lee et al. introduced a handy method for forming a stable SEI layer on the Se cathode.8 They found that an electrochemical pretreatment at a potential as low as 0.1 V vs. Li/Li+ results in the formation of a protective SEI on the Se cathode. This can significantly enhance the cyclability of the cathode to maintain at least 90% of its capacity after 100 cycles at 0.2C, while the base electrode without this pretreatment loses 35% of its capacity.
Other selenium batteries
The key feature of Li–Se batteries is the role of the selenium cathode rather than the Li counterion. Therefore, similar selenium batteries can be categorized in this family of rechargeable batteries as they have similar structures and mechanisms. Sodium–selenium (Na–Se)3,9,25,63–65 and magnesium–selenium (Mg–Se)63,66 batteries have been considered as promising members of this family. Although potassium-ion, potassium–sulfur, and potassium–oxygen batteries have been widely considered as promising alternatives to the lithium counterparts,67 there is no report on a potassium–selenium (K–Se) battery.The Na–Se battery is fundamentally similar to the Li–Se battery, having a comparable electrochemical performance. The redox potential of the sodiation/desodiation of the Se cathode is only 0.3 V lower than that of the lithiation/delithiation. However, the Se cathode stability is much lower for the sodiation/desodiation because of the larger size of the Na atoms causing massive volume changes in the course of charging/discharging.24 With a Se@CNF/CNT cathode, the specific capacity was almost the same as in Li–Se and Na–Se batteries, but the cyclability of the Na–Se battery was significantly poorer.50 For a similar reason, the structural change during the first cycle was much more significant, as can be judged by a significantly different cyclic voltammogram during the first cycle (Fig. 6).
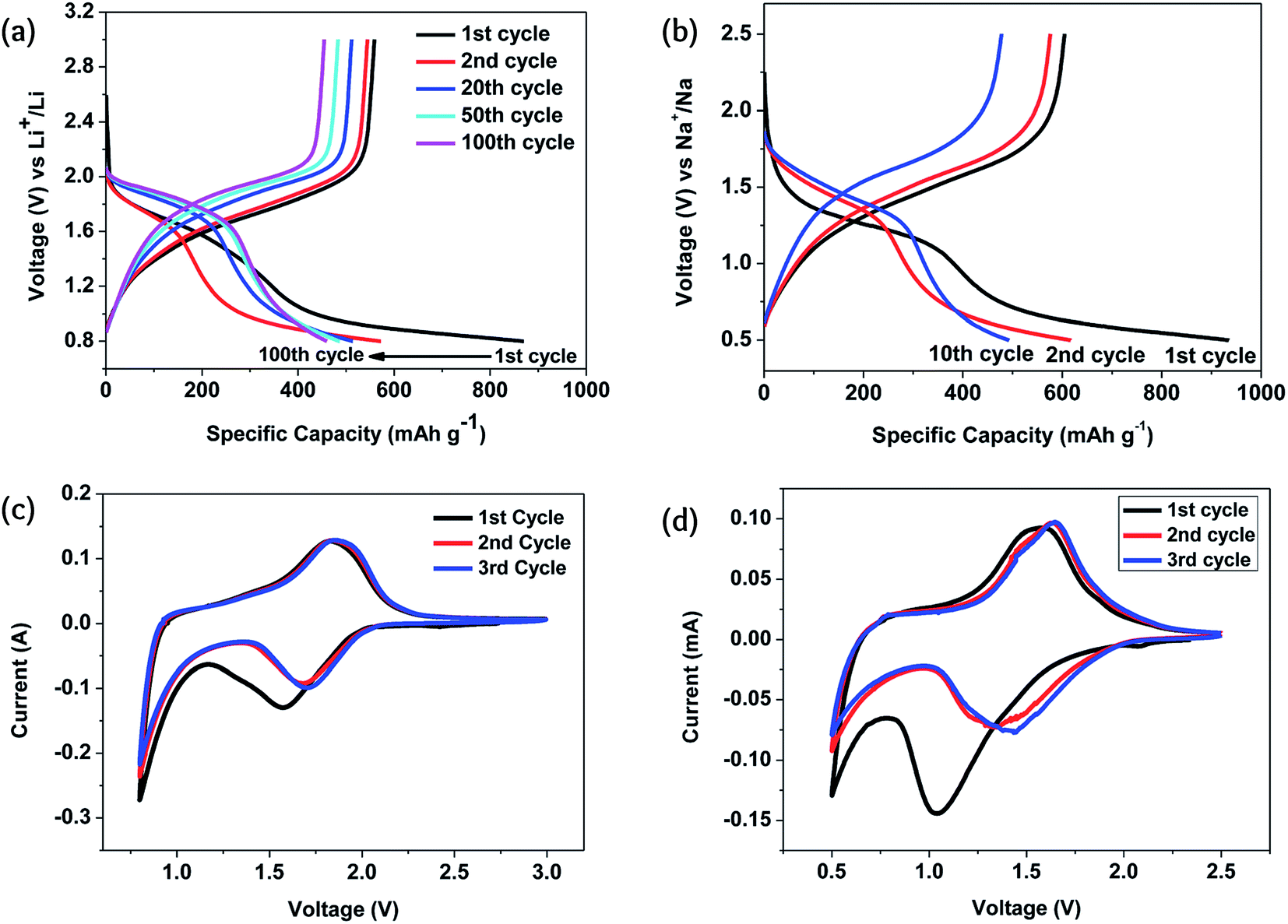 | ||
Fig. 6 Electrochemical performance of the C/Se composite formed in situ: (a and b) the galvanostatic charge–discharge in the corresponding Li and Na electrolytes; (c and d) cyclic voltammograms for the corresponding Li and Na electrolytes. The current density and the potential scan rates were 0.1 A g−1 and 0.1 mV s−1. The electrolyte was composed of a mixture of ethylene carbonate/diethyl carbonate (EC–DEC, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) and 1 M LiPF6 and 1 M NaClO4 were used as the alkali metal salts. Reproduced with permission.24 Copyright 2015, Royal Society of Chemistry. :1 by volume) and 1 M LiPF6 and 1 M NaClO4 were used as the alkali metal salts. Reproduced with permission.24 Copyright 2015, Royal Society of Chemistry. | ||
In situ transmission electron microscopic studies revealed that the sodiation process is composed of three distinguishable steps in which an amorphous phase of Na0.5Se is initially formed.64 The next step is the transformation of this amorphous phase to polycrystalline Na2Se2, and finally, the formation of the Na2Se phase. This three-step process is surprisingly in favor of the battery performance, as the overall rate of these reactions is faster than lithiation. On the other hand, as shown earlier, the electrochemical performance of the Na–Se battery is well-defined, delivering a constant voltage during the discharging process. In fact, these three steps occur at a narrow range of potential and thus, cause no harm to the battery performance.
Zhao-Karger built a Mg–Se battery by coupling a Mg anode and Se confined in an OMC cathode in a non-nucleophilic electrolyte.66 Although the initial volumetric discharge capacity was as large as 1689 mA h cm−1, the battery could deliver a reversible capacity of 480 mA h cm−1 at a charge/discharge rate of 2C. Moreover, the rate capability was noticeably good, as there was less than 25% capacity loss when increasing the charge/discharge rate from 0.2C to 2C. In any case, this battery is still far from being a practical energy storage system because over 80% of the capacity fades during the first 50 cycles. The cathode composition in fully discharged condition was determined to be Mg0.7Se.
Lithium–(sulfur/selenium) batteries
Despite the serious problems of Li–S batteries, they are still promising electrochemical systems for energy storage. In comparison to Li–Se batteries, they are cheaper and have a higher specific capacity. A subtle approach is to gain the advantages of selenium to address the essential disadvantages of Li–S batteries. First of all, the electrical conductivity of selenium can substantially improve that of a S cathode. Furthermore, the well-defined electrochemical performance of selenium, due to the one-step reaction with lithium, can also improve the battery performance of the S cathode, which is accompanied by the formation of a series of intermediates before the final products of Li2S, or vice versa, during the charging process.This possibility was practically considered after the first report of a successful performance of SeS2/CNT cathode made by commercial SeS2, which could deliver a discharge capacity of 512 mA h g−1 at an applied current of 50 mA g−1 after 30 cycles.3 Since then, various selenium sulfides have been employed as potential cathode materials. For instance, a SeS7/CNT cathode delivered a specific capacity of 883 mA h g−1.68 Wang and coworkers prepared a SeS0.7/carbon composite by carbonization of polyacrylonitrile at 600 °C under vacuum.54 Confinement of the SeS0.7 molecules by the N-doped carbon could improve the charge transfer across the carbonaceous matrix while reducing the shuttle effect because of the strong bonds between the carbon atoms and SeS0.7. The resulting cathode could maintain a discharge capacity of 780 mA h g−1 after 1200 cycles at 0.6 A g−1. In similar work on a Se/C composite prepared from polyacrylonitrile, the N content was measured to be about 2.5 wt% while the Se and carbon contents were about 55 and 43 wt%.45
Zeng et al. employed a polyacrylonitrile/CNT fiber composite, prepared by electrospinning, as the initial precursor.50 They reported that the presence of carbon nanotubes could enhance the discharge capacity by 10% and increase the cell voltage by 0.1 V. However, the main difference appeared in the rate capability, as the CNT-based cathode could retain 90% of its capacity when increasing the charge/discharge rate by an order of magnitude (from 0.1 to 1 A g−1), while the other cathode showed a negligible capacity at fast discharge rates.
The discharge capacity values reported in the literature suggest that there is no linear dependency between the specific capacity and the Se/S ratio. Although the specific capacity should be increased by increasing the amount of S, due to its lower mass, the positive effects of Se on the improved capacity are enough to compensate for the overall specific capacity. Qian and coworkers used a small amount of Se in the composition of the S0.94Se0.6/C composite and obtained a specific capacity of 910 mA h g−1 at 1 A g−1;55 compared with a pure S cathode, this small amount of Se could substantially improve the battery performance. Fig. 7 compares the battery performance of the S0.94Se0.6/C and S/C cathodes. Not only was the initial irreversible capacity of the S cathode increased by the presence of a small amount of selenium, but the capacity fading during the first two cycles was substantially reduced. On the other hand, the S0.94Se0.06 cathode showed much better battery performance, characterized by a flat plateau. While the S0.94Se0.06 cathode delivered a constant voltage of 1.9–1.6 V (which was exactly similar to that of Li–Se batteries as described earlier), the cell voltage of the Li–S battery was spread over a wide range from 2.4 to 0.8 V.
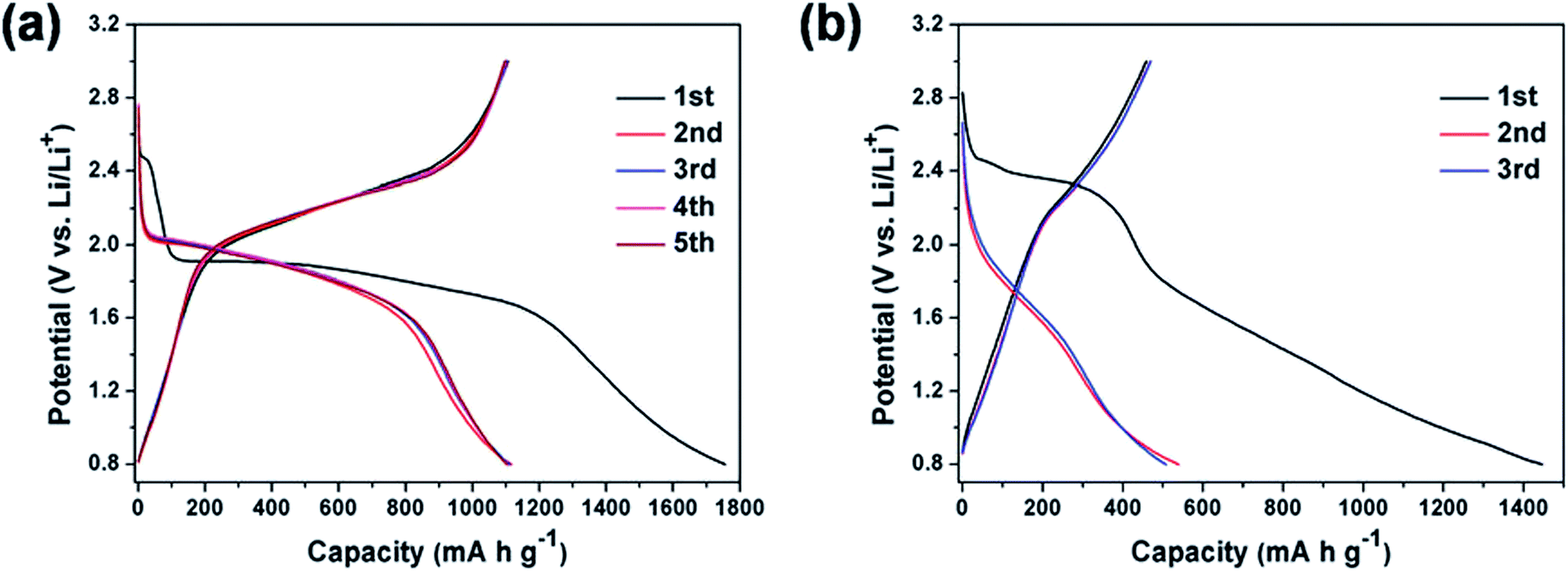 | ||
| Fig. 7 (a and b) Typical galvanostatic discharge–charge curves of the cell with S0.94Se0.06/C and S/C electrodes in the potential region of 0.8–3 V versus Li/Li+ at 0.2 A g−1, respectively. Reproduced with permission.55 Copyright 2015, Royal Society of Chemistry. | ||
The interesting point is the existence of a synergistic effect for the sulfur/selenium electrode, as the electrochemical performance of a SeS2 electrode is even more well-defined than a pristine Se electrode.68 The preparation of selenium sulfide electrode materials is quite easy and facile. In a typical synthesis, the elemental Se and S are mixed by ball milling, and the chemical transformation to selenium sulfide can be completed by a thermal treatment at a temperature close to the melting point of selenium, at which both components are fairly miscible. The importance of the formation of a selenium sulfide crystalline structure is that the sulfur atoms are attached to the chain-like structure of the selenium lattice instead of the S8 ring; this chemical structure can cause the lithium reaction to undergo a different mechanism in which larger polysulfides are formed.55 Due to the lower solubility of large polysulfides, this electrode can also be employed in carbonate-based electrolytes, which is indeed a serious disadvantage of Li–S batteries.
Toxicity of selenium
When considering Li–Se as an alternative to Li–S, one may think of the possible toxicity of selenium. The toxicity of selenium has been subject to investigations since the industrial application of selenium and following the detection of its toxicity.69 Selenium is among the trace elements that are required in human body metabolism, but it is well-known that it is toxic in excess. However, selenium is not considered a highly toxic element compared to others, which is the reason it has attracted considerable attention for medical applications.70–73 As a matter of fact, Se nanoparticles are among the promising candidates for cancer treatment. Therefore, the toxicity of selenium is usually considered in medical applications rather than industrial energy storage systems, and the focus is usually on the toxicity of Se nanoparticles.74,75The toxicity of selenium is obviously greater than that of sulfur, but note that the selenium is supposed to be utilized in the sealed cells. The leakage from lithium batteries has always been a major issue in this class of energy storage devices, but the toxicities of the available electrolytes and electrode materials are not less than that of selenium. Therefore, the selenium toxicity cannot be considered as a serious obstacle to the development of Li–Se batteries. An important point is that the toxicity of elemental selenium is less than its soluble compounds.74,75 This is in agreement with the requirement for the development of Li–Se batteries, as Se solubility is accompanied by an unfavorable capacity fading. Hence, reducing the cathode solubility (i.e., avoiding the undesirable shuttle effect) means less toxicity of the selenium cell.
Perspective
The Li–Se battery is a new member of the large family of electrochemical energy storage systems, with a practical history of only four years. However, during this short period, it has shown promising results and can compete fairly well with its counterparts, particularly its sister system, Li–S batteries. Although the current Li–Se batteries are still struggling with the same issues as the Li–S batteries, such as shuttle effect, there are considerable advantages that make them different. In fact, the extensive research done on Li–S batteries can guide the development of Li–Se batteries, but new opportunities provided by Li–Se batteries need a different research strategy.Se has excellent electrical conductivity while S is practically an insulator. Although there is no chance of using a pure S electrode, the standalone Se electrode can be successfully utilized. Of course, the shuttle effect is a major issue, but it can be overcome by a protective layer. Carbon nanomaterials are employed as the host matrix for the confinement of Se electroactive materials, but they are essentially required for S cathodes to address the low electrical conductivity. Therefore, since there is not a considerable amount of work on Li–S batteries utilizing a pure S cathode, the active topic of research should be Li–Se batteries.
The majority of the studies on Li–S batteries are in ether-based electrolytes due to the chemical reactivity of polysulfides in carbonate-based electrolytes. For practical reasons, the conventional electrolytes of the LIBs are normally carbonate-based, but the popular electrolytes of Li–S batteries are now ether-based, which is indeed a disadvantage for the Li–S batteries. Since the Li–Se batteries are considered an alternative to the Li–S batteries, much work has been conducted on Li–Se batteries in ether-based electrolytes, though carbonate-based electrolytes are practically favorable. In fact, there are a few experiences with the performance of this class of rechargeable batteries in carbonate-based electrolytes, and this is an exclusive area of research for the Li–Se batteries.
Studies of the Li–Se batteries are usually along the same pathway as the Li–S batteries with a focus on the practical potentials; however, our knowledge about this system is very limited. Hence, it is vital to inspect the battery performance by in situ and ex situ techniques to fully understand the conversion mechanism. For instance, the conversion mechanism in carbonate-based electrolytes seems to be one-step, compared to the multi-step conversion in ether-based electrolytes, but it should be involved in several sub-steps. This complexity is even greater for the promising case of the SeS electrode in which the chemical bonds between Se and S should be inspected throughout the charge/discharge process. In addition to in situ techniques, further inspections can provide invaluable information on how the Se cathode is subjected to chemical and structural changes during charge/discharge and long-term cycling. Despite the similarities of the conversion mechanisms for the Li/Se and Li/S systems, the structure of a Se electrode should be significantly different from that of S in the electrochemical performance.
In conclusion, the Li–Se batteries cannot be simply considered as a version of the Li–S batteries in which the S cathode is simply replaced by its neighbor element, Se. Despite essential similarities, there are some unique features of the Li–Se batteries that need exclusive attention. Based on the experimental results reported in the literature and a reasonable deduction, one can see the potential of the Li–Se batteries in the future of energy storage (at least as strong as the popular Li–S batteries). Surely, Se is more expensive than S, and the specific energy is less; but the invaluable advantages make it an attractive option, compared to similar systems.
References
- Batteries: Li–Se on the Way, Chem. Eng. News, 1970, 48, 9, DOI:10.1021/cen-v048n009.p009a.
- Y. Yin, S. Xin, Y. Guo and L. Wan, Lithium–Sulfur Batteries: Electrochemistry, Materials, and Prospects, Angew. Chem., 2013, 52, 13186–13200, DOI:10.1002/anie.201304762.
- A. Abouimrane, D. Dambournet, K. W. Chapman, P. J. Chupas, W. Weng and K. Amine, A New Class of Lithium and Sodium Rechargeable Batteries Based on Selenium and Selenium–Sulfur As a Positive Electrode, J. Am. Chem. Soc., 2012, 134, 4505–4508, DOI:10.1021/ja211766q.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Structure and Compatibility of a Magnesium Electrolyte with a Sulphur Cathode, Nat. Commun., 2011, 2, 427, DOI:10.1038/ncomms1435.
- Y. Liu, L. Si, X. Zhou, X. Liu, Y. Xu, J. Bao and Z. Dai, A Selenium-confined Microporous Carbon Cathode for Ultrastable Lithium–selenium Batteries, J. Mater. Chem. A, 2014, 2, 17735–17739, 10.1039/c4ta03141e.
- Y. Liu, L. Si, Y. Du, X. Zhou, Z. Dai and J. Bao, Strongly Bonded Selenium/Microporous Carbon Nanofibers Composite As a High-Performance Cathode for Lithium–Selenium Batteries, J. Phys. Chem. C, 2015, 119, 27316–27321, DOI:10.1021/acs.jpcc.5b09553.
- J. T. Lee, H. Kim, M. Oschatz, D. Lee, F. Wu, H. Lin, B. Zdyrko, W. I. Cho, S. Kaskel and G. Yushin, Micro- and Mesoporous Carbide-Derived Carbon–Selenium Cathodes for High-Performance Lithium Selenium Batteries, Adv. Energy Mater., 2015, 5, 1400981, DOI:10.1002/aenm.201400981.
- J. T. Lee, H. Kim, N. Nitta, K. Eom, D. Lee, F. Wu, H. Lin, B. Zdyrko, W. I. Cho and G. Yushin, Stabilization of Selenium Cathodes Via in situ Formation of Protective Solid Electrolyte Layer, J. Mater. Chem. A, 2014, 2, 18898–18905, 10.1039/c4ta04467c.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. Wang, Selenium@Mesoporous Carbon Composite with Superior Lithium and Sodium Storage Capacity, ACS Nano, 2013, 7, 8003–8010, DOI:10.1021/nn403108w.
- C. Yang, S. Xin, Y. Yin, H. Ye, J. Zhang and Y. Guo, An Advanced Selenium-Carbon Cathode for Rechargeable Lithium–Selenium Batteries, Angew. Chem., 2013, 52, 8363–8367, DOI:10.1002/anie.201303147.
- Y. J. Hong and Y. C. Kang, Selenium-impregnated Hollow Carbon Microspheres As Efficient Cathode Materials for Lithium–Selenium Batteries, Carbon, 2017, 111, 198–206, DOI:10.1016/j.carbon.2016.09.069.
- W. Yuan, Y. Feng, A. Xie, X. Zhang, F. Huang, S. Li, X. Zhang and Y. Shen, Nitrogen-Doped Nanoporous Carbon Derived from Waste Pomelo Peel As a Metal-free Electrocatalyst for the Oxygen Reduction Reaction, Nanoscale, 2016, 8, 8704–8711, 10.1039/c6nr00764c.
- K. Sun, H. Zhao, S. Zhang, J. Yao and J. Xu, Selenium/pomelo Peel-derived carbon nanocomposite as advanced cathode for lithium–selenium batteries, Ionics, 2015, 21, 2477–2484, DOI:10.1007/s11581-015-1451-x.
- Z. Li and L. Yin, MOF-Derived, N-Doped, Hierarchically Porous Carbon Sponges As Immobilizers to Confine Selenium As Cathodes for Li–Se Batteries with Superior Storage Capacity and Perfect Cycling Stability, Nanoscale, 2015, 7, 9597–9606, 10.1039/c5nr00903k.
- T. Liu, C. Dai, M. Jia, D. Liu, S. Bao, J. Jiang, M. Xu and C. M. Li, Selenium Embedded in Metal–Organic Framework Derived Hollow Hierarchical Porous Carbon Spheres for Advanced Lithium–Selenium Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 6924–6927, DOI:10.1021/acsami.6b04060.
- J. Zhang, Z. Zhang, Q. Li, Y. Qu and S. Jiang, Selenium Encapsulated into Interconnected Polymer-Derived Porous Carbon Nanofiber Webs As Cathode Materials for Lithium–Selenium Batteries, J. Electrochem. Soc., 2014, 161, A2093, DOI:10.1149/2.0451414jes.
- C. Zhao, L. Xu, Z. Hu, S. Qiu and K. Liu, Facile Synthesis of Selenium/Potassium Tartrate Derived Porous Carbon Composite As an Advanced Li–Se Battery Cathode, RSC Adv., 2016, 6, 47486–47490, 10.1039/c6ra07837k.
- S. Jiang, Z. Zhang, Y. Lai, Y. Qu, X. Wang and J. Li, Selenium Encapsulated into 3D Interconnected Hierarchical Porous Carbon Aerogels for Lithium–Selenium Batteries with High Rate Performance and Cycling Stability, J. Power Sources, 2014, 267, 394–404, DOI:10.1016/j.jpowsour.2014.05.116.
- K. Han, Z. Liu, J. Shen, Y. Lin, F. Dai and H. Ye, A Free-Standing and Ultralong-Life Lithium–Selenium Battery Cathode Enabled by 3D Mesoporous Carbon/Graphene Hierarchical Architecture, Adv. Funct. Mater., 2015, 25, 455–463, DOI:10.1002/adfm.201402815.
- Z. Li, L. Yuan, Z. Yi, Y. Liu and Y. Huang, Confined Selenium Within Porous Carbon Nanospheres As Cathode for Advanced Li–Se Batteries, Nano Energy, 2014, 9, 229–236, DOI:10.1016/j.nanoen.2014.07.012.
- Y. Qu, Z. Zhang, S. Jiang, X. Wang, Y. Lai, Y. Liu and J. Li, Confining selenium in nitrogen-containing hierarchical porous carbon for High-rate rechargeable lithium–selenium batteries, J. Mater. Chem. A, 2014, 2, 12255–12261, 10.1039/c4ta02563f.
- J. Zhang, L. Fan, Y. Zhu, Y. Xu, J. Liang, D. Wei and Y. Qian, Selenium/interconnected Porous Hollow Carbon Bubbles Composites As the Cathodes of Li–Se Batteries with High Performance, Nanoscale, 2014, 6, 12952–12957, 10.1039/c4nr03705g.
- L. Liu, Y. Wei, C. Zhang, C. Zhang, X. Li, J. Wang, L. Ling, W. Qiao and D. Long, Enhanced Electrochemical Performances of Mesoporous Carbon Microsphere/Selenium Composites by Controlling the Pore Structure and Nitrogen Doping, Electrochim. Acta, 2015, 153, 140–148, DOI:10.1016/j.electacta.2014.11.199.
- C. Luo, J. Wang, L. Suo, J. Mao, X. Fan and C. Wang, In Situ Formed Carbon Bonded and Encapsulated Selenium Composites for Li–Se and Na–Se Batteries, J. Mater. Chem. A, 2015, 3, 555–561, 10.1039/c4ta04611k.
- H. Wang, S. Li, Z. Chen, H. K. Liu and Z. Guo, A Novel Type of One-dimensional Organic Selenium-containing Fiber with Superior Performance for Lithium–Selenium and Sodium–Selenium Batteries, RSC Adv., 2014, 4, 61673–61678, 10.1039/c4ra10967h.
- Z. Zhang, X. Yang, Z. Guo, Y. Qu, J. Li and Y. Lai, Selenium/Carbon-Rich Core–Shell Composites As Cathode Materials for Rechargeable Lithium–Selenium Batteries, J. Power Sources, 2015, 279, 88–93, DOI:10.1016/j.jpowsour.2015.01.001.
- X. Wang, Z. Zhang, Y. Qu, G. Wang, Y. Lai and J. Li, Solution-Based Synthesis of Multi-walled Carbon Nanotube/Selenium Composites for High Performance Lithium–Selenium Battery, J. Power Sources, 2015, 287, 247–252, DOI:10.1016/j.jpowsour.2015.04.052.
- M. Jia, C. Mao, Y. Niu, J. Hou, S. Liu, S. Bao, J. Jiang, M. Xu and Z. Lu, A Selenium-confined Porous Carbon Cathode from Silk Cocoons for Li–Se Battery Applications, RSC Adv., 2015, 5, 96146–96150, 10.1039/c5ra19000b.
- H. Zhang, F. Yu, W. Kang and Q. Shen, Encapsulating Selenium into Macro-/Micro-porous Biochar-based Framework for High-Performance Lithium–selenium Batteries, Carbon, 2015, 95, 354–363, DOI:10.1016/j.carbon.2015.08.050.
- X. Li, J. Liang, Z. Hou, W. Zhang, Y. Wang, Y. Zhu and Y. Qian, A New Salt-Baked Approach for Confining Selenium in Metal Complex-Derived Porous Carbon with Superior Lithium Storage Properties, Adv. Funct. Mater., 2015, 25, 5229–5238, DOI:10.1002/adfm.201501956.
- D. Huang, S. Li, Y. Luo, X. Xiao, L. Gao, M. Wang and Y. Shen, Graphene Oxide-protected Three Dimensional Se As a Binder-free Cathode for Li–Se Battery, Electrochim. Acta, 2016, 190, 258–263, DOI:10.1016/j.electacta.2015.12.187.
- R. Fang, G. Zhou, S. Pei, F. Li and H. Cheng, Localized Polyselenides in a Graphene-coated Polymer Separator for High Rate and Ultralong Life Lithium–Selenium Batteries, Chem. Commun., 2015, 51, 3667–3670, 10.1039/c5cc00089k.
- Y. Lai, F. Yang, Z. Zhang, S. Jiang and J. Li, Encapsulation of Selenium in Porous Hollow Carbon Spheres for Advanced Lithium–Selenium Batteries, RSC Adv., 2014, 4, 39312–39315, 10.1039/c4ra06856d.
- A. Eftekhari and F. Molaei, Carbon Nanotube-assisted Electrodeposition. Part I: Battery Performance of Manganese Oxide Films Electrodeposited at Low Current Densities, J. Power Sources, 2015, 274, 1306–1314, DOI:10.1016/j.jpowsour.2013.10.136.
- A. Eftekhari and F. Molaei, Carbon Nanotube-assisted Electrodeposition. Part II: Superior Pseudo-capacitive Behavior of Manganese Oxide Film Electrodeposited at High Current Densities, J. Power Sources, 2015, 274, 1315–1321, DOI:10.1016/j.jpowsour.2013.10.144.
- K. Mi, Y. Jiang, J. Feng, Y. Qian and S. Xiong, Hierarchical Carbon Nanotubes with a Thick Microporous Wall and Inner Channel As Efficient Scaffolds for Lithium–Sulfur Batteries, Adv. Funct. Mater., 2016, 26, 1571–1579, DOI:10.1002/adfm.201504835.
- A. Ghosh and Y. H. Lee, Carbon-Based Electrochemical Capacitors, ChemSusChem, 2012, 5, 480–499, DOI:10.1002/cssc.201100645.
- Y. Jiang, X. Ma, J. Feng and S. Xiong, Selenium in Nitrogen-doped Microporous Carbon Spheres for High-performance Lithium–Selenium Batteries, J. Mater. Chem. A, 2015, 3, 4539–4546, 10.1039/c4ta06624c.
- A. Eftekhari and Z. Fan, Ordered Mesoporous Carbon and Its Applications for Electrochemical Energy Storage and Conversion, Materials Chemistry Frontiers, 2017 10.1039/c6qm00298f.
- X. Ji, K. T. Lee and L. F. Nazar, A Highly Ordered Nanostructured Carbon–Sulphur Cathode for Lithium–Sulphur Batteries, Nat. Mater., 2009, 8, 500–506, DOI:10.1038/nmat2460.
- A. Eftekhari and P. Jafarkhani, Curly Graphene with Specious Interlayers Displaying Superior Capacity for Hydrogen Storage, J. Phys. Chem. C, 2013, 117, 25845–25851, DOI:10.1021/jp410044v.
- H. Ye, Y. Yin, S. Zhang and Y. Guo, Advanced Se–C Nanocomposites: a Bifunctional Electrode Material for Both Li–Se and Li-ion Batteries, J. Mater. Chem. A, 2014, 2, 13293–13298, 10.1039/c4ta02017k.
- X. Peng, L. Wang, X. Zhang, B. Gao, J. Fu, S. Xiao, K. Huo and P. K. Chu, Reduced Graphene Oxide Encapsulated Selenium Nanoparticles for High-power Lithium–Selenium Battery Cathode, J. Power Sources, 2015, 288, 214–220, DOI:10.1016/j.jpowsour.2015.04.124.
- J. Guo, Q. Wang, C. Qi, J. Jin, Y. Zhu and Z. Wen, One-step Microwave Synthesized Core–Shell Structured Selenium@Carbon Spheres As Cathode Materials for Rechargeable Lithium Batteries, Chem. Commun., 2016, 52, 5613–5616, 10.1039/c6cc00638h.
- J. Guo, Z. Wen, Q. Wang, J. Jin and G. Ma, A Conductive Selenized Polyacrylonitrile Cathode Material for Re-chargeable Lithium Batteries with Long Cycle Life, J. Mater. Chem. A, 2015, 3, 19815–19821, 10.1039/c5ta04510j.
- J. He, Y. Chen, W. Lv, K. Wen, P. Li, Z. Wang, W. Zhang, W. Qin and W. He, Three-Dimensional Hierarchical Graphene–CNT@Se: a Highly Efficient Freestanding Cathode for Li–Se Batteries, ACS Energy Lett., 2016, 1, 16–20, DOI:10.1021/acsenergylett.6b00015.
- J. Zhang, Y. Xu, L. Fan, Y. Zhu, J. Liang and Y. Qian, Graphene-Encapsulated Selenium/Polyaniline Core–Shell Nanowires with Enhanced Electrochemical Performance for Li–Se Batteries, Nano Energy, 2015, 13, 592–600, DOI:10.1016/j.nanoen.2015.03.028.
- D. Kundu, F. Krumeich and R. Nesper, Investigation of Nano-fibrous Selenium and Its Polypyrrole and Graphene Composite As Cathode Material for Rechargeable Li-Batteries, J. Power Sources, 2013, 236, 112–117, DOI:10.1016/j.jpowsour.2013.02.050.
- F. Wu, J. T. Lee, Y. Xiao and G. Yushin, Nanostructured Li2Se Cathodes for High Performance Lithium–Selenium Batteries, Nano Energy, 2016, 27, 238–246, DOI:10.1016/j.nanoen.2016.07.012.
- L. Zeng, X. Wei, J. Wang, Y. Jiang, W. Li and Y. Yu, Flexible one-dimensional carbon–selenium composite nanofibers with superior electrochemical Performance for Li–Se/Na–Se batteries, J. Power Sources, 2015, 281, 461–469, DOI:10.1016/j.jpowsour.2015.02.029.
- Y. Qu, Z. Zhang, Y. Lai, Y. Liu and J. Li, A Bimodal Porous Carbon with High Surface Area Supported Selenium Cathode for Advanced Li–Se Batteries, Solid State Ionics, 2015, 274, 71–76, DOI:10.1016/j.ssi.2015.03.007.
- D. Babu and K. Ramesha, Constraining Polyselenide Formation in Ether Based Electrolytes Through Confinement of Se in Microporous Carbon Matrix for Li–Se Batteries, Electrochim. Acta, 2016, 219, 295–304, DOI:10.1016/j.electacta.2016.10.026.
- Z. Yi, L. Yuan, D. Sun, Z. Li, C. Wu, W. Yang, Y. Wen, B. Shan and Y. Huang, High-performance lithium–selenium batteries promoted by heteroatom-doped microporous carbon, J. Mater. Chem. A, 2015, 3, 3059–3065, 10.1039/c4ta06141a.
- C. Luo, Y. Zhu, Y. Wen, J. Wang and C. Wang, Carbonized Polyacrylonitrile-Stabilized SeS Cathodes for Long Cycle Life and High Power Density Lithium Ion Batteries, Adv. Funct. Mater., 2014, 24, 4082–4089, DOI:10.1002/adfm.201303909.
- X. Li, J. Liang, K. Zhang, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, Amorphous S-rich S Se/C (x ≤ 0.1) Composites Promise Better Lithium–Sulfur Batteries in a Carbonate-based Electrolyte, Energy Environ. Sci., 2015, 8, 3181–3186, 10.1039/c5ee01470k.
- Y. Xu, Y. Wen, Y. Zhu, K. Gaskell, K. A. Cychosz, B. Eichhorn, K. Xu and C. Wang, Confined Sulfur in Microporous Carbon Renders Superior Cycling Stability in Li/S Batteries, Adv. Funct. Mater., 2015, 25, 4312–4320, DOI:10.1002/adfm.201500983.
- T. Yim, M. Park, J. Yu, K. J. Kim, K. Y. Im, J. Kim, G. Jeong, Y. N. Jo, S. Woo, K. S. Kang, I. Lee and Y. Kim, Effect of Chemical Reactivity of Polysulfide Toward Carbonate-based Electrolyte on the Electrochemical Performance of Li–S Batteries, Electrochim. Acta, 2013, 107, 454–460, DOI:10.1016/j.electacta.2013.06.039.
- J. Gao, M. A. Lowe, Y. Kiya and H. D. Abruña, Effects of Liquid Electrolytes on the Charge–Discharge Performance of Rechargeable Lithium/Sulfur Batteries: Electrochemical and In Situ X-ray Absorption Spectroscopic Studies, J. Phys. Chem. C, 2011, 115, 25132–25137, DOI:10.1021/jp207714c.
- S. S. Zhang, Liquid Electrolyte Lithium/Sulfur Battery: Fundamental Chemistry, Problems, and Solutions, J. Power Sources, 2013, 231, 153–162, DOI:10.1016/j.jpowsour.2012.12.102.
- K. Han, Z. Liu, H. Ye and F. Dai, Flexible self-standing graphene–Se@CNT composite film as a binder-free cathode for rechargeable Li–Se batteries, J. Power Sources, 2014, 263, 85–89, DOI:10.1016/j.jpowsour.2014.04.027.
- A. Eftekhari, Y. Liu and P. Chen, Different Roles of Ionic Liquids in Lithium Batteries, J. Power Sources, 2016, 334, 221–239, DOI:10.1016/j.jpowsour.2016.10.025.
- Z. Zhang, Z. Zhang, K. Zhang, X. Yang and Q. Li, Improvement of Electrochemical Performance of Rechargeable Lithium–Selenium Batteries by Inserting a Free-Standing Carbon Interlayer, RSC Adv., 2014, 4, 15489–15492, 10.1039/c4ra00446a.
- C. Yang, Y. Yin and Y. Guo, Elemental Selenium for Electrochemical Energy Storage, J. Phys. Chem. Lett., 2015, 6, 256–266, DOI:10.1021/jz502405h.
- Q. Li, H. Liu, Z. Yao, J. Cheng, T. Li, Y. Li, C. Wolverton, J. Wu and V. P. Dravid, Electrochemistry of Selenium with Sodium and Lithium: Kinetics and Reaction Mechanism, ACS Nano, 2016, 10, 8788–8795, DOI:10.1021/acsnano.6b04519.
- J. Ding, H. Zhou, H. Zhang, T. Stephenson, Z. Li, D. Karpuzov and D. Mitlin, Exceptional Energy and New Insight with Sodium–Selenium Battery Based on Carbon Nanosheet Cathode and Pseudographite Anode, Energy Environ. Sci., 2017, 10, 153–165, 10.1039/c6ee02274j.
- Z. Zhao-Karger, X. Lin, C. B. Minella, D. Wang, T. Diemant, R. J. Behm and M. Fichtner, Selenium and Selenium–Sulfur Cathode Materials for High-Energy Rechargeable Magnesium Batteries, J. Power Sources, 2016, 323, 213–219, DOI:10.1016/j.jpowsour.2016.05.034.
- A. Eftekhari, Z. Jian and X. Ji, Potassium Secondary Batteries, ACS Appl. Mater. Interfaces, 2017 DOI:10.1021/acsami.6b07989.
- Y. Cui, A. Abouimrane, J. Lu, T. Bolin, Y. Ren, W. Weng, C. Sun, V. A. Maroni, S. M. Heald and K. Amine, (De)Lithiation Mechanism of Li/SeS (=0–7) Batteries Determined by In Situ Synchrotron X-ray Diffraction and X-ray Absorption Spectroscopy, J. Am. Chem. Soc., 2013, 135, 8047–8056, DOI:10.1021/ja402597g.
- E. P. Painter, The Chemistry and Toxicity of Selenium Compounds, with Special Reference to the Selenium Problem, Chem. Rev., 1941, 28, 179–213, DOI:10.1021/cr60090a001.
- L. Kong, Q. Yuan, H. Zhu, Y. Li, Q. Guo, Q. Wang, X. Bi and X. Gao, The Suppression of Prostate LNCaP Cancer Cells Growth by Selenium Nanoparticles Through Akt/Mdm2/AR Controlled Apoptosis, Biomaterials, 2011, 32, 6515–6522, DOI:10.1016/j.biomaterials.2011.05.032.
- Y. Huang, L. He, W. Liu, C. Fan, W. Zheng, Y. Wong and T. Chen, Selective Cellular Uptake and Induction of Apoptosis of Cancer-targeted Selenium Nanoparticles, Biomaterials, 2013, 34, 7106–7116, DOI:10.1016/j.biomaterials.2013.04.067.
- B. Yu, T. Liu, Y. Du, Z. Luo, W. Zheng and T. Chen, X-ray-responsive Selenium Nanoparticles for Enhanced Cancer Chemo-radiotherapy, Colloids Surf., B, 2016, 139, 180–189, DOI:10.1016/j.colsurfb.2015.11.063.
- S. Chaudhary, A. Umar and S. Mehta, Selenium Nanomaterials: an Overview of Recent Developments in Synthesis, Properties and Potential Applications, Prog. Mater. Sci., 2016, 83, 270–329, DOI:10.1016/j.pmatsci.2016.07.001.
- J. Zhang, H. Wang, X. Yan and L. Zhang, Comparison of Short-term Toxicity Between Nano-Se and Selenite in Mice, Life Sci., 2005, 76, 1099–1109, DOI:10.1016/j.lfs.2004.08.015.
- J. Zhang, X. Wang and T. Xu, Elemental Selenium at Nano Size (Nano-Se) As a Potential Chemopreventive Agent with Reduced Risk of Selenium Toxicity: Comparison with Se-Methylselenocysteine in Mice, Toxicol. Sci., 2007, 101, 22–31, DOI:10.1093/toxsci/kfm221.
- L. Liu, Y. Hou, X. Wu, S. Xiao, Z. Chang, Y. Yang and Y. Wu, Nanoporous Selenium As a Cathode Material for Rechargeable Lithium–Selenium Batteries, Chem. Commun., 2013, 49, 11515–11517, 10.1039/c3cc46943c.
- H. Youn, J. H. Jeong, K. C. Roh and K. Kim, Graphene–Selenium Hybrid Microballs As Cathode Materials for High-performance Lithium–Selenium Secondary Battery Applications, Sci. Rep., 2016, 6, 30865, DOI:10.1038/srep30865.
- K. Balakumar and N. Kalaiselvi, Selenium Containing Tube-in-Tube Carbon: a One Dimensional Carbon Frame Work for Selenium Cathode in Li–Se Battery, Carbon, 2017, 112, 79–90, DOI:10.1016/j.carbon.2016.10.097.
| This journal is © The Royal Society of Chemistry 2017 |