
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect heteroarylation polymerization: guidelines for defect-free conjugated polymers†
Thomas
Bura‡
a,
Serge
Beaupré‡
a,
Marc-André
Légaré
b,
Jesse
Quinn
c,
Etienne
Rochette
b,
J. Terence
Blaskovits
a,
Frédéric-Georges
Fontaine
 b,
Agnieszka
Pron
d,
Yuning
Li
c and
Mario
Leclerc
*a
b,
Agnieszka
Pron
d,
Yuning
Li
c and
Mario
Leclerc
*a
aCanada Research Chair on Electroactive and Photoactive Polymers, Department of Chemistry, Université Laval, Quebec City, Quebec G1V 0A6, Canada. E-mail: Mario.Leclerc@chm.ulaval.ca
bDepartment of Chemistry, Université Laval, Quebec City, Quebec G1V 0A6, Canada
cDepartment of Chemical Engineering, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada
dMerck Chemicals Ltd, Chilworth Technical Centre, SO16 7QD, UK (A Subsidiary of Merck KGaA, Darmstadt, Germany)
First published on 20th March 2017
Abstract
Direct (hetero)arylation polymerization (DHAP) has emerged as a valuable and atom-economical alternative to traditional cross-coupling methods for the synthesis of low-cost and efficient conjugated polymers for organic electronics. However, when applied to the synthesis of certain (hetero)arene-based materials, a lack of C–H bond selectivity has been observed. To prevent such undesirable side-reactions, we report the design and synthesis of new, bulky, phosphine-based ligands that significantly enhance selectivity of the DHAP process for both halogenated and non-halogenated electron-rich and electron-deficient thiophene-based comonomers. To better understand the selectivity issues, density functional theory (DFT) calculations have been performed on various halogenated and non-halogenated electron-rich and electron-deficient thiophene-based comonomers. Calculations showed that the presence of bromine atoms decreases the energy of activation (Ea) of the adjacent C–H bonds, allowing undesirable β-defects for some brominated aromatic units. Both calculations and the new ligands should lead to the rational design of monomers and methods for the preparation of defect-free conjugated polymers from DHAP.
1. Introduction
Conjugated polymers are the object of considerable attention from both academic and industrial laboratories. They often combine the excellent electrical and optical properties of metals and inorganic semiconducting materials with the mechanical flexibility, simple processing, and low production cost of synthetic polymers. Their good solubility facilitates large-scale processing techniques such as roll-to-roll printing, which makes low-cost printed electronic device fabrication possible. Polymer solar cells (PSCs)1–4 and organic field-effect transistors (OFETs)5–9 are among the most promising applications for conjugated polymers. With power conversion efficiencies having surpassed 10%,10–12 lifetimes of nearly 15 years13,14 and hole mobilities up to 36 cm2 V−1 s−1,15 conjugated polymers seem to have now reached the requirements for commercial applications.16,17The development of plastic electronics relies on innovative, robust and versatile coupling methods, such as Ziegler–Natta,18,19 Migita–Stille,20 Kumada,21 Heck,22 Miyaura–Suzuki,23 Negishi24 and olefin-metathesis25–27 to produce well-defined conjugated polymers. However, production cost, presence of impurities, batch-to-batch variations and industrial scalability are still major issues for the implementation of plastic electronics. For instance, Migita–Stille cross-coupling polymerization techniques, which involve the use of organostannanes (synthesized via multistep organometallic processes that require demanding purification steps), lead to stoichiometric quantities of toxic by-products such as Me3SnBr. Clearly, cheaper and more atom-efficient methodologies are highly desirable for the preparation of semiconducting organic polymers and their large-scale applications.
For these reasons, direct (hetero)arylation polymerization (DHAP) is generating great interest among the polymer science community and is now considered an important asset for polymer chemists despite the early stage of its development.28–34 Tracing its origins to the synthesis of small organic molecules by direct (hetero)arylation, this polymerization method facilitates C–C bond formation between (hetero)arenes and (hetero)aryl halides (most commonly I and Br) catalyzed by palladium complexes. Unlike the Migita–Stille cross-coupling polymerization, only benign by-products are generated during the DHAP (mainly CsHCO3 and CsBr). Up to now, this polymerization method has been successfully applied to the synthesis of many conjugated polymers based on key monomers such as 3-alkylthiophene,35–39 benzodithiophene (BDT),40–42 5-alkylthieno[3,4-c]pyrrole-4,6-dione (TPD)43–46 and 3,6-bis(thiophen-2-yl)-2,5-bis(2-alkyl)pyrrolo[3,4-c]-pyrrole-1,4-dione (DPP)47–54 derivatives.
Despite recent achievements, significant challenges, mainly for some thiophene-based derivatives that feature several active C–H bonds, remain to be overcome, namely: (1) α-α homocouplings side-reactions and (2) regioselectivity issues (α,β activated hydrogen atoms) that lead to β-defects (see Fig. 1). Mechanistically speaking, direct (hetero)arylation involves the oxidative addition of an (hetero)arylhalide substrate to a palladium catalyst followed by the heterolytic cleavage of a C–H bond of a second (hetero)aryl substrate, leading to the formation a new C–C bond via reductive elimination. Most (hetero)arenes undergo a concerted metalation-deprotonation (CMD) process in which the co-ligand directly participates in the proton transfer.30 Presently, a limited number of phosphine-based ligands yield high reactivity and selectivity for DHAP. Among them, P(o-NMe2(C6H4))3 has proven itself to be suitable to the preparation of highly regioregular poly(alkylthiophene)s while P(o-OMe(C6H4))3 seems adequate for the copolymerization of bromoarenes with heteroarenes.55 Inspired by the pioneering work done by Buchwald et al.56 on the design of specific phosphine ligands (SPhos, XPhos, DavePhos, etc.) for palladium-catalyzed formation of C–C, C–N and C–O bonds and the studies by Thompson et al.,57 Bura et al.32 and Dubnik et al.58 demonstrating the positive influence of bulky carboxylic acid additives on the DHAP selectivity, we report new phosphine-based ligands closely related to P(o-OMe(C6H4))3 (LREF, Fig. 2). The phosphines were made bulkier at the ortho-position (L1–L5) using a low cost, simple and efficient methodology in the hope that they will enhance DHAP selectivity. The effect of the phosphine-based ligands was investigated for the polymerization of various brominated and non-brominated electron-rich and electron-poor thiophene-based monomers (Fig. 2). The influence of the nature of the substituents was also analyzed. Finally, density functional theory (DFT) calculations were used to rationalize the selectivity issues observed for some halogenated and non-halogenated electron-rich and electron-deficient benzo[1,2-b:4,5-b′]dithiophene (BDT), pyrrolo[3,4-c]-pyrrole-1,4-dione (DPP) and thiadiazolo[3,4-e]isoindole-5,7-dione (TID) derivatives. DFT calculations were also utilized to explain why some well-defined and nearly defect-free conjugated polymers reported in literature were successfully prepared by DHAP.
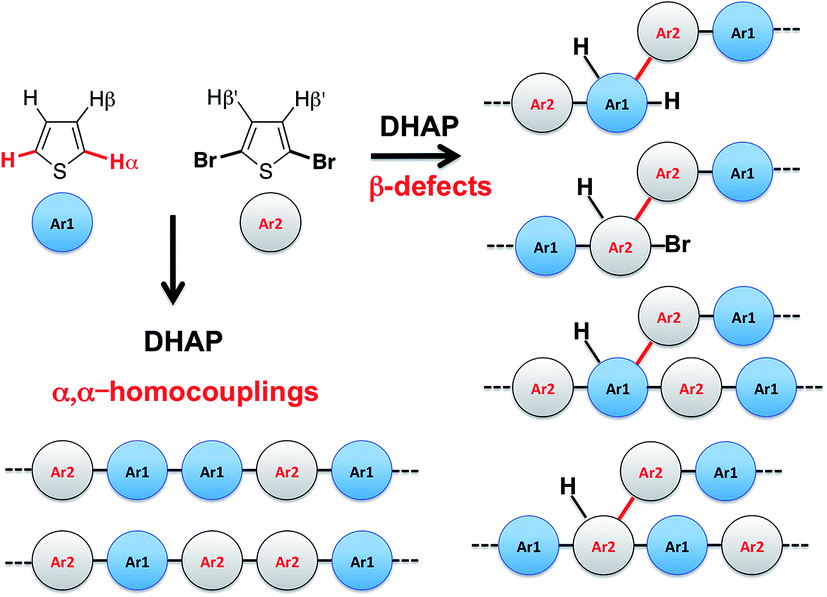 | ||
| Fig. 1 Most probable structural defects upon direct (hetero)arylation polymerization (DHAP). | ||
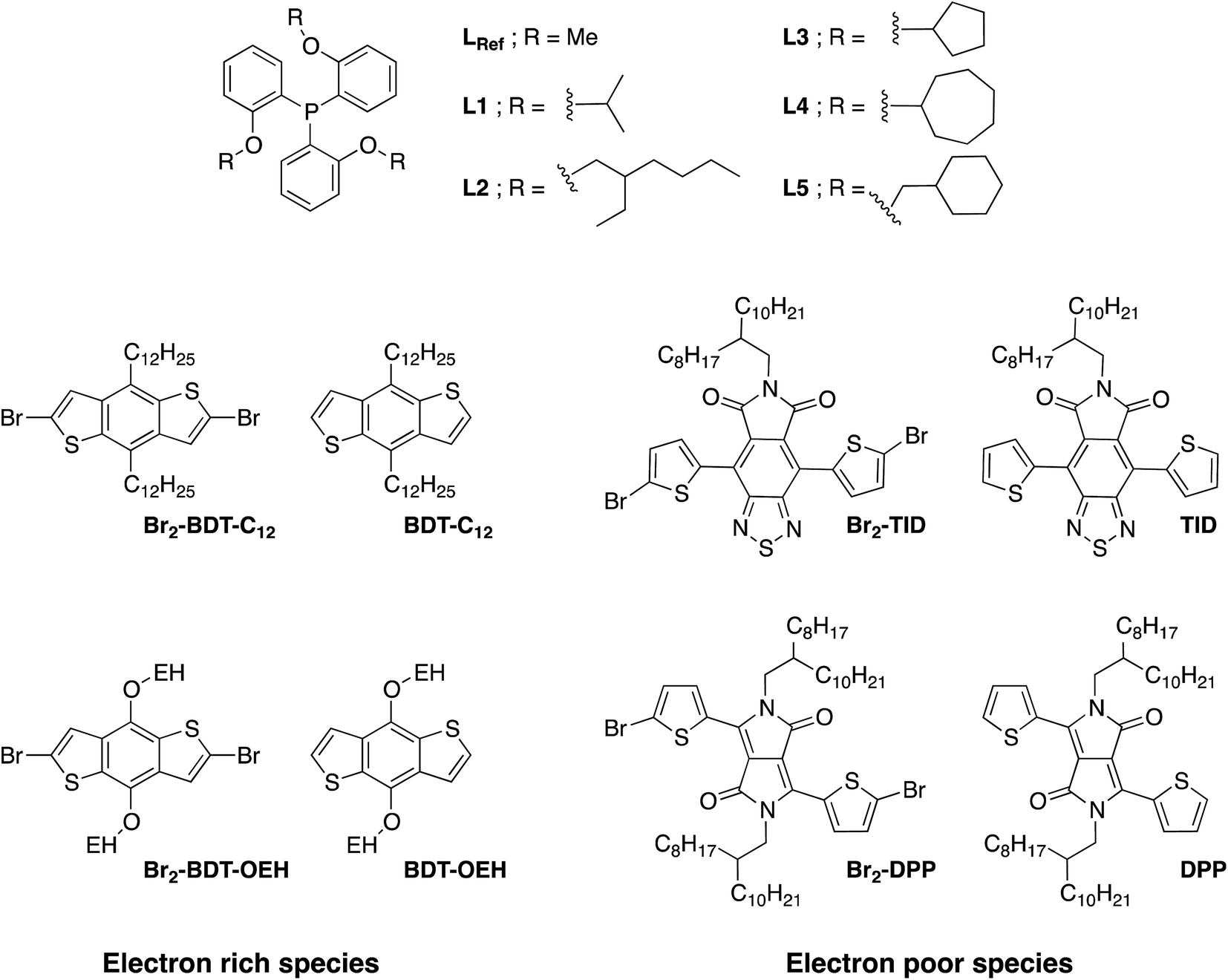 | ||
| Fig. 2 The chemical structures of the phosphines and comonomers studied in this work. | ||
2. Results and discussion
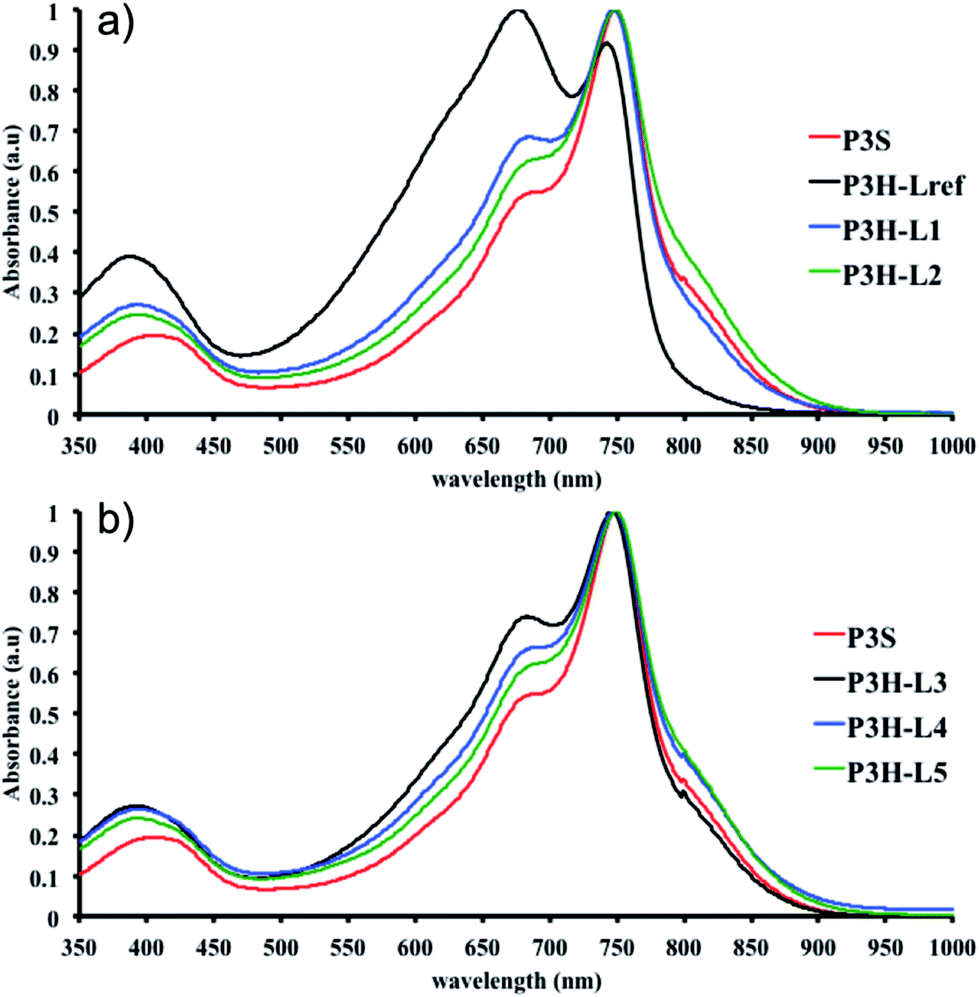
As reported in the ESI,† the reference copolymers were first synthesized using the well-established Migita–Stille cross-coupling polymerization using the following synthetic procedure: Pd2dba3/ligand (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and equimolar amounts of comonomers in toluene at 110 °C led to polymers P1S to P4S. As shown in Table S2,† high number-average molecular weight
:4) and equimolar amounts of comonomers in toluene at 110 °C led to polymers P1S to P4S. As shown in Table S2,† high number-average molecular weight 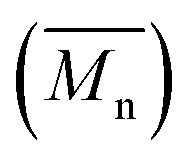 of 56 and 57 kg mol−1 was obtained for P1S and P3S when triphenyl arsine (AsPh3) was used as ligand for the Stille cross-coupling polymerization.59 Unlike P1S and P3S, the same catalytic system led to P2S and P4S with lower molecular weights. The molecular weight of P2S is limited due to its poor solubility (linear side chain on BDT). Indeed, regardless of the polymerization conditions, precipitation of the growing polymer chain occurs within few minutes. On the other hand, high molecular weight P4S (alkoxy side chain on BDT) can be obtained when P(o-Tolyl)3 is used as ligand instead of AsPh3. Indeed,
of 56 and 57 kg mol−1 was obtained for P1S and P3S when triphenyl arsine (AsPh3) was used as ligand for the Stille cross-coupling polymerization.59 Unlike P1S and P3S, the same catalytic system led to P2S and P4S with lower molecular weights. The molecular weight of P2S is limited due to its poor solubility (linear side chain on BDT). Indeed, regardless of the polymerization conditions, precipitation of the growing polymer chain occurs within few minutes. On the other hand, high molecular weight P4S (alkoxy side chain on BDT) can be obtained when P(o-Tolyl)3 is used as ligand instead of AsPh3. Indeed, 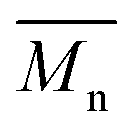 up to 150 kg mol−1 was reached, evidencing both the better efficiency of Pd2dba3/P(o-Tolyl)3 catalytic system and the effect of the side chain on the of the BDT unit (alkoxy vs. alkyl). However the lack of solubility of the higher molecular weight batches led to broad and featureless 1H NMR spectra. For comparison purposes in terms of molecular weights, Pd2dba3/AsPh3 catalytic system was therefore used as reference. All polymers (soluble fraction after Soxhlet extractions) obtained by Migita–Stille cross-coupling polymerization are soluble in chlorinated solvents such as chloroform, o-dichlorobenzene, 1,2,4-trichlorobenzene and 1,1,2,2-tetrachloroethane. In order to implement new DHAP protocols, the new phosphine-based ligands were easily prepared from a modified two-step synthetic procedure derived from Takeda et al.60 using commercially available 2-bromophenol. Simple recrystallization of the crude reaction mixture using an ethyl ether/methanol solution led to the isolation of pure crystalline phosphines with overall yields of 40–60% (see ESI†). The chemical structures of these new phosphines have been determined by 1H, 13C and 31P nuclear magnetic resonance spectroscopies and X-ray crystallography (for L4 and L5) (see ESI†).
up to 150 kg mol−1 was reached, evidencing both the better efficiency of Pd2dba3/P(o-Tolyl)3 catalytic system and the effect of the side chain on the of the BDT unit (alkoxy vs. alkyl). However the lack of solubility of the higher molecular weight batches led to broad and featureless 1H NMR spectra. For comparison purposes in terms of molecular weights, Pd2dba3/AsPh3 catalytic system was therefore used as reference. All polymers (soluble fraction after Soxhlet extractions) obtained by Migita–Stille cross-coupling polymerization are soluble in chlorinated solvents such as chloroform, o-dichlorobenzene, 1,2,4-trichlorobenzene and 1,1,2,2-tetrachloroethane. In order to implement new DHAP protocols, the new phosphine-based ligands were easily prepared from a modified two-step synthetic procedure derived from Takeda et al.60 using commercially available 2-bromophenol. Simple recrystallization of the crude reaction mixture using an ethyl ether/methanol solution led to the isolation of pure crystalline phosphines with overall yields of 40–60% (see ESI†). The chemical structures of these new phosphines have been determined by 1H, 13C and 31P nuclear magnetic resonance spectroscopies and X-ray crystallography (for L4 and L5) (see ESI†).
 | ||
| Scheme 1 Synthetic routes for P1H to P4H and P1H′ to P3H′ by DHAP. | ||
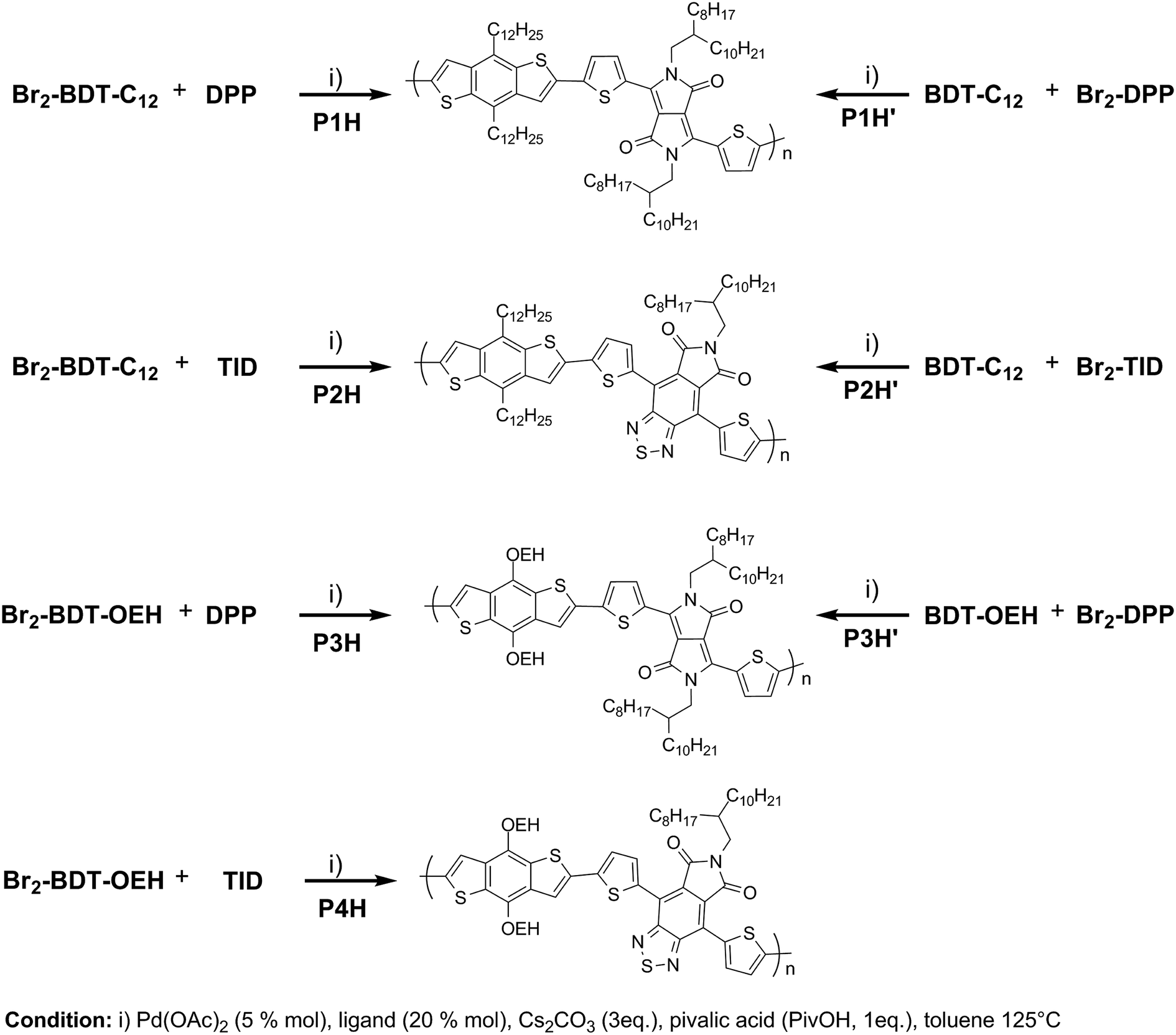
We first evaluated the polymerization of Br2-BDT-C12 (a halogenated electron-rich thiophene-based derivative with chemically reactive β-C–H bonds) with DPP (a non-halogenated electron-poor thiophene-based derivative with reactive α and β C–H bonds), two highly-studied and efficient building blocks in organic electronics (Scheme 1). γ-C–H bonds on DPP are inactive to C–H activation due to steric hindrance.47,61 The copolymerization of these units yielded P1H. On the basis of previous studies32,62 and as a reference point for comparison purposes, we kept the following polymerization conditions constant throughout this study: to a 0.5 mol L−1 solution of the monomers in toluene were added 5 mol% of Pd(OAc)2, 20 mol% of phosphine, 3 equivalents of Cs2CO3, and 1 equivalent of pivalic acid (PivOH). The mixture was reacted at 125 °C (see Experimental details in ESI†). Polymerization using tris(o-methoxyphenyl)phosphine (LREF in Fig. 2) was used as a control because of its generally satisfactory catalytic activity and selectivity in the DHAP (see ESI† for initial screening of DHAP conditions, Table S1†). The polymerization data are reported in Table S3† and the UV-Vis absorption spectra (in solution) of the resulting materials are shown in Fig. 3. For each ligand (L1–L5), the polymerization proceeds as rapidly as when P1H-LREF was utilized, and in order to limit unwanted side-reactions, each polymerization was stopped after one hour due to gelation of the reaction mixture.62 The yields of polymerization (obtained for the soluble fraction collected after Soxhlet extraction) are around 75% for each entry. As reported in Table S3,†P1H-LREF has lower number-average molecular weight (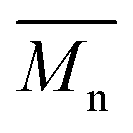 = 21 kg mol−1) than P1S (
= 21 kg mol−1) than P1S (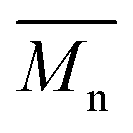 = 56 kg mol−1, obtained by Migita–Stille cross-coupling). Despite optimization of the polymerization parameters, the molecular weights of P1H-LREF were systematically lower than that of P1S. In addition to large discrepancies between
= 56 kg mol−1, obtained by Migita–Stille cross-coupling). Despite optimization of the polymerization parameters, the molecular weights of P1H-LREF were systematically lower than that of P1S. In addition to large discrepancies between 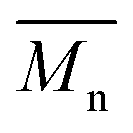 , the shape of the UV-Vis absorption spectrum of P1H-LREF (λmax = 753 nm) also differs from that of P1S (λmax = 754 nm) (Fig. 3). More importantly, the absorption spectra for the P1H series show no sign of a shoulder near 820 nm. This shoulder was already observed by Janssen et al. for certain DPP-based polymers and was attributed to α-α homocoupling of DPP units.63 One may take this absence of a shoulder in the UV-Vis spectra for P1H-L1 to L5 as indirect proof that this catalytic system reduces or inhibits the formation of such defects.64 The UV-Vis absorption spectra also show that the vibronic bands centered at 690 nm and the broad shoulder near 630 nm do not have the same intensity for P1H-LREF as for P1S. These spectroscopic observations suggest that while DHAP using tris(o-methoxyphenyl)phosphine (P(o-OMe3(C6H4)3)) may prevent homocoupling between DPP units, other side reactions such as BDT–BDT homocoupling or β-defects may still occur. However, those defects are difficult to assess even with the most advanced spectroscopic techniques and unfortunately, the NMR analyses performed for P1H series are ineffective for determining and characterizing the defects (β-defects or α-α homocouplings) embedded in the polymer due to broad and featureless signals. From Fig. 3, one can see that the replacement of the methyl group (LREF) on the phosphine ligand by an isopropyl group (L1) has no positive effect on the catalytic activity (Table S3†): while the UV-Vis absorption spectra of P1H-L1 is similar to P1H-LREF, the molecular weight of P1H-L1 is lower than that of both P1H-LREF and P1S. However, increasing the steric bulkiness of the phosphines (L2–L5) led to a significant improvement of both the molecular weight and regioregularity of the BDT–DPP copolymers (Fig. 3 and Table S3†).
, the shape of the UV-Vis absorption spectrum of P1H-LREF (λmax = 753 nm) also differs from that of P1S (λmax = 754 nm) (Fig. 3). More importantly, the absorption spectra for the P1H series show no sign of a shoulder near 820 nm. This shoulder was already observed by Janssen et al. for certain DPP-based polymers and was attributed to α-α homocoupling of DPP units.63 One may take this absence of a shoulder in the UV-Vis spectra for P1H-L1 to L5 as indirect proof that this catalytic system reduces or inhibits the formation of such defects.64 The UV-Vis absorption spectra also show that the vibronic bands centered at 690 nm and the broad shoulder near 630 nm do not have the same intensity for P1H-LREF as for P1S. These spectroscopic observations suggest that while DHAP using tris(o-methoxyphenyl)phosphine (P(o-OMe3(C6H4)3)) may prevent homocoupling between DPP units, other side reactions such as BDT–BDT homocoupling or β-defects may still occur. However, those defects are difficult to assess even with the most advanced spectroscopic techniques and unfortunately, the NMR analyses performed for P1H series are ineffective for determining and characterizing the defects (β-defects or α-α homocouplings) embedded in the polymer due to broad and featureless signals. From Fig. 3, one can see that the replacement of the methyl group (LREF) on the phosphine ligand by an isopropyl group (L1) has no positive effect on the catalytic activity (Table S3†): while the UV-Vis absorption spectra of P1H-L1 is similar to P1H-LREF, the molecular weight of P1H-L1 is lower than that of both P1H-LREF and P1S. However, increasing the steric bulkiness of the phosphines (L2–L5) led to a significant improvement of both the molecular weight and regioregularity of the BDT–DPP copolymers (Fig. 3 and Table S3†). 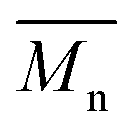 over 39 kg mol−1 were obtained for P1H-L2 to L5 series which are nearly twice the values obtained for P1H-LREF and P1H-L1. UV-visible absorption spectra of P1H-L2 to P1H-L5 also differ from P1S, P1H-LREF and P1H-L1 suggesting that the bulky phosphines can influence the resulting chemical structure of the polymers obtained (Fig. 3a and b). A bathochromic shift (up to 4 nm) of the absorption maxima was observed for P1H-L2 to P1H-L5 when compared to P1S, P1H-LREF and P1H-L1. Since
over 39 kg mol−1 were obtained for P1H-L2 to L5 series which are nearly twice the values obtained for P1H-LREF and P1H-L1. UV-visible absorption spectra of P1H-L2 to P1H-L5 also differ from P1S, P1H-LREF and P1H-L1 suggesting that the bulky phosphines can influence the resulting chemical structure of the polymers obtained (Fig. 3a and b). A bathochromic shift (up to 4 nm) of the absorption maxima was observed for P1H-L2 to P1H-L5 when compared to P1S, P1H-LREF and P1H-L1. Since 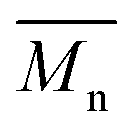 values of P1H-L2 to P1H-L5 are lower than that of P1S, one may conclude that the slight bathochromic shift observed may originate from an increased regioregularity along the conjugated backbone. These results clearly show that when the bulkier phosphines are used, the vibronic band (690 nm) has the same intensity as that observed for P1S, while the shoulder centered at 820 nm (ascribed to DPP–DPP homocoupling) is absent. Despite the lack of clear and well defined NMR spectra, these first experiments indicate that increasing the steric hindrance of the phosphine alkoxy chains can improve the selectivity over the widely used P(o-OMe(C6H4))3 for the copolymerization of a halogenated electron-rich thiophene-based derivative (Br2-BDT-C12) with a non-halogenated electron-poor thiophene-based moiety (DPP).
values of P1H-L2 to P1H-L5 are lower than that of P1S, one may conclude that the slight bathochromic shift observed may originate from an increased regioregularity along the conjugated backbone. These results clearly show that when the bulkier phosphines are used, the vibronic band (690 nm) has the same intensity as that observed for P1S, while the shoulder centered at 820 nm (ascribed to DPP–DPP homocoupling) is absent. Despite the lack of clear and well defined NMR spectra, these first experiments indicate that increasing the steric hindrance of the phosphine alkoxy chains can improve the selectivity over the widely used P(o-OMe(C6H4))3 for the copolymerization of a halogenated electron-rich thiophene-based derivative (Br2-BDT-C12) with a non-halogenated electron-poor thiophene-based moiety (DPP).
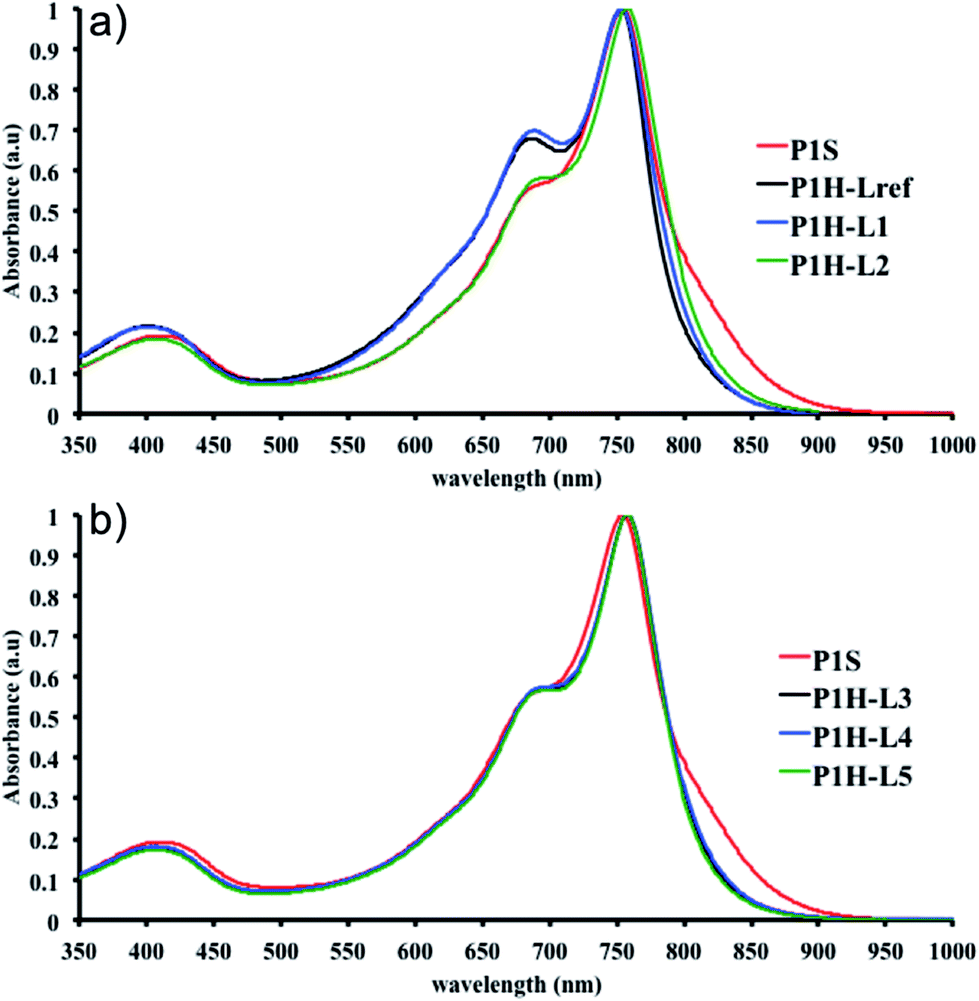 | ||
| Fig. 3 UV-Vis spectra of P1S and P1H series (solution in CHCl3). | ||
In the absence of well-defined NMR data, organic field-effect transistors (OFETs) (see ESI†) were utilized to probe electrical properties of polymers. Indeed, while the performance of such devices depend on tedious processing optimization steps and while OFETs measurements will not allow the accurate identification or quantification of the defects (β-defects or α-α homocouplings), the charge mobility in the devices is still expected to be affected by defects in the polymer (Table 1). In this study, P1S was used as a benchmark (even though the presence of DPP–DPP homocoupling within the main chain is suspected) and was compared to P1H-LREF, P1H-L3 and P1H-L4. All polymers showed ambipolar behavior. As reported in Table 1, the OFET results are in good agreement with the UV-Vis absorption data for the P1H polymer series. Based on the above, P1H-LREF (low molecular weight and different UV-Vis absorption spectra as compared to P1S) is the least efficient material. On the other hand, both P1H-L3 and P1H-L4 (higher molecular weights and displaying a good overlap of the UV-Vis absorption spectra without the shoulder at 820 nm) led to average hole and electron mobilities at least as good as than those measured for P1S and better than those of P1H-LREF. It is worth noting that the average hole mobilities obtained for the P1H series are in the same range as those reported by Morse et al.65 All these results suggest that bulky phosphine ligands result in well-defined BDT–DPP copolymers with fewer defects than those obtained by Migita–Stille polymerization.
| P type | N type | |
|---|---|---|
| Average (±standard deviation) hole mobilitya, cm2 V−1 s−1 | Average (±standard deviation) electron mobilitya, cm2 V−1 s−1 | |
| a The average (±standard deviation) mobilities were obtained in the saturation regime from at least five devices for each condition. | ||
| P1S | 3.8 (±0.8) × 10−2 | 4.0 (±0.2) × 10−3 |
| P1H-LREF | 4.3 (±0.1) × 10−3 | 1.9 (±0.1) × 10−3 |
| P1H-L3 | 1.5 (±0.9) × 10−2 | 1.0 (±0.4) × 10−3 |
| P1H-L4 | 9.0 (±0.2) × 10−3 | 8.3 (±0.3) × 10−3 |
| P2S | 3.1 (±0.7) × 10−2 | 2.0 (±0.1) × 10−3 |
| P2H-LREF | 1.2 (±0.1) × 10−2 | 1.5 (±0.2) × 10−3 |
| P2H-L3 | 1.3 (±0.3) × 10−2 | 3.7 (±0.3) × 10−4 |
| P2H-L4 | 2.5 (±0.2) × 10−2 | 1.4 (±0.1) × 10−3 |
| P2H-L5 | 2.0 (±0.3) × 10−2 | 2.4 (±0.5) × 10−3 |
P3S (High 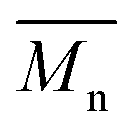 ) ) |
1.4 (±0.2) × 10−3 | 2.9 (±0.5) × 10−4 |
P3S (Low 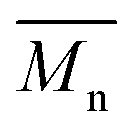 ) ) |
8.0 (±0.9) × 10−4 | 9.4 (±0.3) × 10−5 |
| P3H-LREF | 1.2 (±0.1) × 10−4 | 1.2 (±0.1) × 10−3 |
| P3H-L4 | 2.0 (±0.4) × 10−4 | 4.3 (±0.1) × 10−5 |
| P3H-L5 | 6.2 (±0.1) × 10−5 | 8.0 (±0.1) × 10−5 |
| P4S-High | 2.5 (±0.9) × 10−4 | 4.5 (±1.1) × 10−4 |
| P4H-L2 | 8.7 (±1.0) × 10−5 | 2.3 (±1.1) × 10−4 |
| P4H-L4 | 7.5 (±2.5) × 10−5 | 1.6 (±0.6) × 10−4 |
| P4H-L5 | 5.4 (±0.7) × 10−5 | 5.6 (±2.8) × 10−4 |
To validate this hypothesis, we then applied this catalytic system to the synthesis of a copolymer containing a promising electron-deficient moiety studied in organic electronics: bis(thiophen-2-yl)-6-(2-octyldodecyl)-[1,2,5]thiadiazolo[3,4-e]isoindole-5,7-dione (TID) (Fig. 2).66–68 The experimental conditions in this study were the same as those used for the P1H series. The polymerization data are summarized in Table S4† and the UV-Vis absorption spectra are shown in Fig. 4. The effect of the bulkiness of the phosphine for P2H was the same as P1H. Both polymerization times and yields (around 75%) are similar for most of the ligands except for P2H-LREF where the yield of the soluble fraction was reduced to 39%. As observed for the P1H series, the bulkiest phosphine (L4) led to the highest number-average molecular weight (P2H-L4, 21 kg mol−1). Fig. 4 shows that the UV-Vis absorption spectra of both P2H-LREF and P2H-L1 are quite different from that of P2S. While the band gaps (Eg = 1.48 eV) are similar, the vibronic bands of the polymer prepared by the different polymerization methods do not overlap one another. Although P2H-LREF and P2H-L1 have the same number-average molecular weight (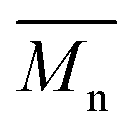 = 14 kg mol−1), the overall shape of the UV-Vis absorption spectra are also different. A substantial shift of the absorption maximum (up to 8 nm) was observed for P2H-L1 relative to P2H-LREF. This behaviour indicates improved regioregularity of P2H-L1. Furthermore, L3, L4 and L5 led to better molecular weights and regioregularity of the BDT–TID copolymer (Table S4;†Fig. 4). P2H-L3, P2H-L4 and P2H-L5 show a small bathochromic shift of the maximum of absorption (up to 2 nm) when compared to P2S. As with the P1H series, since the molecular weight of each entry is comparable, one can assume that the slight bathochromic shift observed for P2H-L3 to L5 to be the result of a more regioregular structure.
= 14 kg mol−1), the overall shape of the UV-Vis absorption spectra are also different. A substantial shift of the absorption maximum (up to 8 nm) was observed for P2H-L1 relative to P2H-LREF. This behaviour indicates improved regioregularity of P2H-L1. Furthermore, L3, L4 and L5 led to better molecular weights and regioregularity of the BDT–TID copolymer (Table S4;†Fig. 4). P2H-L3, P2H-L4 and P2H-L5 show a small bathochromic shift of the maximum of absorption (up to 2 nm) when compared to P2S. As with the P1H series, since the molecular weight of each entry is comparable, one can assume that the slight bathochromic shift observed for P2H-L3 to L5 to be the result of a more regioregular structure.
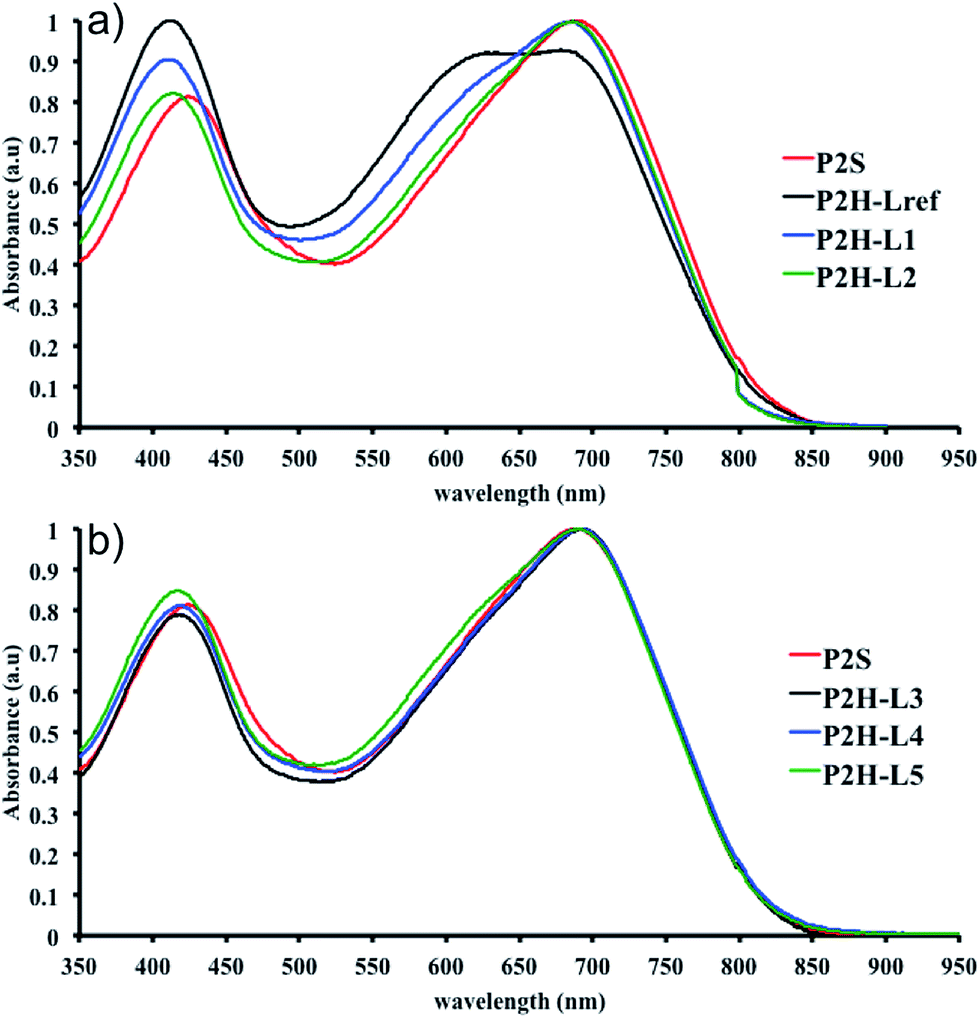 | ||
| Fig. 4 UV-Vis spectra of P2S and P2H series (solution in CHCl3). | ||
Fortunately, well-defined 1H NMR spectra were obtained for this series. The 1H NMR spectra of each monomer and polymer were obtained in deuterated 1,1,2,2-tetrachloroethane (C2D2Cl4) at 90 °C using the same acquisition parameters. In order to assign the signals and to identify defects (i.e. α-α homocouplings) homopolymers of BDT and TID were prepared as model compounds (see ESI†). Fig. 5 highlights the aromatic region of the 1H NMR spectra of P2S, P2H-LREF, P2H-L2 and P2H-L4. For each polymer, the main peaks A, B and C correspond to the protons of the main chain while the residual signals (a–h) are attributed to end groups or α-α homocoupling units (see ESI†). These spectra show for P2H-L2 and P2H-L4, a significant drop of the intensity of the peak at 8.21 ppm (a) (related TID–TID homocoupling) and of the shoulder near 7.7 ppm (c) (related to BDT–BDT homocoupling). For these two polymers, similar UV-Vis absorption spectra were obtained, indicating that the bulkier phosphines limit side reactions, particularly α-α homocouplings. As with the P1H series, OFET measurements were used to evaluate the performance of the P2H series, with P2S being used as a benchmark (Table 1). With the exception of the lower electron mobility observed of P2H-L3, the overall performance of the P2H series is comparable to that of the reference polymer P2S. The overall OFET performance of the P2H series combined with the UV-Vis and 1H NMR spectra tend to show once again that the bulkier phosphine leads to better materials with fewer defects when compared to P2S.
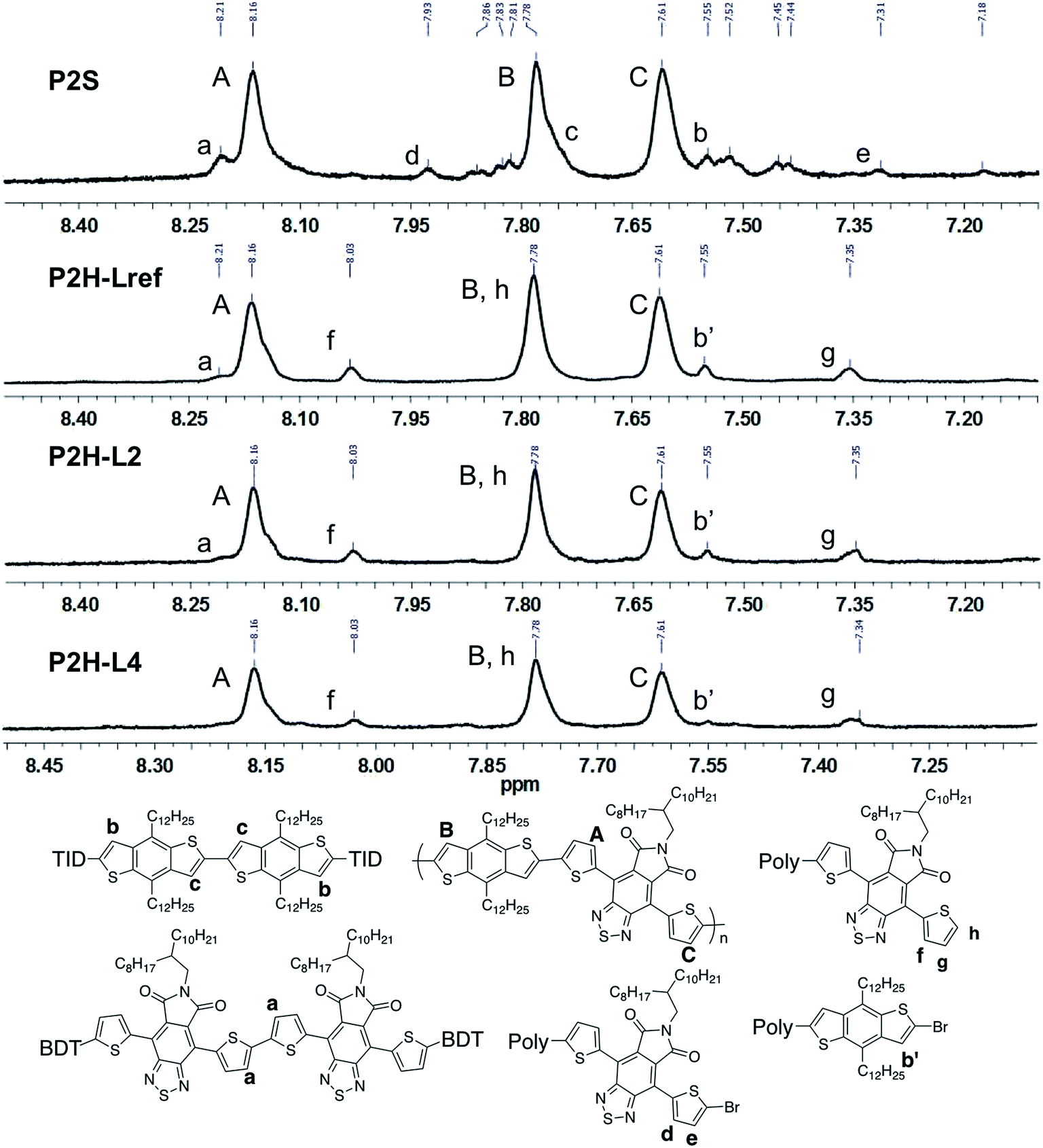 | ||
| Fig. 5 1H NMR spectra of P2S, P2H-LREF, P2H-L2 and P2H-L4. | ||
To push further our investigation, we then applied our catalytic system to the polymerization of Br2-BDT-OEH and DPP. As mentioned in the Introduction, the main objective was to evaluate if the electronic structure of the BDT side chain affects the reactivity and selectivity of the DHAP. The polymerization data are summarized in Table S5† and UV-Vis absorption spectra are shown in Fig. 6. Unlike P1H and P2H series, both 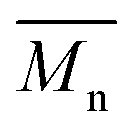 and polymerization yields (about 30%, soluble fraction recovered after Soxhlet extractions) of P3H series were lower than those obtained for P3S (Table S5†). For each entry, the polymerization was terminated after 2 hours due to gelation of the reaction mixture. Longer polymerization times did not lead to higher molecular weights. Moreover, as shown in Fig. 6, UV-Vis absorption spectra clearly demonstrate that the P3H series, regardless of the ligand, are not the same materials as P3S and the discrepancy between the UV-Vis spectra cannot be explained by the lower molecular weights alone. While no DPP–DPP homocoupling can be observed for P3H-LREF (indicated by the absence of a shoulder at 820 nm), the shift of the absorption maximum (centered at 660 nm) indicates that the chemical structure of the conjugated skeleton differs from that of P3S and may be a result of lower DHAP selectivity between Br2-BDT-OEH and DPP. Upon increasing the bulkiness of the alkoxy chain on the phosphine, the overall shape of the UV-Vis spectra approached that of P3S (Fig. 6). A maximum of absorption was found at 677 nm with a vibronic band at 740 nm for P3H-LREF, while absorption maxima at 748 and 750 nm were observed for P3HL1-L2 and P3HL3-L5, respectively, with a vibronic band at 685 nm. Based on these results, it seems that the alkoxy side chains on BDT modify the electronic properties of the monomer and affect the reactivity compared to P1H series. 1H NMR spectra were fruitless in determining the nature of the defects present in the P3S and P3H series due to broad and featureless signals. As with P1H and P2H, OFET measurements were performed. P3S was used as a benchmark and compared to P3H-LREF, P3H-L4 and P3H-L5 (Table 1). With the exception of the electron mobility of P3H-LREF (the least well-defined polymer as determined from absorption spectra), the overall performance of the P3H series were one to two orders of magnitude lower than the reference polymer.
and polymerization yields (about 30%, soluble fraction recovered after Soxhlet extractions) of P3H series were lower than those obtained for P3S (Table S5†). For each entry, the polymerization was terminated after 2 hours due to gelation of the reaction mixture. Longer polymerization times did not lead to higher molecular weights. Moreover, as shown in Fig. 6, UV-Vis absorption spectra clearly demonstrate that the P3H series, regardless of the ligand, are not the same materials as P3S and the discrepancy between the UV-Vis spectra cannot be explained by the lower molecular weights alone. While no DPP–DPP homocoupling can be observed for P3H-LREF (indicated by the absence of a shoulder at 820 nm), the shift of the absorption maximum (centered at 660 nm) indicates that the chemical structure of the conjugated skeleton differs from that of P3S and may be a result of lower DHAP selectivity between Br2-BDT-OEH and DPP. Upon increasing the bulkiness of the alkoxy chain on the phosphine, the overall shape of the UV-Vis spectra approached that of P3S (Fig. 6). A maximum of absorption was found at 677 nm with a vibronic band at 740 nm for P3H-LREF, while absorption maxima at 748 and 750 nm were observed for P3HL1-L2 and P3HL3-L5, respectively, with a vibronic band at 685 nm. Based on these results, it seems that the alkoxy side chains on BDT modify the electronic properties of the monomer and affect the reactivity compared to P1H series. 1H NMR spectra were fruitless in determining the nature of the defects present in the P3S and P3H series due to broad and featureless signals. As with P1H and P2H, OFET measurements were performed. P3S was used as a benchmark and compared to P3H-LREF, P3H-L4 and P3H-L5 (Table 1). With the exception of the electron mobility of P3H-LREF (the least well-defined polymer as determined from absorption spectra), the overall performance of the P3H series were one to two orders of magnitude lower than the reference polymer.
 | ||
| Fig. 6 UV-Vis spectra of P3S and P3H series (solution in CHCl3). | ||
In order to verify that the electronic nature of the side chain installed on BDT moiety does affect the selectivity of the DHAP, we applied this catalytic system to the polymerization of Br2-BDT-OEH and TID (P4H series, Scheme 1). The polymerization data are summarized in Table S6† and UV-Vis absorption spectra are shown in Fig. 7. Molecular weights (from soluble fraction recovered after Soxhlet extraction) of the polymers obtained by the DHAP were lower than those obtained for P4S-High (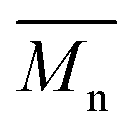 of 30 kg mol−1) except for P4H-L2 (
of 30 kg mol−1) except for P4H-L2 (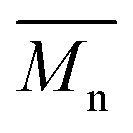 = 27 kg mol−1) and P4H-L4 (
= 27 kg mol−1) and P4H-L4 (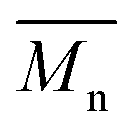 = 30 kg mol−1). Unlike P4S, the kinetics of DHAP was slow and no improvement of molecular weights was observed for longer polymerization times. Moreover, to eliminate the effect of molecular weight discrepancies during polymer comparison experiments, we also prepared P4S-Low, a low molecular weight batch (
= 30 kg mol−1). Unlike P4S, the kinetics of DHAP was slow and no improvement of molecular weights was observed for longer polymerization times. Moreover, to eliminate the effect of molecular weight discrepancies during polymer comparison experiments, we also prepared P4S-Low, a low molecular weight batch (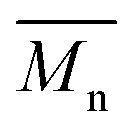 = 16 kg mol−1) similar to those obtained for P4H-L1, L3 and L5. As shown in Fig. 7, the mismatch of all the UV-Vis absorption spectra of P4H series compared to P4S, combined with the shift of the intensity of the two main bands (400 nm and 640 nm) raises concerns as to the nature of the BDT side chain on the DHAP selectivity. P4S-High exhibits two maxima of absorption, one centered at 418 nm and another of higher intensity at 639 nm. These two maxima were observed at 414 nm and 617 nm for P4S-Low, indicating clear relationship between molecular weights and optical properties for P4S. As shown in Fig. 7a, the least comparable polymer was P4H-Lref, with a number-average molecular weight of 10 kg mol−1 and an UV-Vis absorption spectrum with two blue-shifted maxima (407 and 583 nm) compared to P4S-Low (414 and 617 nm). Another striking feature, aside from the hypsochromic shift, is the inversion of the intensity of the two maxima observed for P4H-Lref. This clearly indicates that the chemical structure of the conjugated backbone of P4H-Lref is not the same as P4S (High and Low). Moreover, despite similar number-average molecular weight when compared to P4S-High (
= 16 kg mol−1) similar to those obtained for P4H-L1, L3 and L5. As shown in Fig. 7, the mismatch of all the UV-Vis absorption spectra of P4H series compared to P4S, combined with the shift of the intensity of the two main bands (400 nm and 640 nm) raises concerns as to the nature of the BDT side chain on the DHAP selectivity. P4S-High exhibits two maxima of absorption, one centered at 418 nm and another of higher intensity at 639 nm. These two maxima were observed at 414 nm and 617 nm for P4S-Low, indicating clear relationship between molecular weights and optical properties for P4S. As shown in Fig. 7a, the least comparable polymer was P4H-Lref, with a number-average molecular weight of 10 kg mol−1 and an UV-Vis absorption spectrum with two blue-shifted maxima (407 and 583 nm) compared to P4S-Low (414 and 617 nm). Another striking feature, aside from the hypsochromic shift, is the inversion of the intensity of the two maxima observed for P4H-Lref. This clearly indicates that the chemical structure of the conjugated backbone of P4H-Lref is not the same as P4S (High and Low). Moreover, despite similar number-average molecular weight when compared to P4S-High (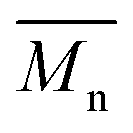 = 30 kg mol−1), P4H-L2 (
= 30 kg mol−1), P4H-L2 (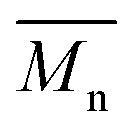 = 27 kg mol−1) and P4H-L4 (
= 27 kg mol−1) and P4H-L4 (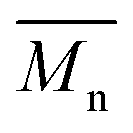 = 30 kg mol−1) also show hypsochromic shifts for both absorption bands, which also indicates the presence of defects within the main chain, as observed for the P3H series. Consistently with the fact that mismatches of the UV-Vis spectra between P4S and P4H series have been observed, lower OFETs performance for P4H series have been measured by experimental data (one order of magnitude lower than the reference polymer, see Table 1).
= 30 kg mol−1) also show hypsochromic shifts for both absorption bands, which also indicates the presence of defects within the main chain, as observed for the P3H series. Consistently with the fact that mismatches of the UV-Vis spectra between P4S and P4H series have been observed, lower OFETs performance for P4H series have been measured by experimental data (one order of magnitude lower than the reference polymer, see Table 1).
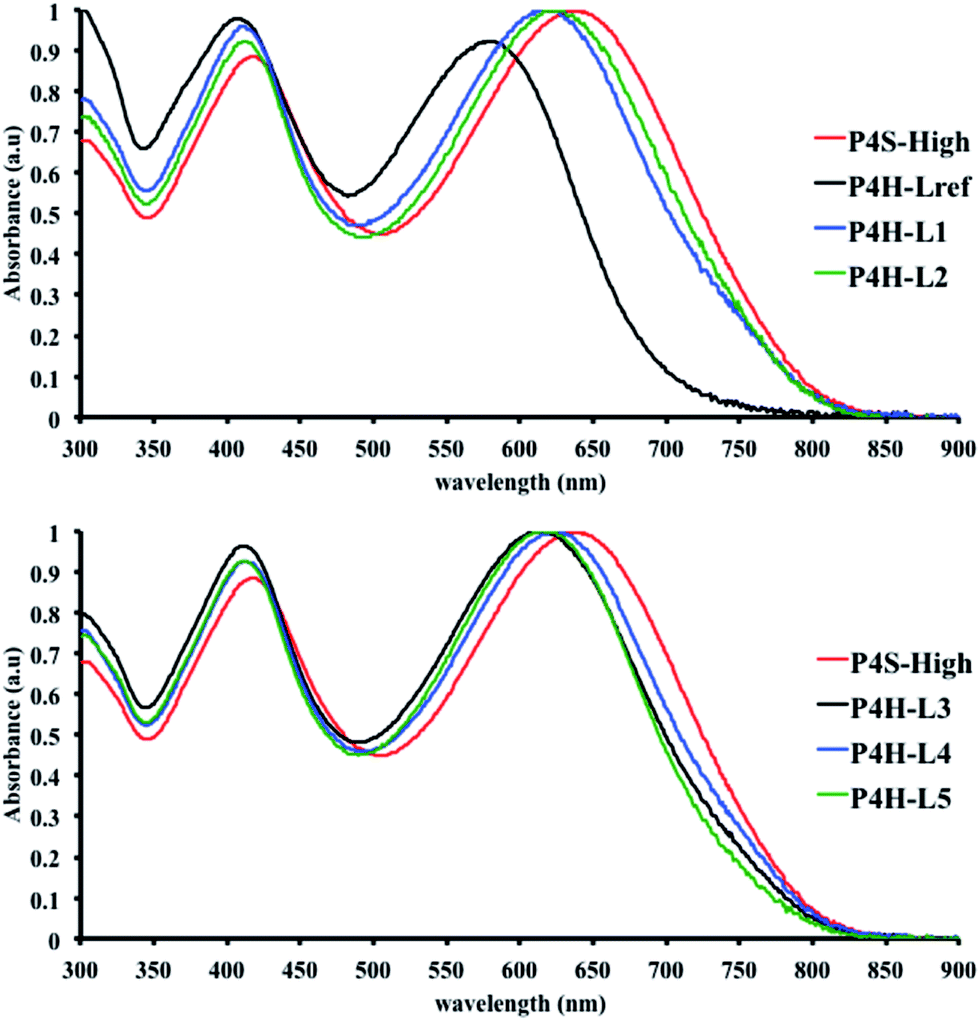 | ||
| Fig. 7 UV-Vis spectra of P4S and P4H series (solution in CHCl3). | ||
Well-defined 1H NMR data were also obtained for P4H. Fig. 8 highlights the aromatic region of the 1H NMR spectra of P4S, P4H-LREF, P4H-L2 and P4H-L4. On the basis of model compounds (see ESI†), the main peaks A, B and C correspond to the protons of the main chain while the residual signals (a–k) are related to end groups or homocoupling units. Although the exact integrations were difficult to determine, the lower intensity of the signal at 8.21 ppm for P4H-LREF compared to the one observed for P4S suggests that the DHAP may lead to lower amounts of TID–TID homocoupling side-reactions. Moreover, for P4H-L2 and P4H-L4, a significant drop of the intensity of the peak at 8.21 ppm (a) (related to the homocoupling of TID) was observed compared to P4S. The other residual peaks (h: 7.77 ppm; f: 8.03 ppm; g: 7.35) for P4H-L2 and P4H-L4 are less intense compared to P4H-LREF due to higher molecular weights.
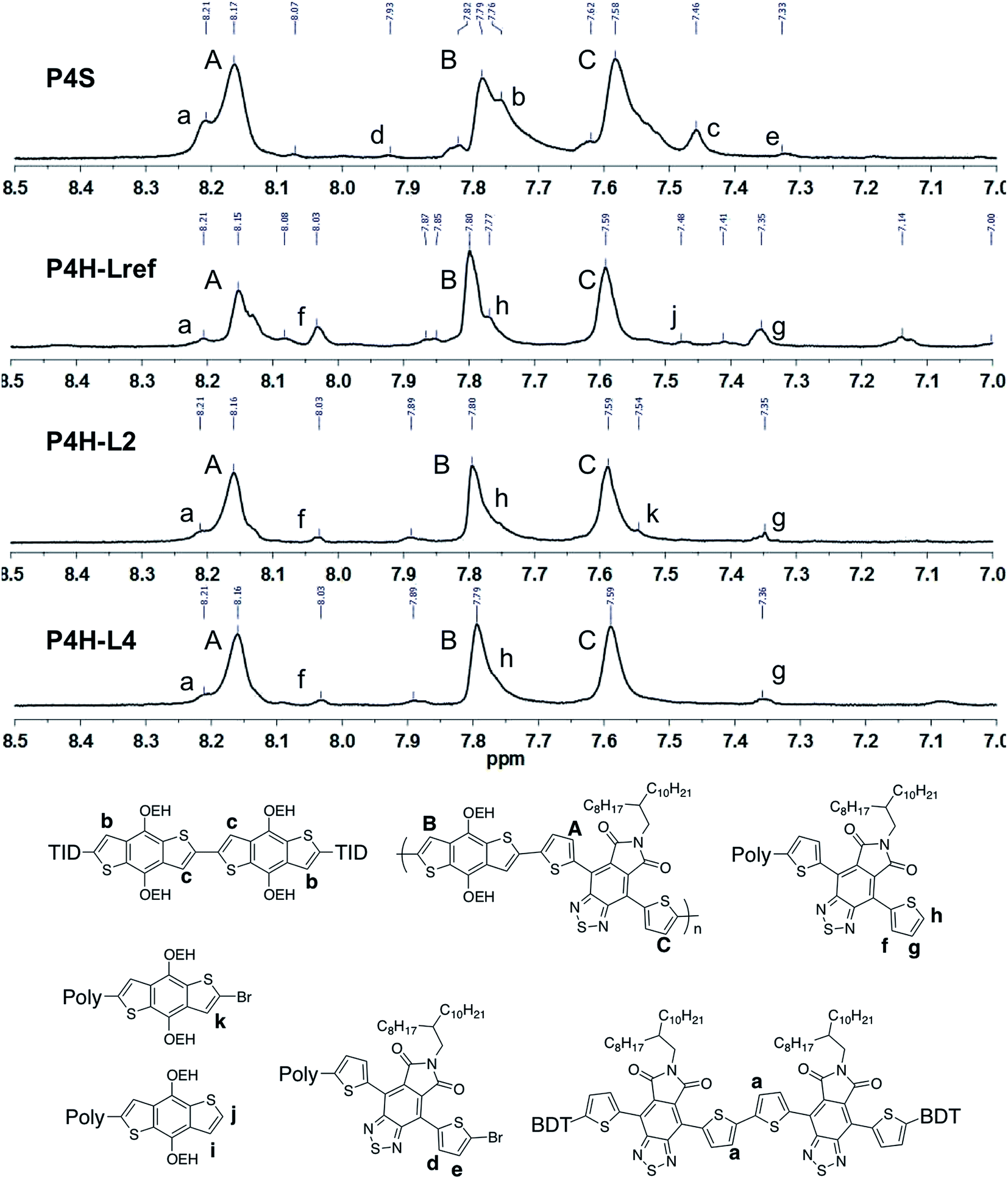 | ||
| Fig. 8 1H NMR spectra of P4S, P4H-LREF, P4H-L2 and P4H-L4. | ||
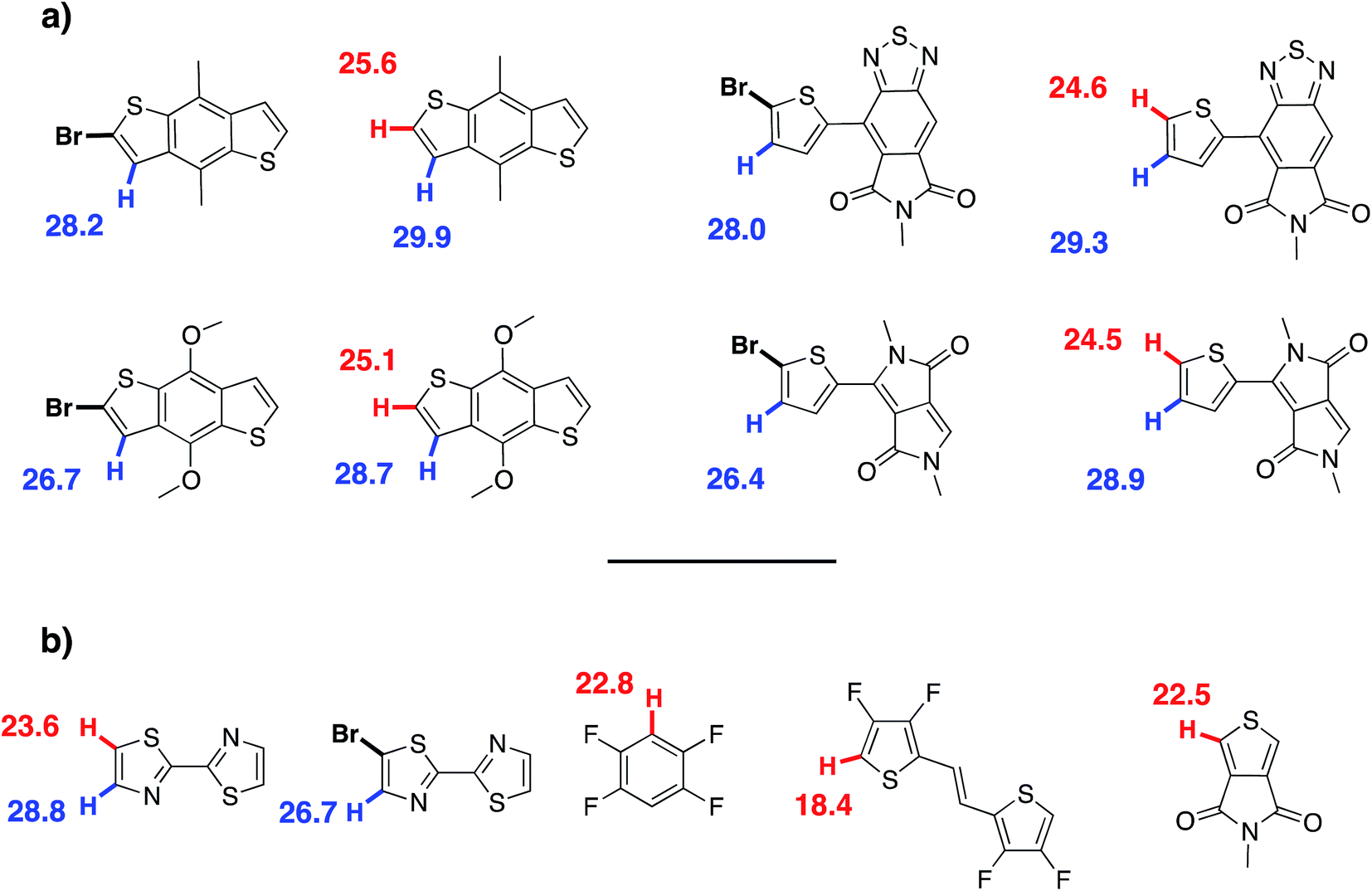
To rationalize this experimental data, DFT calculations at the B3LYP/TZVP (DZVP for palladium) level have been carried out to evaluate the activation barrier of the C–H bond activation of the CMD pathway. This method has been previously successfully applied to the rationalization and prediction of the regioselectivity of direct (hetero)arylation reactions on different arenes and heteroarenes.69 It is worth noting these calculations are relevant to α versus β selectivity for possible β-defects but does not say anything about potential α-α homocouplings. The model palladium catalyst (PMe3)Pd(Ph)(CH3COO–) was used as a platform to calculate the Gibbs free energy of the CMD transition state associated to the activation of the different C–H bonds of each substrate (see Fig. 9 and ESI†). The results obtained using this approach are given in the following test using this notation: the activation energy (Ea) refers to the Gibbs free energy of the CMD transition state referenced to the substrate and model catalyst (ΔG‡298). Thus, in the case of DPP, the difference in the activation energy (ΔEa) between Hα and Hβ for the DPP moiety is found to be 4.4 kcal mol−1 (24.5 vs. 28.9 kcal mol−1, respectively). Accordingly, it is possible to tentatively estimate (using Arrhenius's law) a selectivity ratio of the α-position at 125 °C (the temperature of polymerization). For this system, a ratio of about 250:1 favoring Hα can indeed be calculated for the DPP unit. On the other hand, the Ea of the Hβ of the Br2-BDT-C12 is 28.2 kcal mol−1 whereas the Ea for Hβ on the Br2-BDT-OEH is only 26.7 kcal mol−1. It is worth noting that the theoretical calculations show that the electronic nature of the side-chains installed on BDT unit modifies the energy of activation of both α and β protons. Using these values, the ΔEa between Hα of DPP (24.5 kcal mol−1) and the Hβ of the Br2-BDT-OEH (26.7 kcal mol−1) and Br2-BDT-C12 (28.2 kcal mol−1) are respectively of 2.2 and 3.7 kcal mol−1, giving respective selectivity of about 15:1 and 100:1 at 125 °C. Although the 1.5 kcal mol−1 difference might look small, it indicates that the activation of the Hβ of the Br2-BDT-OEH will occur about one order of magnitude faster than the one of the Hβ of the Br2-BDT-C12 resulting in more β-defects, while the polymer using Br2-BDT-C12 will better behave. This is in fair agreement with the UV-Vis absorption spectra observed for the P1H and P3H series, respectively.
 | ||
| Fig. 9 The Gibbs free energy of the CMD transition state associated to the activation of α,β hydrogen atoms calculated by DFT. | ||
For P4H (Br2-BDT-OEH with TID), the difference in the energy of activation (ΔEa) between Hα and Hβ for the TID moiety is 4.7 kcal mol−1 (29.3–24.6 kcal mol−1). For this system, a ratio of about 400:1 favoring Hα has been calculated. However, the ΔEa between Hα of TID (24.6 kcal mol−1) and the Hβ of the Br2-BDT-OEH (26.7 kcal mol−1) is 2.1 kcal mol−1 while the ΔEa between Hα of TID (24.6 kcal mol−1) and the Hβ of the Br2-BDT-C12 (28.2 kcal mol−1) is 3.6 kcal mol−1. As discussed earlier, one can think that β-defects on BDT moiety can be the main defect found in the P4H series whereas a better selectivity is anticipated for the P2H series.
To continue our investigation, we carried out the polymerization of monomers with the bromide function on the electron-deficient units. 4,8-Bis(didodecyl)benzo[1,2-b:4,5-b′]dithiophene (BDT-C12) or 4,8-bis(2-ethylhexyloxy)benzo[1,2-b:4,5-b′]dithiophene (BDT-OEH) was therefore polymerized with 3,6-bis(5-bromo-thiophen-2-yl)-2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]-pyrrole-1,4-dione (Br2-DPP) or 4,8-bis(5-bromo-thiophen-2-yl)-6-(2-octyldodecyl)-[1,2,5]thiadiazolo[3,4-e]isoindole-5,7-dione (Br2-TID) to obtain the P1H′, P2H′ and P3H′ series (Scheme 1). The polymerization data for P1H′ are reported in Table S7† and the UV-Vis absorption spectra in solution are shown in Fig. 10. The polymerization times were longer and the yields (about 25–30%) of the soluble fraction of the polymers in chloroform were consistently lower relative to both the P1H series and P1S, regardless of the nature of the ligand. DHAP polymerizations were run for 12 hours for all examples. After Soxhlet extractions, a large quantity of insoluble material was recovered indicating possible side reactions. Despite several attempts, both 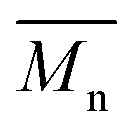 and polymerization yield values were lower than those of the reference polymer P1S. As shown in Fig. 10, the UV-Vis absorption spectra of the P1H′ series were different from the benchmark P1S. Clearly, this catalytic system did not yield the targeted polymers as opposed to the P1H series.
and polymerization yield values were lower than those of the reference polymer P1S. As shown in Fig. 10, the UV-Vis absorption spectra of the P1H′ series were different from the benchmark P1S. Clearly, this catalytic system did not yield the targeted polymers as opposed to the P1H series.
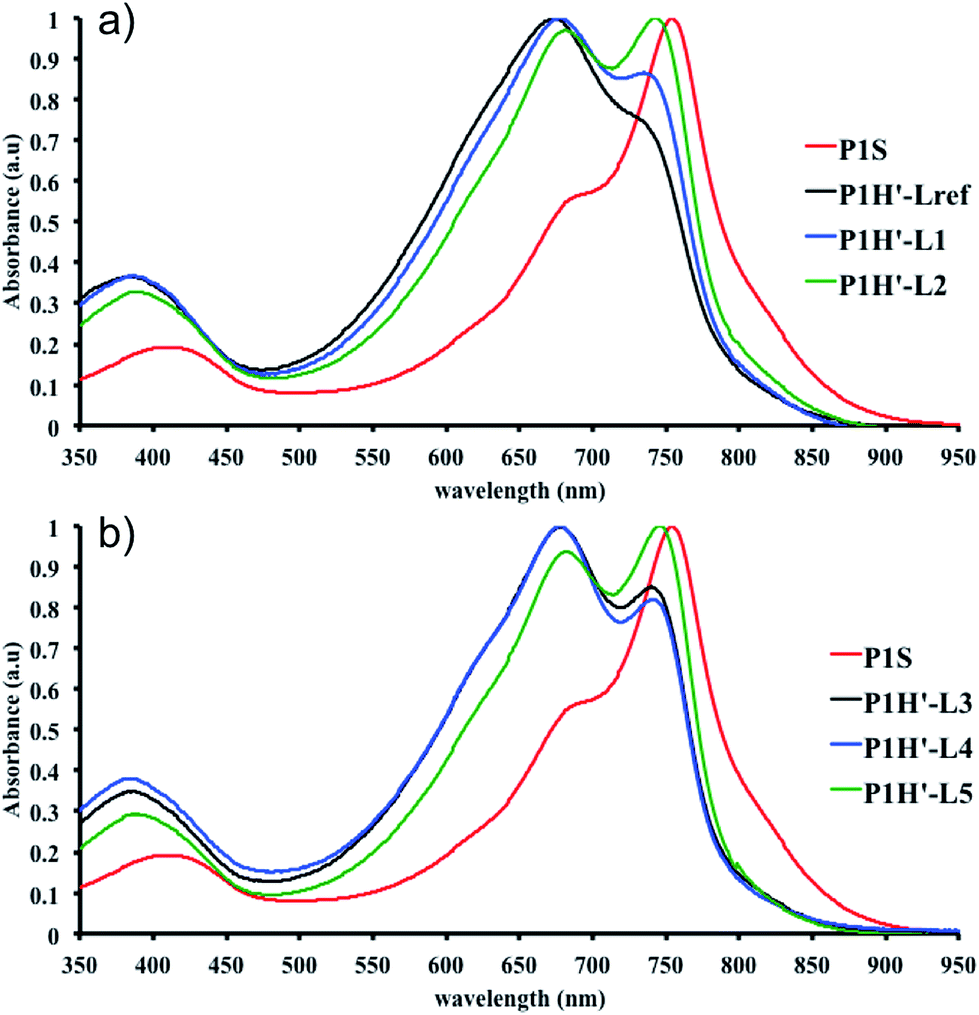 | ||
| Fig. 10 UV-Vis spectra of P1S and P1H′ series (solution in CHCl3). | ||
We then applied the catalytic system for the polymerization of 4,8-bis(didodecyl)benzo[1,2-b:4,5-b′]dithiophene (BDT-C12) and 4,8-bis(5-bromo-thiophen-2-yl)-6-(2-octyldodecyl)-[1,2,5]thiadiazolo[3,4-e]isoindole-5,7-dione (Br2-TID) P2H′ (Scheme 1). The polymerization data for P2H′ are reported in Table S8,† while the UV-Vis absorption spectra in solution are shown in Fig. 11. The polymerization times were generally longer and the yields (soluble fraction of the polymer in chloroform) lower compared to the P2H series and P2S. Like P1H′ series, the UV-Vis absorption spectra obtained for P2H′ were different from the reference, indicating, once again, that the targeted polymers were not obtained with this catalytic system.
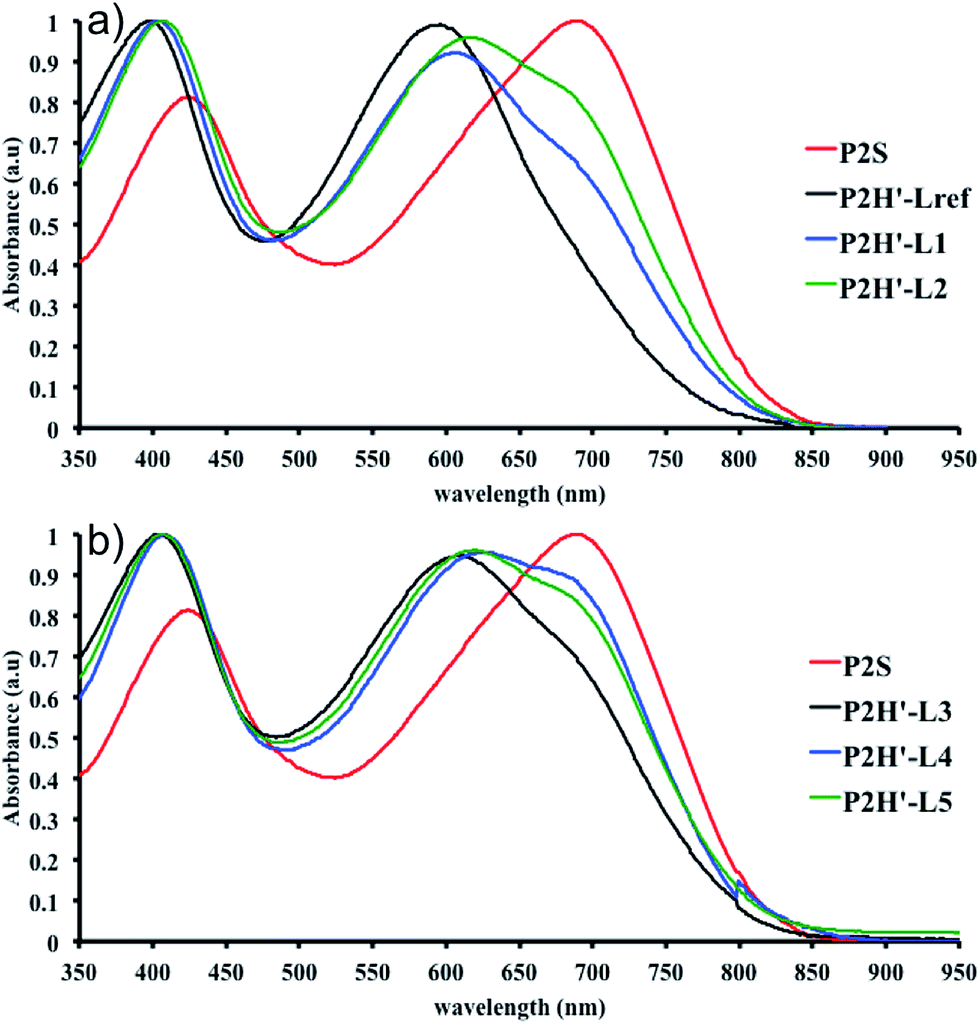 | ||
| Fig. 11 UV-Vis spectra of P2S and P2H′ series (solution in CHCl3). | ||
As for the P2H series, well-defined 1H NMR data were obtained for the P2H′ samples in 1,1,2,2-tetrachloroethane (C2D2Cl4) at 90 °C (see ESI, Section 4†). The shape of the UV-Vis absorption spectra of the P2H′ series combined with 1H NMR analyses confirm the presence of defects within the conjugated main chain. The same trend was observed for the copolymerization of BDT-OEH with Br2-DPP (P3H′ series, see Scheme 1). Lower molecular weights and yields were obtained with all phosphines (Table S9†). As with P1H′, a large quantity of insoluble material was recovered after Soxhlet extractions and the UV-Vis absorption spectra of the soluble fraction did not match that obtained for P3S (Fig. 12). In the light of the result obtained for the P1H′–P3H′ series, we did not synthesize the P4H′ series, i.e. the equivalent polymers to P4H with the bromide moiety on the electron-poor unit.
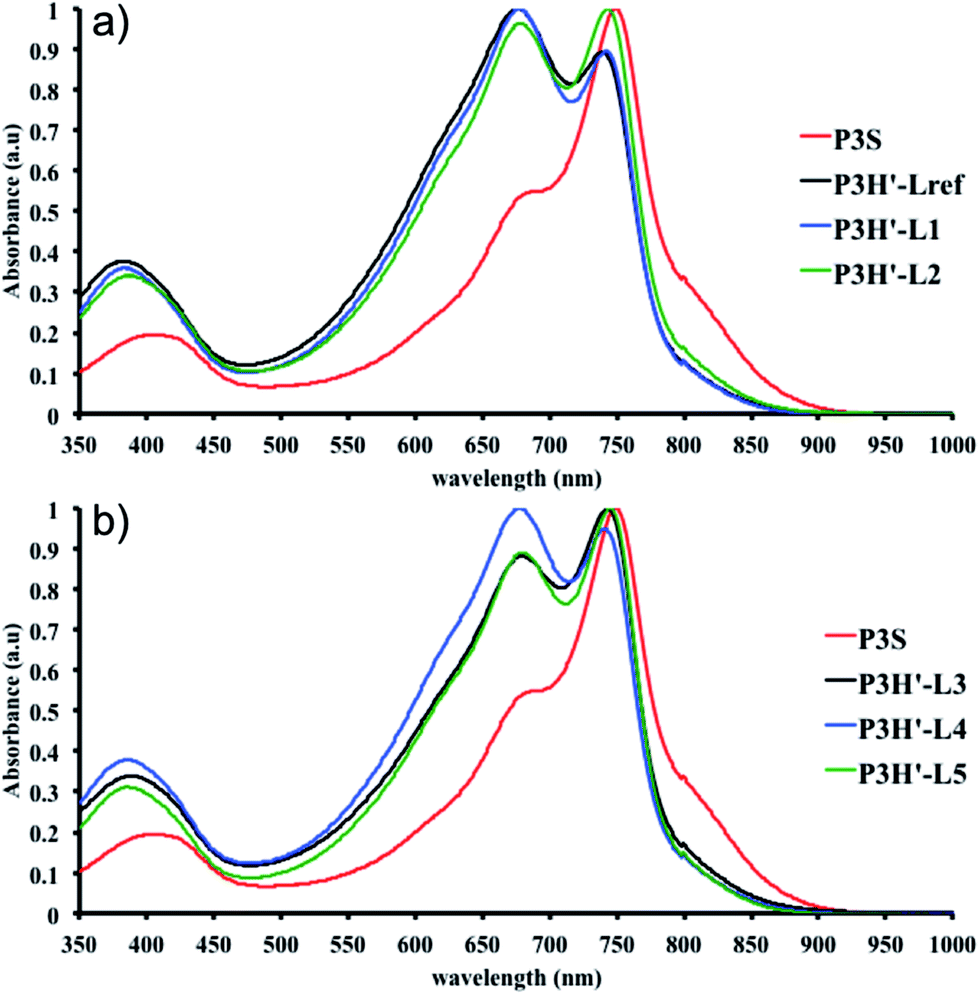 | ||
| Fig. 12 UV-Vis spectra of P3S and P3H′ series (solution in CHCl3). | ||
DFT calculations performed for P1H′ (BDT-C12 with Br2-DPP) helped again to rationalize the experimental data obtained for this series (Fig. 9). Here, the ΔEa between the Hα (25.6 kcal mol−1) and Hβ (29.9 kcal mol−1) of BDT-C12 is 4.3 kcal mol−1, indicating once again an excellent selectivity for the activation of Hα. However, the ΔEa between Hβ of the Br2-DPP (26.4 kcal mol−1) and Hα of the BDT-C12 (25.6 kcal mol−1) is only of 0.8 kcal mol−1, suggesting poor selectivity and significant unwanted DPP–DPP β-defects.
For P2H′ series (BDT-C12 with Br2-TID), the ΔEa between Hα (25.6 kcal mol−1) and Hβ (29.9 kcal mol−1) of BDT-C12 is 4.3 kcal mol−1. On the other hand, the ΔEa between Hβ of the Br2-TID (28.0 kcal mol−1) and Hα of the BDT-C12 (25.6 kcal mol−1) is 2.4 kcal mol−1, leading to a 20:1 selectivity at 125 °C which would induce some TID–TID β-defects. The same trend was observed for the copolymerization between BDT-OEH with Br2-DPP (P3H′ series, see Scheme 1). Although the ΔEa between Hα (25.1 kcal mol−1) and Hβ (28.7 kcal mol−1) of BDT-OEH of 3.6 kcal mol−1 leads to a selectivity of about 100:1, the ΔEa between Hβ of the Br2-DPP (26.4 kcal mol−1) and Hα of the BDT-OEH (25.1 kcal mol−1) of 1.3 kcal mol−1 gives a poor 5:1 selectivity, which would induce possible DPP–DPP β-defects. These observations are in good agreement with the experimental data obtained for P3H′ series. Finally, according to the theoretical calculations, one can think that the synthesis of P4H′ by DHAP (BDT-OEH with Br2-TID) would also suffer from selectivity issues. Indeed, the ΔEa between Hβ of the Br2-TID (28.0 kcal mol−1) and Hα of the BDT-OEH (25.1 kcal mol−1) of 2.9 kcal mol−1 gives a 40:1 selectivity, indicating possible TID–TID β-defects.
On the basis of these experimental-theoretical data, we extended the scope of our DFT calculations to other BDT and DPP-based copolymers recently synthesized by DHAP in order to explain why some of them can be successfully prepared while other cannot. For instance, Li et al. reported that a relatively well-defined copolymer was obtained from the copolymerization of Br2-DPP with bithiazole while poor performance was reported for the same copolymer synthesized using Br2-bithiazole and DPP (Scheme 2a).70 For the first case, the ΔEa between the Hα (23.6 kcal mol−1) and Hβ (28.8 kcal mol−1) of bithiazole moiety is 5.2 kcal mol−1 which means that the activation of the Hα position is highly selective. Meanwhile, a selectivity of about 35:1 is observed when considering the ΔEa value of 2.8 kcal mol−1 between Hβ of the Br2-DPP (26.4 kcal mol−1) and Hα of the bithiazole (23.6 kcal mol−1). On the other hand, for the Br2-bithiazole/DPP system, the ΔEa between Hβ of the Br2-bithiazole (26.7 kcal mol−1) and the Hα of the DPP (24.5 kcal mol−1) is only of 2.2 kcal mol−1 and should lead to less-defined structure. Wang and co-workers have also reported that the synthetic pathways used for the synthesis of DPP–tetrafluorobenzene (TFB) copolymer by DHAP affect the regioregularity of the resulting copolymer (see Scheme 2b) and, once again, the experimental data are in good agreement with the DFT calculations.71 Indeed, for pathway A (DPP and Br2-TFB), the ΔEa between Hα and Hβ for the DPP moiety is 4.4 kcal mol−1 (24.5 vs. 28.9 kcal mol−1, respectively) and leads to a selectivity ratio of about 250:1. For the pathway B (see Scheme 2b), the ΔEa between Hα of TFB (22.8 kcal mol−1) and Hβ on Br2-DPP (26.4 kcal mol−1) is 3.6 kcal mol−1. According to these calculations, this means that some DPP–DPP β-defects might occur in the latter case. Along these lines, Wang et al. have reported the synthesis of PDPP-4FTVT for which experimental data once again are in good agreement with DFT calculations.72 Indeed, 4FTVT comonomer has Ea,Hα of 18.4 kcal mol−1 (see Fig. 9) leading to a ΔHα − Hβ of 8 kcal mol−1 with Hβ of Br2-DPP (26.4 kcal mol−1). With an average hole and electron mobilities of 2.46 and 4.19 cm2 V−1 s−1 respectively, those high values suggest that PDPP-4FTVT is a well-defined material. Finally, Marks et al.58 and Farinola et al.73 recently reported the synthesis of PBDTTPD by DHAP using Br2-BDT-OEH and TPD as comonomers and obtained a polymer with similar properties than that one prepared from Stille coupling. DFT calculations can again explain these results. Indeed, for the TPD monomer with only one C–H bond available, an Ea of 22.5 kcal mol−1 was calculated (see Fig. 9). Thus, the ΔEa between Hα of TPD (22.5 kcal mol−1) and Hβ on Br2-BDT-OEH (26.7 kcal mol−1) is 4.2 kcal mol−1 which should lead to a 200:1 selectivity ratio at 125 °C. All these calculations show that the choice of the comonomers is crucial for DHAP. Indeed, these calculations indicate that the presence of bromine atoms decreases the energy of activation (Ea) of the adjacent C–H bonds, allowing undesirable β-defects for some brominated aromatic units. However, some brominated monomers are not intrinsically inadequate but their utilization is strongly dependent upon the nature and reactivity of the respective comonomer. Finally, DFT calculations reported here show that most non-brominated thiophene-based building blocks have good selectivity at the α-positions.
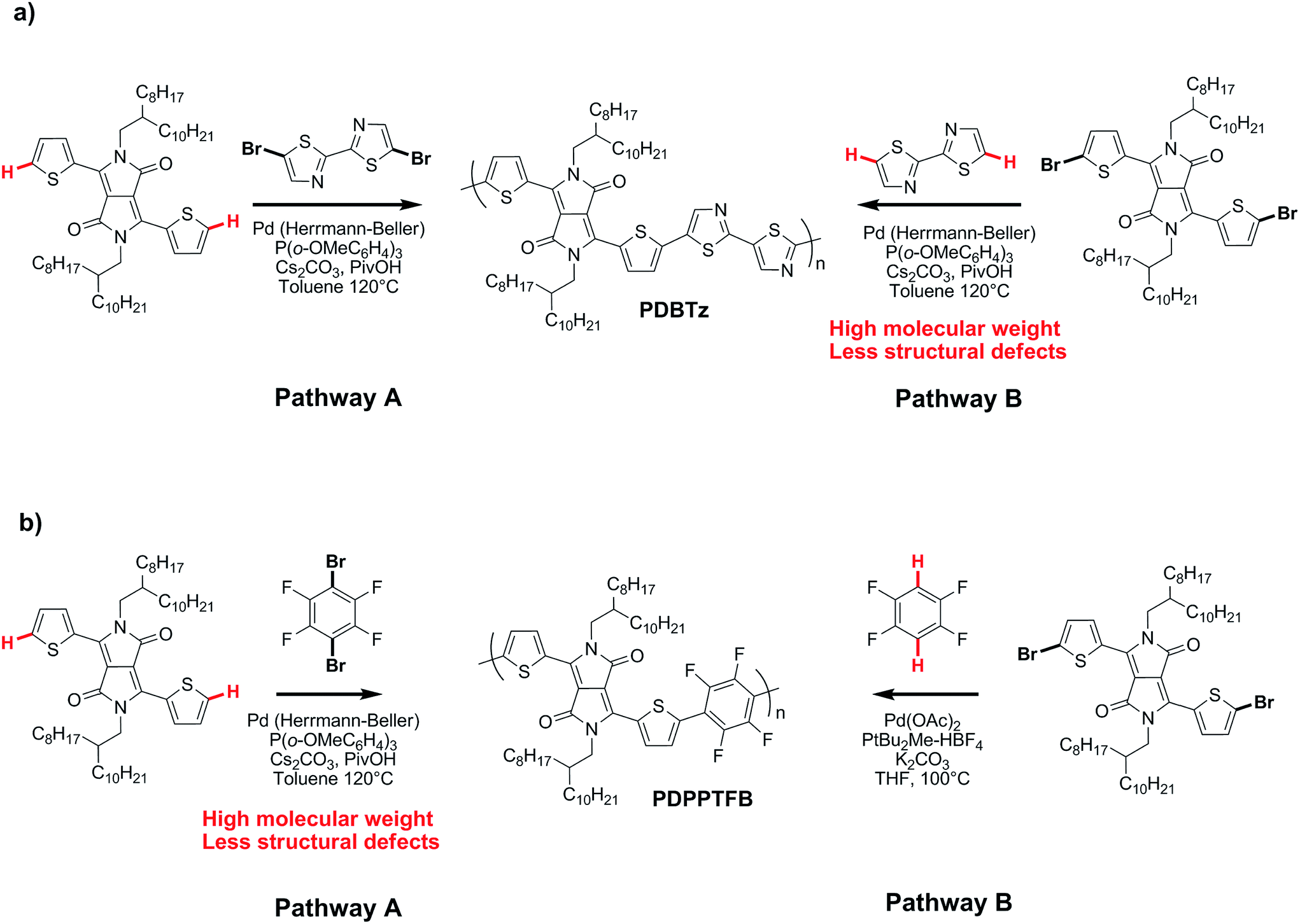 | ||
| Scheme 2 Synthetic routes for (a) PDBTz and (b) PDPPTFB by DHAP.70,71 | ||
3. Conclusion
In this study, we have shown that the selectivity of the direct heteroarylation polymerization (DHAP) of brominated electron-rich thiophene-based units and electron-deficient DPP and TID moieties can be significantly improved through a rational modification of the phosphine-based ligand. Indeed, five new phosphines with different o-substituent moieties (iso-propyl (L1), ethylhexyl (L2), cyclopentane (L3), cycloheptane (L4) and methylcyclohexane (L5)) have been synthesized and compared with tris(o-methoxyphenyl)phosphine. Selectivity enhancement was observed for bulkier phosphines, even preventing homocoupling defects found in the Migita–Stille polymerization. Despite notable selectivity improvements, the utilization of hindered phosphines was not effective with some brominated thiophene-based comonomers. Theoretical calculations using DFT gave an estimation of the activation energies (Ea) for all possible C–H bonds to be activated during the CMD process and those calculations showed that the presence of a bromine atom decreases the energy of activation of the adjacent C–H bonds, potentially leading to undesirable competitive reactions. More importantly, the theoretical calculations tend to show that a careful choice of comonomers might be the key to obtain well-defined materials. This initial experimental-theoretical study is part of a broader interest in mechanistic investigations in our laboratories. With this knowledge, we intend to develop a transition state to rationally design ligands and substrates for site-selective DHAP reactions. Finally, competition experiments on non-polymerizable model compounds could give some quantitative information and be compared to theoretical calculations.Acknowledgements
The authors are grateful to Merck Chemicals Ltd and NSERC for their financial support.References
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- I. Etxebarria, J. Ajuria and R. Pacios, Org. Electron., 2015, 19, 34–60 CrossRef CAS.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- Y. Xu, C. Liu, D. Khim and Y.-Y. Noh, Phys. Chem. Chem. Phys., 2015, 17, 26553–26574 RSC.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- S.-H. Liao, H.-J. Jhuo, P.-N. Yeh, Y.-S. Cheng, Y.-L. Li, Y.-H. Lee, S. Sharma and S.-A. Chen, Sci. Rep., 2014, 4, 6813 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics, 2016, 24, 3–11 Search PubMed.
- C.-H. Peters, I. T. Sachs-Quitana, J. P. Kastrop, S. Beaupré, M. Leclerc and M. D. McGehee, Adv. Energy Mater., 2011, 1, 491–494 CrossRef CAS.
- W. R. Mateker, I. T. Sachs-Quintana, G. F. Burkhard, R. Cheacharoen and M. D. McGehee, Chem. Mater., 2015, 27, 404–407 CrossRef CAS.
- C. Luo, A. K. K. Kyaw, L. A. Perez, S. Patel, M. Wang, B. Grimm, G. C. Bazan, E. J. Kramer and A. J. Heeger, Nano Lett., 2014, 14, 2764–2771 CrossRef CAS PubMed.
- Y. Zhang, E. Bovill, J. Kingsley, A. R. Buckley, H. Yi, A. Iraqi, T. Wang and D. G. Lidzey, Sci. Rep., 2016, 6, 21632 CrossRef CAS PubMed.
- S. Beaupré and M. Leclerc, J. Mater. Chem. A, 2013, 1, 11097–11105 Search PubMed.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1995, 67, 541–547 CrossRef.
- G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708–1710 CrossRef CAS.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- K. Tamao, K. Sumitami and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723–6737 Search PubMed.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3741–3747 CrossRef CAS PubMed.
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- A. Facchetti, L. Vaccaro and M. Assunta, Angew. Chem., Int. Ed., 2012, 51, 3520–3523 CrossRef CAS PubMed.
- S. Kowalski, S. Allard, K. Zilberberg, T. Riedl and U. Scherf, Prog. Polym. Sci., 2013, 38, 1805–1814 CrossRef CAS.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135–147 CrossRef CAS.
- T. Bura, P.-O. Morin and M. Leclerc, Macromolecules, 2015, 48, 5614–5620 CrossRef CAS.
- P.-O. Morin, T. Bura and M. Leclerc, Mater. Horiz., 2016, 3, 11–20 RSC.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056–10071 CrossRef CAS PubMed.
- M. Sévignon, J. Papillon, E. Schulz and M. Lemaire, Tetrahedron Lett., 1999, 40, 5873–5876 CrossRef.
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS PubMed.
- S. Hayashi, Y. Kojima and T. Koizumi, Polym. Chem., 2015, 6, 881–885 RSC.
- A. E. Rudenko, A. A. Latif and B. C. Thompson, Nanotechnology, 2014, 25, 014005 CrossRef PubMed.
- J.-R. Pouliot, M. Wakioka, F. Ozawa, Y. Li and M. Leclerc, Macromol. Chem. Phys., 2016, 217, 1493–1500 CrossRef CAS.
- S. Beaupré, A. Pron, S. H. Drouin, A. Najari, L. Mercier, A. Robitaille and M. Leclerc, Macromolecules, 2012, 45, 6906–6914 CrossRef.
- X. Wang and M. Wang, Polym. Chem., 2014, 5, 5784–5792 RSC.
- G. Marzano, D. Kotowski, F. Badudri, R. Musio, A. Pellegrino, S. Luzzati, R. Po and G. M. Farinola, Macromolecules, 2015, 48, 7039–7048 CrossRef CAS.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068–2071 CrossRef CAS PubMed.
- J. Jo, A. Pron, P. Berrouard, W. L. Leong, J. D. Yuen, J. S. Moon and M. Leclerc, Adv. Energy Mater., 2012, 2, 1397–1403 CrossRef CAS.
- A. Pron, P. Berrouard and M. Leclerc, Macromol. Chem. Phys., 2013, 214, 7–16 CrossRef CAS.
- L. G. Mercier, B. R. Aïch, A. Najari, S. Beaupré, P. Berrouard, A. Pron, A. Robitaille, Y. Tao and M. Leclerc, Polym. Chem., 2013, 4, 5252–5260 RSC.
- J.-R. Pouliot, L. G. Mercier, S. Caron and M. Leclerc, Macromol. Chem. Phys., 2013, 214, 453–457 CrossRef CAS.
- Q. Guo, J. Dong, D. Wan, D. Wu and J. You, Macromol. Rapid Commun., 2013, 34, 522–527 CrossRef CAS PubMed.
- P. Sonar, T. R. B. Foong and A. Dodabalapur, Phys. Chem. Chem. Phys., 2014, 16, 4275–4283 RSC.
- J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, Polym. Chem., 2015, 6, 278–282 RSC.
- S. Broll, F. Nübling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481–7488 CrossRef CAS.
- M. Gruber, S.-H. Jung, S. Schott, D. Venkateshvaran, A. J. Kronemeijer, J. W. Andreasen, C. R. McNeill, W. W. H. Wong, M. Shahid, M. Heeney, J.-K. Lee and H. Sirringhaus, Chem. Sci., 2015, 6, 6949–6960 RSC.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- P. Homyak, Y. Liu, F. Liu, T. P. Russel and E. B. Coughlin, Macromolecules, 2015, 48, 6978–6986 CrossRef CAS.
- E. Iizuka, M. Wakioka and F. Ozawa, Macromolecules, 2015, 48, 2989–2993 CrossRef CAS.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569–575 CrossRef CAS.
- A. S. Dubnik, T. J. Aldrich, N. D. Eastham, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 15699–15709 CrossRef PubMed.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- N. Takeda, T. Tagawa and M. Unno, Heterocycl. Chem., 2014, 25, 628–635 CAS.
- Q. Guo, J. Dong, D. Wan, D. Wu and J. You, Macromol. Rapid Commun., 2013, 34, 522–527 CrossRef CAS PubMed.
- P.-O. Morin, T. Bura, B. Sun, S. I. Gorelsky, Y. Li and M. Leclerc, ACS Macro Lett., 2015, 4, 21–24 CrossRef CAS.
- K. H. Hendriks, W. Li, G. H. L. Heintges, G. W. P. van Pruissen, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 11128–11133 CrossRef CAS PubMed.
- M. Wakioka, S. Ishiki and F. Ozawa, Macromolecules, 2015, 48, 8382–8388 CrossRef CAS.
- G. E. Morse, A. Tournebize, A. Rivaton, T. Chassé, C. Taviot-Gueho, N. Blouin, O. R. Lozman and S. Tierney, Phys. Chem. Chem. Phys., 2015, 17, 11884–11897 RSC.
- H. Li, S. Sun, S. Mhaisalkar, M. T. Zin, U. M. L. Lam and A. C. Grimsdale, J. Mater. Chem. A, 2014, 2, 17925–17933 CAS.
- C. B. Nielsen, R. S. Ashraf, N. D. Treat, B. C. Schroeder, J. E. Donaghey, A. J. P. White, N. Stingelin and I. McCulloch, Adv. Mater., 2015, 27, 948–953 CrossRef CAS PubMed.
- N. Blouin, A. Pron, G. Morse, L. Nanson, M. Krompiec and S. Berny, WO2014/2020184 A1, 2014.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Org. Chem., 2012, 77, 658–668 CrossRef CAS PubMed.
- C. Guo, J. Quinn, B. Sun and Y. Li, Polym. Chem., 2016, 7, 4515–4524 RSC.
- K. Wang, G. Wang and M. Wang, Macromol. Rapid Commun., 2015, 36, 2162–2170 CrossRef CAS PubMed.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- G. Marzano, F. Carulli, F. Babudri, A. Pellegrino, R. Po, S. Luzzati and G. M. Farinola, J. Mater. Chem. A, 2016, 4, 17163–17170 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1507660 and 1507661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00589j |
| ‡ T. Bura and S. Beaupré contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |