
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective single molecule nanopore sensing of proteins using DNA aptamer-functionalised gold nanoparticles†
Xiaoyan
Lin
 ,
Aleksandar P.
Ivanov
* and
Joshua B.
Edel
*
,
Aleksandar P.
Ivanov
* and
Joshua B.
Edel
*
Department of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: joshua.edel@imperial.ac.uk; alex.ivanov@imperial.ac.uk
First published on 20th March 2017
Abstract
Single molecule detection methods, such as nanopore sensors have found increasing importance in applications ranging from gaining a better understanding of biophysical processes to technology driven solutions such as DNA sequencing. However, challenges remain especially in relation to improving selectivity to probe specific targets or to alternatively enable detection of smaller molecules such as small-sized proteins with a sufficiently high signal-to-noise ratio. In this article, we propose a solution to these technological challenges by using DNA aptamer-modified gold nanoparticles (AuNPs) that act as a molecular carrier through the nanopore sensor. We show that this approach offers numerous advantages including: high levels of selectivity, efficient capture from a complex mixture, enhanced signal, minimized analyte-sensor surface interactions, and finally can be used to enhance the event detection rate. This is demonstrated by incorporating a lysozyme binding aptamer to a 5 nm AuNP carrier to selectively probe lysozyme within a cocktail of proteins. We show that nanopores can reveal sub-complex molecular information, by discriminating the AuNP from the protein analyte, indicating the potential use of this technology for single molecule analysis of different molecular analytes specifically bound to AuNP.
Introduction
Proteins are an essential part of living organisms and the ability to discriminate individual proteins in complex environments is crucial for modern chemical, biological research and medical diagnostics. Typical detection strategies include the use of mass spectroscopy1 and enzyme-linked immunosorbent assays (ELISA).2 However, these are bulk methods relying on ensemble averaging masking any rare events. To circumvent this problem, single molecule methods have been developed based on either optical or electrical readout.3,4One example of a single molecule platform is solid-state nanopore sensing.3,5–9 The operating principle is simple, in a typical experiment, biological molecules are electrophoretically or electro-osmotically driven (translocated) through a nanopore by an externally applied electric field, resulting in a temporal modulation of the measured ionic current.3,6 From these modulations one can extract information on molecular properties such as length, composition, and interactions with other biomolecules.3,10 Although DNA based analysis has been most commonly performed,11–13 more recently there has been a growing interest in protein based nanopore sensing to determine physical parameters such as size,9,14 conformational state,15,16 protein–protein interaction and binding kinetics between protein and antibodies/aptamers.17–25 However, compared with DNA, proteins exhibit a diverse range of sizes, three-dimensional structures and have a non-uniform charge distribution, which introduces experimental challenges. This is particularly true for the detection of small protein molecules, due to their fast translocation through the nanopore detector and low event rates, resulting in the detection of only a small fraction of the proteins, even with high bandwidth electronics.26,27 Although thin and small solid-state nanopores (<10 nm) have been developed for the detection of small proteins, fabrication is complex.28–30 Furthermore, with decreasing protein size, signals become increasingly difficult to detect due to lowering of the signal-to-noise ratio.9,26 Perhaps the greatest challenge in protein nanopore detection, has been the lack of selectivity, where proteins of similar size cannot be differentiated. Specifically, it is exceptionally challenging to extract chemical information from a Coulter counter-like measurement without introducing a strategy to tackle selectivity.17 Low selectivity is a major obstacle in many single molecule methods and especially for nanopores in heterogeneous samples and/or biological fluids.
Incorporation of binding sites in solid-state nanopores to improve selectivity has been previously demonstrated, including anchoring nitrilotriacetic acid for stochastic sensing of His-tagged proteins,22 and encoding of aptamers on glass nanopores.19 However, nanopore functionalization requires labour intensive procedures and could potentially lead to irreversible binding of protein to the receptor19 and subsequent blockage of the nanopore. An alternative strategy is to use DNA as a carrier molecule to isolate and detect proteins, however such carriers requires gene cutting engineering and the synthetic DNA chemistry.17,18
Herein, to address the above limitations, by using DNA aptamer functionalised 5 nm Au nanoparticles (AuNPs) that was used to selectively bind to protein targets in complex mixtures. Aptamers are oligonucleic acids that bind to target molecules with high specificity and affinity, and are selected using SELEX (systematic evolution of ligands by exponential enrichment).31–34 Unlike the methods described above, the attachment of aptamers to AuNPs is more efficient, economical and time-saving. Compared with antibodies, aptamers have a number of advantages including small size, low immunogenicity, low toxicity, and ease of modification.35–37 In our case, the attachment of the aptamers to the AuNPs enables increased efficiency in terms of event rates (3.8-fold) and improved signal-to-noise (from 6.3 to 11.1) due to an increase in the excluded volume and decrease in charge. More importantly, due to the specific binding between aptamer and target protein, selective detection in mixed analyte populations is possible. Furthermore, aptamers show high affinity towards a wide variety of targets, ranging from small molecules, nucleic acids, proteins to cells,32 thus the platform can be applied to single molecule detection of different targets by changing the aptamer sequence. To investigate the capability of our method, lysozyme38 was chosen as a target protein. The small size of lysozyme makes it an excellent analyte to investigate the achievable improvement in signal to noise and event rate.
Results and discussions
Nanopipette characterization
Detection was carried out using nanopipettes (Fig. 1A), a sub-class of solid-state nanopores, which were fabricated from quartz capillaries using a laser-based pipette puller as described in the ESI† and in the literature.39–41 Nanopipettes exhibit a number of advantages over conventional solid-state nanopores including quick and low-cost fabrication, good mechanical stability and low noise.40,42–47 The nanopipettes used had a conical geometry at the tip with a nanopore of 21 ± 4 nm in diameter, as determined by the SEM imaging (Fig. 1B). The current–voltage recording of 10 representative nanopipettes illustrated the rectification behaviour of the nanopipettes with an average rectification ratio I500 mV/I−500 mV of 1.4 ± 0.1 (Fig. S1†), comparable with ratios observed in a negatively charged conical geometry.40 The nanopipettes showed a resistance of 354 ± 18 MΩ as measured in (−0.1 V, 0.1 V) regime. The nanopipettes used in this work had resistance variation within 5%.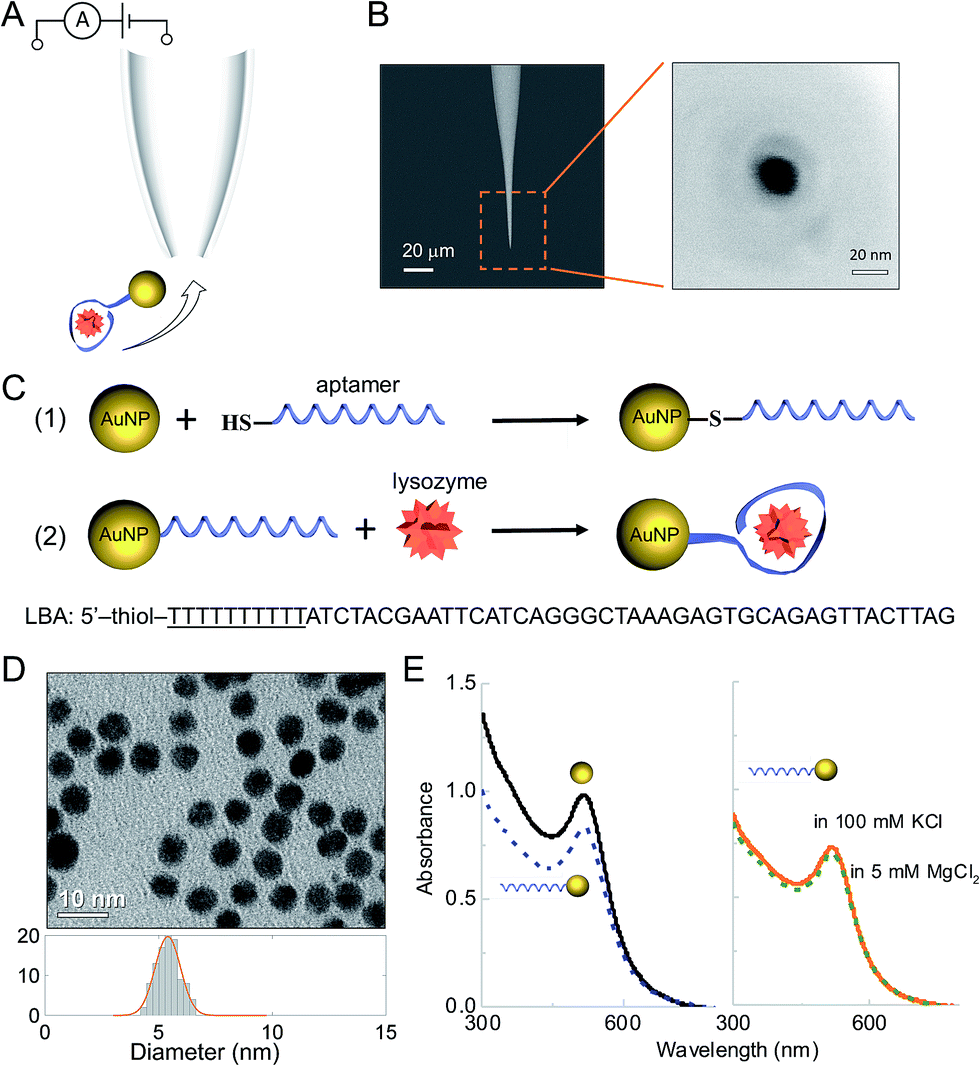 | ||
| Fig. 1 (A) Working principle for single molecule detection of AuNP-LBA/lysozyme complexes through a nanopipette. (B) SEM images of a typical nanopipette (left) and nanopore (right) at the tip of the nanopipette. The scale bars are 20 μm and 20 nm, respectively. (C) Schematic highlighting the strategy for detection of lysozyme using AuNPs functionalised with an LBA aptamer. (D) TEM image and size histogram of the AuNPs used. (E) left: UV-Vis spectra of AuNPs (solid, black) and AuNP-LBAs (dashed, blue); right: UV-Vis spectra of AuNP-LBAs in 100 mM KCl (solid, orange) and 5 mM MgCl2 (dashed, green), showing the stability of AuNPs after surface functionalisation at varying salt concentrations. | ||
Nanoparticle – aptamer engineering and characterization
Thiol-modified aptamers were attached to the surface of the AuNPs via the chemical reaction between thiol and gold.48–50 The sequence of the lysozyme-binding aptamer (LBA)51,52 (shown in Fig. 1C) included a spacer of 10 thymine bases (underlined) to minimize the steric hindrance from AuNPs when LBA bound with lysozyme. The aptamer was bound to the AuNP via thiol modification of the 5′ end. The uniformity and dispersion of the tannic acid stabilized AuNPs was verified using transmission electron microscopy (TEM) (Fig. 1D). From image analysis of over 200 particles, the average AuNP diameter was calculated to be 5.3 ± 0.5 nm. To improve stability and enable better control of the surface aptamer concentration, the AuNPs were modified with PEG-thiol (MW 1000).50 The concentration of both PEG-thiol and the lysozyme binding aptamer (LBA) were optimized to yield a final AuNP to aptamer ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and quantified using a fluorescence based assay (Fig. S2†).49 Therefore, the AuNPs had on average either 0 or 1 aptamer bound. The absolute ratio could be easily controlled by varying the ligand concentrations; however, as the motivation was to achieve single molecule sensitivity it was important to ensure the probability of having 2 or more aptamers bound per AuNP is negligible. As will be shown only the aptamer-modified AuNPs were detected.
:1 and quantified using a fluorescence based assay (Fig. S2†).49 Therefore, the AuNPs had on average either 0 or 1 aptamer bound. The absolute ratio could be easily controlled by varying the ligand concentrations; however, as the motivation was to achieve single molecule sensitivity it was important to ensure the probability of having 2 or more aptamers bound per AuNP is negligible. As will be shown only the aptamer-modified AuNPs were detected.
The AuNPs were well dispersed after surface modification (see Fig. S3†) and UV-Vis confirmed that the AuNP-LBAs did not aggregate (λ = 517–518 nm), Fig. 1E (left). As AuNPs tend to aggregate at higher salt concentrations, it was important to confirm this did not happen using the translocation and reaction buffers (100 mM KCl and 5 mM MgCl2). As shown in Fig. 1E (right), the peak wavelength remained unchanged between 517–518 nm and hence no aggregation was observed.
Single molecule detection with nanopipettes
A comparison between translocation characteristics of controls (i.e. AuNP, AuNP-LBA, lysozyme on its own) and the AuNP-LBA/lysozyme complex is shown in Fig. 2. It is noteworthy to point out that the PEG-modified AuNPs only showed translocation events with small current enhancement. As an example, this is shown in the current–time trace in Fig. 2A taken at an AuNP concentration of 4 nM and applied potential of −600 mV (patch electrode in the external bath and ground electrode inside the nanopipette) where the mean current amplitude was calculated to be 45 ± 2 pA. In comparison the AuNP-LBAs also exhibited current enhancement with a larger amplitude of 77 ± 6 pA at −600 mV and a KCl concentration of 100 mM, Fig. 2B. The change in current amplitude acted as a good indicator of the successful attachment of LBA to the AuNP. The observed current enhancement is in agreement with previous studies where it has been shown that positive counterions loosely attached to the functional groups on the surface of the ANP resulted in current enhancement.53–55 Furthermore, the ionic strength of the solution plays a dominant role and has been documented to directly affect the polarity of translocation events.56–59 For example, it has been shown that for KCl concentrations between 10–300 mM, DNA translocation results in nanopore current enhancement.57 Due to the large surface charge of quartz nanopipettes, at the electrolyte concentrations used, DNA acts as a charge carrier leading to temporary increase in nanopore conductance. Similarly, in this work, the PEG and negatively charged LBA functionalisation on the AuNP surface facilitated charge accumulation on the complex.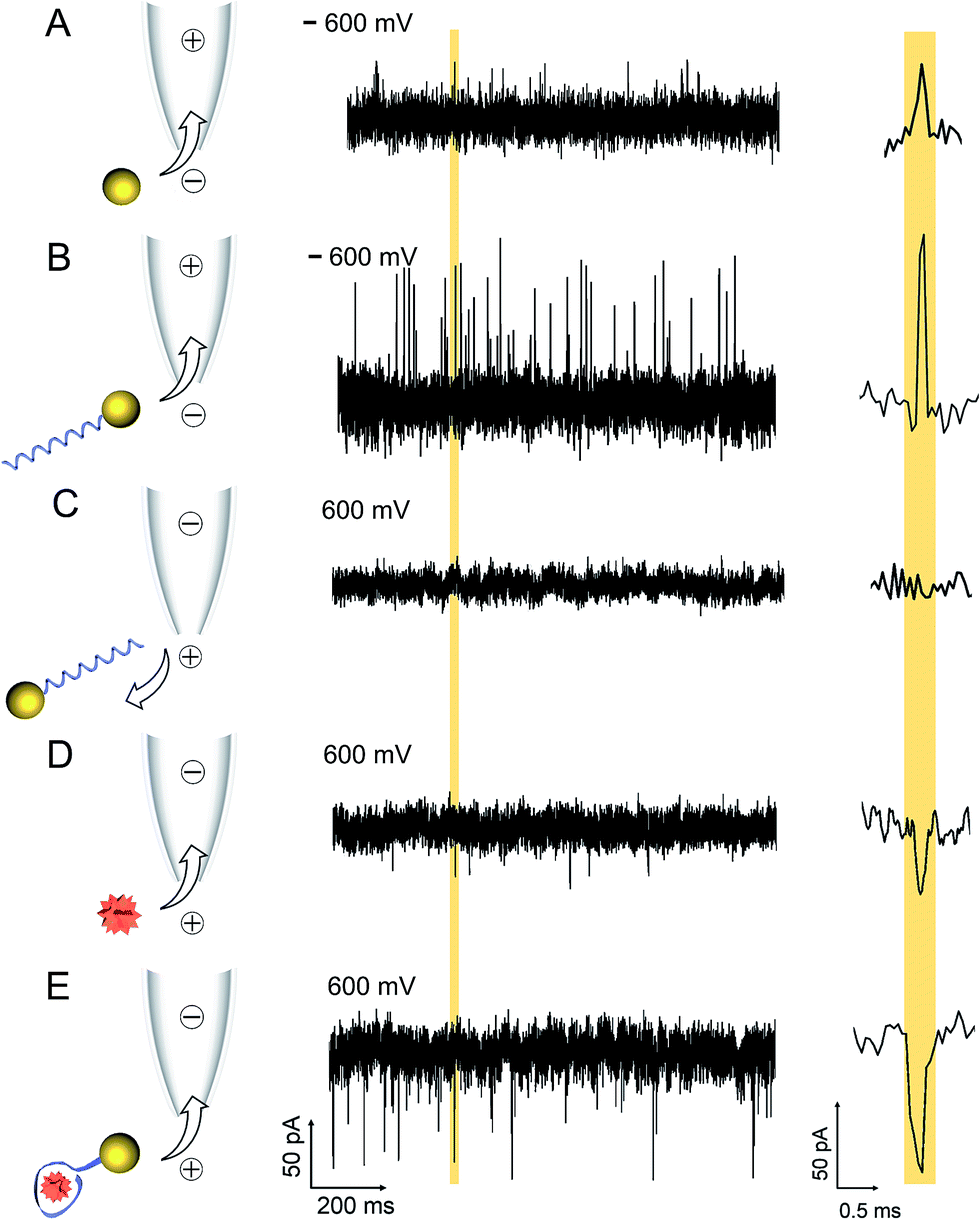 | ||
| Fig. 2 Current–time traces of (A) AuNPs and (B) AuNP-LBA at −600 mV and (C) AuNP-LBA, (D) free lysozyme, and (E) AuNP-LBA/lysozyme complexes at 600 mV with 100 mM KCl. The baseline current was −1230 pA for (A) AuNPs and −1200 pA for (B) AuNP-LBA at −600 mV; 2200 pA for (D) free lysozyme and 2300 pA for (E) AuNP-LBA/lysozyme complexes at 600 mV. AuNPs and AuNP-LBA exhibited current enhancement at negative voltages (events reversed for easier interpretation) while free lysozyme and AuNP-LBA/lysozyme complexes exhibited current blockade at positive voltages. Representative individual events are shown on the right-hand side of each trace. | ||
The translocation of 500 nM lysozyme occurred under a reversed bias due to the +8 charge at pH 7.4. Current blockades were observed with an amplitude of 41 ± 2 pA at 600 mV (Fig. 2D). In contrast, after the binding between 0.5 nM AuNP-LBA and 250 nM lysozyme, the AuNP-LBA/lysozyme complex gave rise to a new class of blocking events with large amplitude (70 ± 9 pA) at 600 mV (Fig. 2E). The signal-to-noise ratio60 increased to 11.1 compared with that of free lysozyme (6.3) at the same voltage. UV-Vis spectra confirmed that there was no aggregation after the binding between AuNP-LBA and lysozyme (Fig. S4†). The choice of lysozyme concentration used was motivated by the Kd (30–600 nM)51,52,61 between LBA and lysozyme, since higher concentration of lysozyme was needed to reach the saturation point. This was comparable to previous study, where anti-digoxigenin was detected (Kd = 3.5 nM) and the signal was saturated at the concentration of 6 nM, approximately at concentration twice the value of Kd.20 To confirm that current transients were caused by the translocation of the AuNP-LBA/lysozyme complex, control experiments were carried out with AuNP-LBA at positive applied bias (600 mV), which showed no detectable events (Fig. 2C). Importantly, this method enabled clear differentiation between AuNP, AuNP-LBA and the AuNP-LBA/lysozyme complex by current amplitudes ranging from current enhancement at negative voltages to blockade at positive voltages. The changing of current polarity from current enhancement to blockade was caused by reduced counterions and increased excluded volume of the AuNP-LBA/lysozyme complexes, both of which resulted in decreased conductance.56,57 Furthermore, control experiments were carried out with LBA + lysozyme, AuNP + lysozyme, respectively, which demonstrated small blocking events at 600 mV (Fig. S5†). Translocation of lysozyme binding with AuNPs functionalised with a greater number of aptamers per particle (average 2) exhibited longer dwell times and a broader distribution, (Fig. S6†). These significant differences between the current–time traces of AuNP-LBA/lysozyme complexes, AuNPs, AuNP-LBAs, free lysozyme, LBA + lysozyme, AuNP + lysozyme verified the successful discrimination of the AuNP-LBA/lysozyme complexes at single molecule level with improved current blockade.
For the AuNP-LBA/lysozyme complex with moderate charge, electroosmotic flow dominates the analyte transport in quartz nanopores due to higher surface charge of quartz compared to silicon nitride.7 We have previously estimated the electroosmotic flow (approximately 20 fL s−1 at 500 mV applied bias) for nanopipettes with the same geometry and at the same electrolyte concentration.40
The larger current signal of the AuNP-LBA/lysozyme complexes relative to free lysozyme at the same voltage can be attributed to the increase in the excluded volume, where the amplitude of the current change is related to the excluded volume of the electrolyte inside the pore.15,62 Free lysozyme in solution has a diameter of around 3 nm38 which results in a relatively small excluded volume and a small current blockade amplitude with a low signal-to-noise ratio. In comparison, for the AuNP-LBA/lysozyme complex: the 52-base LBA is 17 nm in length and 1.5 nm in width when it is fully stretched. After binding with lysozyme, the LBA turns into a folded structure, with an approximate diameter of 6–9 nm. With the addition of the 5 nm AuNP, the diameter of the whole AuNP-LBA/lysozyme complex can be estimated to be around 11 to 14 nm. The larger excluded volume of the AuNP-LBA/lysozyme complexes led to a larger current blockade with a higher signal-to-noise ratio of 11.1, which was substantially easier to detect in a nanopore experiment.
The efficient detection of smaller proteins is often hindered by low event frequency, fast translocation times, and small excluded volume.26 Previous studies have revealed that the rate of detection for proteins is largely discrepant with predictions from the Smoluchowski rate equation (J = 2πcDrp),26 with up to 5 orders of magnitude difference for small proteins less than ∼20 kDa, such as lysozyme (14 kDa). Often what is being detected are translocation events on the tail end of the dwell time distribution (i.e. slower events) whereas most go undetected. Furthermore, the signal of the current blockade or enhancement is often small resulting in a low signal-to-noise ratio. For these reasons the detection of small-sized proteins often requires concentrations significantly higher compared to ones used in the detection of larger proteins.
Strategies have been used to enhance event rates, which include the utilization of high salt gradients,63 high viscosity buffers,64 pressure gradients,65 temperature control66 and dielectrophoretic trapping at the nanopore opening.39 However, most of these studies focused on DNA and when applied to proteins, further experimental challenges could be introduced such as analyte aggregation and denaturation. A unique feature of this work is that the AuNP-LBA acts as a carrier, avoiding such experimental complications, while resulting in a 3.8-fold improvement in the detection rates, Fig. 3B. For example, lysozyme's event frequency was 0.9 ± 0.1 events per s at a concentration of 250 nM. Whilst the event frequency for the AuNP-LBA/lysozyme complex increased to 3.6 ± 0.8 events per s at the same concentration, which was around 100 times of that of ubiquitin at a higher concentration using thin and small solid-state nanopores.30 As discussed previously, the larger excluded volume of the AuNP-LBA/lysozyme complex led to an increased signal-to-noise ratio of 11.1. At the same time, the decrease in the overall charge of the AuNP-LBA/lysozyme complex enabled further slowing down of individual translocations and resulted in improved detection. The combination of increased signal and lower translocation times contributed to the enhanced event rate. The Kd for lysozyme and LBA was 30–600 nM according to previous reports.51,52 Thus, different concentrations of lysozyme, 50 nM, 100 nM, 200 nM, 250 nM were used in the translocation experiment. The event rate increased from 0.8 ± 0.1 events per s at 50 nM to 3.6 ± 0.8 events per s at 250 nM, showing a linear increasing in event rate with the increase of lysozyme concentration (Fig. 3B). The translocation of the AuNP-LBA/lysozyme complexes at different voltages were further studied, showing measurable current blockades at 300 mV–600 mV (100 mV increment). The results verified the binding between LBA and lysozyme was stable even at higher voltages. Voltage-mediated unfolding of proteins has been reported,15 however such phenomenon was not observed in our studies due to the high binding affinity between LBA and lysozyme. The current blockade distribution (Fig. 3C) and mean peak amplitudes (Fig. 3D), rose from 32.9 ± 0.6 pA at 300 mV to 69.6 ± 9.5 pA at 600 mV and the mean dwell time decreased from 0.38 ± 0.03 ms to 0.32 ± 0.05 ms for the same voltages (Fig. 3E and F). Control current–voltage curves were recorded before and after the measurement (Fig. S7†), showing negligible current variation, indicating that there was minimal of any analyte adsorption to the nanopore surface.
 | ||
| Fig. 3 (A) Schematic demonstrating the enhanced detection rate for the AuNP-LBA/lysozyme complex in comparison to that of free lysozyme. (B) The event rate of the AuNP-LBA/lysozyme complex resulted in a 3.8-fold increase in comparison to free lysozyme. (C) Current blockade histogram for the AuNP-LBA/lysozyme complex at applied voltages of 300 mV (grey), 400 mV (orange), 500 mV (blue), 600 mV (green). (D) Mean current blockade of the AuNP-LBA/lysozyme complex as a function of voltage. (E) Dwell time histogram for the AuNP-LBA/lysozyme complex at applied voltages of 300 mV (grey), 400 mV (orange), 500 mV (blue), 600 mV (green). (F) Mean dwell time of the AuNP-LBA/lysozyme complex as a function of voltage, exhibiting a decreasing trend with increasing voltages. | ||
Selective single molecule sensing of lysozyme in complex mixtures
A significant advantage of using aptamers for the detection of proteins with nanopores is in the high binding specificity between the aptamer and its target protein, making the method ideal for sensing in complex mixtures, Fig. 4A. To demonstrate the selectivity of our sensing approach, proteins with similar molecular weight and isoelectric point to lysozyme were used including cytochrome C (MW 12.4 kDa, pI 10.5) and trypsin (MW 23.3 kDa, pI 10.1). First, control experiments were carried out with lysozyme, cytochrome C and trypsin incubated with AuNP-LBAs individually and translocation studies were performed at a concentration of 250 nM and pH = 7.4. Translocation experiments revealed that only lysozyme gave rise to measurable current blockades (Fig. 4B(i)), while no translocations were observed for cytochrome C or trypsin (Fig. 4B(ii) and (iii)), confirming the specific binding of lysozyme to the AuNP-LBA. A comparison with the current–time trace of free lysozyme (Fig. 2D) verified that the events in Fig. 4B(i) were produced by AuNP-LBA/lysozyme complexes rather than free lysozyme. The selectivity was further validated by analysing complex mixtures and varying concentrations (250 nM and 125 nM) as shown in Fig. 4B(iv) and (v). The event rate of the AuNP-LBA in mixed protein population was calculated to be 3.1 ± 0.3 events per s (Fig. 4B(iv)), consistent with that of AuNP-LBA/lysozyme in Fig. 4B(i) (3.2 ± 0.6 events per s), verifying that the AuNP-LBA was able to bind lysozyme specifically in the mixed population without interference from other proteins. With the concentration of lysozyme reduced to 125 nM, the event rate proportionally decreased to 1.1 ± 0.2 events per s (Fig. 4B(v)). Scatter plots of the current blockade against dwell time for AuNP-LBA/lysozyme (Fig. 4C) and AuNP-LBA in mixed protein population (Fig. 4D) demonstrated that the translocation events in both cases had similar current blockade and dwell time distribution, further confirming the applicability of the method to selectively sense lysozyme in protein mixture.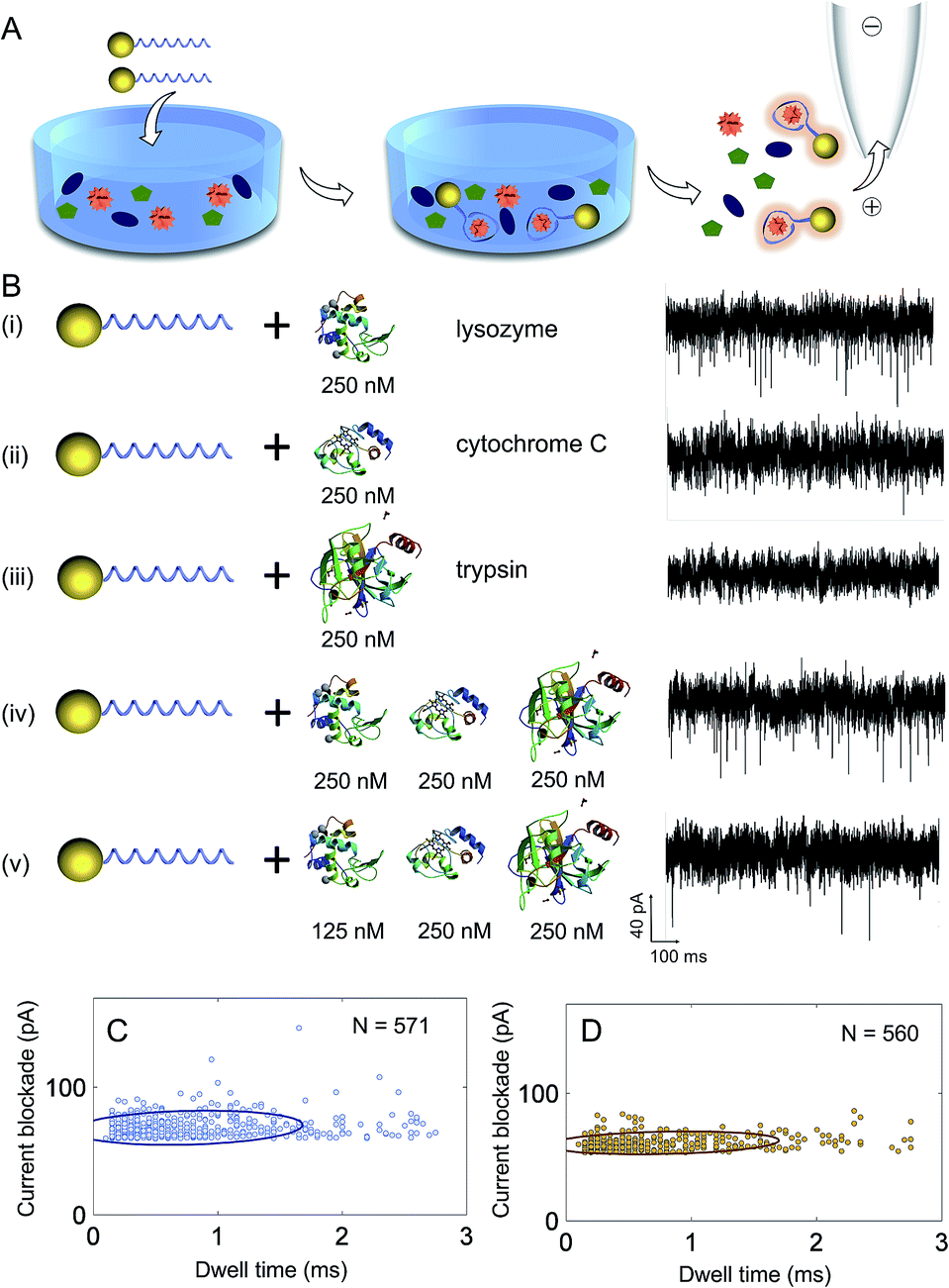 | ||
| Fig. 4 (A) Scheme highlighting the selective single molecule detection of lysozyme in a complex mixture using AuNP-LBAs; (B) the current–time traces of the translocation of (i) AuNP-LBAs with lysozyme, (ii) AuNP-LBAs with cytochrome C, (iii) AuNP-LBAs with trypsin, (iv) AuNP-LBAs with lysozyme + cytochrome C + trypsin, (v) AuNP-LBAs with lysozyme + cytochrome C + trypsin at 600 mV. The X-ray structures of lysozyme, cytochrome C and trypsin are from PDB; scatter plots of the current blockade versus dwell time for the AuNP-LBA/lysozyme (C) and the AuNP-LBA in mixed protein population (D) showing similar distributions of current blockade and dwell time. | ||
Sub-complex molecular information
To elucidate sub-complex molecular information, a high bandwidth amplifier was used (VC 100 Chimera Amplifier, sampling at 1 MHz, with digital filtering at 30–100 kHz) to discriminate between the ionic current signatures of individual components of the AuNP-LBA/lysozyme complexe and to better understand the interaction between AuNP, LBA and lysozyme. Interestingly, distinctive two levels of current blockade were observed: a low current amplitude level was defined as “level 1” and a larger amplitude one, defined as “level 2” (Fig. 5A). At positive voltages, only the AuNP-LBA/lysozyme complexes (not AuNPs or AuNP-LBAs) translocated through the nanopipette and gave rise to such large current blockade. As the amplitude of current change is related to the excluded volume of the molecule that migrates through the nanopore, “level 1” is attributed to the small sized AuNPs, in accordance to the ionic signatures observed in control experiments, while “level 2” was due to the larger LBA/lysozyme complexes, Fig. 5A. The events with dwell time within 0.20–0.45 ms were further analysed and the results showed that ∼65% of the events were in orientation i, while ∼35% were in orientation ii, indicating that a larger proportion of the AuNP-LBA/lysozyme complexes tend to translocate with the AuNP passing firstly through the nanopore, and LBA/lysozyme coming after. These observations can potentially be explained by recent findings from Yusko et al. suggest that proteins with a dipole moment do not rotate and orient randomly under the influence of a high electric fields inside a nanopore.67 Analogues to these findings, the LBA/lysozyme complex also has an inherent dipole moment and likely explains the observed preference in orientation in Fig. 5. Current blockade, dwell time, and equivalent charge distributions (the integrated current area over time) are shown in Fig. 5B and D and Fig. 5C and E for level 1 (AuNP) and level 2 (LBA/lysozyme complex), respectively. The mean current blockade for level 1 (AuNP) and level 2 (LBA/lysozyme complex) was 40 ± 11 pA and 76 ± 5 pA, mean dwell time was 0.10 ± 0.03 ms and 0.22 ± 0.07 ms, and finally the mean charge was 3 ± 1 fAs and 11 ± 4 fAs, respectively. The current blockade, charge and dwell time of level 1 and level 2 were consistent with the size and charge of AuNPs and LBA/lysozyme complex. Perhaps most importantly, the results revealed that the AuNP and LBA/lysozyme can be used to distinguish the individual components relative to the whole complex. This is highly advantageous for applications where further multiplexing can be imagined. For example, it would be possible to distinguish between different analytes bound to an AuNP consisting of multiple aptamer sequences simply by interpreting the current blockade and dwell time.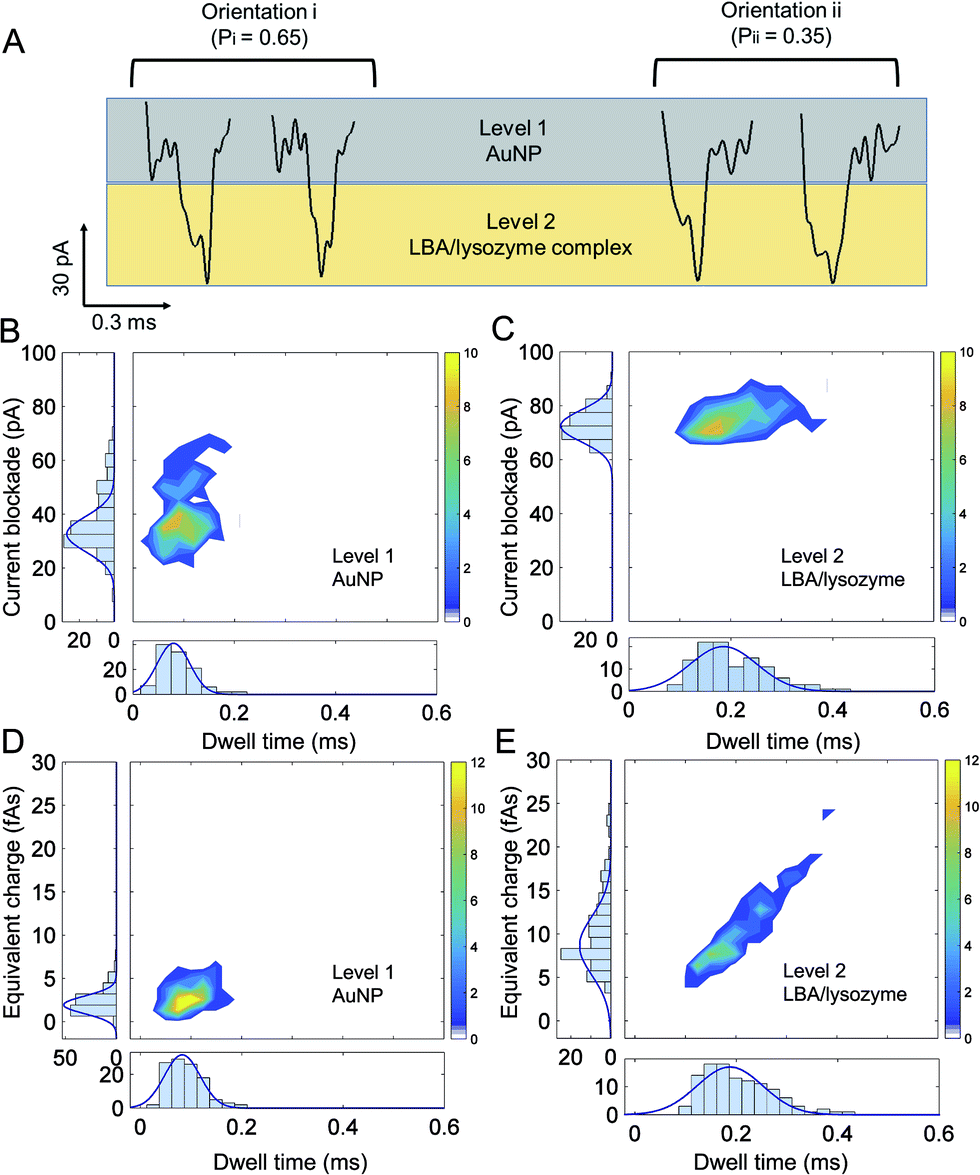 | ||
| Fig. 5 (A) The two orientations of translocation events caused by the AuNP-LBA/lysozyme complex with two different levels of current blockade caused by AuNP (level 1) and LBA/lysozyme complex (level 2) at 400 mV, respectively. The probability for orientation i and ii was 0.65 and 0.35, respectively; heat plots of current blockade against dwell time for level 1 (B) and level 2 (C) with the distribution histograms of current blockade and dwell time on the sides; heat plots of charge against dwell time for level 1 (D) and level 2 (E) with the distribution histograms of charge and dwell time on the sides. | ||
Conclusions
In recent years there has been a substantial progress in the selection and optimisation of aptamers to target specific type of protein or even distinguish between protein isoforms or even to use aptamers as therapeutic agents. In most of the cases, however aptamers target specific proteins in heterogeneous samples, where potential protein biomarkers are often very small fraction of the total protein concentration. It is vital that sensing technologies capable of single molecule detection and discrimination are developed in conjunction with advances in aptamer chemistry. Although this work focuses on technology development, it provides a strategy that enables selective protein detection that is potentially independent of the type of the aptamer used.To demonstrate the feasibility, LBA has been successfully functionalised on the surface of AuNPs and bound at ratios of 1:2. This enabled the detection of lysozyme at the single molecule level by use of nanopore sensing. AuNP-LBAs acted as a carrier enabling more efficient detection of the protein. For example, the larger size and lower charge of the AuNP-LBA/lysozyme complex relative to free lysozyme resulted in larger current blockades and higher event capture rates. The AuNP-LBA/lysozyme complex also reduced the interaction between the negatively charged nanopipette and positively charged lysozyme. The high binding affinity between LBA and lysozyme resulted in the complex being stable at voltages up to 600 mV. It was also possible to discriminate between signal arising from the AuNP and signal arising from LBA/lysozyme binding using a high bandwidth amplifier, establishing the applicability of this method for single molecule detection of proteins in different chemical and biological conditions. This opens the door to the possibility in using this technology to differentiate between different molecular analytes bound to the AuNP (e.g. having multiple aptamers bound the AuNP targeting different proteins).
Due to the high specificity between LBA and lysozyme, the platform exhibited excellent selectivity to discriminate lysozyme from proteins with similar size and charge (as shown with trypsin and cytochrome C). We show that it was possible to detect lysozyme within a complex mixture with no false positives. More typically selectivity is often achieved when using nanopores by functionalising the surface with receptors or alternatively via use of another carrier molecule such as DNA. However, both these methods are either labour intensive or alternatively are not economically viable. Use of AuNPs not only facilitates detection, it is also easy to modify the functionality simply by using single pot thiol–Au chemistry. AuNPs are potential delivery system for therapeutic agents, showing good biocompatibility and low toxicity. Therefore, aptamer-functionalised AuNPs can find promising applications in medical and clinical areas.
Acknowledgements
J. B. E. has been funded in part by an ERC starting and consolidator investigator grant. J. B. E. and A. I. acknowledge receipt of an EPSRC grant. A. I. is funded by an Imperial College Fellowship and X. L. by Imperial College President's PhD Scholarship.References
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- E. Engvall and P. Perlmann, Immunochemistry, 1971, 8, 871–874 CrossRef CAS PubMed.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Dogan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- W. H. Pitchford, H. J. Kim, A. P. Ivanov, H. M. Kim, J. S. Yu, R. J. Leatherbarrow, T. Albrecht, K. B. Kim and J. B. Edel, ACS Nano, 2015, 9, 1740–1748 CrossRef CAS PubMed.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- S. Howorka and Z. Siwy, Chem. Soc. Rev., 2009, 38, 2360–2384 RSC.
- M. Firnkes, D. Pedone, J. Knezevic, M. Doblinger and U. Rant, Nano Lett., 2010, 10, 2162–2167 CrossRef CAS PubMed.
- D. Fologea, B. Ledden, D. S. McNabb and J. Li, Appl. Phys. Lett., 2007, 91, 539011–539013 CrossRef PubMed.
- W. Li, N. A. Bell, S. Hernandez-Ainsa, V. V. Thacker, A. M. Thackray, R. Bujdoso and U. F. Keyser, ACS Nano, 2013, 7, 4129–4134 CrossRef CAS PubMed.
- H. Bayley and C. R. Martin, Chem. Rev., 2000, 100, 2575–2594 CrossRef CAS PubMed.
- H. Chang, F. Kosari, G. Andreadakis, M. A. Alam, G. Vasmatzis and R. Bashir, Nano Lett., 2004, 4, 1551–1556 CrossRef CAS.
- J. L. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, Nat. Mater., 2003, 2, 611–615 CrossRef CAS PubMed.
- R. P. Johnson, A. M. Fleming, L. R. Beuth, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2016, 138, 594–603 CrossRef CAS PubMed.
- L. J. Steinbock, S. Krishnan, R. D. Bulushev, S. Borgeaud, M. Blokesch, L. Feletti and A. Radenovic, Nanoscale, 2014, 6, 14380–14387 RSC.
- K. J. Freedman, S. R. Haq, J. B. Edel, P. Jemth and M. J. Kim, Sci. Rep., 2013, 3, 1638 Search PubMed.
- D. S. Talaga and J. Li, J. Am. Chem. Soc., 2009, 131, 9287–9297 CrossRef CAS PubMed.
- N. A. W. Bell and U. F. Keyser, J. Am. Chem. Soc., 2015, 137, 2035–2041 CrossRef CAS PubMed.
- N. A. W. Bell and U. F. Keyser, Nat. Nanotechnol., 2016, 11, 645–652 CrossRef CAS PubMed.
- S. Ding, C. Gao and L. Q. Gu, Anal. Chem., 2009, 81, 6649–6655 CrossRef CAS PubMed.
- J. L. Kong, N. A. W. Bell and U. F. Keyser, Nano Lett., 2016, 16, 3557–3562 CrossRef CAS PubMed.
- D. Rotem, L. Jayasinghe, M. Salichou and H. Bayley, J. Am. Chem. Soc., 2012, 134, 2781–2787 CrossRef CAS PubMed.
- R. Wei, V. Gatterdam, R. Wieneke, R. Tampe and U. Rant, Nat. Nanotechnol., 2012, 7, 257–263 CrossRef CAS PubMed.
- E. C. Yusko, J. M. Johnson, S. Majd, P. Prangkio, R. C. Rollings, J. L. Li, J. Yang and M. Mayer, Nat. Nanotechnol., 2011, 6, 253–260 CrossRef CAS PubMed.
- O. A. Saleh and L. L. Sohn, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 820–824 CrossRef CAS PubMed.
- J. D. Uram, K. Ke, A. J. Hunt and M. Mayer, Small, 2006, 2, 967–972 CrossRef CAS PubMed.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Nano Lett., 2013, 13, 3445–3445 CrossRef CAS.
- J. Larkin, R. Y. Henley, M. Muthukumar, J. K. Rosenstein and M. Wanunu, Biophys. J., 2014, 106, 696–704 CrossRef CAS PubMed.
- N. Di Fiori, A. Squires, D. Bar, T. Gilboa, T. D. Moustakas and A. Meller, Nat. Nanotechnol., 2013, 8, 946–951 CrossRef CAS PubMed.
- D. J. Niedzwiecki, C. J. Land, G. Shemer, P. S. Cheng, J. G. Seven and M. Drndic, ACS Nano, 2015, 9, 8907–8915 CrossRef CAS PubMed.
- I. Nir, D. Huttner and A. Meller, Biophys. J., 2015, 108, 2340–2349 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- D. H. Bunka and P. G. Stockley, Nat. Rev. Microbiol., 2006, 4, 588–596 CrossRef CAS PubMed.
- C. K. O'Sullivan, Anal. Bioanal. Chem., 2002, 372, 44–48 CrossRef PubMed.
- S. K. Verma, A. Amoah, U. Schellhaas, M. Winterhalter, S. Springer and T. A. Kolesnikova, Adv. Funct. Mater., 2016, 26, 6015–6024 CrossRef CAS.
- C. C. Blake, D. F. Koenig, G. A. Mair, A. C. North, D. C. Phillips and V. R. Sarma, Nature, 1965, 206, 757–761 CrossRef CAS PubMed.
- K. J. Freedman, L. M. Otto, A. P. Ivanov, A. Barik, S. H. Oh and J. B. Edel, Nat. Commun., 2016, 7, 10217 CrossRef CAS PubMed.
- A. P. Ivanov, P. Actis, P. Jonsson, D. Klenerman, Y. Korchev and J. B. Edel, ACS Nano, 2015, 9, 3587–3595 CrossRef CAS PubMed.
- J. Y. Y. Sze, S. Kumar, A. P. Ivanov, S. H. Oh and J. B. Edel, Analyst, 2015, 140, 4828–4834 RSC.
- P. Actis, A. C. Mak and N. Pourmand, Bioanal Rev., 2010, 1, 177–185 CrossRef PubMed.
- N. A. Bell, V. V. Thacker, S. Hernandez-Ainsa, M. E. Fuentes-Perez, F. Moreno-Herrero, T. Liedl and U. F. Keyser, Lab Chip, 2013, 13, 1859–1862 RSC.
- R. A. Levis and J. L. Rae, Biophys. J., 1993, 65, 1666–1677 CrossRef CAS PubMed.
- L. J. Steinbock, R. D. Bulushev, S. Krishnan, C. Raillon and A. Radenovic, ACS Nano, 2013, 7, 11255–11262 CrossRef CAS PubMed.
- L. J. Steinbock, O. Otto, C. Chimerel, J. Gornall and U. F. Keyser, Nano Lett., 2010, 10, 2493–2497 CrossRef CAS PubMed.
- R. J. White, E. N. Ervin, T. Yang, X. Chen, S. Daniel, P. S. Cremer and H. S. White, J. Am. Chem. Soc., 2007, 129, 11766–11775 CrossRef CAS PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS PubMed.
- S. J. Hurst, A. K. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- J. X. Li, B. Q. Zhu, X. J. Yao, Y. C. Zhang, Z. Zhu, S. Tu, S. S. Jia, R. D. Liu, H. Z. Kang and C. J. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 16800–16807 CAS.
- A. K. Cheng, B. Ge and H. Z. Yu, Anal. Chem., 2007, 79, 5158–5164 CrossRef CAS PubMed.
- J. C. Cox and A. D. Ellington, Bioorg. Med. Chem., 2001, 9, 2525–2531 CrossRef CAS PubMed.
- H. Cai, Y. Wang, Y. Yu, M. V. Mirkin, S. Bhakta, G. W. Bishop, A. A. Joshi and J. F. Rusling, Anal. Chem., 2015, 87, 6403–6410 CrossRef CAS PubMed.
- G. Goyal, K. J. Freedman and M. J. Kim, Anal. Chem., 2013, 85, 8180–8187 CrossRef CAS PubMed.
- Y. X. Wang, K. Kececi, M. V. Mirkin, V. Mani, N. Sardesai and J. F. Rusling, Chem. Sci., 2013, 4, 655–663 RSC.
- R. M. M. Smeets, U. F. Keyser, D. Krapf, M. Y. Wu, N. H. Dekker and C. Dekker, Nano Lett., 2006, 6, 89–95 CrossRef CAS PubMed.
- L. J. Steinbock, A. Lucas, O. Otto and U. F. Keyser, Electrophoresis, 2012, 33, 3480–3487 CrossRef CAS PubMed.
- W. J. Lan, C. Kubeil, J. W. Xiong, A. Bund and H. S. White, J. Phys. Chem. C, 2014, 118, 2726–2734 CAS.
- Y. Qiu, C. Yang, P. Hinkle, I. V. Vlassiouk and Z. S. Siwy, Anal. Chem., 2015, 87, 8517–8523 CrossRef CAS PubMed.
- R. M. M. Smeets, U. F. Keyser, N. H. Dekker and C. Dekker, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 417–421 CrossRef CAS PubMed.
- R. Kirby, E. J. Cho, B. Gehrke, T. Bayer, Y. S. Park, D. P. Neikirk, J. T. McDevitt and A. D. Ellington, Anal. Chem., 2004, 76, 4066–4075 CrossRef CAS PubMed.
- A. Oukhaled, B. Cressiot, L. Bacri, M. Pastoriza-Gallego, J. M. Betton, E. Bourhis, R. Jede, J. Gierak, L. Auvray and J. Pelta, ACS Nano, 2011, 5, 3628–3638 CrossRef CAS PubMed.
- M. Wanunu, W. Morrison, Y. Rabin, A. Y. Grosberg and A. Meller, Nat. Nanotechnol., 2010, 5, 160–165 CrossRef CAS PubMed.
- D. Fologea, J. Uplinger, B. Thomas, D. S. McNabb and J. L. Li, Nano Lett., 2005, 5, 1734–1737 CrossRef CAS PubMed.
- B. Lu, D. P. Hoogerheide, Q. Zhao, H. Zhang, Z. Tang, D. Yu and J. A. Golovchenko, Nano Lett., 2013, 13, 3048–3052 CrossRef CAS PubMed.
- K. R. Mahendran, C. Chimerel, T. Mach and M. Winterhalter, Eur. Biophys. J., 2009, 38, 1141–1145 CrossRef CAS PubMed.
- E. C. Yusko, B. R. Bruhn, O. M. Eggenberger, J. Houghtaling, R. C. Rollings, N. C. Walsh, S. Nandivada, M. Pindrus, A. R. Hall, D. Sept, J. Li, D. S. Kalonia and M. Mayer, Nat. Nanotechnol., 2016 DOI:10.1038/nnano.2016.267.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc00415j |
| This journal is © The Royal Society of Chemistry 2017 |