
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post-polymerization modification reactions of poly(glycidyl methacrylate)s
Ezzah M. Muzammil
a,
Anzar Khan
 *b and
Mihaiela C. Stuparu
*ac
*b and
Mihaiela C. Stuparu
*ac
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371-Singapore. E-mail: mstuparu@ntu.edu.sg
bDepartment of Chemical and Biological Engineering, Korea University, Seoul 02841, Korea. E-mail: anzar@korea.ac.kr; Tel: +82 2 3290 4859
cSchool of Materials Science and Engineering, Nanyang Technological University, 21 Nanyang Link, 637371-Singapore
First published on 8th December 2017
Abstract
Post-polymerisation modification of poly(glycidyl methacrylate) (PGMA) through the nucleophilic ring opening reactions of the pendent epoxide groups allows for the installation of a variety of functionalities onto the reactive scaffold. The primary modification processes involve amine-epoxy, thiol–epoxy, azide-epoxy, acid-epoxy, and hydrolysis reactions. In all cases, sequential post-synthesis modification reactions can also be carried out if multiply-functionalised polymers are required. This, in particular, includes reactions of the hydroxyl group(s) that come into being through the initial oxirane ring-opening reaction. The overall flexibility of these functionalisations, coupled with the commercial availability of glycidyl methacrylate monomer, its controlled polymerisation behaviour through free radical polymerisation methods and high shelf life of the resulting polymers makes PGMA one of the most adaptable reactive scaffolds in polymer chemistry. In this review article, our aim is to discuss the fundamental aspects of the epoxy ring-opening reactions and highlight the utilitarian nature of PGMA by addressing the range of chemistry that has been used to transform this simple structure into a plethora of customised functional polymers.
 Ezzah M. Muzammil | Ezzah M. Muzammil is a Master student from Nanyang Technological University (NTU) in the Division of Chemistry and Biological Chemistry. She received her Bachelor(Hons) degree in Chemistry and Biological Chemistry in 2016 and hopes to pursue a doctorate degree under the mentorship of Prof. Stuparu. Her research interest is to explore corannulene chemistry in the area of organic syntheses to further study this exceptional molecule. |
 Anzar Khan | Anzar Khan carried out his doctoral studies at the Freie Universität Berlin (Germany) under the supervision of Prof. Stefan Hecht. He then joined Prof. Craig Hawker's lab at the University of California at Santa Barbara (USA) as a post-doctoral scholar. Recently, Anzar joined the Department of Biological and Chemical Engineering at Korea University. Here, he is leading a research group with interests ranging from synthesis of multifunctional polymers to fabrication of stimuli-responsive nanostructured soft materials. |
 Mihaiela C. Stuparu | Born in Romania, Mihaiela received her Master degree in Organic Chemistry from the Babes-Bolyai University, Romania, under the guidance of Prof. I. Grosu. She then moved to Freie Universität Berlin, Germany, and finally to ETH-Zürich, Switzerland, to obtain her doctoral degree in the group of Prof. A. D. Schlüter. After a one-year postdoctoral stay in the group of Prof. H. K. Hall Jr at the University of Arizona at Tucson (USA), Mihaiela returned to Zürich as a member of the Prof. J. Siegel's group at the University of Zürich. In 2009, Mihaiela received the Ambizione fellowship from the Swiss National Science Foundation. Under the mentorship of Prof. Siegel, Mihaiela's research efforts at the University of Zürich were directed towards creating a research program focused on introducing the corannulene chemistry to the macromolecular science. The University of Zürich recognized Mihaiela's research efforts in this direction by bestowing upon her the title of ‘Privat-Dozent’. Since 2014, Mihaiela is a Nanyang Assistant Professor at NTU Singapore. Here, her group is investigating utility of the corannulene motif for the preparation of functional soft materials. |
Introduction
Poly(glycidyl methacrylate) (PGMA) is an interesting polymer. At first glance, its highly strained three-membered epoxide side chains would suggest instability resulting in a short shelf-life. One could also imagine that such a reactive polymer might be difficult to access synthetically especially as a homopolymer. However, reality is far from such assumptions. In fact, PGMA can be easily synthesised from its commercially available and inexpensive monomer, glycidyl methacrylate, by free radical polymerization processes (Scheme 1).1–16 | ||
| Scheme 1 Synthesis of polyglycidyl methacrylate by free radical polymerisation methods. | ||
Stability-wise, the purified homopolymer can be stored at room temperature and under ambient conditions for years without compromising its structural integrity. In this way, it differs from other reactive scaffolds like alkene and alkyne-carrying which can crosslink during polymerisation if the concentration of the active-site-carrying monomer is high and the active site is unprotected.
PGMA, therefore, is a unique structure that can be accessed with a considerable degree of freedom over the synthesis method, purified, and stored in its homopolymer structure. Of course, co-polymerization is possible with any other acrylate or methacrylate monomer to further diversify the range of structures and applications of the final materials.1–16 When required, the epoxy side chains of PGMA can be subjected to a nucleophilic ring-opening reaction under a variety of conditions to afford functionalised polymers (Scheme 2). The wide range of chemistry available allows one to freely choose the nature of the functional group and the manner of the ring-opening reaction. It is this versatility that establishes PGMA as a general and powerful reactive scaffold in polymer chemistry.
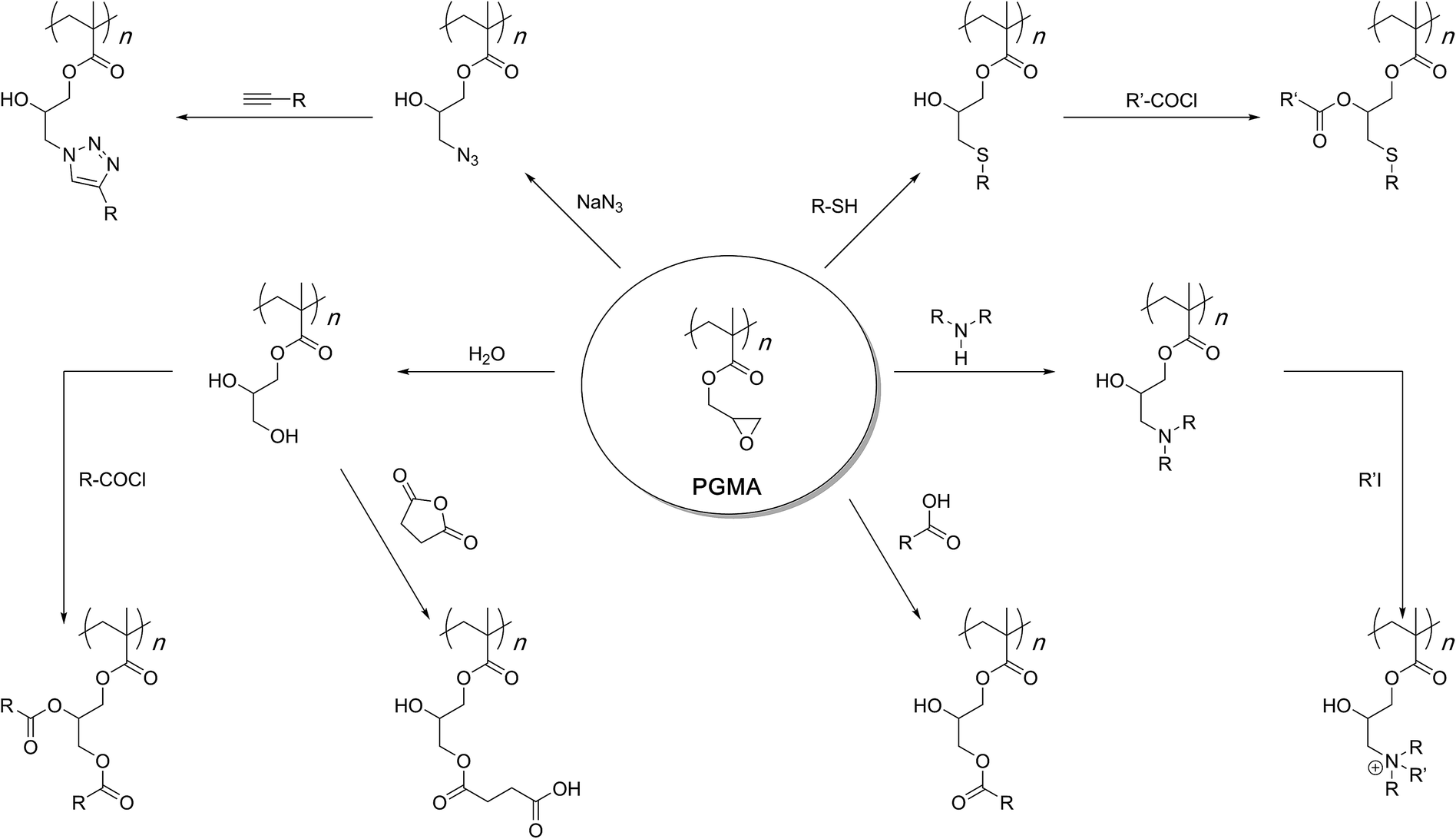 | ||
| Scheme 2 Post-polymerisation modification reactions of polyglycidyl methacrylate. Sequential multiple functionalisations are possible in most cases as the hydroxyl group(s) formed through the ring-opening reaction can be utilised to install a second functionality. Alternatively, the first functionality can create a new reactive site for subsequent modifications. | ||
Scope of the review
Our goal is to illustrate the adaptable nature of PGMA by discussing selected and early examples that reveal its multifaceted chemistry through preparation and characterisation of molecularly well-defined polymers. On this basis, the discussion does not exhaust the entire literature and in particular does not examine surface functionalisations and particulate materials.17–29 Furthermore, cross-linked systems are not covered.30–34 The synthesis of the backbone itself is not discussed and the reader is referred to a number of important publications in this regard.1–16 Other polymer backbones that carry a side-chain epoxide functionality are important as is seen in the works of Harth, Dove, He, Siegwert, and Theato.35–40 However, these backbones are outside the scope of the present review as we limit ourselves to PGMA polymers only. Finally, assembly and applications of the PGMA-derived materials are not discussed as a recent review article has examined these aspects comprehensively.41General considerations
How stable is PGMA?
Although epoxides are inherently strained three-membered rings, an acid catalyst or a strong nucleophile – typically created through use of a base catalyst, is essential for the ring-opening reaction. In the absence of either, epoxy derivatives remain perfectly stable under ambient conditions. It is for this reason that polymers of epoxides such as PGMA can be stored for years without any detection of crosslinking or degradation.What will be the regio-chemistry of functionalisation?
Let us first consider the case of acid-catalysed ring-opening reaction of epoxides (Scheme 3). Here, the first step is the protonation of the oxygen atom which is stabilised by inductive effect from the most substituted (i.e. most electron rich) carbon atom. This renders the tertiary carbon atom electrophilic and susceptible to a nucleophilic attack by a mild nucleophile such as water or methanol that would otherwise not be able to force the ring-opening reaction if acting alone. | ||
| Scheme 3 Acid-catalysed oxirane ring-opening reaction. The positive charge builds on the most substituted carbon atom that is most suitable for stabilising it through inductive effect based on the substituents. | ||
In the absence of an acid activating the molecule, the ring-opening reaction can only proceed if a strong nucleophile is available to eject the oxygen atom so that the oxygen atom may form a favourable leaving group (Scheme 4). In this case, since a positive charge is absent, steric rather than electronic factors determine the outcome of the reaction and the second possible regio-isomer forms exclusively in which the nucleophile is attached to the least substituted and least hindered carbon atom. A model-compound-based systematic study has recently confirmed this notion.42
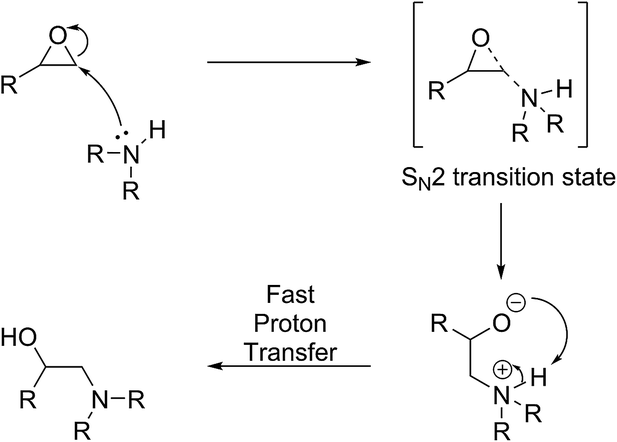 | ||
| Scheme 4 Base-catalysed oxirane ring-opening reaction. Strong nucleophiles approach the least hindered carbon atom, forming a new thio-ether linkage. Although intramolecular proton transfer reaction is depicted, intermolecular transfer processes can also occur to quench the alkoxide anion. | ||
Can alkoxide anion not initiate oxirane ring-opening polymerization?
Under acidic conditions, the oxygen atom is already protonated before ring-opening and poses no threat for further reaction. Under basic conditions, an alkoxide anion is formed (Scheme 5). Polymer chemists would envisage that such a structure would quickly bring about anionic ring-opening polymerisation of an epoxide monomer. However, no such polymerisation occurs because the alkoxide anion (pKa = 17) is a very strong base and is rapidly quenched by a proton transfer reaction. The proton can be abstracted from multiple sources within a reaction mixture. For instance, weakly acidic protons associated with the nucleophile, often employed aqueous/alcohol medium, or intentionally added proton sources such as ammonium chloride to the reaction, all possess pKa values below 17 (see Scheme 5). It is a simple acid–base reaction that causes the quenching of the alkoxide ion by the mildly acidic protons present in the system, therefore impeding a ring-opening polymerisation.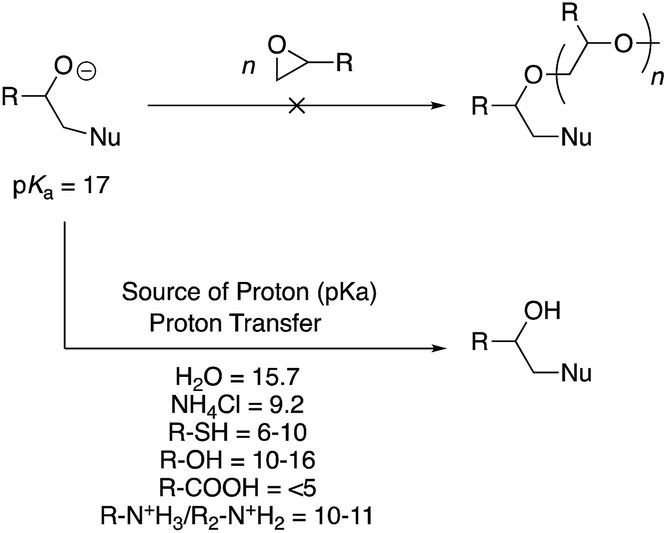 | ||
| Scheme 5 The alkoxide anion is a strong base and is quenched by weak acidic protons present in the system. Therefore, it cannot act as an initiator for ring-opening polymerisation of the pendent epoxide groups. The bottom shows pKa of various functional groups, solvents, or additives that are typically present in the PGMA functionalisation reactions. Nu stands for nucleophile. | ||
Post-polymerisation modifications
Post-synthesis modification of reactive polymeric scaffolds is an important route to functional polymers.43–53 An advantage of this route is that a general scaffold can lead to various structures that differ in the nature of the attached functionality but the molecular weight attributes remain the same within a functionalised polymer family. Such a system allows for an unambiguous development of structure–property relationships. PGMA is such a general reactive scaffold which can be turned into a functionalised polymer family through a range of chemical reactions. The following is a discussion of its chemistry arranged based on the nature of the functionalisation reaction.Functionalisation with amines
In 2010, Leroux and co-workers described the synthesis of linear and branched GMA polymers and their post-polymerization modifications with butylmethylamine, propylamine, methylpropylamine, and trimethylethylenediamine (Scheme 6).54 The ring-opening reactions were carried out at 90 °C, overnight in an argon atmosphere. Ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:1, 1:0.5 of the amine to the epoxy group were used in separate experiments. Interestingly, water was added to the reaction mixture and a second reflux was carried out at the same temperature for the same period of time. Elemental analysis indicated that sterically demanding trimethylethylenediamine led to a lower degree of functionalisation (72–81%) whereas smaller amines exhibited better efficiencies (88–98%). Most of the functionalised structures were, however, found to be insoluble in water. It is likely that the post-polymerization reaction did not stop at the formation of secondary/tertiary amines but proceeded to the higher levels of tertiary/quaternary structures especially under the employed reaction conditions. Such intermolecular crosslinks can explain the reduced solubility of the functionalised polymers. Earlier, Huck's group had described such crosslinking processes in polymer brushes grown onto silicon substrates.55 Furthermore, Li and co-workers, while working with oligoethylene amines at 70 °C noticed the same issue of intermolecular crosslinking and decreased functionalisation efficiency with increasing bulk of the amine group.56 The issue of intermolecular crosslinking could be solved by reducing the reaction temperature (55–37 °C) and/or by using a large (at least 10 fold) excess of the amine.57–61 Remarkably, Tang and co-workers have shown that the functionalisation reaction can be carried out at room temperature for 24 h with near quantitative efficiencies.62 Such mild conditions are certainly useful to avoid unwanted further reaction of the secondary/tertiary amines and crosslinking.
:2, 1:1, 1:0.5 of the amine to the epoxy group were used in separate experiments. Interestingly, water was added to the reaction mixture and a second reflux was carried out at the same temperature for the same period of time. Elemental analysis indicated that sterically demanding trimethylethylenediamine led to a lower degree of functionalisation (72–81%) whereas smaller amines exhibited better efficiencies (88–98%). Most of the functionalised structures were, however, found to be insoluble in water. It is likely that the post-polymerization reaction did not stop at the formation of secondary/tertiary amines but proceeded to the higher levels of tertiary/quaternary structures especially under the employed reaction conditions. Such intermolecular crosslinks can explain the reduced solubility of the functionalised polymers. Earlier, Huck's group had described such crosslinking processes in polymer brushes grown onto silicon substrates.55 Furthermore, Li and co-workers, while working with oligoethylene amines at 70 °C noticed the same issue of intermolecular crosslinking and decreased functionalisation efficiency with increasing bulk of the amine group.56 The issue of intermolecular crosslinking could be solved by reducing the reaction temperature (55–37 °C) and/or by using a large (at least 10 fold) excess of the amine.57–61 Remarkably, Tang and co-workers have shown that the functionalisation reaction can be carried out at room temperature for 24 h with near quantitative efficiencies.62 Such mild conditions are certainly useful to avoid unwanted further reaction of the secondary/tertiary amines and crosslinking.
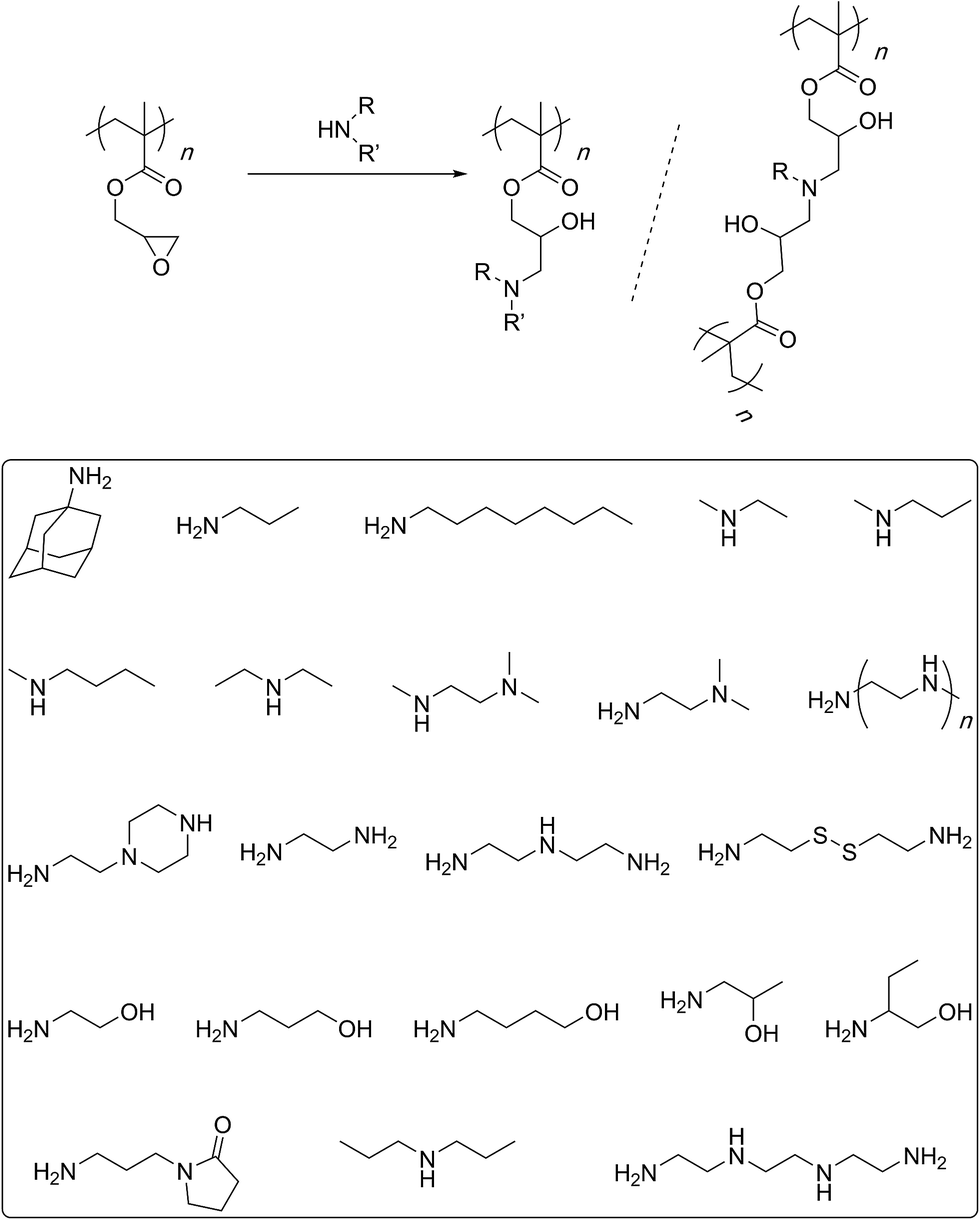 | ||
| Scheme 6 Amine-epoxy reaction for the post-polymerisation modification of the PGMA backbone. The bottom shows the amine structures used by various researchers for the functionalisation process. | ||
The amine functionalisation strategy has been heavily used in the preparation of amphiphilic copolymers, star polymers, and graft copolymers that have found applications mainly in the arena of polyplexes formation and gene delivery.63–67 In some cases, the tertiary amines were further quaternised through the use of an alkyl halide to give access to side-chain cationic polymers.68,69
In most cases so far, atom transfer radical polymerization (ATRP) was used for the preparation of the PGMA scaffold. Haddleton and co-workers showed that synthesis by a cobalt-catalysed chain transfer polymerisation could lead to the polymer chains displaying a methacrylate chain-end. In such cases, therefore, difference in reactivity of the chain-end (methacrylate group) as compared to the side-chain (oxirane group) can allow for sequential modifications and preparation of doubly-functionalised structures (Scheme 7).70 This was demonstrated in two different fashions. In case of primary amines being the nucleophiles designed to open the side chains, the acrylate group was consumed first through a thiol nucleophile. This is because primary amines can add to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond via a Michael-type of addition reaction.71 In case of secondary amines, the post-polymerisation modification of the side chain epoxides could be achieved first and the methacrylate was modified in a subsequent step. Both reaction sequences led to the formation of dual-functional polymers.
C bond via a Michael-type of addition reaction.71 In case of secondary amines, the post-polymerisation modification of the side chain epoxides could be achieved first and the methacrylate was modified in a subsequent step. Both reaction sequences led to the formation of dual-functional polymers.
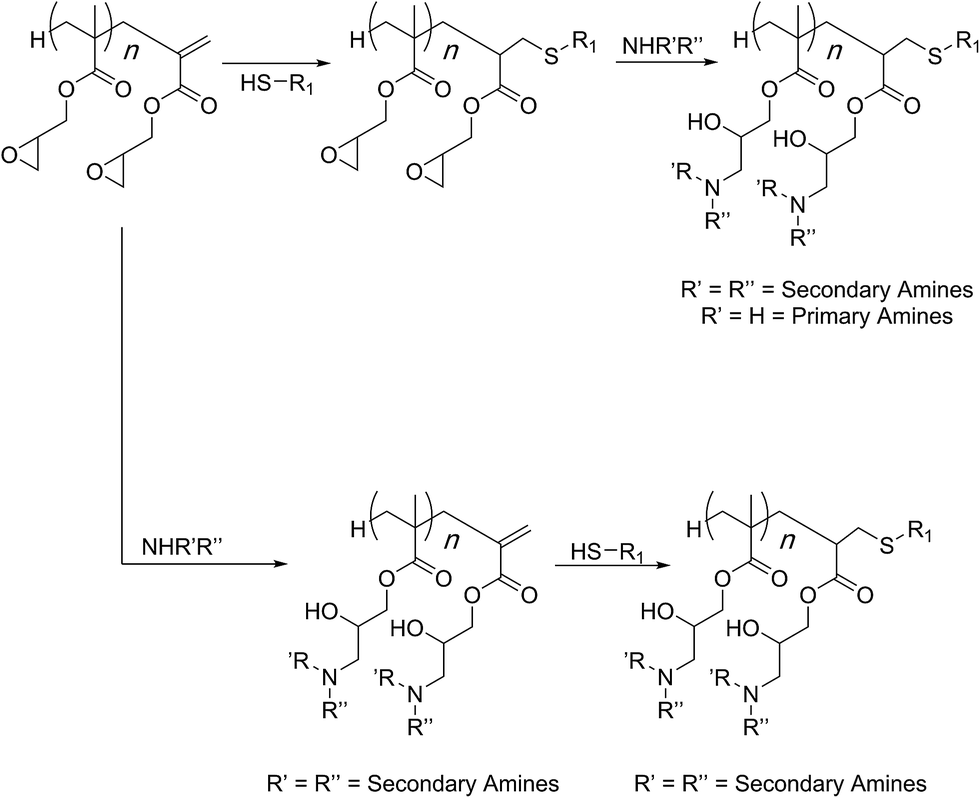 | ||
| Scheme 7 Orthogonal reactivity of functional groups in the PGMA scaffold functionalisations as established by Haddleton and co-workers. | ||
Functionalisation with thiols
The first study that established the use of thiol nucleophiles72–80 for post-polymerisation modifications of the GMA polymers was published in 2012 (Scheme 8).81 It utilised conventional free radical polymerisation process for the synthesis of the reactive scaffold. Therefore, molecular weights were very high and the degree of polymerization was estimated to be about 12000 based on the number average molecular weight (Pn). The study showed that even at room temperature, such high molecular weight polymers could be completely functionalised with a number of thiol nucleophiles. Furthermore, the generated secondary hydroxyl groups could be modified through an esterification reaction with a variety of acid chlorides.
 | ||
| Scheme 8 First application of the thiol-based ring-opening reaction for modification of the PGMA scaffold. In this study, bi-functional structures were also synthesised through esterification of the generated secondary hydroxyl groups. The top shows the general synthetic scheme and the bottom shows various singly and doubly functionalised structures. | ||
In a follow up study, copolymerization of GMA monomer with polyethylene glycol-based macro-initiator or a macro-monomer through ATRP was shown to give water soluble reactive scaffolds that can be transformed into bi-functionalised structures (Scheme 9).82 A later study investigated the optimised reaction conditions and quantified the results of the bifunctionalisation strategy using thiol–epoxy and esterification reactions (Scheme 10).42 This study suggested that the choice and amount of a catalyst heavily influenced the outcome of the thiol–epoxy reaction. Tetrabutylammonium fluoride (TBAF) in tetrahydrofuran (THF), and lithium hydroxide in aqueous THF (10% water) were found to be good catalyst choices for reactions involving aliphatic and aromatic thiols. In the case of TBAF, >20 mol% catalyst loading was required to achieve complete conversion of the epoxy groups in a reaction time of 3 h. Lithium hydroxide proved to be a better catalyst as full conversions could be obtained at a catalyst loading of 1–4 mol% and in a reaction time of 1–3 hours. Triethylamine (TEA), when used in THF, failed to provide any ring-opening reaction. However, it could be used in the case of aromatic thiols. Nonetheless, high catalyst loading (>34%) and long reaction times (∼12 h) were required for quantitative conversions at room temperature. Interestingly, a change of reaction medium to dimethylsulfoxide (DMSO) could lead to the ring-opening reaction while using TEA as a base and an aliphatic thiol as a reactant. Similarly in this case, longer reaction times, high catalyst loading, higher temperature, and higher thiol content were required to observe >85% epoxy group conversion.
 | ||
| Scheme 9 Application of the thiol–epoxy and esterification reactions-based sequential modifications strategy to prepare poly(ethylene glycol)-based doubly modified water soluble copolymers. | ||
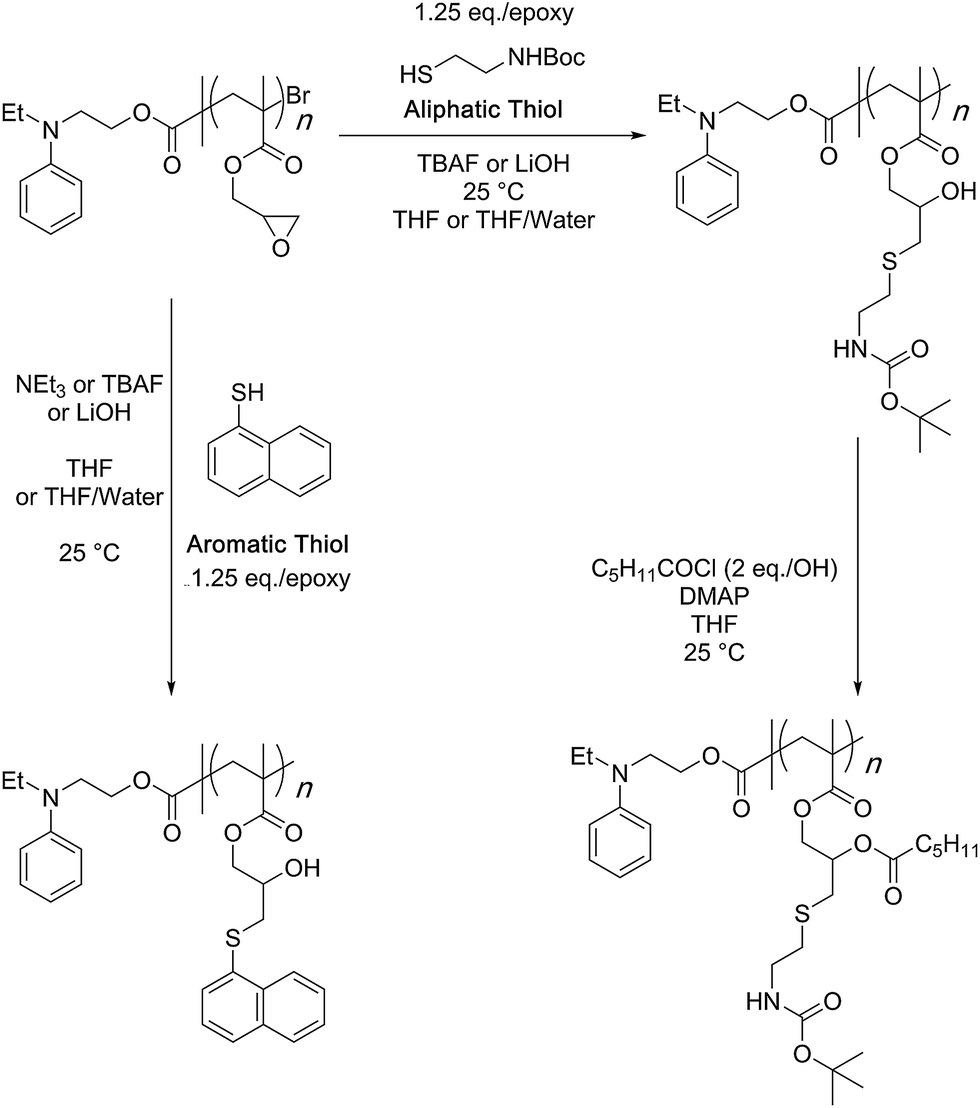 | ||
| Scheme 10 Examination of the optimum reaction conditions and quantification of the PGMA bifunctionalisation strategy was done using the shown synthetic scheme. | ||
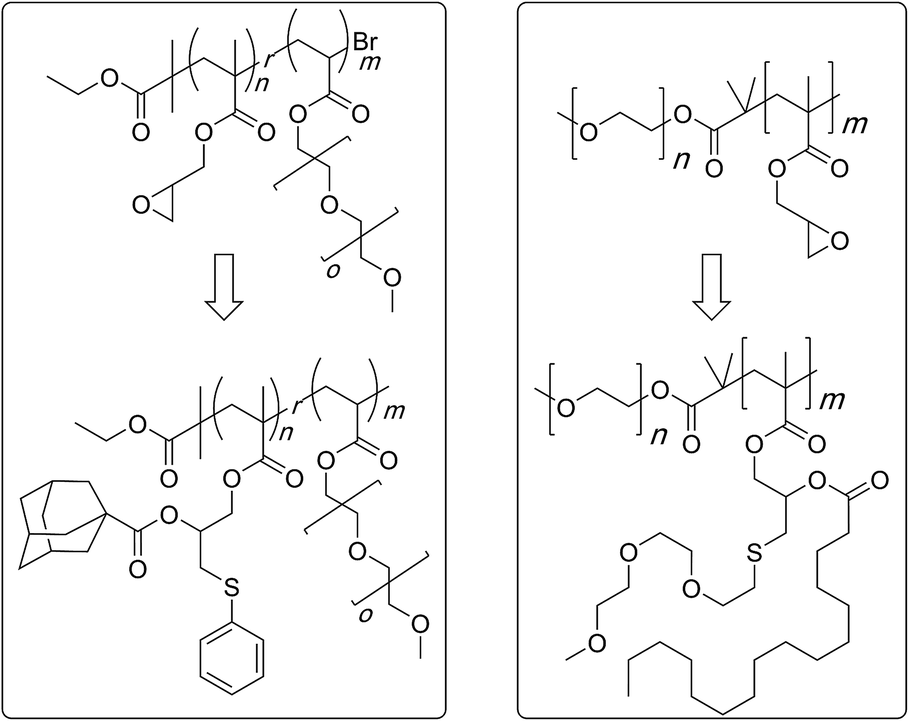
In a related study, sterically demanding and thiol-terminated poly(ethylene glycol) (PEG) polymers (0.18, 0.8, and 2 kDa) were attached to the PGMA backbone (25 and 46 kDa) (Scheme 11).83 This polymer–polymer coupling reaction resulted in the formation of water-soluble bottlebrush copolymers with molecular weights ranging from 50–426 kDa. When low molecular weight PEG polymers with relatively low steric demand were used as the side-chain precursor, high grafting densities were observed irrespective of the length of the PGMA backbone (96–97%). The grafting density, however, decreased with an increase in the length and steric demand of the PEG side-chain (88–95%). In all cases discussed above, a second modification through an esterification reaction was shown to give doubly-functionalised structures.84–87 Finally, this strategy was applied in the preparation of an amphipathic homopolymer library and shown to have siRNA delivery capacity (Fig. 1).88 Benagali and co-workers showed that the secondary hydroxyl group could also be modified through urethane bond formation with small molecular isocyanates.89
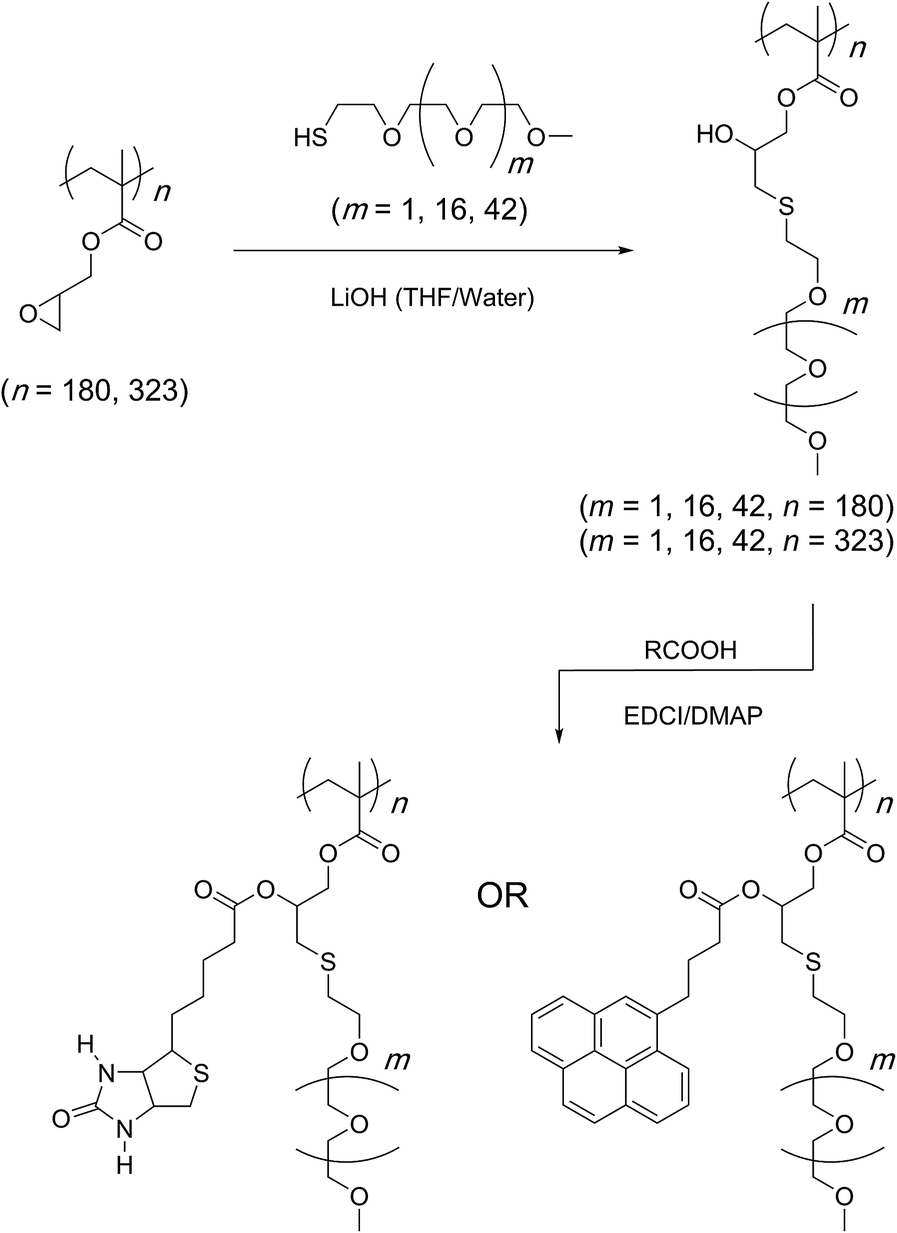 | ||
| Scheme 11 Preparation of bivalent molecular bottlebrushes through the use of a sequential thiol–epoxy and esterification reactions. | ||
 | ||
| Fig. 1 Amphipathic homopolymer family synthesized for siRNA delivery applications. | ||
Stepping away from linear PGMA structures, Gao and co-worker showed that segmented hyperbranched polymers with high density of functional groups could be prepared by coupling with a number of thiol molecules. In some cases, the thiol molecule carried another functionality such as an olefin bond. Therefore, a second functionalisation reaction using the thiol–ene ‘click’ reaction could be carried out to install a different functionality onto the hyperbranched scaffold in a sequential manner.90
In another impressive example of creating complex and sophisticated segmented yet linear structures, Haddleton and co-workers utilized the orthogonal reactivity of the epoxy group and the alkyne functionality to gain access to multiply-reactive and multiply-functional sequence regulated polymers (Scheme 12).91
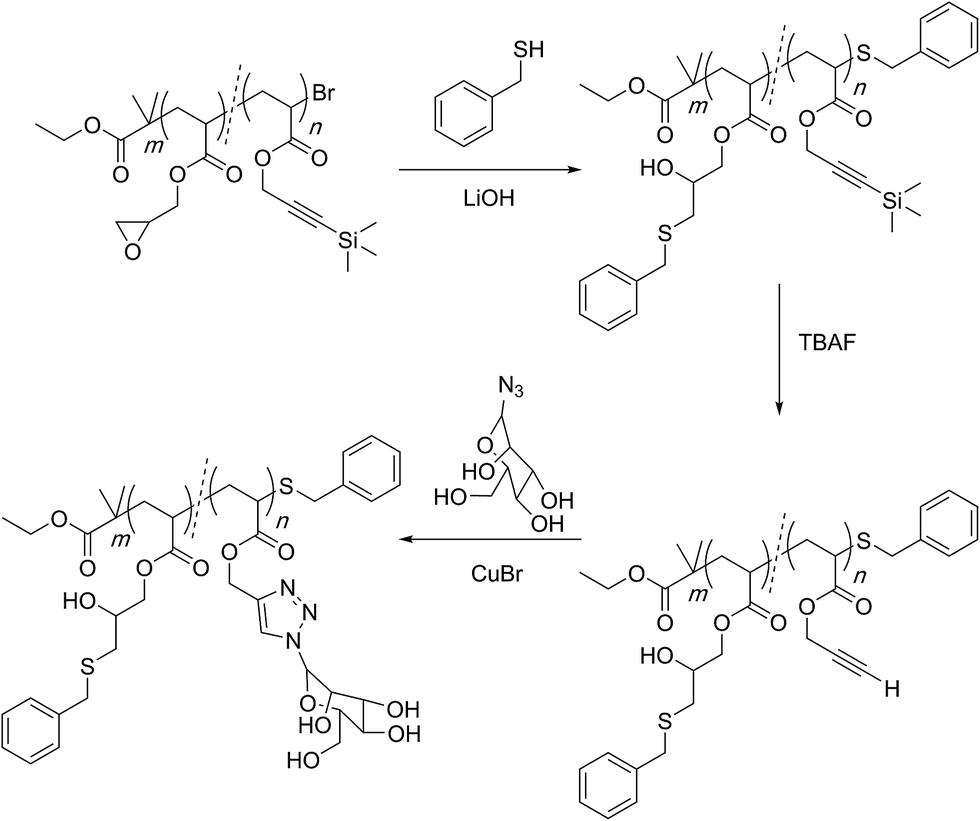 | ||
| Scheme 12 Multi-block orthogonal reactivity concept established by Haddleton and co-workers for the preparation of sequence-regulated multifunctional polymers. | ||
Functionalization with azide
Matyjaszewski and co-workers have established this concept through use of copolymers carrying GMA and methyl methacrylate monomers (Scheme 13).92 The epoxide groups were opened with sodium azide at 50 °C and in the presence of ammonium chloride. As mentioned earlier, ammonium chloride is a proton source to quench the alkoxide anion and stop an anionic ring-opening polymerisation from proceeding. NMR and IR spectroscopies were employed for the characterisation of the produced materials. It was shown that the ring-opening reaction was quantitative. The prepared polymer was further subjected to a copper-catalysed click reaction with polyethylene glycol (2000 g per mol) terminated with an acetylene functionality to finally afford graft copolymers.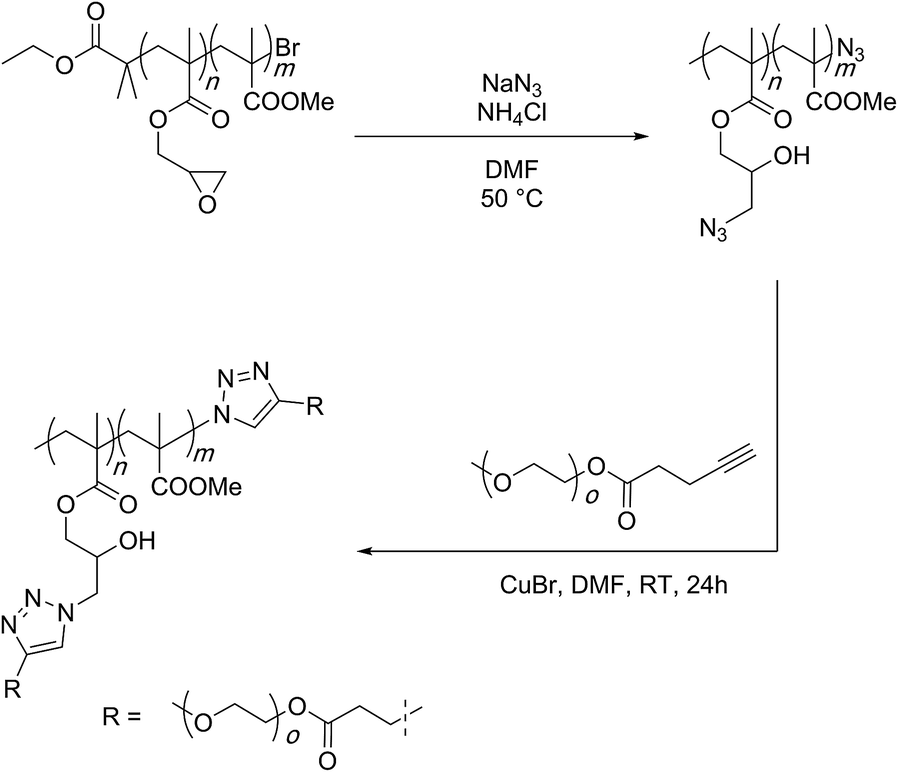 | ||
| Scheme 13 Epoxy-azide reaction for the functionalisation of the PGMA scaffold as established by Matyazweski and co-workers. | ||
In an alternative fashion, Zhao and co-workers showed that a small molecular acetylene appended with a RAFT agent can be attached to the azide group of the polymer and a subsequent RAFT polymerisation of NIPAM monomer could then lead to the graft copolymer structures (Scheme 14).93 The formed secondary hydroxyl group could be subjected to an esterification reaction with a pyrene-based acid molecule. In aqueous solution, these polymers assembled into vesicular structures in which the hydrophilic NIPAM chains protected the hydrophobic pyrene groups. Replacing pyrene with a polyethylene glycol chain and carrying out the RAFT polymerization with styrene and NIPAM monomers in a sequential manner afforded even more complex amphiphilic structures also capable of assembling into vesicles in water.94
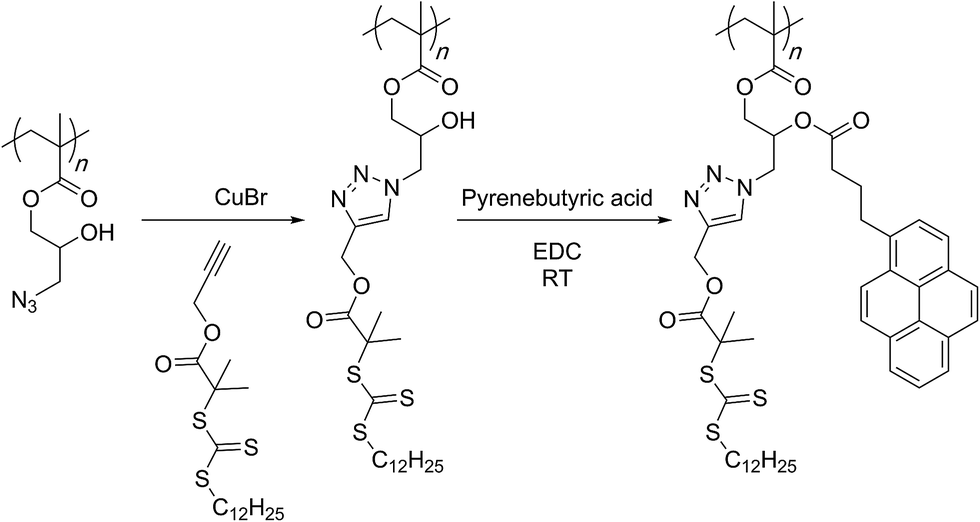 | ||
| Scheme 14 Grafting-from approach through use of RAFT polymerization for the preparation of bivalent graft copolymers by Zhao and co-workers. | ||
This theme of preparing molecular bottlebrushes through the azidation strategy of PGMA is further seen in the works of Chen and co-workers who prepared a large family of graft copolymers based upon the ethylene-oxide, ester, acrylate, methacrylate, acrylamide and styrenic polymers (Fig. 2).95,96
 | ||
| Fig. 2 Chemical structure of the glycopolymer synthesised through use of azide-epoxy reaction on the PGMA scaffold. | ||
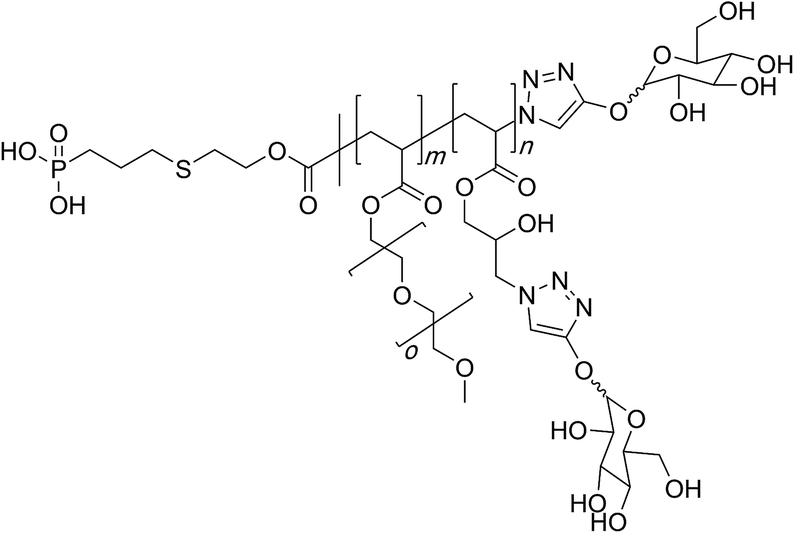
In a completely different manner, Gao and co-workers employed reversible addition–fragmentation chain transfer to synthesise PGMA-based highly branched architectures. The authors then utilised the azide-epoxy reaction to generate polymeric azides that could be modified in a subsequent copper-catalysed ‘click’ chemistry step with small molecular alkynes to access a myriad of functionalised polymers. In some cases, the hydroxyl group was also modified either prior or after the azide–alkyne reaction.97
Finally, glycopolymers could be prepared through azide–alkyne coupling reaction between the polymer and free-acetylene carrying sugar molecules for use in synthesis of bio-imaging probes (Fig. 3).98
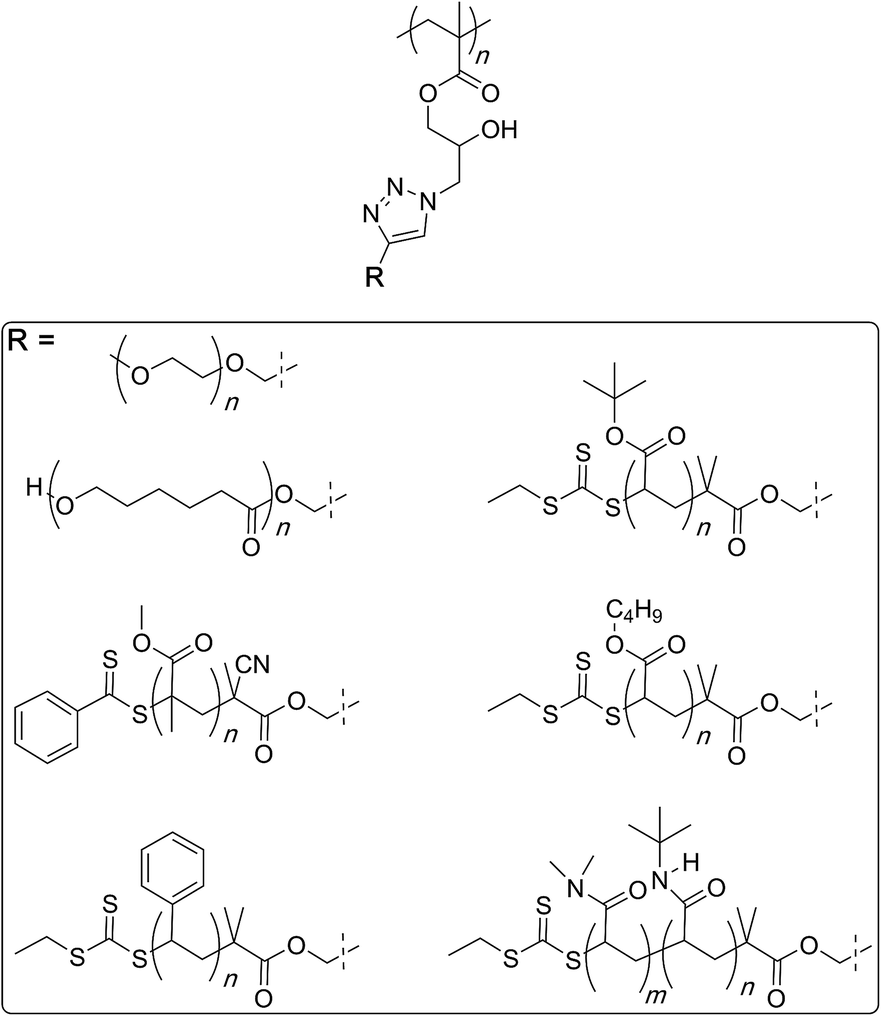 | ||
| Fig. 3 Synthesis of a family of molecular bottlebrushes through the polymer azide route by Chen and co-workers. | ||
Functionalisation with water
Hydrolysis of PGMA can be achieved using acid or base catalysis. Some of the early examples however rely only on thermal activation at temperatures of 120–140 °C for long periods of time (24 hours) (Scheme 15).99,100 The high temperatures can be avoided by using sulfuric acid as a catalyst that allows for the hydrolysis to occur smoothly at ambient conditions.101,103 In terms of regio-chemistry, irrespective of the method, the final hydrolysis product is the same diol structure that can further be functionalized. For this, esterification of the hydroxyl groups with fatty acid is shown to allow the polymer chain to form reverse micelles that could trap and release peptides and proteins. | ||
| Scheme 15 Hydrolysis of PGMA-based 4-arm star polymers by Leroux and co-workers. Only one arm is shown for simplicity. | ||
Gao and co-workers developed a different strategy for utilizing the diol functionalities. Exploiting higher reactivity of the primary hydroxyl group, they attached acid groups through ring-opening reaction with succinic anhydride (Scheme 16).102 The secondary hydroxyl groups were then used to form intermolecular polyurethane crosslinks to furnish polymer nanoparticles that could trap and release the anticancer drug doxorubicin.103
 | ||
| Scheme 16 Second functionalisation of hydrolyzed PGMA through ring-opening reaction with succinic anhydride by Gao and co-workers. | ||
Functionalization with carboxylic acids
Minko and Luzinov described the use of PGMA as a means to prepare fluorescent core–shell type of nanoparticles while still having the reactive epoxy groups on the particle surface (Scheme 17).104 To achieve this, PGMA was first functionalised with rhodamine-B through an acid–epoxy reaction and later deposited on silica particles. This functionalisation required a relatively high temperature of 80 °C. Through elemental analysis, it was confirmed that the extent of reaction was such that one epoxy group out of 311 reacted to yield functionalised polymer chains (0.3% efficiency). This synthetic strategy was used by Liu and co-workers to prepare polymers capable of emissive or self-healing properties.105,106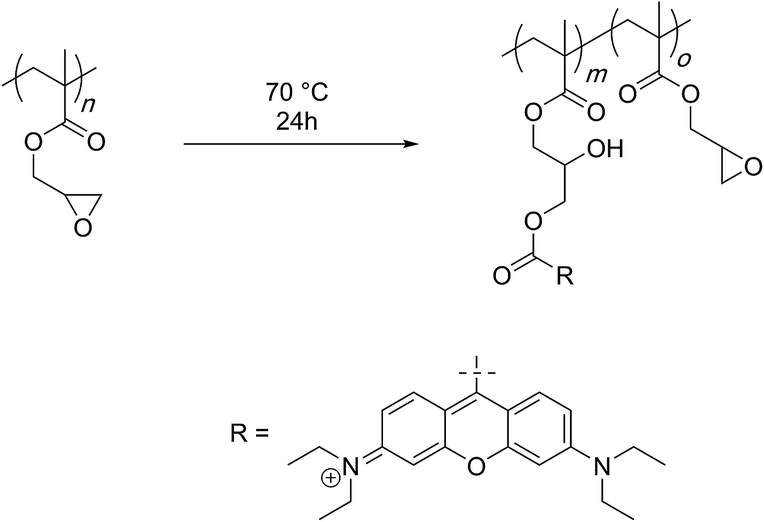 | ||
| Scheme 17 Oxirane ring-opening reaction with rhodamine-B by Minko and Luzinov. | ||
Conclusions
Following are the conclusions that can be drawn from the aforementioned discussion.1. PGMA scaffold can be modified through the use of amines, thiols, azides, carboxylic acids, and water as nucleophiles. In all cases, the epoxide ring-opening reaction generates a second reactive site, a secondary hydroxyl group. This hydroxyl group can also be modified in a second post-polymerisation modification step. In case of hydrolysis, a diol forms and can be modified either selectively at the primary hydroxyl group due to its higher reactivity or at both hydroxyl groups. Alternatively, the initially installed functionality may be able to react further as in the case of amines. In many cases, multiply functionalised structures have been obtained through sequential post-polymerization modification reactions.
2. While using amines for modification purposes, at least a 10-fold excess of the amine reactant must be used and the reaction temperature must be lowered to 35 °C or room temperature. These two factors can avoid inter and intramolecular crosslinking reactions from occurring. These crosslinking reactions originate from the desire of the primary amine to become secondary, secondary to tertiary, and tertiary to quaternary amine. This is due to the fact that amine reactivity increases as a result of electron-donating nature of the alkyl groups to the nitrogen atom making each product more nucleophilic than its predecessor.
3. Amine functionalisations require long reaction times (24 hours) for completion at room temperature.
4. Functionalisation with thiols is regio-selective, quantitative, and fast. The functionalisation reactions can be performed at room temperature and requires 1–3 hours for completion even if very high molecular weights (Pn = 12000) of the PGMA scaffold is used. Sterically demanding thiols can also be used. However, the degree of grafting decreases with increasing molecular weight (>2 kDa) of the thiol used.
5. Reaction with sodium azide (50 °C) requires a mild acid such as ammonium chloride as the proton source for quenching the alkoxide anion. The reaction generates polymeric azides in high efficiency. A subsequent modification of the azide groups through copper-catalysed ‘click’ reaction then gives access to functionalised polymers. The utility and efficiency of this second post-polymerisation modification is shown through preparation of a number of graft copolymers with high grafting densities.
6. Carboxylic acids represent the least active and least efficient system towards functionalisation of the PGMA scaffold even when relatively high temperatures (80 °C) and relatively long reaction times (12 h) are employed.
In general, the PGMA scaffold can be transformed into various functionalised structures through the opening of side-chain epoxide rings with a number of nucleophiles. This particular synthetic strategy to functional polymers bears the hallmark of simplicity and adaptability. This is evident from a commercial access to the monomer, polymerisability with robust and tolerant free radical polymerisation protocols and functionalizations with a number of nucleophiles in the presence of air and moisture and sometimes, under ambient conditions. This strategy has been put to use by many researchers and the final materials have often found bio-relevant applications. In most cases, a single nucleophile is used for the first functionalisation of the PGMA scaffold.
If we examine the modification reactions in terms of the reaction speed, efficiency of functionalisation and reaction conditions, it becomes clear that distinct differences in reactivity exists and relate to the nature of the nucleophile. This presents an opportunity to modify one scaffold in a one-pot or sequential manner with different nucleophiles to generate multiply functionalised polymers. Overall, it seems that the single-nucleophile modifications will still dominate the research scene but new materials based on the (orthogonal) reactivity concepts are likely to add new dimensions to this historic chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
EMM and MCS are thankful for financial support from NTU Singapore (M4081566). This work was by National Research Foundation of Korea grant funded by the Korea government (MSIP) (NRF-2015R1D1A1A01057796).Notes and references
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- P. F. Cañamero, J. L. de la Fuente, E. L. Madruga and M. Fernández-García, Macromol. Chem. Phys., 2004, 205, 2221–2228 CrossRef.
- H. Yin, H. Zheng, L. Lu, P. Liu and Y. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5091–5102 CrossRef CAS.
- K. Sha, D. Li, Y. Li, X. Liu, S. Wang, J. Guan and J. Wang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5037–5049 CrossRef CAS.
- A. Hayek, Y. Xu, T. Okada, S. Barlow, X. Zhu, J. H. Moon, S. R. Marder and S. Yang, J. Mater. Chem., 2008, 18, 3316 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- B. Zhang, Y. Li, P. Ai, Z. Sa, Y. Zhao, M. Li, D. Wang and K. Sha, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5509–5526 CrossRef CAS.
- R. París, M. Liras and I. Quijada-Garrido, Macromol. Chem. Phys., 2011, 212, 1859–1868 Search PubMed.
- A. Moayeri, B. Lessard and M. Maric, Polym. Chem., 2011, 2, 2084 RSC.
- S. Höhne and P. Uhlmann, J. Polym. Sci., Part A: Polym. Chem., 2014, 53, 675–684 CrossRef.
- Q. Yang, S. Balverde, F. Dumur, J. Lalevee and J. Poly, Polym. Chem., 2016, 7, 6084 RSC.
- X. Yan, A. Sivignon, N. Barnich, S. G. Gouin, J. Bouckaert, E. Fleury and J. Bernard, Polym. Chem., 2016, 7, 2674 RSC.
- X. Zhang, Q. Zhang, C. Xie, A. Gao, Z. Chang, J. Kwon Oh, P. Yang and P. Li, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1396–1408 CrossRef CAS.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856 RSC.
- C. A. Follit, S. R. Woodruff, P. D. Vogel, J. G. Wise and N. V. Tsarevsky, J. Drug Delivery Sci. Technol., 2017, 39, 324–333 CrossRef CAS.
- F. J. Xu, Q. J. Cai, Y. L. Li, E. T. Kang and K. G. Neoh, Biomacromolecules, 2005, 6, 1012–1020 CrossRef CAS PubMed.
- R. Iwata, R. Satoh, Y. Iwasaki and K. Akiyoshi, Colloids Surf., B, 2008, 62, 288–298 CrossRef CAS PubMed.
- F. J. Xu, Z. H. Wang and W. T. Yang, Biomaterials, 2010, 31, 3139–3147 CrossRef CAS PubMed.
- W. Yuan, C. Li, C. Zhao, C. Sui, W.-T. Yang, F.-J. Xu and J. Ma, Adv. Funct. Mater., 2012, 22, 1835–1842 CrossRef CAS.
- D. Damiron, J. Mazzolini, F. Cousin, C. Boisson, F. D'Agosto and E. Drockenmuller, Polym. Chem., 2012, 3, 1838–1845 RSC.
- M. Lillethorup, K. Shimizu, N. Plumeré, S. U. Pedersen and K. Daasbjerg, Macromolecules, 2014, 47, 5081–5088 CrossRef CAS.
- I. Álvarez-Martos, R. Alonso-Bartolomé, V. M. Hernández, A. Anillo, A. Costa-García, F. J. G. Alonso and M. T. Fernández-Abedul, React. Funct. Polym., 2016, 100, 89–96 CrossRef.
- H. Zhu, Q. Liu and Y. Chen, Langmuir, 2007, 23, 790–794 CrossRef CAS PubMed.
- W. L. Zhang, S. H. Piao and H. J. Choi, J. Colloid Interface Sci., 2013, 402, 100–106 CrossRef CAS PubMed.
- Y. Zhu, L. Fan, B. Yang and J. Du, ACS Nano, 2014, 8, 5022–5031 CrossRef CAS PubMed.
- S. B. Rahane, R. M. Hensarling, B. J. Sparks, C. M. Stafford and D. L. Patton, J. Mater. Chem., 2012, 22, 932–943 RSC.
- Y. Sun, H. Hu, B. Yu and F.-J. Xu, Bioconjugate Chem., 2016, 27, 2744–2754 CrossRef CAS PubMed.
- S. Shibasaki, Y. Sasaki, K. Nakabayashi and H. Mori, React. Funct. Polym., 2016, 107, 11–19 CrossRef CAS.
- R. B. Grubbs, J. M. Dean, M. E. Broz and F. S. Bates, Macromolecules, 2000, 33, 9522–9534 CrossRef CAS.
- G. Kocak, G. Solmaz and V. Bütün, Macromol. Chem. Phys., 2016, 217, 2744–2754 CrossRef CAS.
- I. Perez-Baena, F. Barroso-Bujans, U. Gasser, A. Arbe, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 775–779 CrossRef CAS.
- M. Mauri, N. Tran, O. Prieto, T. Hjertberg and C. Müller, Polymer, 2017, 111, 27–35 CrossRef CAS.
- J. Steinkoenig, H. Rothfuss, A. Lauer, B. T. Tuten and C. Barner-Kowollik, J. Am. Chem. Soc., 2017, 139, 51–54 CrossRef CAS PubMed.
- A. E. V. D. Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef PubMed.
- C.-F. Wang, Y.-X. Lin, T. Jiang, F. He and R.-X. Zhuo, Biomaterials, 2009, 30, 4824–4832 CrossRef CAS PubMed.
- D. M. Stevens, S. Tempelaar, A. P. Dove and E. Harth, ACS Macro Lett., 2012, 1, 915–918 CrossRef CAS PubMed.
- Y. Yan and D. J. Siegwart, Polym. Chem., 2014, 5, 1362–1371 RSC.
- P. K. Kuroishi, M. J. Bennison and A. P. Dove, Polym. Chem., 2016, 7, 7108–7115 RSC.
- T. Krappitz, P. Feibusch, C. Aroonsirichock, V. P. Hoven and P. Theato, Macromolecules, 2017, 50, 1415–1421 CrossRef CAS.
- Q.-L. Li, W.-X. Gu, H. Gao and Y.-W. Yang, Chem. Commun., 2014, 50, 13201–13215 RSC.
- M. C. Stuparu, A. Khan and I. Gadwal, Polym. Chem., 2015, 6, 1393–1404 RSC.
- P. Theato and H.-A. Klok, Functional Polymers by Post-Polymerization Modification, John Wiley & Sons, Weinheim, Germany, 2013 Search PubMed.
- K. A. Günay, P. Theato and H. A. Klok, History of post-polymerization modification, in Functional Polymers by Post-Polymerization Modification: Concepts, Guidelines, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 1st edn. 2013 Search PubMed.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2012, 51, 1–28 CrossRef.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2008, 48, 48–58 CrossRef PubMed.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- H. Durmaz, A. Sanyal, G. Hizal and U. Tunca, Polym. Chem., 2012, 3, 825–835 RSC.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 4103–4120 CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66–80 RSC.
- H. Gao, M. Elsabahy, E. V. Giger, D. Li, R. E. Prud’homme and J.-C. Leroux, Biomacromolecules, 2010, 11, 889–895 CrossRef CAS PubMed.
- S. Edmondson and W. T. S. Huck, J. Mater. Chem., 2004, 14, 730 RSC.
- M. Ma, F. Li, F.-J. Chen, S.-X. Cheng and R.-X. Zhuo, Macromol. Biosci., 2010, 10, 183–191 CrossRef CAS PubMed.
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS PubMed.
- H. Gao, X. Lu, Y. Ma, Y. Yang, J. Li, G. Wu, Y. Wang, Y. Fan and J. Ma, Soft Matter, 2011, 7, 9239 RSC.
- F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7, 3131–3140 CrossRef CAS PubMed.
- Y. Hu, Y. Zhu, W. T. Yang and F. J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 703–712 CAS.
- Y. Hu, M. Y. Chai, W. T. Yang and F. J. Xu, Bioconjugate Chem., 2013, 24, 1049–1056 CrossRef CAS PubMed.
- Y. Tang, L. Liu, J. Wu and J. Duan, J. Colloid Interface Sci., 2013, 397, 24–31 CrossRef CAS PubMed.
- X. C. Yang, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, Bioconjugate Chem., 2012, 23, 618–626 CrossRef CAS PubMed.
- R. Q. Li, Y. L. Niu, N. N. Zhao, B. R. Yu, C. Mao and F. J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 3969–3978 CAS.
- L. Wang, Y.-W. Yang, M. Zhu, G. Qiu, G. Wu and H. Gao, RSC Adv., 2014, 4, 6478 RSC.
- R. Q. Li, Y. Hu, B. R. Yu, N. N. Zhao and F. J. Xu, Bioconjugate Chem., 2014, 25, 155–164 CrossRef CAS PubMed.
- X.-C. Yang, Y.-L. Niu, N.-N. Zhao, C. Mao and F.-J. Xu, Biomaterials, 2014, 35, 3873–3884 CrossRef CAS PubMed.
- X. B. Dou, M. Y. Chai, Y. Zhu, W. T. Yang and F. J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 3212–3218 CAS.
- Z. Liang, X. Wu, Y.-W. Yang, C. Li, G. Wu and H. Gao, Polym. Chem., 2013, 4, 3514 RSC.
- K. A. McEwan, S. Slavin, E. Tunnah and D. M. Haddleton, Polym. Chem., 2013, 4, 2608 RSC.
- C. Hoffmann, M. C. Stuparu, A. Daugaard and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2014, 53, 745–749 CrossRef.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- A. Brändle and A. Khan, Polym. Chem., 2012, 3, 3224 RSC.
- I. Gadwal and A. Khan, Polym. Chem., 2013, 4, 2440 RSC.
- N. Cengiz, J. Rao, A. Sanyal and A. Khan, Chem. Commun., 2013, 49, 11191 RSC.
- I. Gadwal, S. Binder, M. C. Stuparu and A. Khan, Macromolecules, 2014, 47, 5070–5080 CrossRef CAS.
- S. Binder, I. Gadwal, A. Bielmann and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2040–2046 CrossRef CAS.
- I. Gadwal and A. Khan, RSC Adv., 2015, 5, 43961–43964 RSC.
- I. Gadwal, T. Eom, J. Hwang, Y. Choe, J. Bang and A. Khan, RSC Adv., 2017, 7, 19439–19447 RSC.
- J. Hwang, Y. Choe, J. Bang and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3381–3386 CrossRef CAS.
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130 RSC.
- S. De, C. Stelzer and A. Khan, Polym. Chem., 2012, 3, 2342 RSC.
- I. Gadwal, J. Rao, J. Baettig and A. Khan, Macromolecules, 2014, 47, 35–40 CrossRef CAS.
- F. S. Gungor and B. Kiskan, React. Funct. Polym., 2014, 75, 51–55 CrossRef CAS.
- B. Hanbeyoglu, B. Kiskan and Y. Yagci, Macromolecules, 2013, 46, 8434–8440 CrossRef CAS.
- A. Musa, B. Kiskan and Y. Yagci, Polymer, 2014, 55, 5550–5556 CrossRef CAS.
- A. Saha, S. De, M. C. Stuparu and A. Khan, J. Am. Chem. Soc., 2012, 134, 17291–17297 CrossRef CAS PubMed.
- C. Buerkli, S. H. Lee, E. Moroz, M. C. Stuparu, J.-C. Leroux and A. Khan, Biomacromolecules, 2014, 15, 1707–1715 CrossRef CAS PubMed.
- M. Benaglia, A. Alberti, L. Giorgini, F. Magnoni and S. Tozzi, Polym. Chem., 2013, 4, 124–132 RSC.
- S. Li, J. Han and C. Gao, Polym. Chem., 2013, 4, 1774–1787 RSC.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilson and D. M. Haddleton, Polym. Chem., 2014, 5, 3876 RSC.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
- C. Zhao, D. Wu, X. Lian, Y. Zhang, X. Song and H. Zhao, J. Phys. Chem. B, 2010, 114, 6300–6308 CrossRef CAS PubMed.
- X. Lian, D. Wu, X. Song and H. Zhao, Macromolecules, 2010, 43, 7434–7445 CrossRef CAS.
- P. Zhao, Y. Yan, X. Feng, L. Liu, C. Wang and Y. Chen, Polymer, 2012, 53, 1992–2000 CrossRef CAS.
- Y. Yan, Y. Shi, W. Zhu and Y. Chen, Polymer, 2013, 54, 5634–5642 CrossRef CAS.
- S. Li and C. Gao, Polym. Chem., 2013, 4, 4450 RSC.
- J. S. Basuki, L. Esser, H. T. T. Duong, Q. Zhang, P. Wilson, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Chem. Sci., 2014, 5, 715–726 RSC.
- M.-C. Jones, P. Tewari, C. Blei, K. Hales, D. J. Pochan and J.-C. Leroux, J. Am. Chem. Soc., 2006, 128, 14599–14605 CrossRef CAS PubMed.
- H. Gao, M.-C. Jones, P. Tewari, M. Ranger and J.-C. Leroux, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2425–2435 CrossRef CAS.
- C. S. Gudipati, M. B. H. Tan, H. Hussain, Y. Liu, C. He and T. P. Davis, Macromol. Rapid Commun., 2008, 29, 1902–1907 CrossRef CAS.
- Y. Ma, H. Gao, W. Gu, Y.-W. Yang, Y. Wang, Y. Fan, G. Wu and J. Ma, Eur. J. Pharm. Sci., 2012, 45, 65–72 CrossRef CAS PubMed.
- W. Gu, Y. Ma, C. Zhu, B. Chen, J. Ma and H. Gao, Eur. J. Pharm. Sci., 2012, 47, 556–563 CrossRef CAS PubMed.
- V. Tsyalkovsky, V. Klep, K. Ramaratnam, R. Lupitskyy, S. Minko and I. Luzinov, Chem. Mater., 2008, 20, 317–325 CrossRef CAS.
- S.-C. Chang, S.-J. Chiu, C.-Y. Hsu, Y. Chang and Y.-L. Liu, Polymer, 2012, 53, 4399–4406 CrossRef CAS.
- T.-W. Chuo, T.-C. Wei and Y.-L. Liu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3395–3403 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |