
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual modal imaging agents based on chromophore-bearing DTPA analogues†
CuhaWijay Sathiyajith ,
Andrew J. Hallett,
Angelo J. Amoroso* and
Peter G. Edwards*
,
Andrew J. Hallett,
Angelo J. Amoroso* and
Peter G. Edwards*
School of Chemistry, Cardiff University, Main Building, Park Place, CF10 3AT, Cardiff, UK. E-mail: AmorosoAJ@cardiff.ac.uk
First published on 4th August 2017
Abstract
Two new DTPA analogues, centrally (L1) and terminally (L2) functionalised with a 1,8-naphthalimide chromophore, have been successfully prepared and fully characterized. Their Gd(III) complexes have also been prepared and evaluated for their ability to act as dual modal contrast agents (MRI/OI). The highly reproducible R1 relaxivity of L1 (8.10 ± 0.21 mM−1 s−1, 25 °C and 30 MHz) is markedly higher than other DTPA based contrast agents. The Gd(III) complexes of both L1 and L2 have been evaluated as luminescence probes; the ligand based fluorescence is not quenched upon complexation.
Introduction
Diethylenetriaminepentaacetic acid (DTPA) is one of the most well-known ligands and yields highly stable complexes with hard metal ions. DTPA and its derivatives have been used in a vast array of applications of medical interest, such as contrast agents for medical imaging,1–5 radiotherapy, radiolabeling of antibodies and peptides,6–9 radiopharmaceuticals and chelation therapy.10–12 In particular, the gadolinium complex of DTPA has long been successful as a contrast agent, ever since it was approved by the FDA in 1988.13However, the perceived disadvantage of Gd-DTPA is that the chelate has been prepared in hypertonic solutions, where issues related to high osmolarity become significant. The investigations have been intensified on the synthesis and characteristics of novel Gd(III) complexes to minimize toxicity issues while ensuring relaxation rate enhancement. The biocompatibility of the multidentate DTPA derivatives makes it an ideal choice. DTPA also offers the advantage of incorporating a well-defined structural molecule, to a chosen biological molecule (which could be proteins in the blood, antibodies or similar biological macromolecules) due to their multidentate feature.
Several strategies have been utilised in the synthesis of substituted DTPA frameworks. These approaches include (i) the attachment of substituents via an amide linkage to a carboxylate group of the preformed DTPA ligand or (ii) the direct bonding of the substituent to the diethyltriamine core.
The attachment of substituents to DTPA via an amide link is a useful strategy as it is a simple and convenient method that can give compounds in high yields and good purity. There are numerous reported methods to carry this reaction out.14–25 Some methods involve the use of coupling reagents to couple the amine containing substituent to the DTPA,26 though the simplest method involves the conversion of DTPA to the bisanhydride analogue, which may then be directly reacted with a suitable amine.27–29 While this is an extremely effective approach, the method has some drawbacks in that, despite reports of the synthesis on mono substituted analogues,30 such reactions are difficult and often an impure product is obtained which requires extensive efforts to purify. Secondly, the substituent is bound by an amide group which results in a lower thermodynamic stability and potentially increased toxicity.
For the direct attachment of a moiety to the diethyltriamine framework, typically, (i) a single moiety may be attached to the central nitrogen,31 (ii) two identical moieties may be attached to the terminal nitrogens32,33 or (iii) the ethylene spacers of the framework may be substituted.34,35
The dialkylation of a primary amine using di-tert-butyl 2,2′-((2-bromoethyl)azanediyl)diacetate (2) will result in the formation of a DTPA framework. Amino acids, such as L-cysteine,36 L-phenylalanine,37 L-glutamic acid and lysine,38 were previously utilised as primary amines in the construction of such DTPA analogues. In addition, there is a further report of an aliphatic alkyl amine31 being used as the central moiety of a constructed DTPA-like ligand.
Further, bi-substituted DTPA analogues have been constructed from diethylene triamine starting material using two equivalents of 2,2′-bipyridine-5-carboxaldehyde,32 while Artali et al.33 reported an analogous approach to incorporate the 2-hydroxymethyl pyridine group. However, it should be noted, that reports of mono substituted terminal nitrogens of a DTPA frame-work are scarce.39–42
Furthermore, reproducible relaxivity measurements reported for L1 in this report is on the higher side. Most of the polyamino polycarboxylate scaffolds used as commercial contrast agents have relaxivities in the range of 4–5 mM−1 s−1 (at 30 MHz, 25 °C). Acyclic DTPA analogues reported to date also have comparable relaxivities to those of commercially available contrast agents based on polyamino polycarboxylates (Fig. 1). Exceptionally higher relaxivities were reported by Raymond et al.43 for their HOPO based contrast agents (7–13 mM−1 s−1). Very recently, piperadine based rigidified DTPA analogues have been reported to exhibit relaxivity in the range of 5.2–6.0 mM−1 s−1 (at 20 MHz, 25 °C).
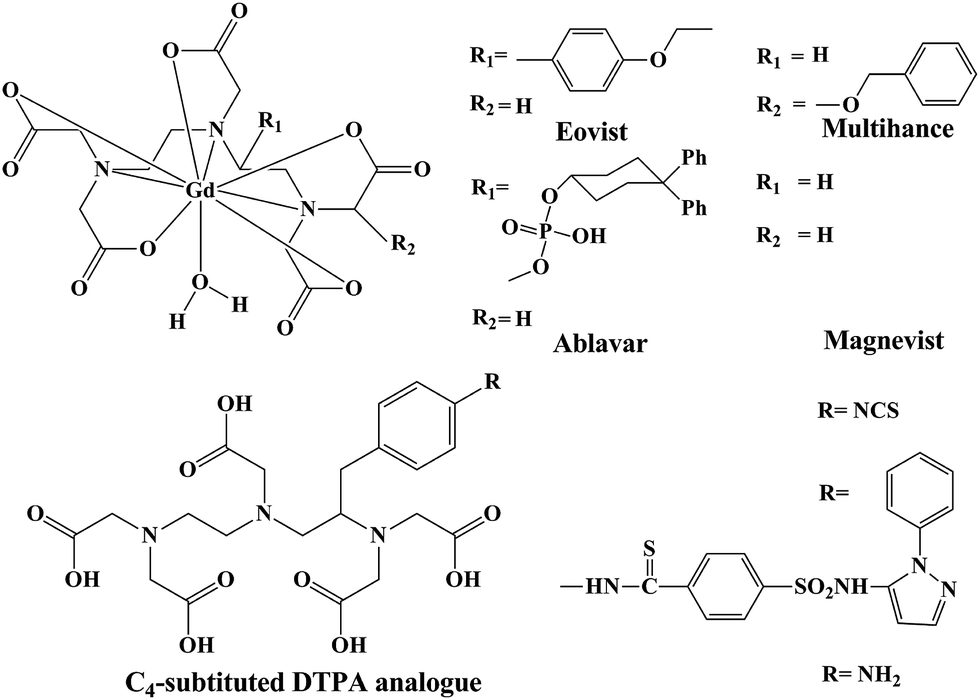 | ||
| Fig. 1 Schematic representation of commercial contrast agents and some selected C4-substitued DTPA analogues. | ||
DTPA based amide derivatives bearing chromophores: a novel approach
Herein we report the synthesis of two potential dual mode imaging agents. The synthesised ligand will contain a single organic luminescent moiety which has been directly alkylated to the nitrogen atom of a diethylene triamine. Two possibilities exist, with the lumophore being alkylated to the central nitrogen or to one of the two terminal nitrogens. The first approach utilises 2 for the dialkylation of the appropriate amine. This approach will produce a symmetric ligand (Fig. 2, type A). The appropriate choice of amine will yield a ligand with a lumophore bonded to the central amine. The second approach utilises a monosubstituted diethylene triamine species which may be alkylated to yield an asymmetrically monosubstituted DTPA ligand (type B). The lumophore is incorporated into these ligand frameworks as the naphthalimide moiety (Fig. 2).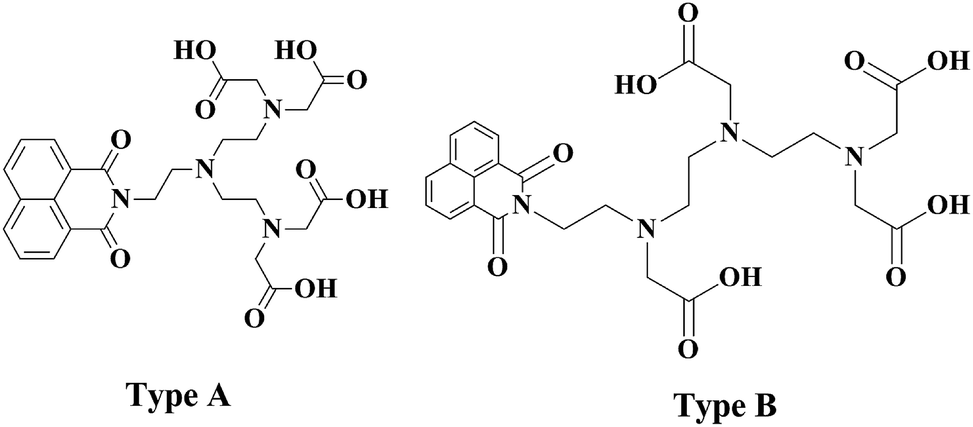 | ||
| Fig. 2 Depicting the novel chromophore bearing centrally substituted and terminally substituted DTPA analogues. | ||
Experimental
General experimental
Reagents were obtained from commercial sources and used as received unless otherwise noted. Solvents were dried and purified under N2 by using standard methods and were distilled immediately before use, specifically dimethlyformamide, was distilled using anhydrous calcium hydride. All compounds were prepared under N2 unless otherwise mentioned. The NMR spectra were obtained using a Bruker ARX 400 at 20 °C in CDCl3 unless otherwise noted. Mass spectra were performed on a micromass platform II system, operating in flow injection analysis mode, with the electrospray method. Infrared spectra were recorded with a JASCO FTIR-410 spectrometer, between 4000 and 500 cm−1 as KBr pellets. UV/vis spectra and measurements were recorded with a JASCO V-570 spectrophotometer.The synthesis of 2-(2-aminoethyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione (3) was carried out following the reported literature procedure.36b
Luminescence measurements
The photophysical data for luminescence were obtained on a JobinYvon-Horiba Fluorolog spectrometer fitted with a JYTBX picosecond photo detection module. Water insoluble DTPA analogues were dissolved in chloroform and as required water-soluble lanthanide complexes of DTPA analogues were dissolved in water. Although all of the new ligands and complexes prepared were air stable both in the solid state and solution, some of the ligands and complexes were found to be hygroscopic, when left in the open atmosphere for prolonged periods of time.1H NMRD acquisition
The 1/T1 NMRD profiles were obtained on a Stellar Spin master FFC-2000 relaxometer, typically covering a continuum of magnetic fields from 2.4 × 10−4 to 0.72 T (corresponding to a proton Larmor frequency range 0.01–30 MHz). A Spin master Variable Temperature Controller (VCT) allowed the setting and monitoring of the sample temperature at two different temperatures, 25 °C and 37 °C with a resolution up to 0.1 °C. Each sample was allowed to acclimatise to the desired temperature for 15 minutes prior to data collection. The reproducibility in T1 measurements was within ±1%.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl ether, 5:1). The product was obtained as a colourless oil. Yield 2.0 g, 28%; 1H NMR (400 MHz; CDCl3) δH (ppm) 3.45 (s, 4H, NCH2COOC (CH3)3), 3.41 (t, 2H, J = 7.4 Hz, BrCH2), 3.11 (t, 2H, J = 7.4 Hz, BrCH2CH2N), 1.44 (s, 18H, COOC (CH3)3).:DCM mixture). The pure product was obtained as a yellow oil. Yield 0.62 g, 54%; 1H NMR (400 MHz, CDCl3): δH (ppm) 8.50 (d, 2H, J = 7.27 Hz, ArH), 8.10 (d, 2H, J = 8.13 Hz, ArH), 7.70 (m, 2H, ArH), 4.30 (m, 4H), 3.40 (m, 8H), 2.85 (br s, 8H), 1.40 (s, 36H); 13C NMR (400 MHz; D2O): δC 170.7, 163.4, 133.2, 130.6, 127.2, 121.5, 80.9, 45.4, 52.4, 55.1, 27.1; IR (KBr disc) (cm1): 3440(br), 2977(s), 2931(w), 1736(s), 1702(w), 1661(vs), 1628(w), 1591(s); ESI-MS (+ion): found m/z 783.4578, calc. 783.4544 for [(4) H]+.
:diethyl ether, 5:1). The product was obtained as a colourless oil. Yield 2.0 g, 28%; 1H NMR (400 MHz; CDCl3) δH (ppm) 3.45 (s, 4H, NCH2COOC (CH3)3), 3.41 (t, 2H, J = 7.4 Hz, BrCH2), 3.11 (t, 2H, J = 7.4 Hz, BrCH2CH2N), 1.44 (s, 18H, COOC (CH3)3).:DCM mixture). The pure product was obtained as a yellow oil. Yield 0.62 g, 54%; 1H NMR (400 MHz, CDCl3): δH (ppm) 8.50 (d, 2H, J = 7.27 Hz, ArH), 8.10 (d, 2H, J = 8.13 Hz, ArH), 7.70 (m, 2H, ArH), 4.30 (m, 4H), 3.40 (m, 8H), 2.85 (br s, 8H), 1.40 (s, 36H); 13C NMR (400 MHz; D2O): δC 170.7, 163.4, 133.2, 130.6, 127.2, 121.5, 80.9, 45.4, 52.4, 55.1, 27.1; IR (KBr disc) (cm1): 3440(br), 2977(s), 2931(w), 1736(s), 1702(w), 1661(vs), 1628(w), 1591(s); ESI-MS (+ion): found m/z 783.4578, calc. 783.4544 for [(4) H]+.Complexation of L1 with Gd(III) chloride
L1 (28 mg, 0.05 mmol) and GdCl3. 6H2O (18.0 mg 0.05 mmol) were added to two different vials. The ligand was dissolved in ethanol 10 ml, and heated slightly to ensure complete dissolution. Then GdCl3 was dissolved in distilled H2O, and again heated to ensure complete dissolution. Thereafter the vial containing the metal salt solution was continuously stirred, and the ligand solution was added dropwise. The instantaneous formation of the precipitate was observed. After the complete addition of the ligand to the metal, the mixture was stirred for two days in the dark. The solvent was evaporated to give a yellow precipitate of the complex. Yield 84%; IR (KBr disc, cm−1): 3425(br), 1729(w), 1625(s), 1408(s); ESI-MS (−ion): found m/z 712.0898, calc. 712.0890 for [(L1) Gd]. UV/vis [λmax, nm (ε, M−1 cm−1)] in H2O: 235(17658), 274(4128), 344(6279). The complexation reaction of L1 with Eu(III) chloride, Yb(III) chloride and Nd(III) chloride were carried out as described for complexation with Gadolinium chloride. However in the case of Nd(III) chloride it had to be dissolved in DMF, instead of water.
:10 dichloromethane/methanol); the product was obtained from the eluents by evaporation of the solvent mixture. Yield 0.25 g, 20%; 1H NMR (CDCl3, 400 MHz) δH 8.50 (d, 2H, J = 7.13, ArHa), 8.10 (d, 2H, J = 7.98, ArHc), 7.70 (t, 2H, J = 7.73, ArHb), 4.2 (m, 2H), 3.4 (s, 8H), 2.9 (m, 2H), 2.8 (m, 2H), 2.7 (m, 2H), 2.6 (m, 4H), 1.4 (s, 36H); 13C NMR (400 MHz, CDCl3): δC 169.0, 163.1, 132.8, 130.6, 130.2, 125.9, 121.7, 79.8, 55.0, 54.8, 51.9, 51.8, 50.9, 37.2, 27.1, 27.0, 27.1, 26.9; IR (KBr disc) (cm−1): 3467(br), 2973(s), 2360(s), 1733(vs), 1648(s), 1595(s); ESI-MS (+ion): found m/z 783.45, calc. 783.45 for [(6) H]+.Complexation of L2 with Gd(III) chloride
Complexation was carried out in a similar manner described for L1. Yield 85%; IR (KBr disc) (cm1): 3404(br), 1728(w), 1644(s), 1408(s); ESI-MS (−ion): found m/z 714.23, calc. 714.11 for [(L2) Gd]. UV/vis [λmax, nm (εM, M−1 cm−1)] in H2O: 235(6752), 267(1679), 345(2309).Results and discussion
General synthesis
Symmetric ligand, L1.
(a) Synthesis of 2. The synthesis of 2 following the reported procedure gave low yields in our hands. However a small modification to the procedure allowed us to synthesise 2 in improved yield and purity. The phosphonium bromide was first prepared by the portionwise addition of NBS to the dissolved solution of triphenyl phosphine in dichloromethane (successful preparation of the phosphonium bromide is indicated by the formation of sticky, semi solid-triphenylphosphine oxide).44 The prepared phosphonium bromide was then added dropwise to the solution of 1 in dichloromethane. The rest of the procedure was followed as reported by Anelli et al.38
(b) Alkylation of 3. Initially, compound 3 was added together with bromide 2 in the stoichiometric ratio of 1
:2 in anhydrous DMF (Fig. 3). The reaction mixture was heated to 65 °C and allowed to reflux for 48 h. Purification by thin layer chromatography (TLC) was carried out with 5% methanol/dichloromethane used as the eluent. The purified protected form of the ligand thus obtained, subsequently underwent deprotection by refluxing the protected ligand in 2 M HCl for two hours according to the procedure reported by Vogt et al.30 The ligand L1 was isolated in good yield (90%).
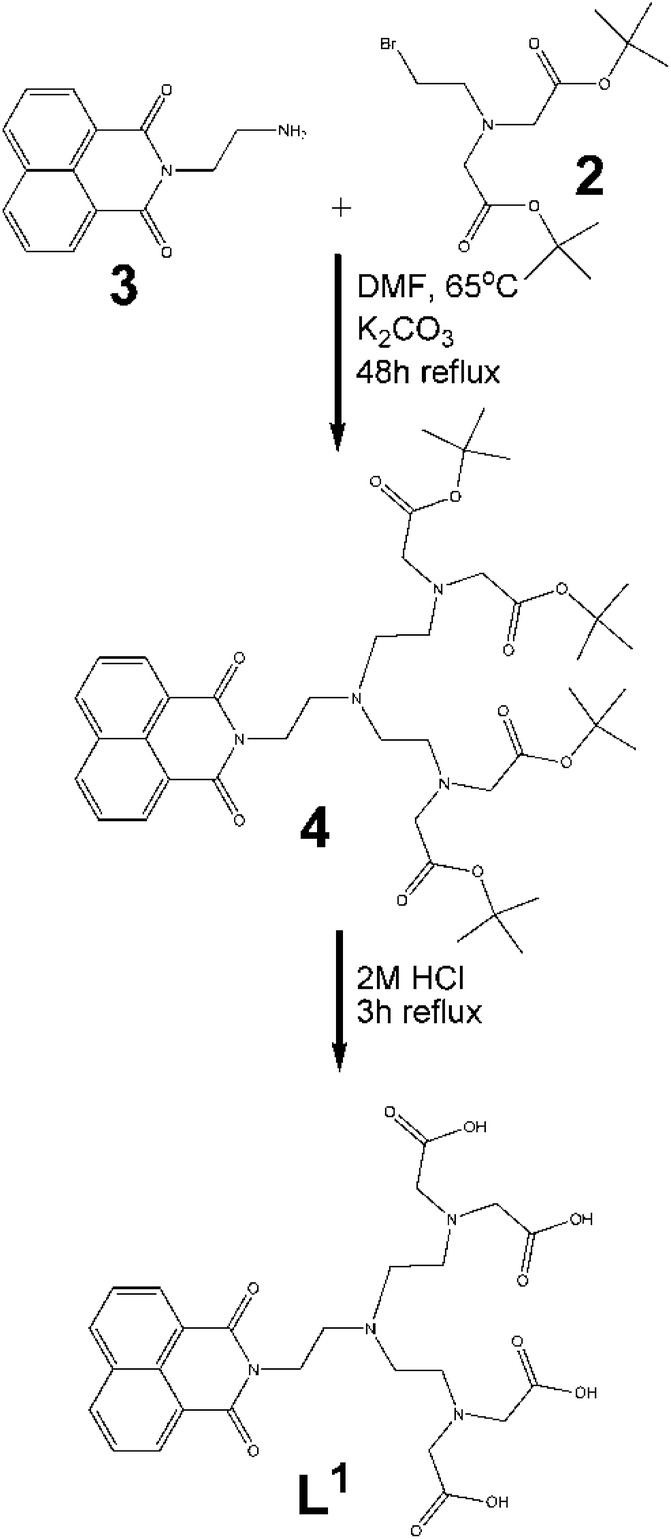 | ||
| Fig. 3 Schematic representation of the synthesis of symmetric ligand (L1). | ||
Asymmetric ligand, L2. Triethylenetetramine (TRIEN) was reacted with 1,8-naphthalic anhydride to produce the new ligand 5. The 1H NMR spectrum of the crude mixture confirmed the reaction had occurred with a significant change to the aliphatic protons consistent with a lowering of the symmetry of the TRIEN core.
The alkylation of this product was carried out using the same conditions as for the symmetric ligand, using stoichiometric amounts of alkylation agent (Fig. 4). With resulting low yields, a 1:5 molar ratio was used to try to improve the amount of product and its purity, however 1H NMR still indicated an the excess of compound 5. The reaction time was varied from 2 to 7 days, with little effect on the yield. Similarly, increasing the reaction temperature did not appreciably improve the yield. Purification of the reaction mixture was carried out by chromatography (95:5 dichloromethane/methanol) giving a yield of <20% isolated product.
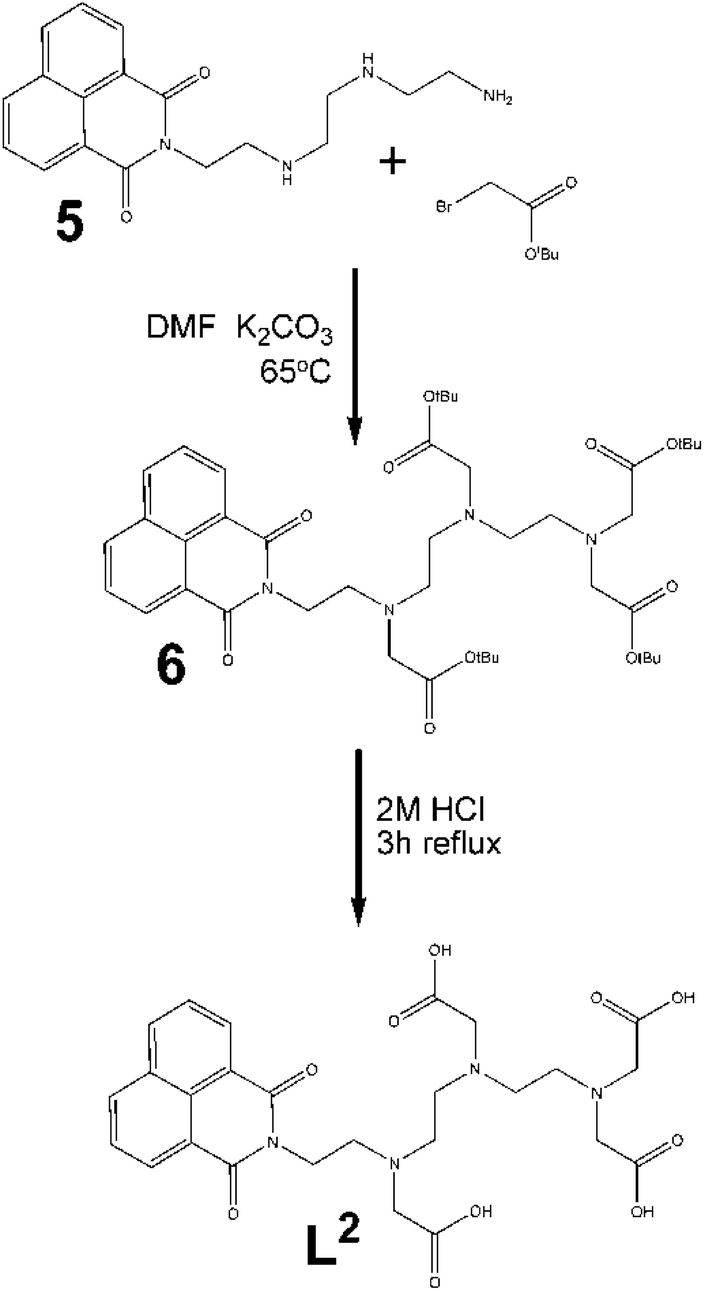 | ||
| Fig. 4 Schematic representation of the synthesis of asymmetric ligand (L2). | ||
Characterisation
In 4, the four equivalent pendant carboxylate CH2-protons appear as a broad singlet at 2.85 ppm indicating a strong upfield shift due to alkylation and the CH2-protons are subsequently shifted to 3.40 ppm upon deprotection, in L1. Curiously, a similar singlet was visible in the protected form of L2 (6) at 3.40 ppm, suggesting a less intensive inductive effect by adjacent t-butyl groups.
| Compound | ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) amide O) amide |
ν(CO) ester |
ν(CO) acid |
ν(O–H) | ν(N–H) |
|---|---|---|---|---|---|
| a 1a–tert-butyl bromoacetate. | |||||
| 2 | 1737 | ||||
| 3 | 1662 | 3346 | |||
| 4 | 1654 | 1739 | 3314 | ||
| L1 | 1655 | 1695 | 3429 | ||
| Gd-L1 | 1623 | 1729 | 3424 | ||
| 1a | 1731 | ||||
| 5 | 1659 | 3416 | |||
| 6 | 1648 | 1732 | 3414 | ||
| L2 | 1654 | 1702 | 3427 | ||
| Gd-L2 | 1644 | 1728 | 3403 | ||
Evaluation of DTPA analogues
Both centrally and terminally substituted DTPA analogues have been exploited for their use in MRI applications.47 As of today, most of the DTPA analogues tested and recognized as contrast agents belong to the terminally-substituted category,48 but investigations related to direct substitution of a chromophoric moiety in the central nitrogen, remains scarce.13,44,46Similarly, terminally substituted and C-substituted (on ethylenic carbon) analogues comprise the majority of DTPA analogues (including the commercially available contrast agents, namely Eovist® and Multihance®). The relaxivity of the new contrast agent based on L1 at relevant magnetic field strengths (8.10 mM−1 s−1 at 30 MHz and 25 °C) is considerably higher than that of the commercially available contrast agents Eovist®, Multihance® and Ablavar™ (previously known as Vasovist® or MS-325) (Table 2).
In contrast to L1, the poor yield of L2 limited analogous investigations of relaxivity.
DTPA analogues have also been exploited for luminescence based applications. Examples include DTPA functionalised with Carbostyril-124 (CS-124),52 CS-124 covalently bound to trimethoprim,53 amine functionalised reactive groups such as isothiocynates and thiol-reactive groups such as pridyldithiol, maleimide, methanethiosulfonate and haloacetyl acetamide.54 Further, DTPA analogues have been investigated for their usage in dissociation enhanced fluoroimmunoassay (DELFIA).55
Fluorescence on binding with lanthanides
In the following experiments we have not concerned ourselves with the lanthanide phosphorescence spectrum. Instead, we are interested by changes in the organic fluorescence spectrum on lanthanide binding. Primarily, we wish to show that the organic lumophore is not totally quenched upon lanthanide binding and any shift in the spectrum is small.The emission profiles of the protected form of the symmetric ligand, 4, compared to its deprotected form, L1, exhibited only slight differences, with only a slight red shift occurring in the excitation wavelength. The formation of the gadolinium complex, confirmed through the use of xylenol orange, results in little change to the luminescence spectrum, with no difference noted in the excitation wavelength and no change observed in the emission spectrum, compared to L1 (Table 3 and Fig. 5). Similarly, the formation of the gadolinium complex of L2 did not result in a significant quenching of the luminescence signal of the free ligand (Fig. 6), nor did it shift the excitation and emission wavelengths to a significant degree.
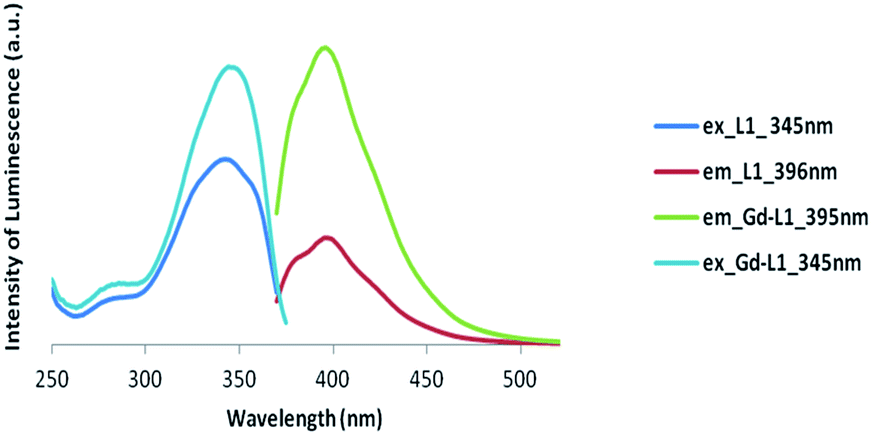 | ||
| Fig. 5 Luminescence spectra of Gd complex of symmetric ligand (Gd-L1) (excitation at 345 nm and emission at 395 nm). | ||
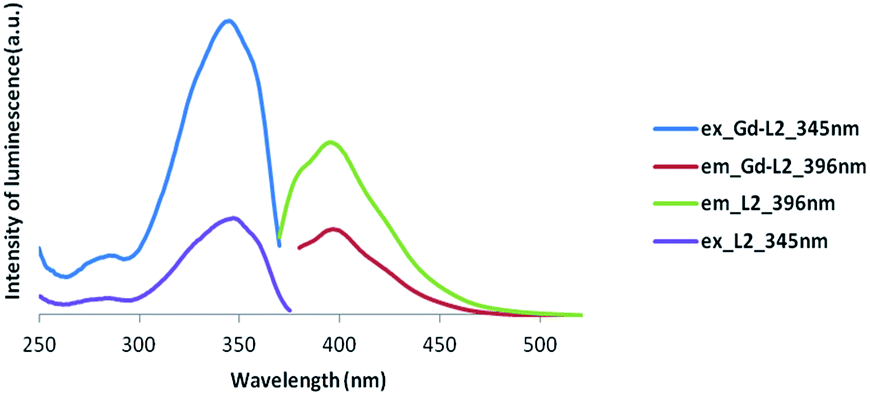 | ||
| Fig. 6 Luminescence spectra of Gd complex of asymmetric ligand (Gd-L2) (excitation at 345 nm and emission at 396 nm). | ||
Clearly, the luminescence spectroscopy indicates that it may be possible to utilize the gadolinium complexes of L1 and L2 as luminescent probes, though their efficiency in such an application remains to be determined.
Relaxivity measurements
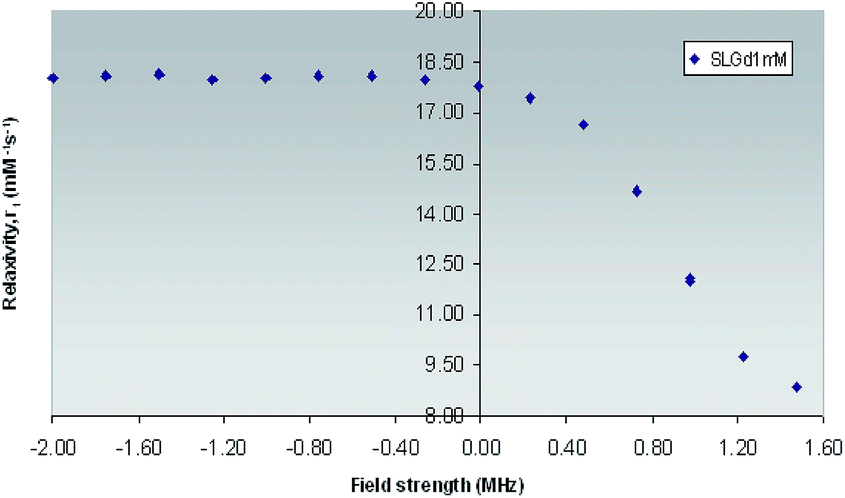 | ||
| Fig. 7 1H NMRD Profile of Gd(III)-L1 complex obtained across field strengths ranging from 0.01–30 MHz (expressed in logarithmic scale for purpose of clarity) at 25 °C. | ||
The enhancement relaxivity of water protons results from dipolar interaction of the electron magnetic moment of the paramagnetic metal ion with that of the nuclear magnetic moment of solvent nuclei. This interaction comprises contributions from water molecules that belong to the inner, outer and secondary coordination spheres. It has been reported, that for low molecular weight Gd(III) based mono aqua hydrophilic complexes, the inner-sphere and outer-sphere contributions are comparable, giving rise to relaxivity values which lie in the range of 4–5 mM−1 s−1 at 25 °C and 20 MHz,1 while the contribution from secondary-sphere water molecules has not been as extensively studied.56
However, the observed relaxivity was very much on the higher side for Gd(III)-L1 (8.65 mM−1 s−1) could be related to the concentration of the contrast agent by the following equation.
| R1obs = R1W + [Gd(III)-L1]r1p |
| (1/T1) = qPm[1/(T1m + τM)] |
Conclusions
Two new DTPA analogues have been synthesized. A chromophore bearing, central nitrogen substituted, DTPA analogue has been synthesized distinguishing these new ligands from commercial contrast agents and incorporating the naphthalimide moiety into the DTPA backbone. The Gd(III) complexes exhibit ligand-based luminescence and Gd-L1 has a relatively high relaxivity. Further investigations are necessary to determine if these reagents are toxic and the relaxivity is maintained in vivo.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank CITER (Cardiff Institute of Tissue Engineering and Repair) for infrastructure support (JobinYvon-Horiba Fluorolog spectrometer and Stellar Spin master FFC-2000 relaxometer).References
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- C. Paul-Roth and K. N. Raymond, Inorg. Chem., 1995, 34, 1408–1412 CrossRef CAS.
- H. S. Chong, K. Garmestani, L. H. Bryant and M. W. Brechbiel, J. Org. Chem., 2001, 66, 7745–7750 CrossRef CAS PubMed.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- M. W. Brechbiel, Q. J. Nucl. Med. Mol. Imag., 2008, 52, 166–173 CAS.
- J. D. G. Correia, A. Paulo, P. D. Raposinho and I. Santos, Dalton Trans., 2011, 40, 6144–6167 RSC.
- Y. F. Liu and C. C. Wu, Pure Appl. Chem., 1991, 63, 427–463 CrossRef CAS.
- G. L. Griffiths, D. M. Goldenberg, A. L. Jones and H. J. Hansen, Bioconjugate Chem., 1992, 3, 91–99 CrossRef CAS PubMed.
- Y. Liu, G. Liu and D. J. Hnatowich, Materials, 2010, 3, 3204–3217 CrossRef CAS.
- R. E. Weiner and M. L. Thakur, Appl. Radiat. Isot., 2002, 57, 749–763 CrossRef CAS PubMed.
- T. Storr, K. H. Thompson and C. Orvig, Chem. Soc. Rev., 2006, 35, 534–544 RSC.
- V. C. Pierre, M. J. Allen and P. Caravan, J. Biol. Inorg Chem., 2014, 19, 127–131 CrossRef CAS PubMed.
- D. W. Zhang, Z. Y. Yang, B. D. Wang, S. P. Zhang and R. D. Yang, Chem. Pharm. Bull., 2006, 54, 1203–1206 CrossRef CAS PubMed.
- Z. Jászberényi, A. Sour, É. Tóth, M. Benmelouka and A. E. Merbach, Dalton Trans., 2005, 2713–2719 RSC.
- J. Feng, G. Sun, F. Pei and M. Liu, Bioorg. Med. Chem., 2003, 11, 3359–3366 CrossRef CAS PubMed.
- L. V Elst, A. Roch, P. Gillis, S. Laurent, F. Botteman, J. W. M. Bulte and R. N. Muller, Magn. Reson. Med., 2002, 47, 1121–1130 CrossRef PubMed.
- Y. M. Wang, S. T. Lin, Y. J. Wang and R. S. Sheu, Polyhedron, 1998, 17, 2021–2028 CrossRef CAS.
- H. Imura, G. R. Choppin, W. P. Cacheris, L. A. de Learie, T. J. Dunn and D. H. White, Inorg. Chim. Acta, 1997, 258, 227–236 CrossRef CAS.
- C. F. G. C. Geraldes, A. M. Urbano, M. C. Alpoim, A. D. Sherry, K. T. Kuan, R. Rajagopalan, F. Maton and R. N. Muller, Magn. Reson. Imaging, 1995, 13, 401–420 CrossRef CAS PubMed.
- S. Aime, M. Botta, W. Dastru, M. Fasano, M. Panero and A. Arnelli, Inorg. Chem., 1993, 32, 2068–2071 CrossRef CAS.
- C. F. G. C. Geraldes, A. M. Urbano, M. C. Alpoim, M. A. Hoefnagel and J. A. Peters, J. Chem. Soc., Chem. Commun., 1991, 656–658 RSC.
- W. P. Cacheris, S. C. Quay and S. M. Rocklage, Magn. Reson. Imaging, 1990, 8, 467–481 CrossRef CAS PubMed.
- A. D. Sherry, W. P. Cacheris and K.-T. Kuan, Magn. Reson. Med., 1988, 8, 180–190 CrossRef CAS PubMed.
- L. Frullano and P. Caravan, Curr. Org. Synth., 2011, 8, 535–565 CrossRef CAS PubMed.
- V. Darras, M. Nelea, F. M. Winnik and M. D. Buschmann, Carbohydr. Polym., 2010, 80, 1137–1146 CrossRef CAS.
- S. W. A. Bligh, A. H. M. S. Chowdhury, D. Kennedy, C. Luchinat and G. Parigi, Magn. Reson. Med., 1999, 41, 767–773 CrossRef CAS PubMed.
- Y. M. Wang, T. H. Cheng, G. C. Liu and R. S. Sheu, J. Chem. Soc., Dalton Trans., 1997, 833–838 RSC.
- M. S. Konings, W. C. Dow, D. B. Love, K. N. Raymond, S. C. Quay and S. M. Rocklage, Inorg. Chem., 1990, 29, 1488–1491 CrossRef CAS.
- T. N. Parac-Vogt, K. Kimpe, S. Laurent, C. Piérart, L. V. Elst, R. N. Muller and K. Binnemans, Eur. J. Inorg. Chem., 2004, 2004, 3538–3543 CrossRef.
- T. Storr, B. R. Cameron, R. A. Gossage, H. Yee, R. T. Skerlj, M. C. Darkes, S. P. Fricker, G. J. Bridger, N. A. Davies, M. T. Wilson, K. P. Maresca and J. Zubieta, Eur. J. Inorg. Chem., 2005, 2005, 2685–2697 CrossRef.
- J. M. Couchet, J. Azéma, P. Tisnès and C. Picard, Inorg. Chem. Commun., 2003, 6, 978–981 CrossRef CAS.
- R. Artali, M. Botta, C. Cavallotti, G. B. Giovenzana, G. Palmisano and M. Sisti, Org. Biomol. Chem., 2007, 5, 2441–2447 CAS.
- M. W. Brechbiel and O. A. Gansow, Bioconjugate Chem., 1991, 2, 187–194 CrossRef CAS PubMed.
- C. H. Cummins, E. W. Rutter and W. A. Fordyce, Bioconjugate Chem., 1991, 2, 180–186 CrossRef CAS PubMed.
- (a) K. Choi, Y. D. Hong, M. S. Pyun and S. J. Choi, Bull. Korean Chem. Soc., 2006, 27, 1194–1198 CrossRef CAS; (b) M. Licchelli, A. O. Biroli, A. Poggi, D. Sacchi, C. Sangermani and M. Zema, Dalton Trans., 2003, 4537–4545 RSC.
- M. A. Williams and H. Rapoport, J. Org. Chem., 1993, 58, 1151–1158 CrossRef CAS.
- P. L. Anelli, F. Fedeli, O. Gazzotti, L. Lattuada, G. Lux and F. Rebasti, Bioconjugate Chem., 1999, 10, 137–140 CrossRef CAS PubMed.
- Y. Arano, T. Uezono, H. Akizawa, M. Ono, K. Wakisaka, M. Nakayama, H. Sakahara, J. Konishi and A. Yokoyama, J. Med. Chem., 1996, 39, 3451–3460 CrossRef CAS PubMed.
- S. Laurent, F. Botteman, L. V. Elst and R. N. Muller, Helv. Chim. Acta, 2004, 87, 1077–1089 CrossRef CAS.
- J. F. W. Keana and J. S. Mann, J. Org. Chem., 1990, 55, 2868–2871 CrossRef CAS.
- D. A. Westerberg, P. L. Carney, P. E. Rogers, S. J. Kline and D. K. Johnson, J. Med. Chem., 1989, 32, 236–243 CrossRef CAS PubMed.
- A. Datta and K. N. Raymond, Acc. Chem. Res., 2009, 42, 938–947 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- (a) M. S. Pyun, K. H. Choi, Y. D. Hong and S. J. Choi, Bull. Korean Chem. Soc., 2009, 30, 1187–1189 CrossRef CAS; (b) H. R. Maecke, A. Riesen and W. Ritter, J. Nucl. Med., 1989, 30, 1235–1239 CAS.
- R. Krüger, K. Braun, R. Pipkorn and W. D. Lehmann, J. Anal. At. Spectrom., 2004, 19, 852–857 RSC.
- S. Laurent, C. Henoumont, L. V. Elst and R. N. Muller, Eur. J. Inorg. Chem., 2012, 2012, 1889–1915 CrossRef CAS.
- P. Lebdušková, A. Sour, L. Helm, É. Tóth, J. Kotek, I. Lukeš and A. E. Merbach, Dalton Trans., 2006, 3399–3406 RSC.
- K.-H. Jung, H.-K. Kim, G. H. Lee, D.-S. Kang, J.-A. Park, K. M. Kim, Y. Chang and T.-J. Kim, J. Med. Chem., 2011, 54, 5385–5394 CrossRef CAS PubMed.
- S. Laurent, L. V. Elst and R. N. Muller, Contrast Media Mol. Imaging, 2006, 1, 128–137 CrossRef CAS PubMed.
- F. Uggeri, S. Aime, P. L. Anelli, M. Botta, M. Brocchetta, C. de Haeen, G. Ermondi, M. Grandi and P. Paoli, Inorg. Chem., 1995, 34, 633–643 CrossRef CAS.
- P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275–302 CrossRef CAS PubMed.
- D. R. Reddy, L. E. Pedró Rosa and L. W. Miller, Bioconjugate Chem., 2011, 22, 1402–1409 CrossRef CAS PubMed.
- P. Ge and P. R. Selvin, Bioconjugate Chem., 2004, 15, 1088–1094 CrossRef CAS PubMed.
- J. Peuralahti, L. Meriö, V.-M. Mukkala, K. Blomberg and J. Hovinen, Bioorg. Med. Chem. Lett., 2006, 16, 4760–4762 CrossRef CAS PubMed.
- S. Aime, M. Botta and E. Terreno, Adv. Inorg. Chem., 2005, 57, 173–237 CrossRef CAS.
- J. Costa, É. Tóth, L. Helm and A. E. Merbach, Inorg. Chem., 2005, 44, 4747–4755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06946d |
| This journal is © The Royal Society of Chemistry 2017 |