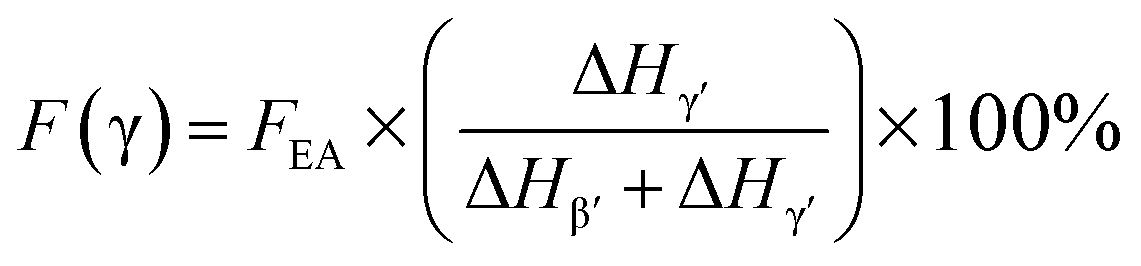DOI:
10.1039/C7RA01267E
(Paper)
RSC Adv., 2017,
7, 15382-15389
A critical analysis of the α, β and γ phases in poly(vinylidene fluoride) using FTIR†
Received
30th January 2017
, Accepted 28th February 2017
First published on 7th March 2017
Abstract
Poly(vinylidene fluoride) (PVDF) has been widely utilized in scientific research and the manufacturing industry for its unique piezoelectric properties. In the past few decades, the vibrational spectra of PVDF polymorphic polymers via FTIR (Fourier transform infrared spectroscopy) have been extensively investigated and documented. However, reports on the analysis of α, β and γ phases often have conflicting views based on measured data. In this work, we analyze the FTIR vibrational bands of PVDF materials fabricated by different processes with detailed XRD (X-ray diffraction) characterization to identify the structural α, β and γ phases. By examining the results in this work and extensively reviewing published research reports in the literature, a universal phase identification procedure using only the FTIR results is proposed and validated. Specifically, this procedure can differentiate the three phases by checking the bands around 763 and/or 614, 1275, and 1234 cm−1 for the α, β and γ phases, respectively. The rule for assignment of the 840* and 510* cm−1 bands is provided for the first time and an integrated quantification methodology for individual β and γ phase in mixed systems is also demonstrated.
Introduction
Since the discovery of piezoelectricity in poly(vinylidene fluoride) (PVDF),1 strong interest has been focused on the processing of the semi-crystalline polymer2–6 and at least five distinct polymorphs, α (TGTG′, form II), β (TTTT, form I), γ (T3GT3G′, form III), δ and ε have been constructed depending on the molecular chain conformation during the fabrication processes. Processing parameters, such as mechanical, thermal, electrical and chemical treatments all affect the final PVDF properties, including but not limited to electroactivity, dielectric and mechanical properties, antifouling behavior, and evoking cellular behavior.7–12 Although FTIR has been widely used in characterizing PVDF,13–23 reports in the existing literature (sometimes by the same authors) have many conflicting characterization results based on FTIR results (Table 1), especially in the electroactive β and γ phases. Two main reasons have resulted in this divergence. First, many authors directly assigned the FTIR bands at around 840 and 510 cm−1 to the β and/or γ phases without providing sufficient evidence.12,16–18,24–33 Second, several bands exclusive to the β and γ phases in the wavenumber range of 400–1500 cm−1 (or smaller range) have seldom been taken into consideration previously.
Table 1 Divergence in the assignment of some typical bandsa
| Wave number (cm−1) |
Crystalline phase |
Reference |
| Note: the assignment of some other bands such as 833 and 1431 cm−1 is also in dispute. |
| 431 |
β |
23, 36–38 |
| γ |
4, 18, 22, 24, 29, 39–41 |
| β + γ |
28 |
| 482 |
β |
42–45 |
| γ |
14–15, 17, 20, 22, 23 and 46 |
| α |
47–49 |
| 510 |
β |
12, 14, 16, 17, 19, 28, 40, 50–52 |
| γ |
24, 53–56 |
| β + γ |
35, 39, 41, 42, 57–60 |
| α |
47 and 48 |
| 840 |
β |
12, 16, 17, 24, 26, 27, 51, 52, 61–67 |
| γ |
15, 68–72 |
| β + γ |
4, 9, 15, 18, 28, 35, 39, 57, 58, 60, 73–76 |
| 881 |
β |
77 |
| γ |
48, 51 and 72 |
| β + γ |
60 and 78 |
| α + β |
62 |
| α + β + γ |
73 |
| α |
79 |
| 1071 |
β |
14, 79–84 |
| β + γ |
60 |
| α + β + γ |
21, 22, 46, 65, 73, 74 and 85 |
| α |
47, 48, 86–88 |
| 1176 |
β |
34, 52, 62 and 86 |
| γ |
42 |
| β + γ |
14 and 60 |
| 1234 |
β |
36, 37, 89–94 |
| γ |
9, 15, 19, 21, 22, 57, 65, 73, 76, 95–99 |
| β + γ |
18, 42 and 58 |
| 1275 |
β |
14–15, 19, 22, 23, 42, 73, 76, 94, 96–98 and 100 |
| γ |
71 and 72 |
| 1401 |
β |
77, 84, 86–88 |
| γ |
48 |
| β + γ |
60 |
| α |
79, 101 and 102 |
Furthermore, the relative amounts of the electroactive β and γ phases have been quantified in the works of Gregorio et al.24 and Lopes et al.,34 for samples containing only two-phase of α and β; or α and γ without considering the cases for three phases (α, β, and γ), or the β and γ two-phase systems. Although a recent report has proposed the quantification of β and γ phases system,35 the procedure is rather complex. In the present contribution, the procedure for the identification of α, β and γ phases using the FTIR vibrational spectrum is proposed and demonstrated with an integrated quantification methodology for individual β and γ phase for PVDF materials made of various α, β and γ phase compositions. Both FTIR and XRD data have been utilized to validate and identify the phases of various PVDF polymeric systems, because the nonelectroactive α phase and the electroactive β phase can be clearly identified by FTIR and XRD, respectively.4,5,20
Procedure for phase identification
By sorting out more than 100 prior publications, the FTIR absorption peaks for the three main α, β and γ polymorphs of PVDF can be classified into three major categories: (1) common peaks that appear in all three phases; (2) exclusive peaks that only appear in one of the three phases; and (3) dual peaks that could come from two different phases. In general, spectrum peaks around 881, 1071, 1176 and 1401 cm−1 with high intensity were previously used to characterize the crystal phases in some papers (see Table 1). However, peaks in the range of 876–885, 1067–1075, 1171–1182 and 1398–1404 cm−1 have similar characteristics in samples of α-, β- and γ-phases or other mixed systems.19,21,22,46,65,66,73,74,100,103 In other words, these are common peaks that appear in all three phases. The exclusive peaks can be used to identify the corresponding crystalline phases, whereas the dual peaks may be the results of experimental uncertainty for small wavenumber shifts (typically within 2 cm−1), or truely different phases depending on experimental conditions. Specifically, the exclusive peaks for the α phase (characteristic bands of the α phase) are around at 410, 489, 532, 614, 763, 795, 854, 975, 1149, 1209, 1383 and 1423 cm−1; the exclusive peaks for the β phase are around at 445, 473 and 1275 cm−1; and the exclusive peaks for the γ phase are around at 431, 482, 811 and 1234 cm−1. In contrast, the peaks in the range of 837–841 and 508–512 cm−1, although may appear in many different samples, the absorbance for the β- and γ-phase is much stronger than that of the α-phase. These two peaks (below using 840* and 510* cm−1 for simplicity) can therefore be used to characterize the electroactive β and/or γ phases (detailed in assignment of the 840* and 510* cm−1 bands). Although the β and γ phases show very close peaks in the range of 1428–1432 cm−1, various results support that the peaks at 1431 and 1429 cm−1 can be used as the characteristic bands of the β and γ phases, respectively.20,80,85,104,105 The 600 cm−1 band that was previously assigned to the β-phase by some authors,17,38,101,106 should not be used to characterize PVDF as this band is commonly showed up in many samples, including α-phase ones, due to the other intensive peak around at 613 cm−1.11,12,16,18,20,29,31,46,51,107,108 It is noted that the 776 and 833 cm−1 bands exclusively shown in high temperature crystallization γ-phase,18,68,109 are rather seldom observed in other γ-phase domination samples.14,19,40,42,55,69,70,85
Since the amounts of the three phases can be in any percentage, establishing a universal but simplified procedure to trace these phases is therefore of great significance. It is found that the bands around at 763 and/or 614, 1275, and 1234 cm−1 can be consistently used to differentiate and identify the α, β and γ phases, respectively. Therefore, the procedure for the identification of α, β and γ phases can be summarized as illustrated in Fig. 1.
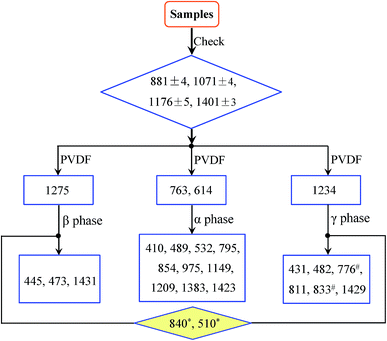 |
| | Fig. 1 Flow diagram for the identification of α, β and γ phases; 840* and 510* represents bands in the range of 837–841 and 508–512 cm−1, respectively; 776# and 833# reflects possible variations based on specific processes. | |
Experimental
Polymer solutions
PVDF (D692, from Shanghai Sensure Chemical Co. Ltd, with a molecular weight of 625![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) powders were dissolved in N-methyl-2-pyrrolidinone (NMP) and acetone to prepare the polymer solutions comprising 8 and 12 wt% PVDF with various NMP/acetone volume ratios (VNMP/Vacetone). All chemicals and solvents were used directly without further purification. Experimentally, the polymer solution was obtained by continuously stirring the mixture at 50 °C for a few hours in a Parafilm-sealed frosted glass bottle until transparent, and then degassed to remove bubbles for the membrane preparation.
000) powders were dissolved in N-methyl-2-pyrrolidinone (NMP) and acetone to prepare the polymer solutions comprising 8 and 12 wt% PVDF with various NMP/acetone volume ratios (VNMP/Vacetone). All chemicals and solvents were used directly without further purification. Experimentally, the polymer solution was obtained by continuously stirring the mixture at 50 °C for a few hours in a Parafilm-sealed frosted glass bottle until transparent, and then degassed to remove bubbles for the membrane preparation.
Membrane preparation
PVDF nanofibrous membranes were fabricated by electrospinning technique reported elsewhere.110 Non-nanofibrous membranes were prepared either by casting or spin coating onto RCA-cleaned silicon wafers at room temperature and some of them were further treated by a stretching process. Specifically, electrospinning experiments were performed in indoor atmosphere with fixed tip-to-collector distance of 10 cm and applied voltage of 7.5 kV, while casting and spin coating experiments were done in a clean room. All polymer membranes were dried at room temperature or in a temperature-controlled oven for the subsequent characterizations.
Characterization techniques
Infrared spectra of the above polymer membranes and the raw PVDF powder were taken via a Thermo Scientific Nicolet iS50 FTIR spectrometer in the range of 400–1500 cm−1 with a resolution of 2 cm−1, where the transmission mode was adopted for most samples except the cast membranes in the ATR (attenuated total reflection) mode. XRD results were obtained using a PANalytical X'Pert PRO X-ray diffractometer with Cu-Kα radiation (wavelength 0.154 nm) operated at 40 kV and 30 mA. The samples were scanned in the 2θ range of 10° to 45° with a step interval of 0.0167°.
Results and discussion
XRD and FTIR analyses of raw PVDF powder
It is well known that in typical PVDF samples the crystalline content is no more than 50–60%. However, there is still no concrete information how the amorphous part of PVDF influences the XRD and IR spectra. Therefore, the discussion in this work concentrates on the crystalline phase, especially on the most common α, β, and γ phases. As shown in the XRD characterization in Fig. 2(A), the PVDF powder is mainly made of the α phase, as evidenced by two intensive diffraction peaks at 18.4 and 20.0° and a medium peak at 26.6°, corresponding to 020, 110 and 021 reflections of the monoclinic α-phase crystal, respectively.20,111–113 It is noted from Fig. 2(A) that the powder also presents a peak at 20.6° (inset) corresponding to 110/200 reflection of the orthorhombic β-phase and four weak peaks at 33.2, 35.9, 38.8, and 41.1° corresponding to 130, 200, 002, and 111 reflection of the monoclinic α-phase, respectively.94,112,114 The corresponding FTIR spectrum shown in Fig. 2(B) is in good agreement with the XRD results to indicate strong α-phase crystal based on the scheme in Fig. 1. Specifically, there are two intensive peaks at 763 and 614 cm−1 (characteristics of the α phase) with a weak peak at 1275 cm−1 exclusive for the β phase and the absence of the peak at 1234 cm−1 exclusive for the γ phase (inset). Hence, it can be concluded that strong α-phase with some traces of β phase coexists in the neat PVDF powder as evidenced by the small peaks at 1275, 841 and 510 cm−1 for the β phase.
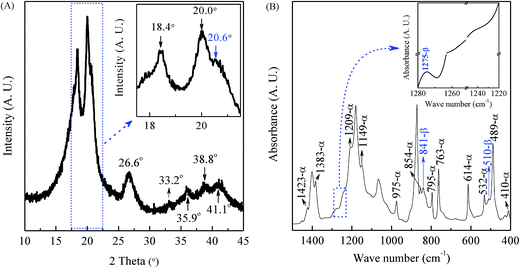 |
| | Fig. 2 XRD pattern (A) and FTIR spectrum (B) of the raw PVDF powders. | |
Electrospun nanofibrous membranes
As demonstrated in previous reports, PVDF nanofibers produced by the electrospinning technique are in favor of β phase due to the large mechanical elongation and strong electrical field during this process.64,101,115 However, different phases can also be constructed by tuning electrospinning parameters as evidenced from the XRD characterizations in Fig. 3(A) with enlarged views of two key areas of 17–22° and 35–42° as shown in Fig. 3(B), where three types of electrospun membranes have been constructed: (a) 12 wt% PVDF, VNMP/Vacetone = 9/1, under a flow rate of 60 μL h−1 and collected at 60 °C (black curves); (b) 12 wt% PVDF, VNMP/Vacetone = 9/1, under a flow rate of 2000 μL h−1 and collected at room temperature (red curves); and (c) 12 wt% PVDF, VNMP/Vacetone = 5/5, under a flow rate of 60 μL h−1 and collected at room temperature (blue curves). It is found that the type (a) membrane is mainly in α phase as indicated by the presence of two intensive peaks at 18.4 and 20.0° with two weak peaks at 26.6 and 35.9°, similar to that of the raw PVDF powder in Fig. 2(A), corresponding to 020, 110, 021 and 200 reflections of the monoclinic α-phase crystal.20,111,112,114 The crystalline phase of the type (b) membrane is mainly in γ phase as XRD exhibits a strong peak at 20.3° and two medium peaks around 18.5 and 39.0°, which are diffraction peaks on planes (110/101), (020), and (211) of monoclinic γ-phase crystal, respectively. Several prior reports have also described the formations of γ phase by using the electrospinning process.19,20,61,64,74 On the other hand, the type (c) membrane shows a very strong diffraction peak at 20.6° and a weak peak at 36.3° and can be categorized as mainly dominated in β phase.20,111,112,114
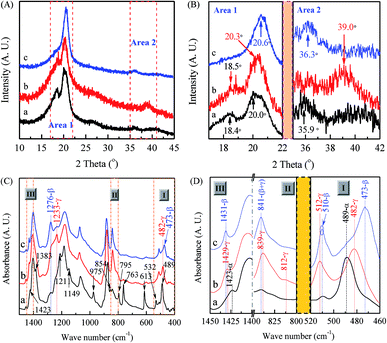 |
| | Fig. 3 XRD patterns (A) & (B) and FTIR spectra (C) & (D) of three types of electrospun PVDF membranes: curve ‘a’ (black color) is sample made from 12 wt% PVDF, VNMP/Vacetone = 9/1, under a flow rate of 60 μL h−1 and collected at 60 °C; curve ‘b’ (red color) is sample made from 12 wt% PVDF, VNMP/Vacetone = 9/1, under a flow rate of 2000 μL h−1 at room temperature; curve ‘c’ (red color) is sample made from 12 wt% PVDF, VNMP/Vacetone = 5/5, under a flow rate of 60 μL h−1 at room temperature. | |
Fig. 3(C) shows spectra results from FTIR and Fig. 3(D) shows the enlarged views of three areas around 510, 840, and 1430 cm−1. Specifically, if one follows the scheme in Fig. 1, the black curve for membrane (a) shows strong α phase signal as it has intensive peaks at 763, and 614 cm−1 (characteristics of the α phase) without clear peaks at 1276 cm−1 (exclusive for the β phase) and 1233 cm−1 (exclusive for the γ phase). The red curve for membrane (b) shows strong γ phase signal as it has the clear peak at 1233 cm−1 without peaks at 614/763 cm−1 (exclusive for α phase) or 1276 cm−1 (exclusive for β phase). Furthermore, one can also identify some of the characteristic bands of the γ phase at 482, 812, and 1429 cm−1 as labeled in Fig. 3(D). The blue curve for membrane (c) shows strong β phase signal as it has the clear peak at 1276 cm−1 (exclusive for β phase) without peaks at 614/763 cm−1 (exclusive for α phase) or 1233 cm−1 (exclusive for γ phase). Furthermore, one can also identify some of the characteristic bands of β phase are at 473 and 1431 cm−1 (Fig. 3(D)). It is not surprising that the 1233 cm−1 band also appears weakly since the relaxation process (β → γ) normally occurs in the formation of electrospun PVDF samples.116 It is noted that characteristic bands of α, β and γ phases in the 400–460 cm−1 range have many experimental uncertainties, thus these bands have been seldom reported in the characterizations of electrospun PVDF previously.19,101,115
Solution-cast membranes
Unlike electrospun membranes, the crystalline phase of membranes prepared by the solution casting method without experiencing any mechanical stretching is closely related to the crystallization rate of the solution. Numerous works have demonstrated that low-temperature solution crystallization (T < 70 °C) resulted in α or γ phase or their mixture, depending on the type of solvents,13,14,22,39,69,85,96,102,112 while some other works reported β phase under similar conditions.18,24,28 Here, a large number of casting experiments have been conducted by changing the solution concentration, solvent component, and the crystallization temperature with detailed characterizations.
The XRD patterns and FTIR spectra of membranes cast from 8 wt% PVDF solution with VNMP/Vacetone at 9/1 but dried and processed at different temperatures are shown in Fig. 4. It is found that the samples dried at 150 (black curves) and 40 °C (red curves) are dominated by the α and γ phases, respectively, as evidenced by the corresponding two intensive diffraction peaks at 18.4 and 19.9° and a remarkable peak at 26.6° (black curve in Fig. 4(A)), and the corresponding two dull peaks at 18.5 and 20.2° and a remarkable peak at 39.0° (red curve in Fig. 4(A)). On the other hand, the sample dried at 40 °C and further mechanically drawn at 80 °C with a draw ratio of 4 is dominated by the β phase, as revealed by a very strong diffraction peak at 20.6° (inset in the blue curve in Fig. 4(A)). The draw velocity used was 3 mm min−1 and the draw ratio was the ratio of the final and initial sample length. Using the scheme in Fig. 1, one can also use the FTIR spectra in Fig. 4(B) to characterize these films. First, the black curve in Fig. 4(B) has strong peaks at 614 and 763 cm−1 and should be strong in α phase; the red curve has a good peak at 1232 cm−1 and should be strong in γ phase; and the blue curve has a good peak at 1275 cm−1 and should be strong in β phase – these correspond very well with the XRD results. One can also check some of the characteristic bands in the three curves in Fig. 4(B) following the scheme in Fig. 1 for α, β and γ phase, respectively, with good agreements. For example, the 447, 472 and 1431 cm−1 bands all show up in the blue curve c in Fig. 4(B) as the strong supporting evidence for the β phase crystal. It is noted that the 1234 cm−1 band also appears as a tiny peak in this blue curve in Fig. 4(B), since the relaxation process (β → γ) often occurs in stretched PVDF samples.116 Interestingly, it is also noted that the spin-coated PVDF films have very similar XRD and FTIR spectra with those of the cast films (Part I in ESI†).
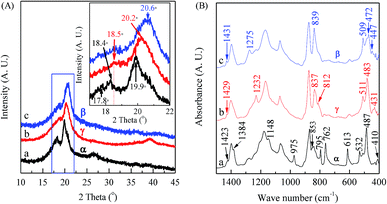 |
| | Fig. 4 XRD patterns (A) and FTIR spectra (B) of membranes cast from 8 wt% PVDF solution with VNMP/Vacetone at 9/1: curve ‘a’ (black) for the sample dried at 150 °C; curve ‘b’ (red) for the sample dried at 40 °C; curve ‘c’ (blue) for the sample dried at 40 °C and drawn at 80 °C with a draw ratio of ca. 4. | |
Assignment of the 840* and 510* cm−1 bands
From the literature survey, the 840* and 510* cm−1 bands have often yielded conflicting conclusions in assigning crystal phases. Experimental results from this work and the scheme in Fig. 1 are utilized to illustrate the correct assignment procedures. For example, the 839 cm−1 band is observed not only in the γ-phase electrospun membrane (red curve in Fig. 3(D)), but also in the β-phase cast membrane (blue curve c in Fig. 4(B)). If no exclusive bands in 400–1500 cm−1 are referred as illustrated in Fig. 1, the arbitrary assignment of the 839 cm−1 band to one of the electroactive phase becomes the source of confusions.
To further examine these two bands, a series of experiments have been conducted using electrospun membranes from 12 wt% PVDF under different flow rates and ratios of NMP and acetone (VNMP/Vacetone) and examined using FTIR as shown in Fig. 5: curve ‘a’ (black) for 60 μL h−1 and VNMP/Vacetone = 8/2; curve ‘b’ (red) for 2000 μL h−1 and VNMP/Vacetone = 8/2; curve ‘c’ (orange) for 60 μL h−1 and VNMP/Vacetone = 7/3; and curve ‘d’ (blue) for 500 μL h−1 and VNMP/Vacetone = 7/3. The electrospinning was performed at room temperature with the applied voltage of 7.5 kV and tip-to-collector distance of 10 cm. Using the scheme in Fig. 1 and based on the exclusive bands of the three phases, the crystalline phase of the fabricated membrane can be characterized as: α and β (α + β), α and γ (α + γ), β and γ (β + γ), and α, β and γ (α + β + γ) phases for curves ‘a’, ‘b’, ‘c’, and ‘d’, respectively. In this example, it is observed that several bands of β phase at 445, 473 and 1431 cm−1 and the bands of γ phase at 431, 482, 812 and 1429 cm−1, are not observed in the samples containing α + β phases (curve ‘a’) and β + γ phases (curve ‘c’), respectively, indicating that these bands cannot be used exclusively for β and γ phase characterizations. They are good “supporting” evidences and there are only two exclusive bands of 1275 cm−1 for the β phase and 1234 cm−1 for the γ phase. As such, in the presence of the 1275 cm−1 band and absence of the 1234 cm−1 band, the 840* and 510* cm−1 bands are considered as the β phase; on the contrary, the two bands (840* and 510* cm−1) are considered as the γ phase. If both 1275 and 1234 cm−1 bands appear together, the two bands (840* and 510* cm−1) are considered as both the β and γ phases; in such a case by taking a higher resolution measurement one can split the 840* or 510* cm−1 band into another two distinct bands for the β and γ phases, respectively.17 The above assignment of the 840* and 510* cm−1 bands is also supported by past prior work where the 840 cm−1 band has been assigned to β-only, γ-only or β and γ phases in PVDF samples manufactured by different ways.18
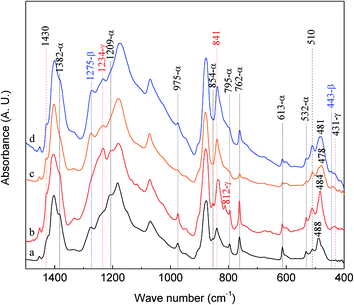 |
| | Fig. 5 FTIR spectra of electrospun membranes from 12 wt% PVDF under different flow rates and ratios of NMP and acetone (VNMP/Vacetone): curve ‘a’ (black) for 60 μL h−1 and VNMP/Vacetone = 8/2; curve ‘b’ (red) for 2000 μL h−1 and VNMP/Vacetone = 8/2; curve ‘c’ (orange) for 60 μL h−1 and VNMP/Vacetone = 7/3; curve ‘d’ (blue) for 500 μL h−1 and VNMP/Vacetone = 7/3. | |
Integrated quantification of individual β and γ phase
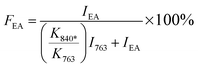
Since the 840* cm−1 band can be assigned to the β, γ, or both phases based on other band information, the relative fraction of the electroactive β and γ phases (FEA) in terms of crystalline components in any samples, such as a sample containing only two phases (α + β, α + γ, or β + γ) or three phases (α + β + γ), can be quantified as follows:| |
 | (1) |
where, IEA and I763 are the absorbencies at 840* and 763 cm−1, respectively; K840* and K763 are the absorption coefficients at the respective wave numbers, whose values are 7.7 × 104 and 6.1 × 104 cm2 mol−1, respectively.24
It is easy to see from eqn (1) that, when the 840* cm−1 band is assigned to one of the electroactive phases, the FEA is no doubt equivalent to the relative fraction of the corresponding phase. Thus, below is to show how to quantify individual β and γ phases when the 840* cm−1 band is for both phases. Fig. 6 shows the FTIR spectra of electrospun membranes using 12 wt% of PVDF mixed with different VNMP/Vacetone at 3/7, 4/6, 5/5, and 6/4 with curves labeled as ‘a’ (black), ‘b’ (red), ‘c’ (blue) and ‘d’ (orange), respectively. Insets are the magnification of frequency regions in the 1220–1280 cm−1 (left) and 465–490 cm−1 (right) range. The electrospinning was performed at room temperature with the applied voltage of 7.5 kV, flow rate of 500 μL h−1 and tip-to-collector distance of 10 cm. It can be seen from Fig. 6 that the β and γ phases coexist in these samples, as evidenced by the simultaneous presence of the 1275 and 1234 cm−1 bands. According to the inset figures, higher NMP/acetone ratio results in the increment and decrement of the absorption intensities of the 1275 and 1234 cm−1 bands (left inset), respectively. A corresponding red shift from 479 to 475 cm−1 (from the closest γ phase band of 482 cm−1 to the closest β phase band of 473 cm−1) is observed for the peaks in the range of 473–482 cm−1 (right inset). These observations reveal that the higher absorption intensity of the 1275 and 1234 cm−1 bands implies more β and γ crystals, respectively. Hence, the quantification of individual β and γ phases could be performed by using the absorbance (peak area or peak height) of the two bands (1275 and 1234 cm−1). However, a much more preferable method is proposed by calculating the peak-to-valley height ratio (P2VHR) between the peaks around at 1275 and 1234 cm−1 and their nearest valleys (Part II in ESI†), as demonstrated in equations of (2a) and (2b), respectively.
| |
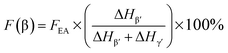 | (2a) |
| |
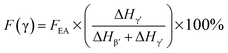 | (2b) |
where, Δ
Hβ′ and Δ
Hγ′ are the height differences (absorbance differences) between the peak around at 1275 cm
−1 and the nearest valley around at 1260 cm
−1, and the peak around at 1234 cm
−1 and the nearest valley around at 1225 cm
−1, respectively.
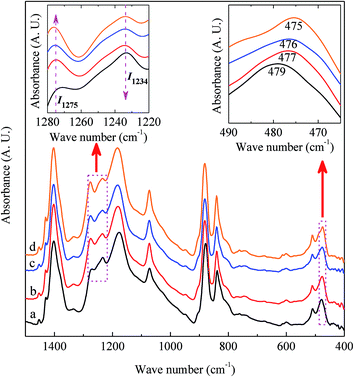 |
| | Fig. 6 FTIR spectra of electrospun membranes using 12 wt% of PVDF mixed with different VNMP/Vacetone at 3/7, 4/6, 5/5, and 6/4 as labeled with ‘a’, ‘b’, ‘c’ and ‘d’, respectively. Insets are the magnification of frequency regions in 1220–1280 cm−1 (left) and 465–490 cm−1 (right). The arrows in the left inset indicate the direction of an increase in absorption. | |
Based on eqn (1), (2a) and (2b), the relative fraction of the electroactive β and γ phases for all samples presented in the main article was calculated (Part III in ESI†). A very high fraction of the γ phase, namely 99.3%, was obtained for sample in Fig. 4(B)-curve b, while a lower but still comparatively high fraction of the β phase, namely 94.3%, was for sample in Fig. 3(C)-curve c. These results indicate that by taking some treatments on PVDF materials, high-performance PVDF membranes with good electroactivities can be achieved for various applications.
Conclusions
The characteristic bands of the α, β and γ phases in the 400–1500 cm−1 range have been determined by comparatively studying and summarizing the FTIR vibrational bands from reports published in the literature as well as new experimental samples characterized in this work. The characteristic bands of α phase are around at 410, 489, 532, 614, 763, 795, 854, 975, 1149, 1209, 1383 and 1423 cm−1, whereas characteristic bands of the β and γ phases are around at 445, 473, 1275 and 1431 cm−1 and around at 431, 482, 811, 1234 and 1429 cm−1, respectively. One can differentiate the three phases by checking the bands around 763 and 614 cm−1 for the α phase; 1275 cm−1 for the β phase, and 1234 cm−1 for the γ phase, respectively. Furthermore, the often confused crystal phase characterizations for the 840* and 510* cm−1 bands in prior publications are clarified by checking the bands around at 1275 and 1234 cm−1 to identify the existence of the β and γ phases, respectively. Finally, an integrated procedure is demonstrated to quantify the content of electroactive phases by using the peak-to-valley height ratio (P2VHR) between the peaks around at 1275 and 1234 cm−1 and their nearest valleys. This investigation provides clear guidelines to solve the long-term problems in the phase characterizations of PVDF using FTIR, but also delivers a simple methodology to quantify individual β and γ phase in PVDF polymers with multiple phases based on the FTIR spectrum.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 61404059 and U1505243), Fujian Natural Science Foundation (No. 2015J01205), and the Project for Distinguished Young Scientists of Fujian Province.
References
- H. Kawai, Jpn. J. Appl. Phys., 1969, 8, 975–976 CrossRef CAS.
- A. Lovinger, Science, 1983, 220, 1115–1121 CAS.
- M. Li, H. J. Wondergem, M.-J. Spijkman, K. Asadi, I. Katsouras, P. W. Blom and D. M. de Leeuw, Nat. Mater., 2013, 12, 433–438 CrossRef CAS PubMed.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- Z. Cui, N. T. Hassankiadeh, Y. Zhuang, E. Drioli and Y. M. Lee, Prog. Polym. Sci., 2015, 51, 94–126 CrossRef CAS.
- F. Liu, N. A. Hashim, Y. Liu, M. R. M. Abed and K. Li, J. Membr. Sci., 2011, 375, 1–27 CrossRef CAS.
- C. Chang, V. H. Tran, J. Wang, Y. K. Fuh and L. Lin, Nano Lett., 2010, 10, 726–731 CrossRef CAS PubMed.
- P. Martins, J. S. Nunes, G. Hungerford, D. Miranda, A. Ferreira, V. Sencadas and S. Lanceros-Mendez, Phys. Lett. A, 2009, 373, 177–180 CrossRef CAS.
- N. Jia, Q. Xing, X. Liu, J. Sun, G. Xia, W. Huang and R. Song, J. Colloid Interface Sci., 2015, 453, 169–176 CrossRef CAS PubMed.
- M. Darestani, H. Coster and T. Chilcott, J. Membr. Sci., 2013, 435, 226–232 CrossRef CAS.
- Y. K. A. Low, N. Meenubharathi, N. D. Niphadkar, F. Y. C. Boey and K. W. Ng, J. Biomater. Sci., Polym. Ed., 2011, 22, 1651–1667 CrossRef CAS PubMed.
- J. Gomes, J. Serrado Nunes, V. Sencadas and S. Lanceros-Méndez, Smart Mater. Struct., 2010, 19, 065010 CrossRef.
- G. Cortili and G. Zerbi, Spectrochim. Acta, Part A, 1967, 23, 285–299 CrossRef.
- M. Kobayashi, K. Tashiro and H. Tadokoro, Macromolecules, 1975, 8, 158–171 CrossRef CAS.
- T. Boccaccio, A. Bottino, G. Capannelli and P. Piaggio, J. Membr. Sci., 2002, 210, 315–329 CrossRef CAS.
- A. Salimi and A. Yousefi, Polym. Test., 2003, 22, 699–704 CrossRef CAS.
- Y. Bormashenko, R. Pogreb, O. Stanevsky and E. Bormashenko, Polym. Test., 2004, 23, 791–796 CrossRef CAS.
- R. Gregorio Jr, J. Appl. Polym. Sci., 2006, 100, 3272–3279 CrossRef.
- J. Zheng, A. He, J. Li and C. Han, Macromol. Rapid Commun., 2007, 28, 2159–2162 CrossRef CAS.
- T. Lei, X. Cai, X. Wang, L. Yu, X. Hu, G. Zheng, W. Lv, L. Wang, D. Wu, D. Sun and L. Lin, RSC Adv., 2013, 3, 24952–24958 RSC.
- D. M. Dhevi, A. A. Prabu and K. J. Kim, J. Mater. Sci., 2016, 51, 3619–3627 CrossRef CAS.
- M. Bachmann, W. Gordon, J. Koenig and J. Lando, J. Appl. Phys., 1979, 50, 6106–6112 CrossRef CAS.
- K. Tashiro, M. Kobayashi and H. Tadokoro, Macromolecules, 1981, 14, 1757–1764 CrossRef CAS.
- R. Gregorio Jr and M. Cestari, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 859–870 CrossRef.
- J. Andrew and D. Clarke, Langmuir, 2008, 24, 670–672 CrossRef CAS PubMed.
- L. Li, M. Zhang, M. Rong and W. Ruan, RSC Adv., 2014, 4, 3938–3943 RSC.
- K. Ravikumar, G. P. Kar, S. Bose and B. Basu, RSC Adv., 2016, 6, 10837–10845 RSC.
- R. Gregorio Jr and D. S. Borges, Polymer, 2008, 49, 4009–4016 CrossRef.
- W. Ma, J. Zhang, S. Chen and X. Wang, Appl. Surf. Sci., 2008, 254, 5635–5642 CrossRef CAS.
- V. Sencadas, R. Gregorio and S. Lanceros-Méndez, J. Macromol. Sci., Part B: Phys., 2009, 48, 514–525 CrossRef CAS.
- S. Yu, W. Zheng, W. Yu, Y. Zhang, Q. Jiang and Z. Zhao, Macromolecules, 2009, 42, 8870–8874 CrossRef CAS.
- P. Martins, C. Costa, M. Benelmekki and G. Botelho, CrystEngComm, 2012, 14, 2807–2811 RSC.
- B. Dutta, E. Kar, N. Bose and S. Mukherjee, RSC Adv., 2015, 5, 105422–105434 RSC.
- A. Lopes, C. Costa, C. Tavares, I. Neves and S. Lanceros-Mendez, J. Phys. Chem. C, 2011, 115, 18076–18082 CAS.
- S. K. Ghosh, M. M. Alam and D. Mandal, RSC Adv., 2014, 4, 41886–41894 RSC.
- M. El Achaby, F. E. Arrakhiz, S. Vaudreuil, E. M. Essassi, A. Qaiss and M. Bousmina, Polym. Eng. Sci., 2013, 53, 34–43 CAS.
- M. El Achaby, F. Arrakhiz, S. Vaudreuil, E. Essassi and A. Qaiss, Appl. Surf. Sci., 2012, 258, 7668–7677 CrossRef CAS.
- T. U. Patro, M. V. Mhalgi, D. Khakhar and A. Misra, Polymer, 2008, 49, 3486–3499 CrossRef CAS.
- W. Prest Jr and D. Luca, J. Appl. Phys., 1978, 49, 5042–5047 CrossRef.
- W. Ma, J. Zhang and X. Wang, J. Mater. Sci., 2008, 43, 398–401 CrossRef CAS.
- B. S. Ince-Gunduz, R. Alpern, D. Amare, J. Crawford, B. Dolan, S. Jones, R. Kobylarz, M. Reveley and P. Cebe, Polymer, 2010, 51, 1485–1493 CrossRef CAS.
- G. Cortili, Spectrochim. Acta, Part A, 1967, 23, 2216–2218 CrossRef CAS.
- K. Tashiro and M. Kobayashi, Phase Transitions, 1989, 18, 213–246 CrossRef CAS.
- M. Buonomenna, P. Macchi, M. Davoli and E. Drioli, Eur. Polym. J., 2007, 43, 1557–1572 CrossRef CAS.
- W. Wang, S. Zhang, L. o. Srisombat, T. R. Lee and R. C. Advincula, Macromol. Mater. Eng., 2011, 296, 178–184 CrossRef CAS.
- T. Lei, P. Zhu, X. Cai, L. Yang and F. Yang, Appl. Phys. A, 2015, 120, 5–10 CrossRef CAS.
- F. Mokhtari, M. Shamshirsaz and M. Latifi, Polym. Eng. Sci., 2016, 56, 61–70 CAS.
- I. Eleshmawi, J. Elastomers Plast., 2008, 107, 96–100 Search PubMed.
- R. H. Upadhyay and R. R. Deshmukh, J. Electrost., 2013, 71, 945–950 CrossRef CAS.
- R. Gregorio Jr and N. de Souza Nociti, J. Phys. D: Appl. Phys., 1995, 28, 432–436 CrossRef.
- A. Salimi and A. Yousefi, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3487–3495 CrossRef CAS.
- N. Jia, Q. Xing, G. Xia, J. Sun, R. Song and W. Huang, Mater. Lett., 2015, 139, 212–215 CrossRef CAS.
- S. Osaki and Y. Ishida, J. Polym. Sci., Polym. Phys. Ed., 1975, 13, 1071–1083 CrossRef CAS.
- S. K. Ghosh, W. Rahman, T. R. Middya, S. Sen and D. Mandal, Nanotechnology, 2016, 27, 215401 CrossRef PubMed.
- S. J. Kang, I. Bae, J. H. Choi, Y. J. Park, P. S. Jo, Y. Kim, K. J. Kim, J. M. Myoung, E. Kim and C. Park, J. Mater. Chem., 2011, 21, 3619–3624 RSC.
- X. Tian and X. Jiang, J. Hazard. Mater., 2008, 153, 128–135 CrossRef CAS PubMed.
- S. Ramasundaram, S. Yoon, K. J. Kim and C. Park, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 2173–2187 CrossRef CAS.
- S. Manna and A. K. Nandi, J. Phys. Chem. B, 2011, 115, 12325–12326 CrossRef CAS PubMed.
- D. Mandal, K. Henkel and D. Schmeißer, Mater. Lett., 2012, 73, 123–125 CrossRef CAS.
- Y. Imai, Y. Kimura and M. Niwano, Appl. Phys. Lett., 2012, 101, 181907 CrossRef.
- L. Yu and P. Cebe, Polymer, 2009, 50, 2133–2141 CrossRef CAS.
- R. Song, G. Xia, X. Xing, L. He, Q. Zhao and Z. Ma, J. Colloid Interface Sci., 2013, 401, 50–57 CrossRef CAS PubMed.
- D. Shah, P. Maiti, E. Gunn, D. F. Schmidt, D. D. Jiang, C. A. Batt and E. P. Giannelis, Adv. Mater., 2004, 16, 1173–1177 CrossRef CAS.
- G. Zhong, L. Zhang, R. Su, K. Wang, H. Fong and L. Zhu, Polymer, 2011, 52, 2228–2237 CrossRef CAS.
- M. M. Abolhasani, F. Zarejousheghani, Z. Cheng and M. Naebe, RSC Adv., 2015, 5, 22471–22479 RSC.
- Y. Sui, W.-T. Chen, J.-J. Ma, R.-H. Hu and D.-S. Liu, RSC Adv., 2016, 6, 7364–7369 RSC.
- S. Roy, P. Thakur, N. A. Hoque, B. Bagchi and S. Das, RSC Adv., 2016, 6, 21881–21894 RSC.
- R. Gregorio and R. Capitao, J. Mater. Sci., 2000, 35, 299–306 CrossRef CAS.
- Y. J. Park, Y. S. Kang and C. Park, Eur. Polym. J., 2005, 41, 1002–1012 CrossRef CAS.
- J. H. Park, N. Kurra, M. AlMadhoun, I. N. Odeh and H. N. Alshareef, J. Mater. Chem. C, 2015, 3, 2366–2370 RSC.
- S. Choi, J. Kim, Y. Ahn, S. Jo and E. Cairns, Chem. Mater., 2007, 19, 104–115 CrossRef CAS.
- M. Kanik, O. Aktas, H. S. Sen, E. Durgun and M. Bayindir, ACS Nano, 2014, 8, 9311–9323 CrossRef CAS PubMed.
- S. Yoon, A. Prabu, K. Kim and C. Park, Macromol. Rapid Commun., 2008, 29, 1316–1321 CrossRef CAS.
- Y. L. Liu, Y. Li, J. T. Xu and Z. Q. Fan, ACS Appl. Mater. Interfaces, 2010, 2, 1759–1768 CAS.
- H. Pan, B. Na, R. Lv, C. Li, J. Zhu and Z. Yu, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1433–1437 CrossRef CAS.
- C.-L. Liang, Q. Xie, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, J. Mater. Sci., 2014, 49, 4171–4179 CrossRef CAS.
- Y. Peng and P. Wu, Polymer, 2004, 45, 5295–5299 CrossRef CAS.
- C.-L. Liang, Z.-H. Mai, Q. Xie, R.-Y. Bao, W. Yang, B.-H. Xie and M.-B. Yang, J. Phys. Chem. B, 2014, 118, 9104–9111 CrossRef CAS PubMed.
- S. Janakiraman, A. Surendran, S. Ghosh, S. Anandhan and A. Venimadhav, Solid State Ionics, 2016, 292, 130–135 CrossRef CAS.
- Y. Xin, X. Qi, H. Tian, C. Guo, X. Li, J. Lin and C. Wang, Mater. Lett., 2016, 164, 136–139 CrossRef CAS.
- H. Zhang, P. Zhang, Z. Li, M. Sun, Y. Wu and H. Wu, Electrochem. Commun., 2007, 9, 1700–1703 CrossRef CAS.
- D. Dhakras, V. Borkar, S. Ogale and J. Jog, Nanoscale, 2012, 4, 752–756 RSC.
- H. Yu, T. Huang, M. Lu, M. Mao, Q. Zhang and H. Wang, Nanotechnology, 2013, 24, 405401 CrossRef PubMed.
- P. Zhang, X. Zhao, X. Zhang, Y. Lai, X. Wang, J. Li, G. Wei and Z. Su, ACS Appl. Mater. Interfaces, 2014, 6, 7563–7571 CAS.
- M. Benz, W. Euler and O. Gregory, Macromolecules, 2002, 35, 2682–2688 CrossRef CAS.
- W. Zhou, X. Jiang, P. Wang and H. Wang, Fibers Polym., 2013, 14, 100–104 CrossRef CAS.
- N. A. Hashim, Y. Liu and K. Li, Chem. Eng. Sci., 2011, 66, 1565–1575 CrossRef.
- N. A. Hashim, Y. Liu and K. Li, Ind. Eng. Chem. Res., 2011, 50, 3035–3040 CrossRef CAS.
- I. B. Rietveld, K. Kobayashi, T. Honjo, K. Ishida, H. Yamada and K. Matsushige, J. Mater. Chem., 2010, 20, 8272–8278 RSC.
- B. Luo, X. Wang, Y. Wang and L. Li, J. Mater. Chem. A, 2014, 2, 510–519 CAS.
- M. Zhang, A. Q. Zhang, B. K. Zhu, C. H. Du and Y. Y. Xu, J. Membr. Sci., 2008, 319, 169–175 CrossRef CAS.
- I. B. Rietveld, K. Kobayashi, H. Yamada and K. Matsushige, Soft Matter, 2009, 5, 593–598 RSC.
- R. A. Amoresi, A. A. Felix, E. R. Botero, N. L. Domingues, E. A. Falcão, M. A. Zaghete and A. W. Rinaldi, Ceram. Int., 2015, 41, 14733–14739 CrossRef CAS.
- D. Okada, H. Kaneko, K. Kato, S. Furumi, M. Takeguchi and Y. Yamamoto, Macromolecules, 2015, 48, 2570–2575 CrossRef CAS.
- M. Sharma, G. Madras and S. Bose, Phys. Chem. Chem. Phys., 2014, 16, 14792–14799 RSC.
- M. Benz and W. B. Euler, J. Appl. Polym. Sci., 2003, 89, 1093–1100 CrossRef CAS.
- X. He and K. Yao, Appl. Phys. Lett., 2006, 89, 112909 CrossRef.
- J. Andrew and D. Clarke, Langmuir, 2008, 24, 8435–8438 CrossRef CAS PubMed.
- Y. Li, J.-Z. Xu, L. Zhu, G.-J. Zhong and Z.-M. Li, J. Phys. Chem. B, 2012, 116, 14951–14960 CrossRef CAS PubMed.
- W. Yee, M. Kotaki, Y. Liu and X. Lu, Polymer, 2007, 48, 512–521 CrossRef CAS.
- A. Baji, Y. W. Mai, Q. Li and Y. Liu, Nanoscale, 2011, 3, 3068–3071 RSC.
- S. Satapathy, S. Pawar, P. Gupta and K. B. R. Varma, Bull. Mater. Sci., 2011, 34, 727–733 CrossRef CAS.
- T. Lei, L. Yu, W. Lingyun, F. Yang and D. Sun, J. Macromol. Sci., Part B: Phys., 2015, 54, 91–101 CrossRef CAS.
- S. K. Karan, D. Mandal and B. B. Khatua, Nanoscale, 2015, 7, 10655–10666 RSC.
- L. T. Beringer, X. Xu, W. Shih, W.-H. Shih, R. Habas and C. L. Schauer, Sens. Actuators, A, 2015, 222, 293–300 CrossRef CAS.
- P. Thakur, A. Kool, B. Bagchi, N. A. Hoque, S. Das and P. Nandy, Phys. Chem. Chem. Phys., 2015, 17, 13082–13091 RSC.
- B. Mohammadi, A. A. Yousefi and S. M. Bellah, Polym. Test., 2007, 26, 42–50 CrossRef CAS.
- Y. K. A. Low, L. Y. Tan, L. P. Tan, F. Y. C. Boey and K. W. Ng, J. Appl. Polym. Sci., 2013, 128, 2902–2910 CrossRef CAS.
- Y. Li, S. Tang, M.-W. Pan, L. Zhu, G.-J. Zhong and Z.-M. Li, Macromolecules, 2015, 48, 8565–8573 CrossRef CAS.
- T. Lei, Z. Zhan, W. Zuo, W. Cheng, B. Xu, Y. Su and D. Sun, Int. J. Nanomanuf., 2012, 8, 294–305 CrossRef CAS.
- D. Esterly and B. Love, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 91–97 CrossRef CAS.
- R. Hasegawa, Y. Takahashi, Y. Chatani and H. Tadokoro, Polym. J., 1972, 3, 600–610 CrossRef CAS.
- X. Cai, X. Huang, Z. Zheng, J. Xu, X. Tang and T. Lei, J. Macromol. Sci., Part B: Phys., 2017, 57, 75–82 CrossRef.
- G. Davis, J. McKinney, M. Broadhurst and S. Roth, J. Appl. Phys., 1978, 49, 4998–5002 CrossRef CAS.
- T. Lei, L. Yu, G. Zheng, L. Wang, D. Wu and D. Sun, J. Mater. Sci., 2015, 50, 4342–4347 CrossRef CAS.
- J. Miao, R. S. Bhatta, D. H. Reneker, M. Tsige and P. L. Taylor, Polymer, 2015, 56, 482–489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01267e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *bc,
Daoheng Sund and
Liwei Linde
*bc,
Daoheng Sund and
Liwei Linde