
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New protocols to access imidazoles and their ring fused analogues: synthesis from N-propargylamines
Esmail Vessally
 *a,
Somayeh Soleimani-Amiri
b,
Akram Hosseinian
c,
Ladan Edjlali
d and
Ahmadreza Bekhradnia
*e
*a,
Somayeh Soleimani-Amiri
b,
Akram Hosseinian
c,
Ladan Edjlali
d and
Ahmadreza Bekhradnia
*e
aDepartment of Chemistry, Payame Noor University, Tehran, Iran. E-mail: vessally@yahoo.com
bDepartment of Chemistry, Karaj Branch, Islamic Azad University, Karaj, Iran
cDepartment of Engineering Science, College of Engineering, University of Tehran, P. O. Box 11365-4563, Tehran, Iran
dDepartment of Chemistry, Tabriz Branch, Islamic Azad University, Tabriz, Iran
ePharmaceutical Sciences Research Center, Department of Medicinal Chemistry, Mazandaran University of Medical Sciences, Sari, Iran. E-mail: reza_bnia@yahoo.com; abekhradnia@mazums.ac.ir
First published on 20th January 2017
Abstract
Imidazole and its derivatives are privileged N-heterocyclic structures present in various natural products and synthetic pharmaceuticals. Despite the numerous methods that have been developed for the synthesis of imidazole cores, it is still challenging to readily achieve high efficiency and regioselectivity in imidazole synthesis. Therefore, synthesis of these compounds using new protocols is always interesting. In this study we discuss the most representative and interesting reports on the synthesis of imidazoles and their fused analogues from N-propargylamines. Mechanistic aspects of the reactions are considered and discussed in detail.
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from the University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now, he is working at Payame Noor University as an Associate Professor. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Somayeh Soleimani-Amiri | Somayeh Soleimani-Amiri was born in Tehran, Iran, in 1975. She received her B.S. degree in Pure Chemistry from Shahid Beheshti University, Tehran, Iran, and her M.S. degree in organic chemistry from Alzahra University, Tehran, Iran, in 2002 under the supervision of Prof. S. H. Abdi Oskooie and Prof. M. M. Heravi. She completed his Ph.D. degree in 2009 under the supervision of Prof. M. Z. Kassaee. Now, she is working at Karaj Branch, Islamic Azad University as an Assistant Professor. Her research interests include Computational Organic Chemistry, Nano Chemistry, and the synthesis of organic compounds. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from the University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her Ph.D. degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now, she is working at the University of Tehran as an Assistant Professor. Her research interests include inorganic and organic synthesis and new methodologies in nanomaterial synthesis. |
 Ladan Edjlali | Ladan Edjlali was born in Tabriz, Iran, in 1960. She received her B.S. degree in Applied Chemistry from the University of Tabriz, Iran, and her M.S. degree in organic chemistry from the University of Tabriz, Tabriz, Iran, in 1993 under the supervision of Prof. Y. Mirzaei. She completed her Ph.D. degree in 2000 under the supervision of Prof. Y. Mirzaei and Prof. S. M. Golabi. Now, she is working at Islamic Azad University, Tabriz Branch, as an Associate Professor. Her research interests include organic synthesis and new methodologies in organic synthesis. |
 Ahmadreza Bekhradnia | Ahmadreza Bekhradnia is an Associated Professor at the Mazandaran University of Medical Sciences, Iran. His current research interests focus on pharmaceutical intermediates, ranging from synthesis to the study of biological and photophysical properties, as well as application of molecular modeling in mechanistic investigations. He received his Ph.D. degree in organic chemistry from Tarbiat Modares University, Tehran, Iran in 2005. During his sabbatical period, he worked on the mechanism of transition metal-catalyzed cross-coupling reactions, both from an experimental and computational viewpoint, under the supervision of Prof. Per-Ola Norrby in Gothenburg University, Sweden (2012–2013). |
1. Introduction
Among all of the nitrogen-containing heterocycles, imidazole is one of the most important motifs in organic and medicinal chemistry. Several drug candidates endowed with potent biological activity against cancers, arrhythmias, convulsions, migraines, bacteria, microbes, and viral infections contain an imidazole moiety (Fig. 1).1 It is also one of the essential cores in a number of ionic liquids and natural products like histidine, histamine, vitamin B12, pilocarpine alkaloids, nucleic acid bases and biotin (Fig. 2).2 Moreover, imidazole derivatives are widely applied as fungicides for plants and their products.3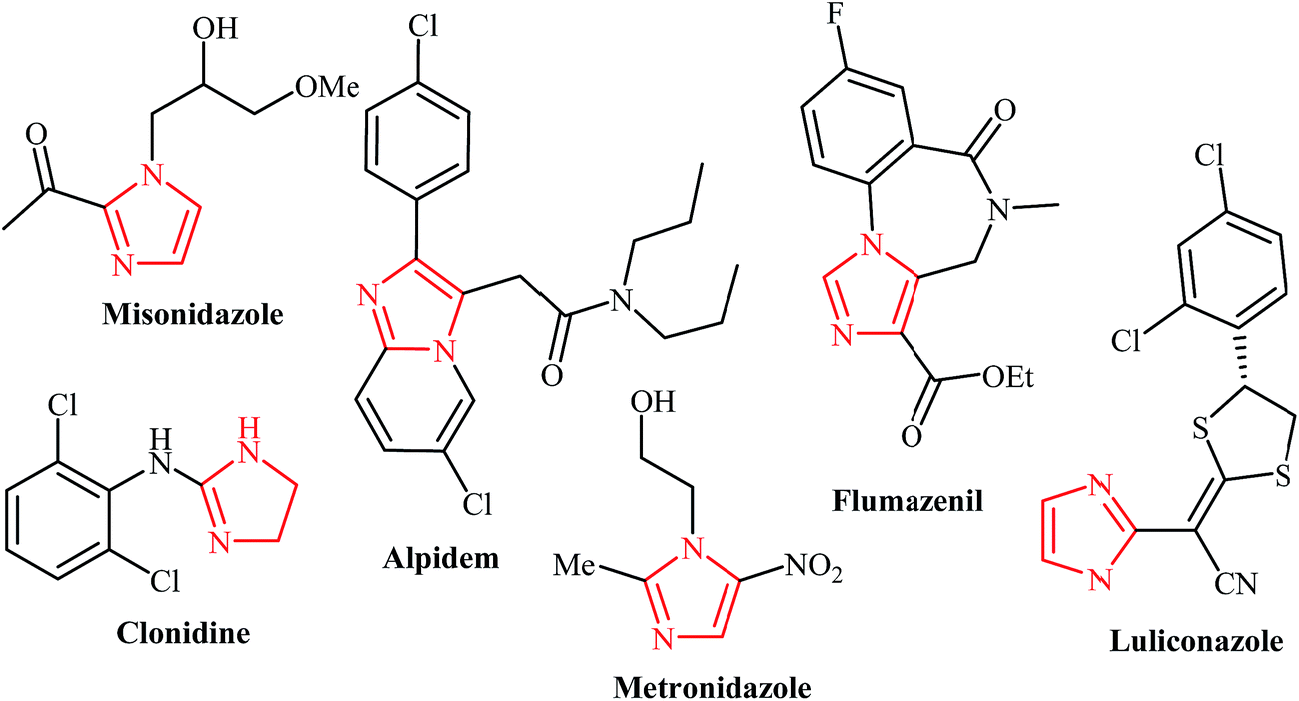 | ||
| Fig. 1 Selected examples of drugs containing an imidazole core. | ||
 | ||
| Fig. 2 Natural sources of imidazoles. | ||
The preparation of the imidazole cores generally depends on classical approaches, such as the Debus,4 Radiszewski,5 Wallach,6 Marckwald,7 and Van Leusen8 methods. Despite their popularity in academic and industrial areas, these methods suffer from several drawbacks, including low regioselectivity, low yields, and poor functional group tolerance.9 In this respect, the design of improved, highly efficient and regioselective approaches for imidazole preparation is of prime importance.
N-Propargylamines are some of the most useful and versatile building blocks in organic synthesis because they have diverse reaction patterns. They have been abundantly used as precursors in the synthesis of various nitrogen-based heterocycles and complex natural products. More recently, we published five review papers that covered the synthesis of pyrrole,10 pyridine,11 quinoline,12 pyrazine,13 1,4-oxazepane, and 1,4-diazepane14 derivatives from N-propargylamines. Over the last ten years, the synthesis of imidazole derivatives from N-propargylamines has been a very active area of research. This new protocol in the imidazole synthetic methods offers several advantages, such as high atom economy, high regioselectivity, and shorter synthetic routes. To the best of our knowledge, a comprehensive review on the synthesis of imidazole derivatives from N-propargylamines has not been reported in the literature. This study is an attempt to summarize the data available from the literature about the synthesis of imidazoles from N-propargylamines (Fig. 3). In this study, we have classified these reactions into two sections. The first section of this review will be focused on the synthesis of substituted imidazoles from N-propargylamines. In the second section, methodologies for the synthesis of fused imidazoles from N-propargylamines will be discussed. The mechanistic aspects of the reactions will be considered and discussed in detail.
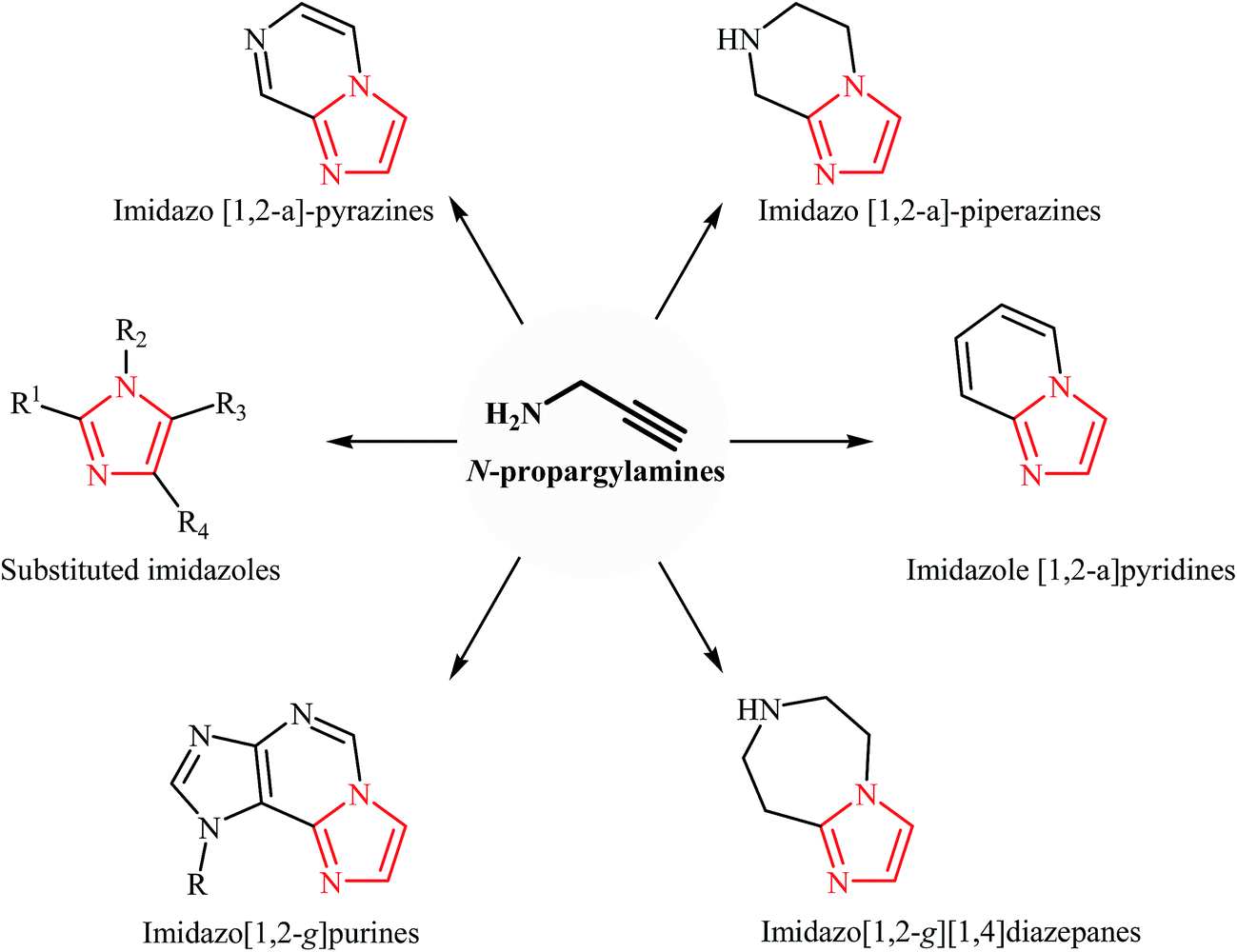 | ||
| Fig. 3 Some important imidazole-based compounds derived from N-propargylamines. | ||
2. Highly substituted imidazoles
One of the earliest reports on the synthesis of the imidazole ring from N-propargylamines appeared in 1974 when N-propargylamine and acetamidic esters were cyclized to their corresponding imidazoles through the intramolecular cyclization of propargylamidine intermediates.15 Since then, the use of N-propargylamines as attractive starting materials for the synthesis of this heterocyclic system has been widely reported.16–20 In 2007, the group of Abbiati showed that a series of 4-substituted-2-phenylimidazoles 3 were formed from easily available N-propargyl-benzamidine 1 and aryl halides 2 through a tandem aminopalladation/reductive elimination/isomerization method employing Pd(PPh3)4 as the catalyst, CuI as the co-catalyst, and K2CO3 as the base, in anhydrous DMF. They found that the presence of a co-catalyst was crucial for the success of this reaction. In the absence of CuI, low reaction yields and long reaction times were observed. Depending on the electronic effects of substituents on the aryl ring, substrates with electron-withdrawing groups gave higher yields than those with electron-donating groups. The reaction also worked well with heteroaryl halides (Scheme 1).21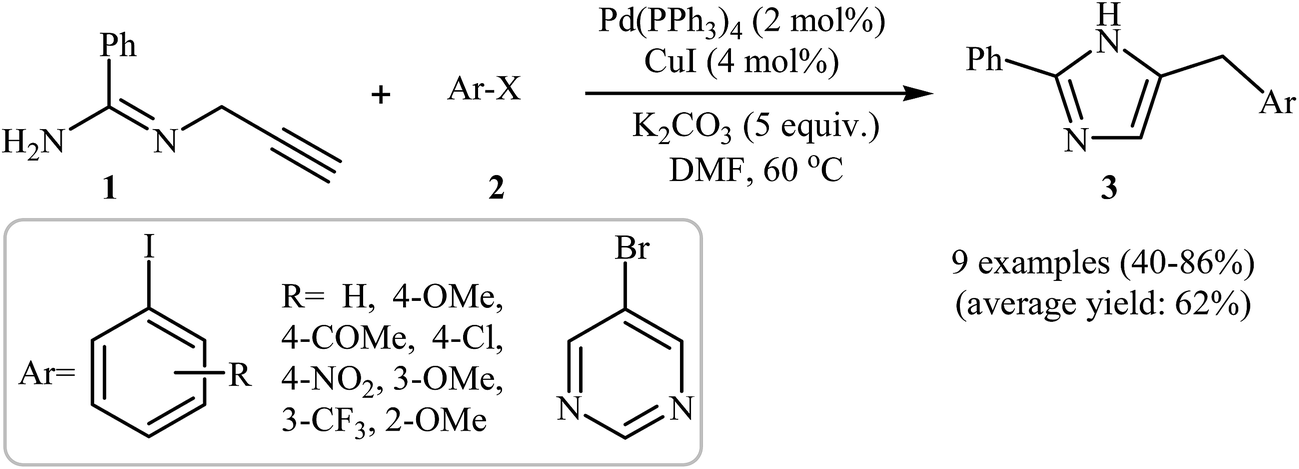 | ||
| Scheme 1 Synthesis of 4-substituted-2-phenylimidazoles 3 via a Pd-catalyzed cascade reaction of N-propargyl-benzamidine 1 and aryl halides 2. | ||
Along this line, Wu and co-workers reported a Pd(II)-catalyzed coupling reaction of fluorinated propargylamidines 4 with aryl iodides 5 into 2-fluoroalkyl-5-benzyl imidazoles 6 (Scheme 2). After studying a number of solvents, catalysts, and bases, a combination of Pd(OAc)2/K2CO3/DMF at 80 °![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22a was found to be optimal with respect to isolated product yield. The optimized protocol tolerated a variety of functional groups, such as chloro, fluoro, methoxy, acetyl, and ester functionalities. However, the substrate with R1 = CF2Br failed to participate in this reaction. In contrast to Abbiati's method, this method was more efficient for aryl halides bearing an electron-donating group.22b
22a was found to be optimal with respect to isolated product yield. The optimized protocol tolerated a variety of functional groups, such as chloro, fluoro, methoxy, acetyl, and ester functionalities. However, the substrate with R1 = CF2Br failed to participate in this reaction. In contrast to Abbiati's method, this method was more efficient for aryl halides bearing an electron-donating group.22b
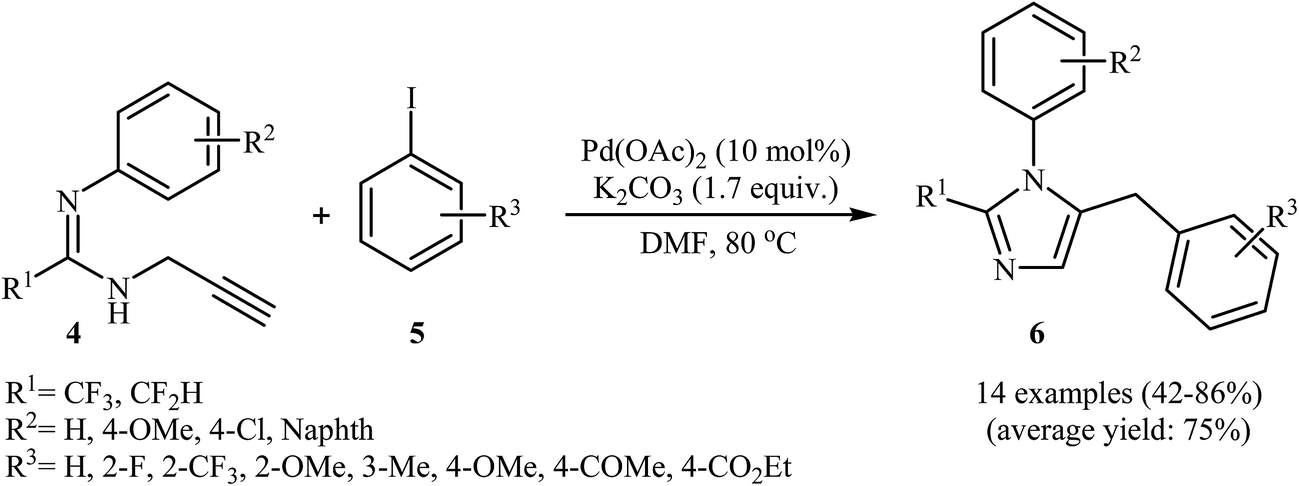 | ||
| Scheme 2 Pd(II)-Mediated synthesis of 2-fluoroalkyl-5-benzyl imidazoles 6 developed by Wu. | ||
An important study on imidazole-5-carbaldehydes 8 was carried out by the same research team, in which propargylamidines 7 were transformed into substituted imidazole-5-carbaldehydes 8 upon treatment with N-iodosuccinimide (NIS) in the presence of Ph3PAuCl/AgBF4/K2CO3 as a catalytic system in acetone at room temperature (Scheme 3a). The nature of the substituent attached to the aryl ring had a small impact on the success of the cyclization. Under optimized conditions, the reaction tolerated both electron-donating and electron-withdrawing substituents on the aryl ring and gave final products in good to excellent yields. However, internal alkynes failed to participate in the reaction. The mechanistic course of this reaction sequence is shown in Scheme 3b, and involves the initial formation of intermediate A from the reaction of activated propargylamidine 7 and NIS. The hydrogen shift of intermediate A gives intermediate B, which undergoes homolytic cleavage at the C–I bond to produce radical intermediate C. Subsequently, reaction of this intermediate with air oxygen gives peroxy-intermediate D. Finally, removal of a hydroxyl radical from D affords the observed imidazole-5-cabaldehydes 8.23,24
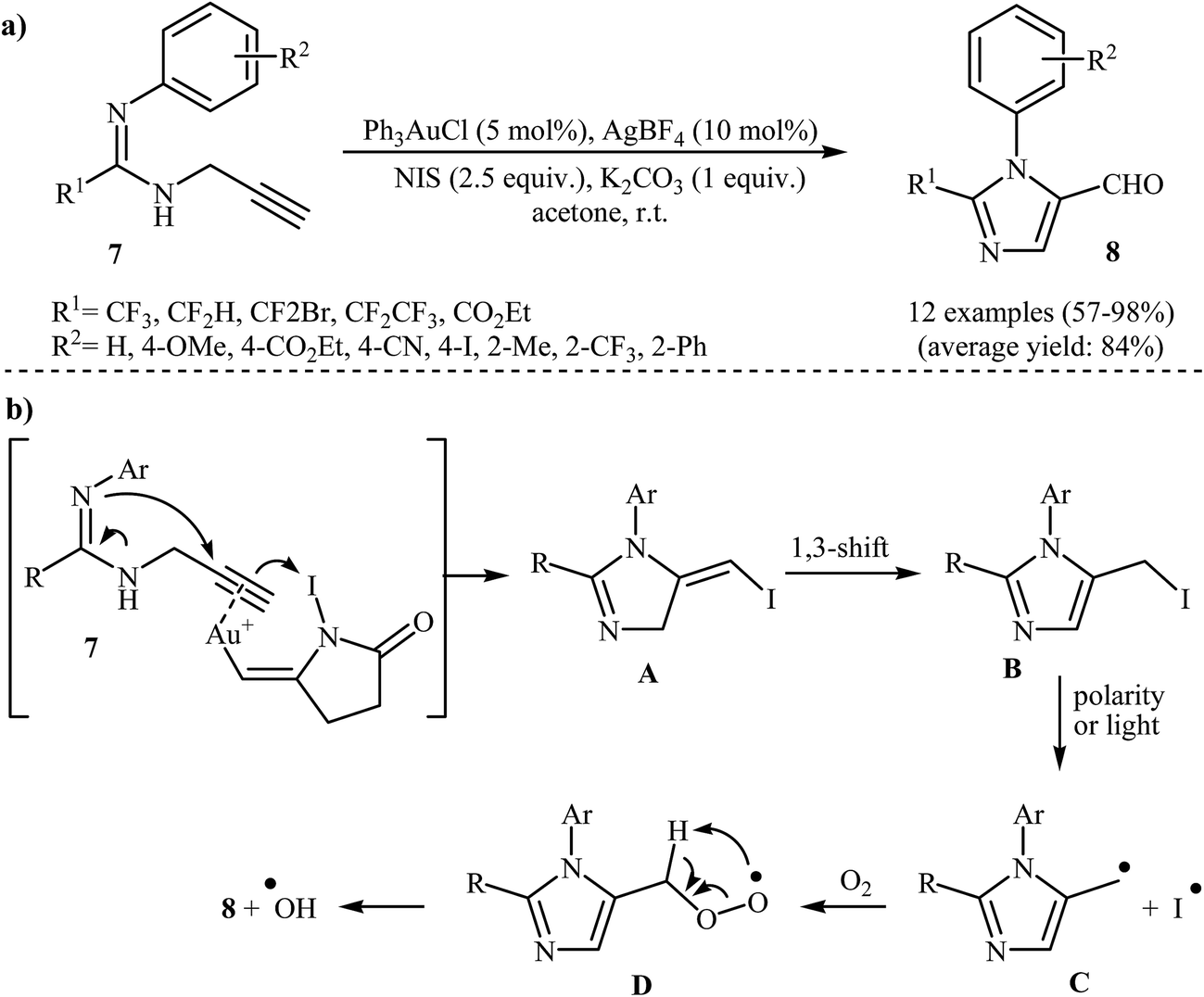 | ||
| Scheme 3 (a) Au(I)-Catalyzed intramolecular hydroamination of the fluorinated N-propargylamidines 7 to imidazole-5-carbaldehydes 8. (b) Proposed mechanism for the formation of 8. | ||
In 2010, Shen and Xie described a general and efficient synthesis of a diverse collection of di- and tri-substituted imidazoles 12 via the Ti-catalyzed regioselective cyclization of the corresponding N-propargylamines 10 with nitriles 11 under reflux conditions in toluene. The results demonstrated that the substrates with an internal alkyne unit gave higher yields than those with a terminal unit, and aryl nitriles were more reactive than alkyl nitriles. It was also found that the electronic character of the aryl nitriles had little to no effect on the yield or outcome of the methodology. The mechanism proposed by the authors to explain this reaction starts with the generation of the titanacarborane amide A via the amine-exchange reaction between [σ:η1:η5 (OCH2)(Me2NCH2)C2B9H9]Ti(NMe2) 9 and N-propargylamine 10. Then, coordination of nitrile 11 to the Ti atom of this intermediate furnishes intermediate B. Subsequently, a migratory insertion gives intermediate C. The formation of intermediate D occurs next, followed by its acid–base reaction with N-propargylamine 10 to give the dihydro-imidazole E with concomitant regeneration of A. Finally, the dehydrative aromatization of E affords the observed products 12 (Fig. 4).25 Recently the authors improved the efficiency of this reaction in terms of the yield and reaction time by performing the process in THF using Sm[N(SiMe3)2]3 as the catalyst.26
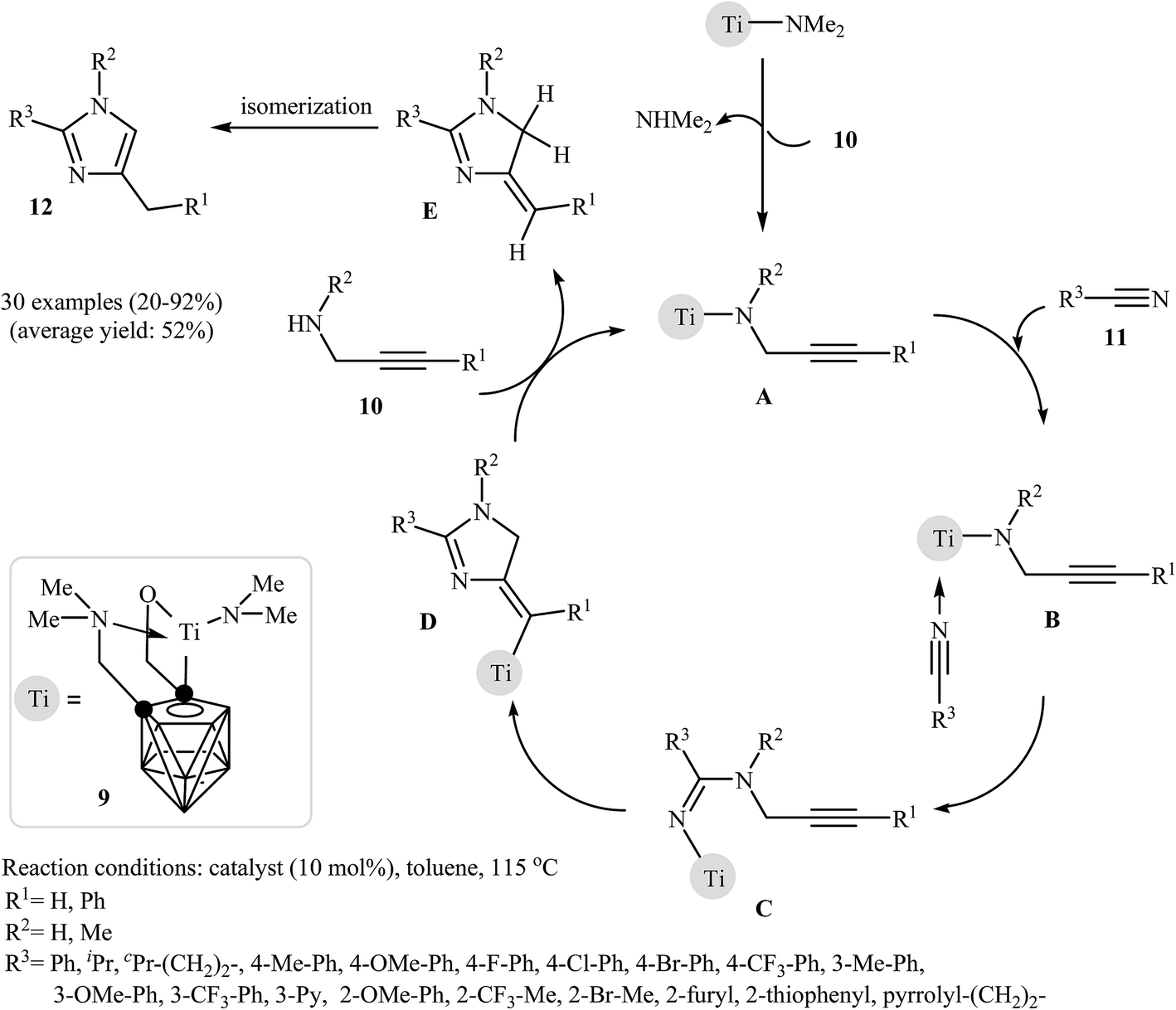 | ||
| Fig. 4 Mechanism proposed to explain the synthesis of substituted imidazoles 12 developed by Shen and Xie. | ||
The same authors were also able to demonstrate that a series of 2-aminoimidazoles 15 could be obtained from the reaction of N-propargylamines 13 with carbodiimides 14 in the presence of 5 mol% titanacarborane monoamide 9 in refluxing toluene (Scheme 4a). The reaction tolerated both terminal and aryl-substituted internal N-propargylamines and gave the corresponding 2-aminoimidazoles in good to excellent yields; however, extension of the reaction to alkyl-substituted internal alkynes failed. The reaction proceeds along a similar mechanistic pathway to that described in Fig. 4. It is worth noting that when secondary propargylamines were used as starting materials, instead of 2-aminoimidazole 15, the corresponding N-(1H-imidazol-2(3H)-ylidene)amines 16 were formed as the final products because they could not undergo aromatization to give imidazoles 15 (Scheme 4b).27
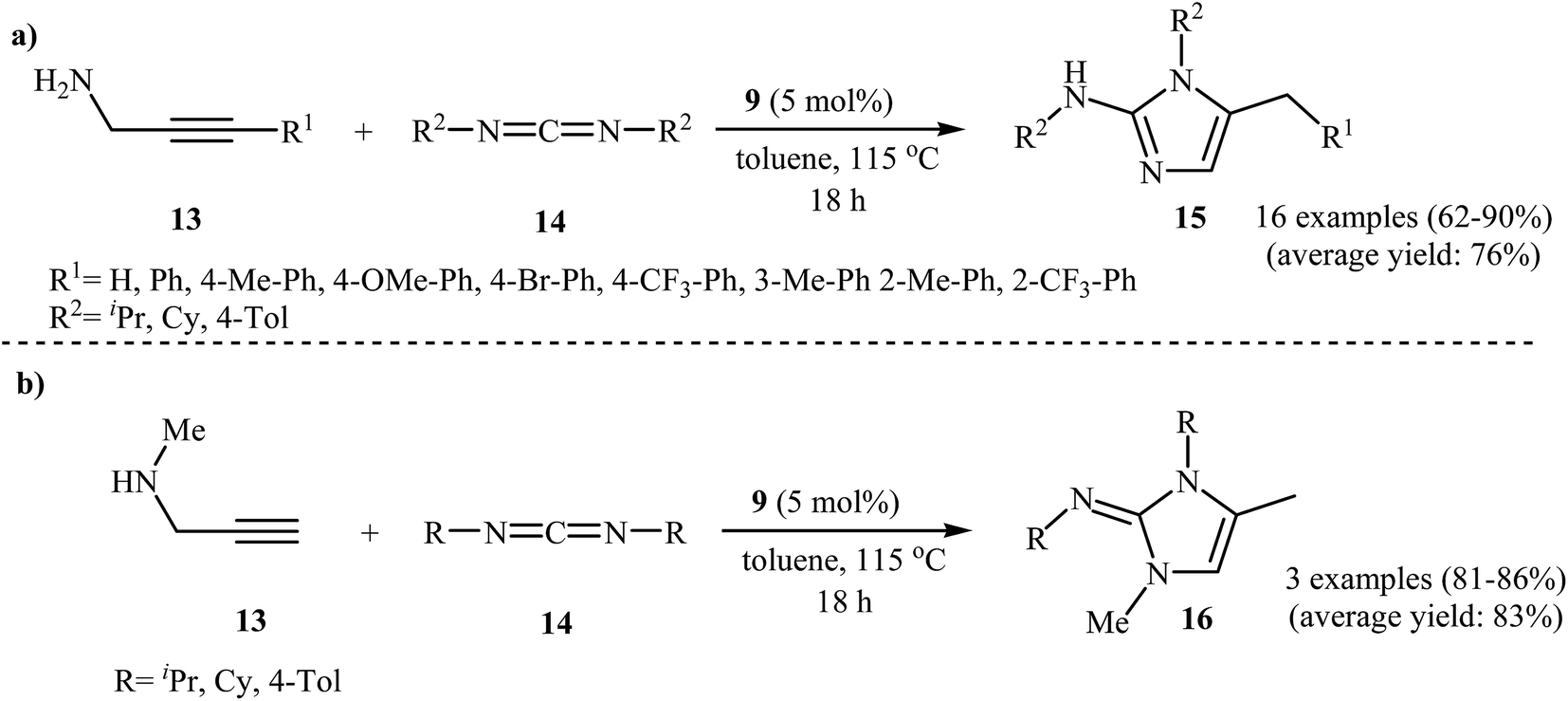 | ||
| Scheme 4 (a) Synthesis of 2-aminoimidazole 15 from the reaction of primary N-propargylamines 13 with carbodiimides 14. (b) Synthesis of N-(1H-imidazol-2(3H)-ylidene)amines 16 from the reaction of secondary N-propargylamines 13 with 14. | ||
More recently, Wang and co-workers studied the possibility of synthesizing imidazoles 19 from N-propargylamines 17 and ketenimines 18 through a silver-catalyzed nucleophilic addition/5-exo-dig cyclization/isomerization sequential process. The optimal reaction conditions developed included stirring of 1 mmol of the propargylamine, 1 equiv. of the ketenimine, 1 equiv. of Et3N and 10 mol% of AgOTf in 10 mL of THF at reflux temperature. The optimized protocol tolerated a variety of functional groups, such as nitro, bromo, ester, and methoxy, and generally provided the corresponding 1,2,5-trisubstituted 1H-imidazoles 19 in good yields (Scheme 5). The author proposed mechanism for this cyclization, which is outlined in Scheme 6.9
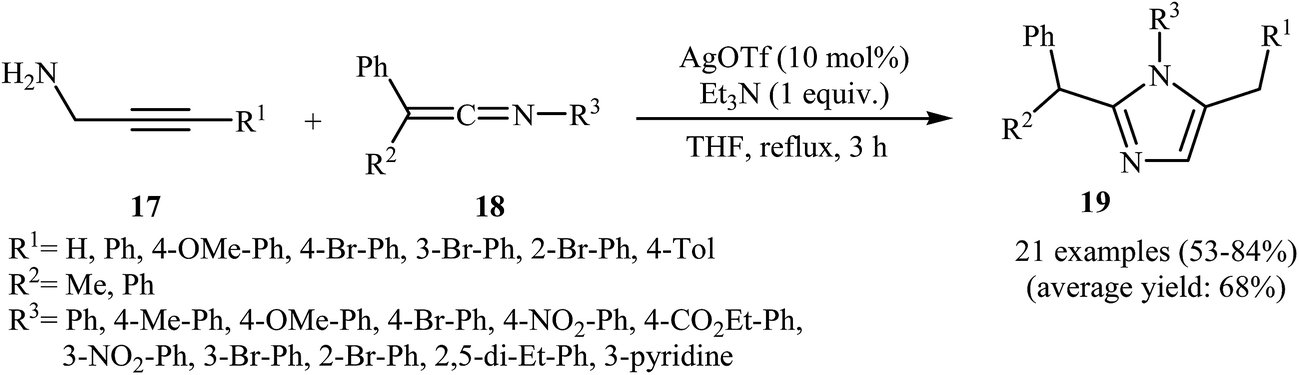 | ||
| Scheme 5 Ag-Catalyzed synthesis of imidazoles 19 from N-propargylamines 17 and ketenimines 18. | ||
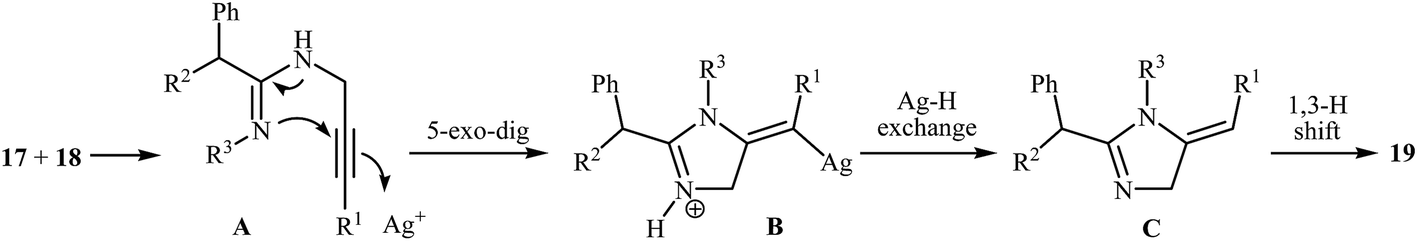 | ||
| Scheme 6 Plausible mechanism for the formation of 19. | ||
An efficient route to afford tetrasubstituted imidazoles 23 has been developed by the same group, which was based on a one-pot, two-step, three-component reaction between N-propargylamines 20, terminal alkynes 21, and N-sulfonyl azides 22 (Scheme 7a). These steps involve: (i) stirring of the starting materials in the presence of CuI as a catalyst and Et3N as a base in acetonitrile under nitrogen atmosphere and (ii) addition of Cs2CO3 and heating of the reaction mixture at 80 °C for 4 h. According to the mechanistic studies, this reaction proceeds through the Cu-catalyzed formation of ketenimine intermediate A, from the starting alkynes 21 and N-sulfonyl azides 22, followed by the nucleophilic addition of N-propargylamine 20 to the central carbon of ketenimine A to give intermediate B, which, in the presence of base, transforms into allene intermediate C. Subsequently the 6π-electrocyclic ring closing of intermediate C gives the ionic intermediate D. After a sulfonyl 1,3-shift, imidazoles 23 are observed (Scheme 7b).28
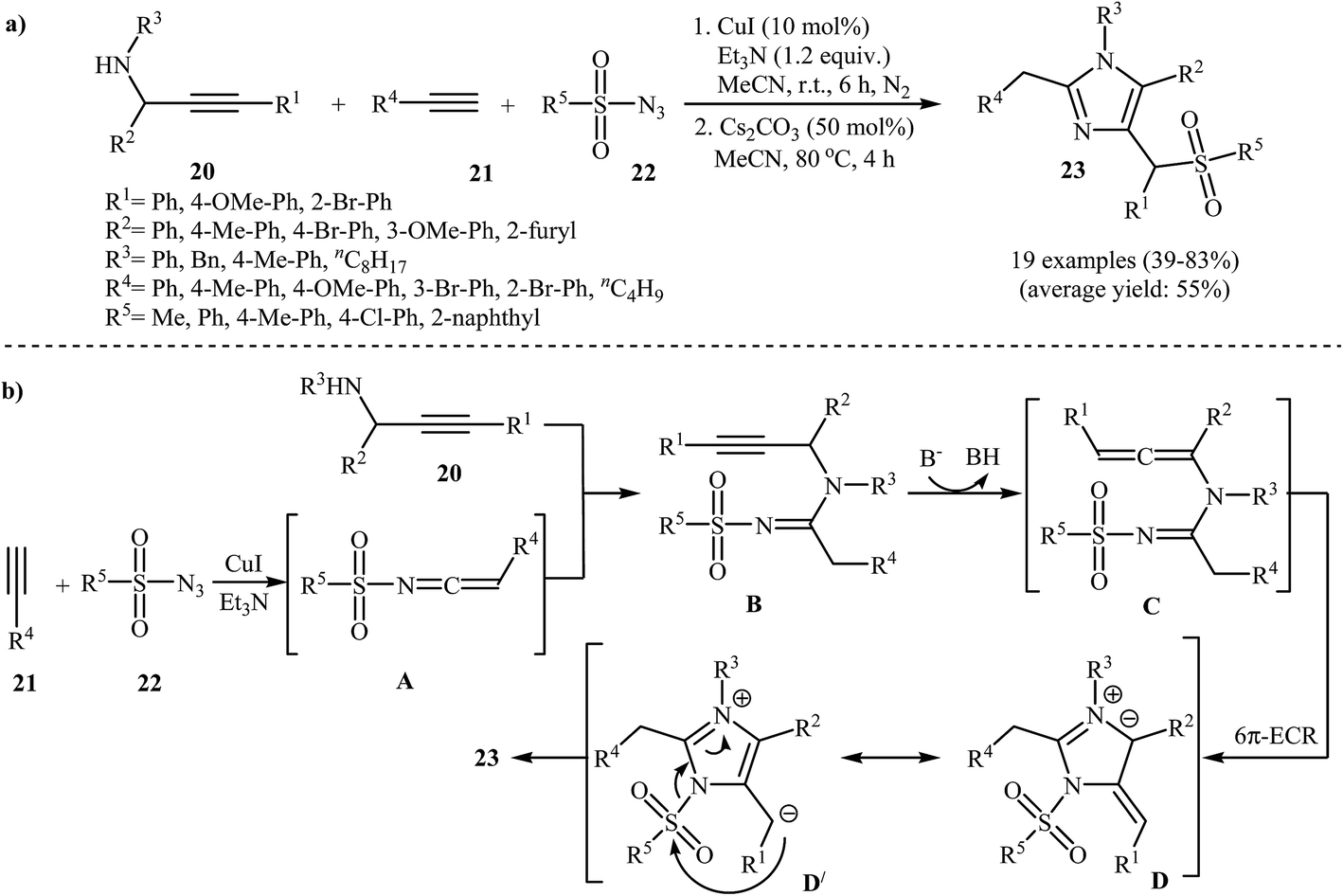 | ||
| Scheme 7 (a) Synthesis of tetrasubstituted imidazoles 23 via a Cu(I)-catalyzed three-component coupling approach. (b) Proposed mechanism for the formation of 23. | ||
3. Fused imidazoles
3.1. Imidazo[1,2-a]pyridines
Despite the fact that intramolecular cyclization of 2-amino N-propargylpyridinium bromides to afford the corresponding imidazo[1,2-a]pyrimidines had been known since 1964,29 it was not until 2008 that Bakherad and co-workers developed the first intramolecular cyclization of 2-amino N-propargylpyridinium bromides to form imidazo[1,2-a]pyridines. They showed that treatment of N-propargylpyridinium bromide 24 with iodobenzenes 25 in the presence of [(PPh3)2PdCl2]/CuI/Et3N as a catalytic system in DMF resulted in the corresponding 2-benzylimidazo[1,2-a]pyridines 26 in good to high yields. However, iodobenzenes bearing an electron-donating group failed to participate in this reaction. This transformation is believed to occur through a tandem Sonogashira coupling/intramolecular cyclization/aromatization process (Scheme 8a).30 Shortly afterwards, the authors improved their methodology in terms of yield and reaction time using polystyrene-supported palladium(II) ethylenediamine complex 27 as the catalyst (Scheme 8b).31 This reaction has been successfully applied as the key strategic step in synthesis of a series of imidazopyridinium analogues 28 as antagonists of neuropeptide S receptor (Scheme 9).32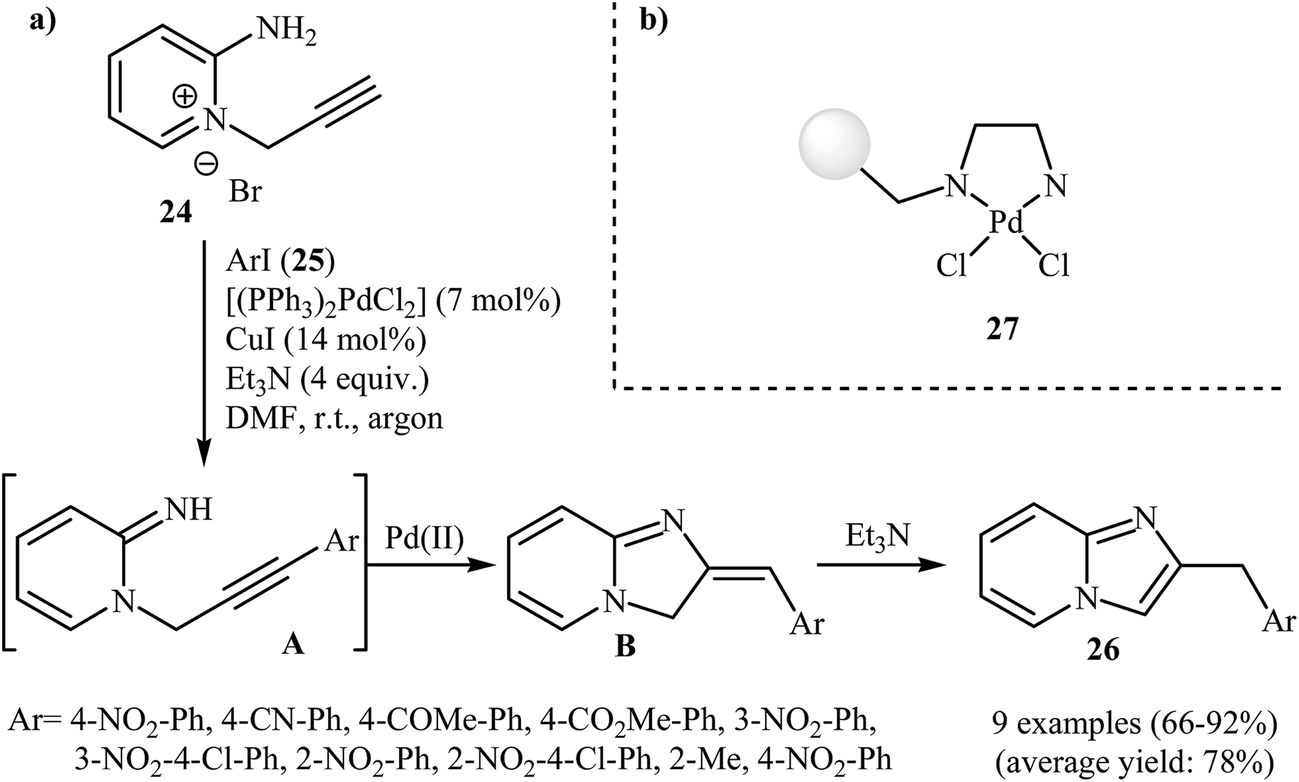 | ||
| Scheme 8 (a) Synthesis of 2-benzylimidazo[1,2-a]pyridines 26 through a tandem Sonogashira coupling/intramolecular cyclization/aromatization sequential process. (b) Chemical structure of polystyrene-supported palladium(II) ethylenediamine complex 27. | ||
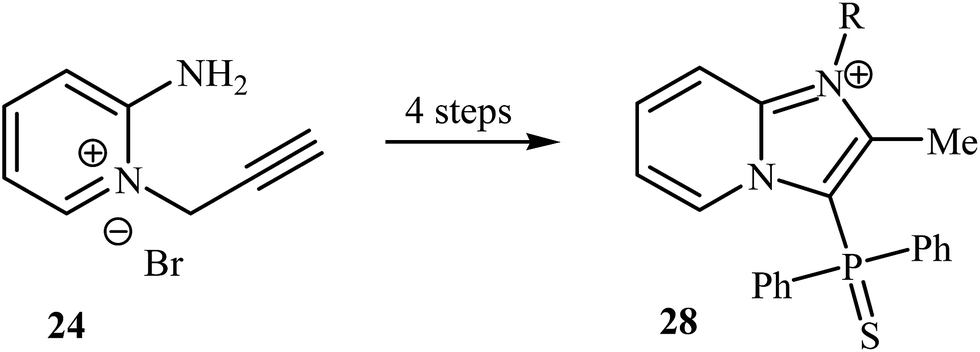 | ||
| Scheme 9 Synthesis of imidazopyridinium analogues 28 as antagonists of neuropeptide S receptor reported by Marugan. | ||
More recently, an impressive and more robust protocol for the synthesis of substituted imidazo[1,2-a]pyridines was introduced by Nguyen et al. They found that readily accessible N-propargylpyridiniums 29, in the presence of NaOH in water, rapidly cyclized and generally produced the corresponding substituted 2-methylH-imidazo[1,2-a]pyridines 30 in almost quantitative yields (Scheme 10). The mechanism of this cyclization was demonstrated via isotopic labeling experiments. The mechanism has been determined to proceed via a tandem alkyne–allene isomerization/Nazarov-type cyclization.33
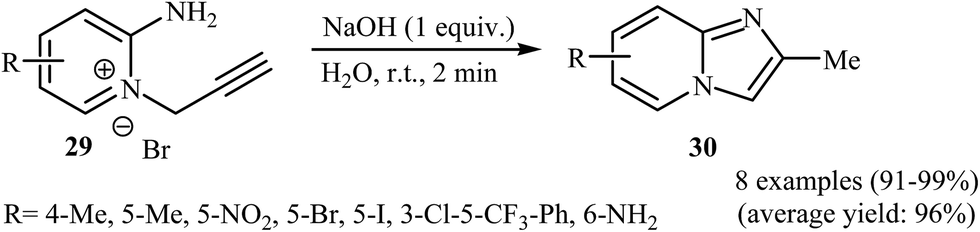 | ||
| Scheme 10 Metal-free and aqueous synthesis of imidazo[1,2-a]pyridine 30 from N-propargylpyridiniums 29. | ||
The possibility of regioselective intramolecular cyclization of N-propargylaminopyridines to pyridine fused imidazoles was first realized by Savic and co-workers, who synthesized a series of mono and disubstituted 3-methylH-imidazo[1,2-a]pyridine derivatives 32 from Boc-protected N-propargylaminopyridines 31 in the presence of tBuOK in THF. As shown in Scheme 11, the reaction showed remarkable flexibility and the desired products were formed in moderate to good yields with both electron-rich and electron-poor N-propargylaminopyridines; however, it could not be extended to 3-bromo substituted pyridines.34 Shortly after, the group of M. Chioua further improved the efficiency of this cyclization using AgOTf as the catalyst in deoxygenated acetonitrile. They also probed the mechanism of the reaction and found that the reaction proceeded via a 5-exo-dig cyclization process.35
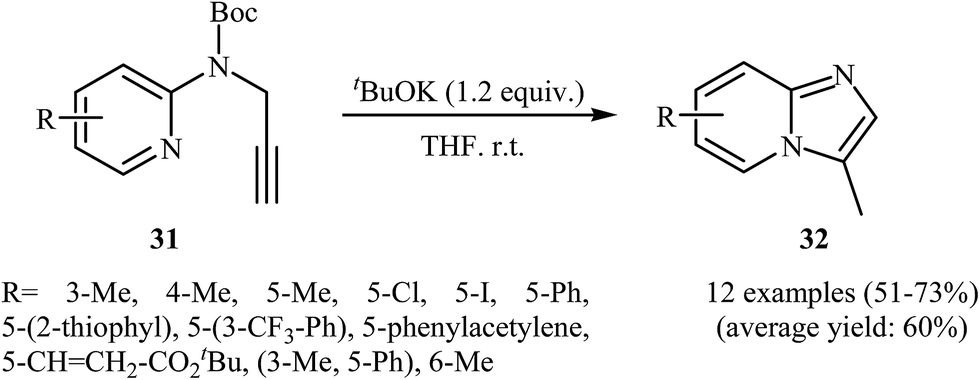 | ||
| Scheme 11 Intramolecular cyclization of N-propargylaminopyridines 31 to afford imidazo[1,2-a]pyridines 32 developed by Savic. | ||
With the objective of designing a greener procedure to pyridine-fused imidazoles through an intramolecular hydroamination strategy, Adimurthy and co-workers were able to demonstrate that a series of substituted imidazole[1,2-a]pyridines 34 could be obtained from N-propargylaminopyridines 33 under metal-free conditions in the most environmentally benign solvent, water. The mechanistic course of this reaction sequence is depicted in Scheme 12 and shows that water, presumably, plays a dual role as solvent and catalyst. It should be mentioned that the reaction failed under an open air atmosphere.36
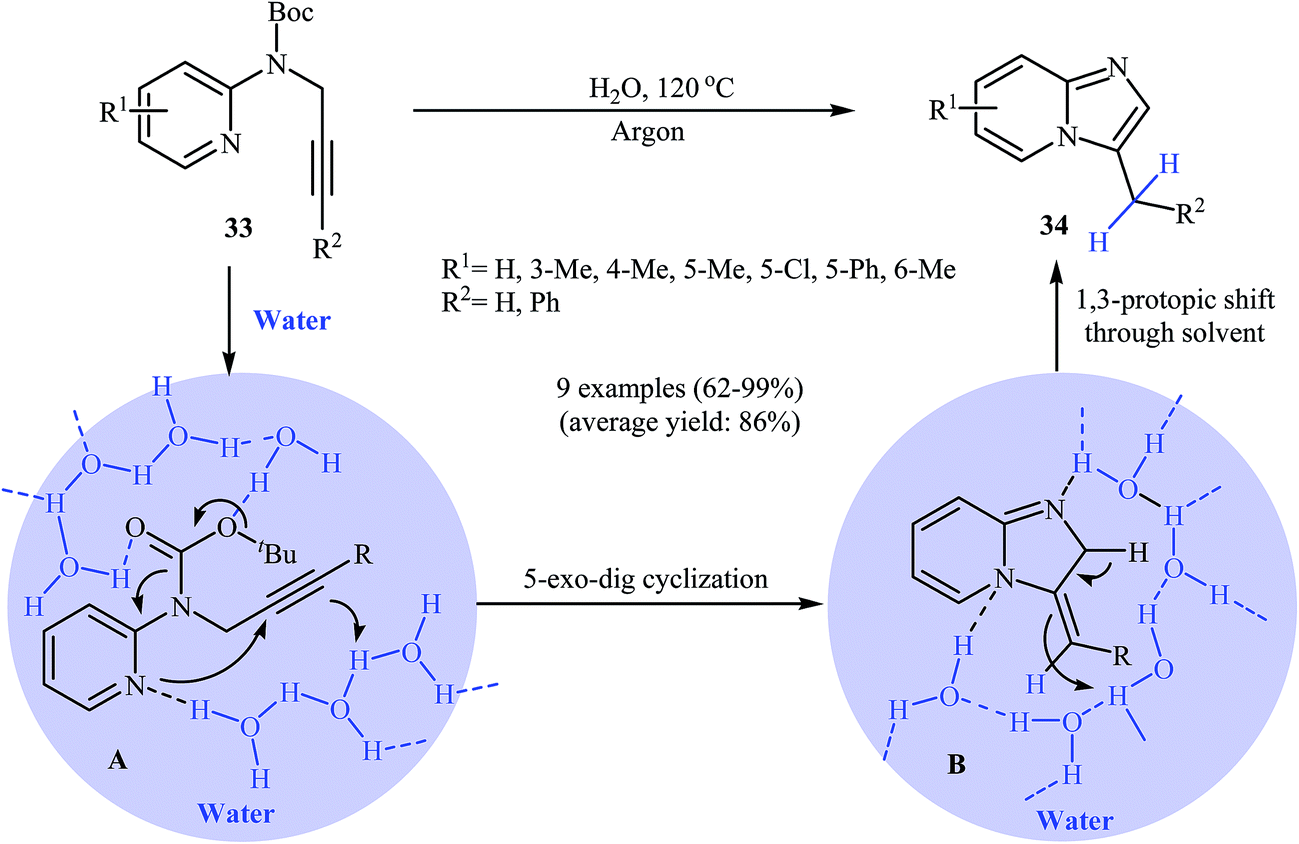 | ||
| Scheme 12 Intramolecular hydroamination of Boc-protected N-propargylaminopyridines 33 in water. | ||
Interestingly, when the abovementioned reaction is performed in the presence of AgNO3/TMEDA/acetonitrile under an oxygen atmosphere, imidazo[1,2-a]-pyridine-3-carbaldehydes 36 are obtained. The mechanism proposed to explain this transformation starts with the generation of π-complex A via coordination of the alkyne moiety with silver. Then, the cyclization of A gives intermediate B. Subsequently, addition of oxygen to intermediate B furnishes organosilver peroxide intermediate C. The formation of intermediate D occurs next, followed by its isomerization to give intermediate E. Finally, the elimination of the silver(I) species affords the observed products 36 (Scheme 13).37 It should be noted that, previously, the research team of Marco-Contelles had reported two examples of imidazo[1,2-a]-pyridine-3-carbaldehydes preparation via the Sandmeyer reaction of N-propargylaminopyridines.38
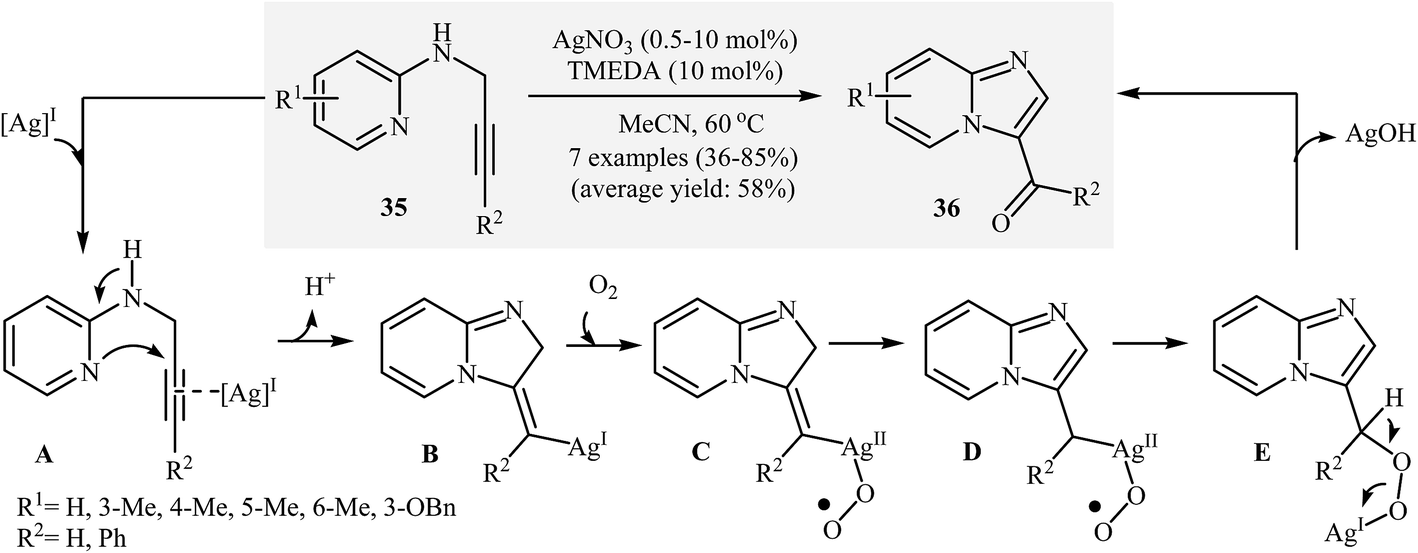 | ||
| Scheme 13 Ag-Catalyzed synthesis of imidazo[1,2-a]-pyridine-3-carbaldehydes 36 described by Adimurthy. | ||
3.2. Imidazo[1,2-a]-pyrazine (piperazine)
An interesting and rare example for the synthesis of piperazine-fused imidazole was reported by McCort and Pascal in 1992. In anhydrous toluene at 100 °C, an intermolecular heterocyclization between 3-ethoxy-tetrahydropyrazine 37 and N-propargylamine 38 furnished imidazo[1,2-a]piperazine 39 in a yield of 65% (Scheme 14a).39 Later, imidazo[1,2-a]quinoxaline 41 was synthesized via an intramolecular reaction (Scheme 14b). 4-Amino-substituted analogues of 41 showed good inhibitory activities against IKK (IκB kinase).40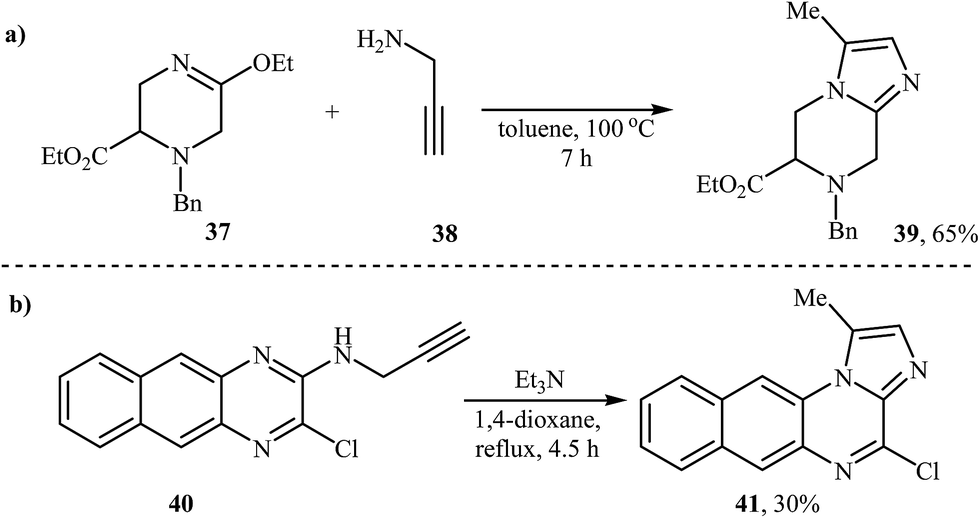 | ||
| Scheme 14 (a) Synthesis of imidazo[1,2-a]piperazine developed by McCort and Pascal. (b) Beaulieu's synthesis of imidazo[1,2-a]quinoxaline. | ||
A new method for the synthesis of substituted benzimidazolopyrazines 44 from N-propargylamine 42 and phenylenediamines 43, based on a tandem imidazole formation/heteroannulation process, has been reported recently. The reaction takes place through a tandem process consisting of an initial imidazole formation, followed by a copper-catalyzed 6-exo-dig cyclization and final isomerization (Scheme 15).41
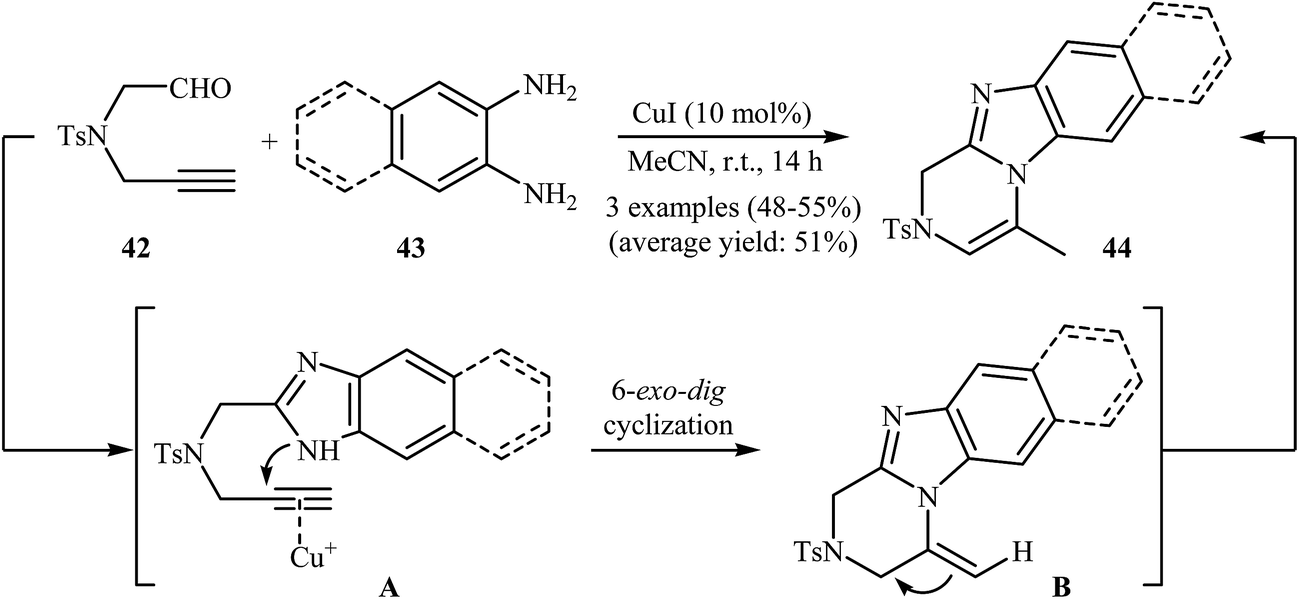 | ||
| Scheme 15 Synthesis of benzimidazolopyrazines 44 from N-propargylamine 42 and phenylenediamines 43 through a tandem imidazole formation/heteroannulation sequential process. | ||
3.3. Imidazo-diazepines
One of the earliest reports on the synthesis of benzoazepine-fused imidazoles from N-propargylamines appeared in 1977 when 4-amino-benzo[b][1,4]diazepinone 45 underwent cyclization with N-propargylamine 46 in the presence of p-toluenesulfonic acid as a catalyst in boiling butanol. The corresponding benzo[b][1,4]diazepinone-fused imidazoles 47 were obtained in moderate to high yields (Scheme 16).42 It is noted that 4-ethylthio-benzo[b][1,4]diazepines also gave the corresponding diazepine-fused imidazoles using the aforementioned reaction conditions.43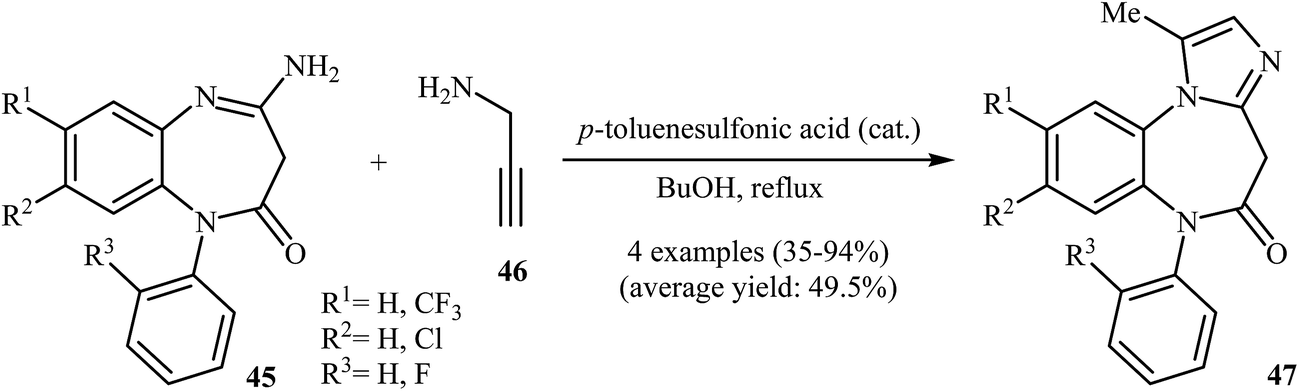 | ||
| Scheme 16 Synthesis of benzo[b][1,4]diazepinone fused imidazoles 47 from 45 and 46. | ||
In 2005, the group of Gracias described a general and efficient synthesis of tetracyclic diazepine-fused imidazoles 52 from N-propargylamines 48, o-azidobenzaldehydes 49, and p-toluenesulfonylmethyl isocyanides 50 through a van Leusen/alkyne–azide cycloaddition sequence. The reaction starts with the formation of van Leusen intermediates 51 using K2CO3 as a catalyst in DMF, and then, in situ intramolecular alkyne–azide cycloaddition of intermediates 50 furnishes the corresponding diazepine-fused imidazoles 52 in good to high yields. In the case of internal alkynes, the intramolecular alkyne–azide cycloaddition step was performed in tBuOH/H2O (1:1), and CuSO4·5H2O/sodium ascorbate was used as the catalytic system (Scheme 17).44
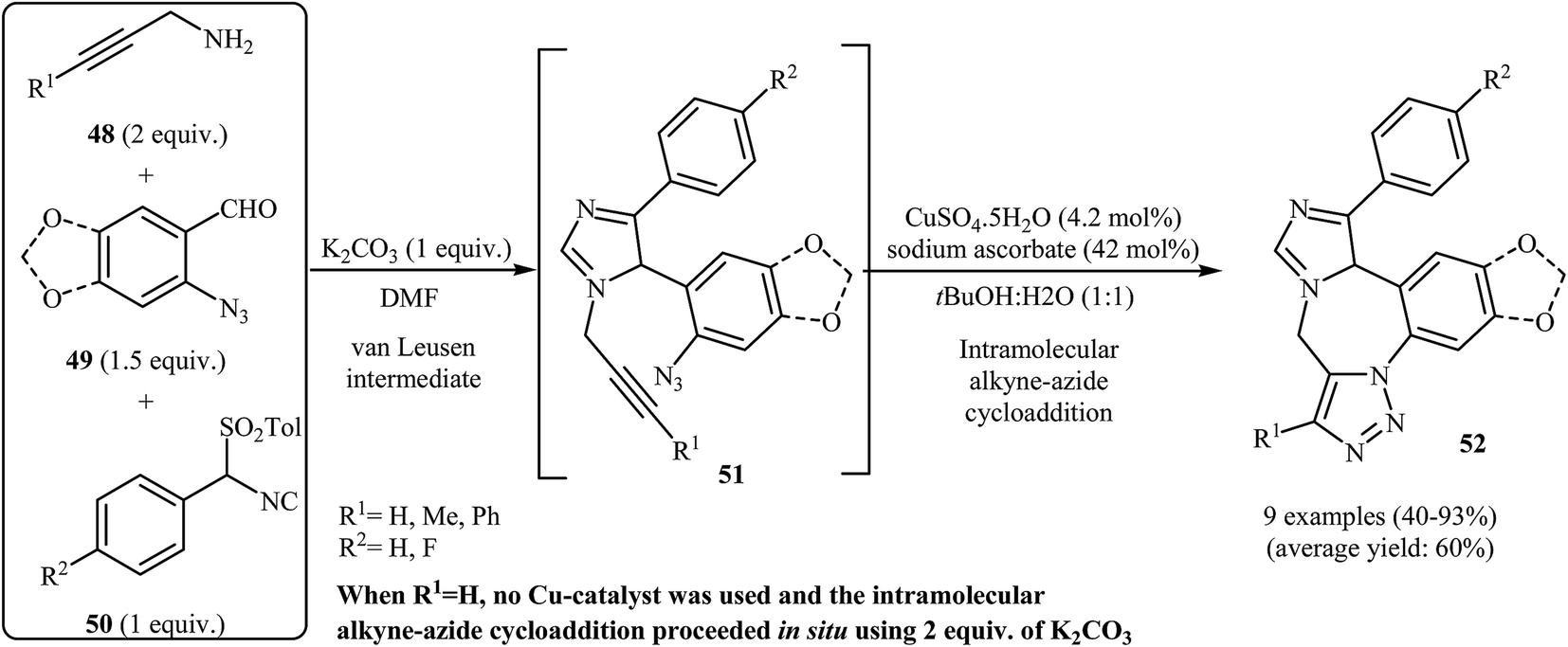 | ||
| Scheme 17 Synthesis of tetracyclic diazepine-fused imidazoles 52 by sequential van Leusen/alkyne–azide cycloaddition reactions. | ||
A straightforward route to 9H-benzo[f]imidazo[1,2-d1,2,3]triazolo[1,5-a1,4]diazepines 56, which were prepared because of their potential pharmaceutical interest, was developed by Kurth and co-workers. This route was based on a one-pot four-component reaction in methanol using o-azidobenzaldehydes 53, α-diketones 54, N-propargylamines 55, and ammonium acetate in the presence of 10 mol% InCl3 as the catalyst (Scheme 18). The mechanism of this cyclization reaction is believed to involve: (i) coordination of InCl3 to the oxygen of the aldehyde, which resulted in the formation of intermediate A; (ii) nucleophilic addition of ammonia to activated aldehyde A to produce imine intermediate B; (iii) reaction of imine B with N-propargylamine 55 to generate (2-azidophenyl)-N-(prop-2-ynyl)methanediamine intermediate C; (iv) intermolecular cyclization of C with activated α-diketone D to give intermediate E; and (v) intramolecular [3 + 2] Huisgen cycloaddition of E produced the corresponding imidazo-diazepines 56. In another possible mechanism, intramolecular [3 + 2] Huisgen cycloaddition of intermediate C affords intermediate F that transforms to the observed product 56 via condensation with activated α-diketone D (Scheme 19).45
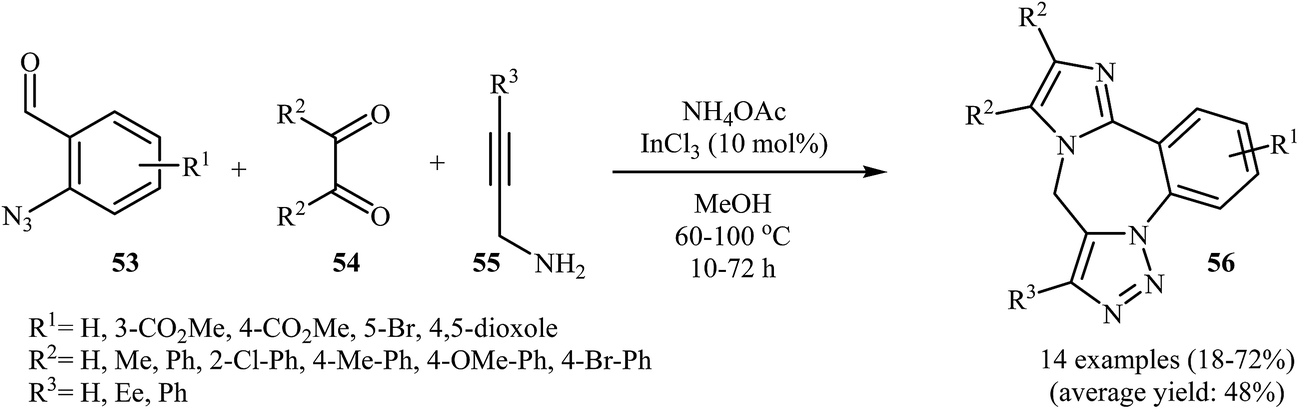 | ||
| Scheme 18 Four-component synthesis of imidazotriazolobenzodiazepanes 56 starting from o-azidobenzaldehydes, α-diketones, N-propargylamines, and ammonium acetate. | ||
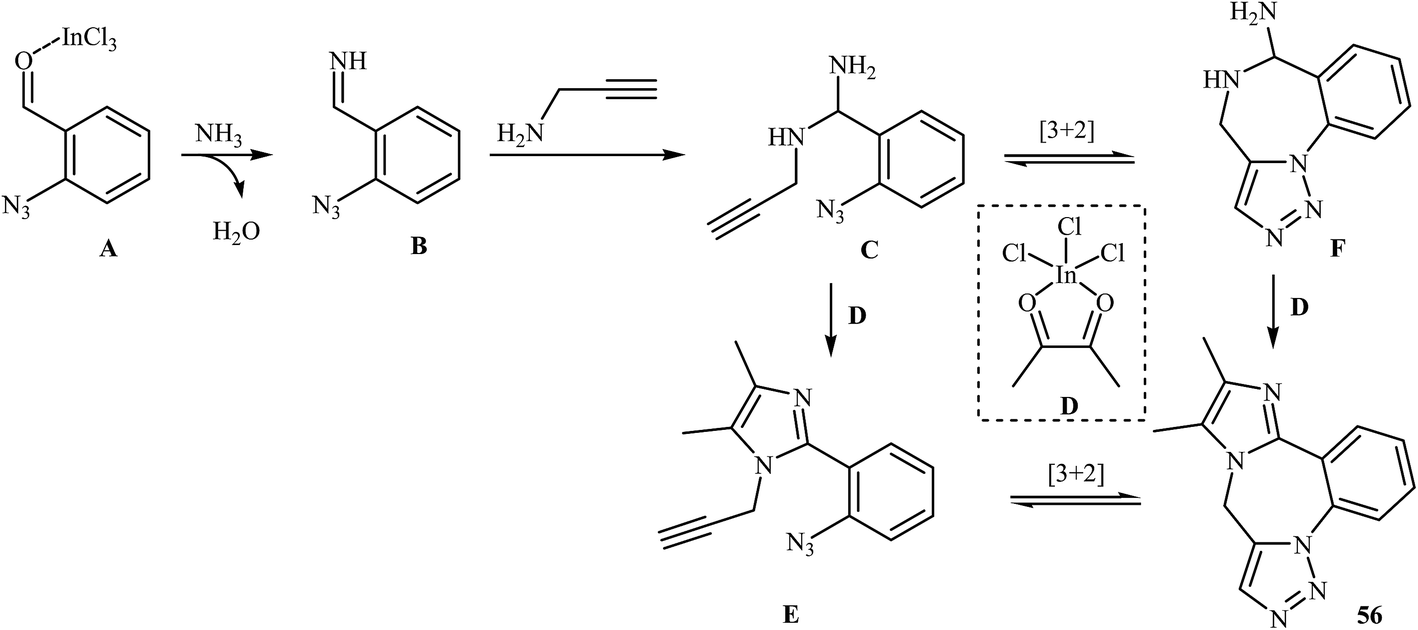 | ||
| Scheme 19 Mechanism that accounts for the formation of 56. | ||
3.4. Purine-fused imidazoles
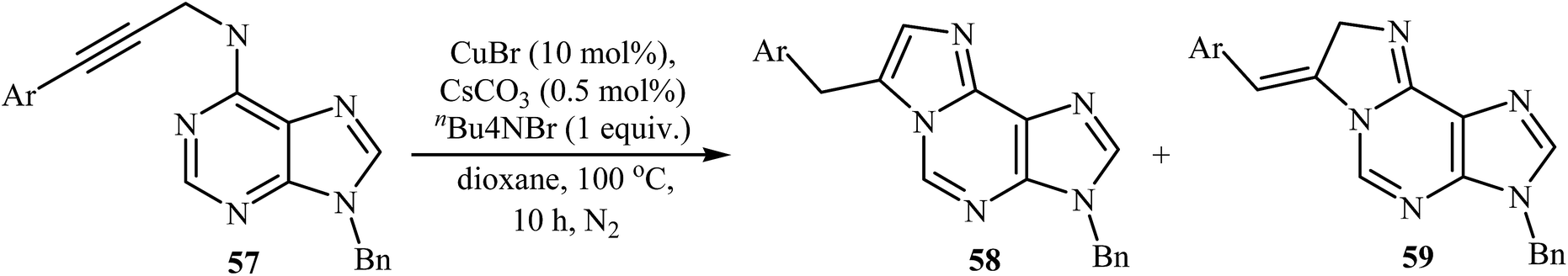
The intramolecular cyclization of N-propargyladenines 57, in the presence of CuBr/CsCO3/n-Bu4NBr as a catalytic system, to give purine-fused imidazoles 58 was described by Qu and co-workers in 2014. They observed that the substituent effects on the intramolecular cyclization afforded a mixture of products. Thus, the terminal alkynes afforded predominantly purine-fused imidazoles 58, but the aryl substituted internal alkynes yielded a mixture of 58 and dihydroimidazoles 59. The substrates that had electron-rich aryl rings in the alkenyl part favored endocyclic double bond products, whereas the substrates bearing electron-poor aryl rings in the alkyne terminus favored exocyclic double bond products (Table 1). According to the proposed mechanism, the reaction proceeds via an alkyne–allene isomerization/tautomerization/cyclization sequential process.46
|
|||
|---|---|---|---|
| Entry | Ar | Yield (%) | 58:59 |
| 1 | Ph | 88 | 99:1 |
| 2 | 2-Me-Ph | 89 | 92:8 |
| 3 | 3-Me-Ph | 87 | 91:9 |
| 4 | 3,5-Di-Me-Ph | 79 | 83:13 |
| 5 | 2-OMe-Ph | 85 | 86:14 |
| 6 | 4-OMe-Ph | 85 | 88:12 |
| 7 | 4-OEt-Ph | 88 | 92:8 |
| 8 | 3-Cl-Ph | 81 | 47:53 |
| 9 | 4-Cl-Ph | 88 | 45:55 |
| 10 | 4-F-Ph | 75 | 43:57 |
| 11 | 2-Thiophyl | 77 | 45:55 |
4. Conclusion
The imidazole scaffold is a core structure of a large number of natural products and synthetic pharmaceuticals that show an impressive variety of biological properties. Such compounds exhibit anticancer, anti-tubercular, anti-parasitic, anti-neuropathic, anti-convulsion, anti-Parkinson's, antifungal, antibacterial, antiviral, antihistaminic, anti-inflammatory, and anti-obesity properties. Several commercially available drugs, including alpidem, zolpidem, clonidine, metronidazole, flumazenil, bretazenil, imidazenil, and midazolam, are derived from imidazo-core entities. Consequently, numerous efforts have been devoted to the design of expedient and efficient synthetic routes to this heterocycle. Some of the most popular methods for their preparation include the Debus, Radiszewski, Wallach, Marckwald, and Van Leusen reactions. Widespread use of these methods is limited by the low yields, low selectivity, or narrow substrate scope.9,35 Thus, new and more efficient synthetic methods are sought. Developing more efficient methods for the generation of fused imidazole cores is particularly interesting.New methods that produce complex molecules from simpler materials in a single operation are important challenges for modern synthetic chemistry. Over the past decade, N-propargylamines have attracted much attention due to the fact that they are versatile and simple synthetic intermediates for the synthesis of many biologically active N-heterocycles. As illustrated, the synthesis of imidazole derivatives from N-propargylamines has gained a great deal of interest in recent years as useful alternative procedures. Shorter synthetic routes, high regioselectivity, and high atom economy are the key features of these reactions. It is anticipated that the insight provided in this account will be beneficial for eliciting further research in this domain.
References
- L. Zhang, X. M. Peng, G. L. Damu, R. X. Geng and C. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed.
- (a) G. K Gupta, V. Kumar and K. Kaur, Nat. Prod. J., 2014, 4, 73–81 Search PubMed; (b) V. Kumar and M. P. Mahajan, Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, pp. 507–533 Search PubMed; (c) S. N. Azizi, P. Shakeri, M. J. Chaichi, A. Bekhradnia, M. Taghavi and M. Ghaemy, Spectrochim. Acta, Part A, 2014, 122, 482–488 CrossRef CAS PubMed; (d) S. Arshadi, A. R. Bekhradnia and A. Ebrahimnejad, Can. J. Chem., 2011, 89, 1403–1409 CrossRef CAS.
- M. B. Kjærstad, C. Taxvig, C. Nellemann, A. M. Vinggaard and H. R. Andersen, Reprod. Toxicol., 2010, 30, 573–582 CrossRef PubMed.
- H. Debus, Justus Liebigs Ann. Chem., 1858, 107, 199–208 CrossRef.
- B. Radzisewski, Chem. Ber., 1882, 15, 2706–2708 CrossRef.
- T. Benincori, E. Brenna and F. Sannicolo, J. Chem. Soc., Perkin Trans. 1, 1993, 675–679 RSC.
- W. Marckwald, Chem. Ber., 1892, 25, 2359–2361 Search PubMed.
- A. M. Vanleusen, J. Wildeman and O. Oldenziel, J. Org. Chem., 1977, 42, 1153–1159 CrossRef CAS.
- X. Zhou, Z. Jiang, L. Xue, P. Lu and Y. Wang, Eur. J. Org. Chem., 2015, 5789–5797 CrossRef CAS.
- E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC.
- E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, Curr. Org. Synth., 2016 DOI:10.2174/1570179413666160818144816.
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC.
- F. Eloy, A. Deryckere and J. Maffrand, Eur. J. Med. Chem., 1974, 9, 602–606 CAS.
- E. C. Taylor, P. Zhou, C. M. Tice, Z. Lidert and R. C. Roemmele, Tetrahedron Lett., 1997, 38, 4339–4342 CrossRef CAS.
- C. M. Tice and L. M. Bryman, Tetrahedron, 2001, 57, 2689–2700 CrossRef CAS.
- A. Jezewski, J. Jurczak, Z. Lidert and C. M. Tice, J. Heterocycl. Chem., 2001, 38, 645–648 CrossRef CAS.
- D. D. Diaz, W. G. Lewis and M. Finn, Synlett, 2005, 2214–2218 CAS.
- A. C. Barrios Sosa, R. T. Williamson, R. Conway, A. Shankar, R. Sumpter and T. Cleary, Org. Process Res. Dev., 2011, 15, 449–454 CrossRef CAS.
- G. Abbiati, A. Arcadi, V. Canevari and E. Rossi, Tetrahedron Lett., 2007, 48, 8491–8495 CrossRef CAS.
- (a) A. Shafee, M. Z. Kassaee and A. R. Bekhradnia, J. Heterocycl. Chem., 2007, 44, 471–474 CrossRef; (b) S. Li, Y. Yuan, Y. Li, Z. Li, L. Zhang and Y. Wu, Org. Biomol. Chem., 2013, 11, 41–43 RSC.
- S. Li, Z. Li, Y. Yuan, D. Peng, Y. Li, L. Zhang and Y. Wu, Org. Lett., 2012, 14, 1130–1133 CrossRef CAS PubMed.
- S. Li, Z. Li, Y. Yuan, Y. Li, L. Zhang and Y. Wu, Chem.–Eur. J., 2013, 19, 1496–1501 CrossRef CAS PubMed.
- H. Shen and Z. Xie, J. Am. Chem. Soc., 2010, 132, 11473–11480 CrossRef CAS PubMed.
- L. Hong, Y. Shao, L. Zhang and X. Zhou, Chem.–Eur. J., 2014, 20, 8551–8555 CrossRef CAS PubMed.
- Y. Wang, H. Shen and Z. Xie, Synlett, 2011, 2011, 969–973 CrossRef.
- Z. Jiang, P. Lu and Y. Wang, Org. Lett., 2012, 14, 6266–6269 CrossRef CAS PubMed.
- I. Iwai and T. Hiraoka, Chem. Pharm. Bull., 1964, 12, 813 CrossRef CAS PubMed.
- M. Bakherad, H. Nasr-Isfahani, A. Keivanloo and N. Doostmohammadi, Tetrahedron Lett., 2008, 49, 3819–3822 CrossRef CAS.
- M. Bakherad, B. Bahramian, H. Nasr-Isfahani, A. Keivanloo and N. Doostmohammadi, J. Heterocycl. Chem., 2009, 46, 100–104 CrossRef CAS.
- S. Patnaik, J. J. Marugan, K. Liu, W. Zheng, N. Southall, S. J. Dehdashti, A. Thorsell, M. Heilig, L. Bell and M. Zook, J. Med. Chem., 2013, 56, 9045–9056 CrossRef CAS PubMed.
- M. R. Chapman, M. H. Kwan, G. E. King, B. A. Kyffin, A. J. Blacker, C. E. Willans and B. N. Nguyen, Green Chem., 2016, 18, 4623–4627 RSC.
- S. Husinec, R. Markovic, M. Petkovic, V. Nasufovic and V. Savic, Org. Lett., 2011, 13, 2286–2289 CrossRef CAS PubMed.
- M. Chioua, E. Soriano, L. Infantes, M. L. Jimeno, J. Marco-Contelles and A. Samadi, Eur. J. Org. Chem., 2013, 35–39 CrossRef CAS.
- D. C. Mohan, N. B. Sarang and S. Adimurthy, Tetrahedron Lett., 2013, 54, 6077–6080 CrossRef.
- D. Chandra Mohan, S. Nageswara Rao and S. Adimurthy, J. Org. Chem., 2013, 78, 1266–1272 CrossRef CAS PubMed.
- D. Sucunza, A. Samadi, M. Chioua, D. B. Silva, C. Yunta, L. Infantes, M. C. Carreiras, E. Soriano and J. Marco-Contelles, Chem. Commun., 2011, 47, 5043–5045 RSC.
- G. A. McCort and J. C. Pascal, Tetrahedron lett., 1992, 33, 4443–4446 CrossRef CAS.
- F. Beaulieu, C. Ouellet, E. H. Ruediger, M. Belema, Y. Qiu, X. Yang, J. Banville, J. R. Burke, K. R. Gregor and J. F. MacMaster, Bioorg. Med. Chem. Lett., 2007, 17, 1233–1237 CrossRef CAS PubMed.
- S. Ramesh, S. K. Ghosh and R. Nagarajan, Org. Biomol. Chem., 2013, 11, 7712–7720 CAS.
- T. Hara, H. Fujimori, Y. Kayama, T. Mori, K. Itoh and Y. Hashimato, Chem. Pharm. Bull., 1977, 25, 2584–2592 CrossRef CAS PubMed.
- M. Di Braccio, G. Grossi, G. Roma, L. Vargiu, M. Mura and M. E. Marongiu, Eur. J. Med. Chem., 2001, 36, 935–949 CrossRef CAS PubMed.
- V. Gracias, D. Darczak, A. F. Gasiecki and S. W. Djuric, Tetrahedron Lett., 2005, 46, 9053–9056 CrossRef CAS.
- H. H. Nguyen, T. A. Palazzo and M. J. Kurth, Org. Lett., 2013, 15, 4492–4495 CrossRef CAS PubMed.
- R.-L. Li, L. Liang, M.-S. Xie, G.-R. Qu, H.-Y. Niu and H.-M. Guo, J. Org. Chem., 2014, 79, 3665–3670 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |