DOI:
10.1039/C7QO00061H
(Research Article)
Org. Chem. Front., 2017,
4, 823-827
Synthesis of largely π-extended naphthalenediimides via C–H activation towards highly soluble and narrow band-gap organic optoelectronic materials†
Received
22nd January 2017
, Accepted 21st March 2017
First published on 22nd March 2017
Abstract
Atom-economic coupling reactions based on C–H activation are generally used to synthesize small conjugated systems, but their applications in constructing largely π-conjugated systems have been rarely reported. Herein, we report the preparation of largely π-extended naphthalenediimides (4a–c, with molecular weights of up to 2800) by using efficient palladium-catalyzed C–H/C–H homocouplings, with the yields of up to 94%. The physicochemical properties of 4a–c were studied and they exhibited excellent solubility, thermal stability, quasi-reversible reduction processes, broad and long-wavelength optical absorptions with thin-film optical gaps <1.4 eV and fine-tuned deep LUMO energies (<−4.2 eV), which together with ambient stable electron-transporting behaviors demonstrate their promising applications in optoelectronic devices.
Introduction
Organic semiconductors (OSCs) with largely π-conjugated scaffolds have recently attracted remarkable attention due to their wide applications in organic optoelectronic devices,1 such as organic photovoltaics (OPVs),2 organic field-effect transistors (OFETs)3 and organic photodetectors.4 Among them, those having electron-deficient characteristics are particularly significant for their use in n-type semiconductors, electron acceptors of OPVs, organic thermoelectricity materials, etc.5 Although great achievements have been made for OSCs in terms of the varieties of chemical structures and high optoelectronic performances,3b their synthetic approaches largely depend on traditional transition-metal-catalyzed coupling techniques, especially the high toxic Stille coupling reactions.6 Typically, traditional coupling reactions involve either coupling of aryl halides with organometallic reagents or homocoupling of two aryl halides or two organometallic reagents. Most of them often require the use of pre-activated aryl halides and/or organometallic substrates, especially high toxic organotin reagents, which are wasteful, time-consuming and even toxic with environmentally risky by-products.7 Recently, palladium-catalyzed coupling reactions based on C–H activation have emerged as a sustainable, atom-economic and environmentally friendly synthetic strategy for the construction of aromatic carbon–carbon (C–C) bonds.8 In particular, the dehydrohalogenative coupling of (hetero)arenes with aryl halides (so called direct arylation) has been deeply investigated and successfully applied in the synthesis of conjugated small molecules and polymers for organic optoelectronic devices.9 Moreover, oxidative dehydrogenative coupling of aromatic C–H bonds represents another simple, efficient and pre-activation free access to small molecular and polymeric π-systems.8a,10 However, the preparation of OSCs, especially the ones with large π-conjugated scaffolds, via this tandem oxidation of C–H bonds has been rarely explored,10 except the homocoupling of oligothiophenes.11 Therefore, the exploration of synthesis of π-extended conjugations by using oxidative dehydrogenative coupling is highly desirable.
In 2014, we reported a strong electron-deficient building block NDI–DTYA2 (Fig. 1), where the naphthalenediimide (NDI) core was fused with two 2-(1,3-dithiol-2-ylidene)acetonitrile (DTYA) units.12 Conjugated copolymers based on NDI–DTYA2 were prepared and exhibited near-infrared (NIR) optical properties as well as the pure electron transport behaviour.12 It should be noted that two vinyl C–H bonds of NDI–DTYA2 (Fig. 1) could be further modified to afford novel π-conjugated structures. In this contribution, we report a green and efficient C–H/C–H homocoupling method for preparing largely π-extended scaffolds (DTYA–NDI–DTYA)2 (Fig. 1) based on NDI–DTYA2 units, and a plausible reaction mechanism of homocoupling is proposed. As shown in Scheme S2,† the homocoupling to achieve 4 proceeds via reductive coupling of bis-vinylpalladium species A, which is formed by the electrophilic C–H substitution of PdCl2(PhCN)2 with 3 and the following disproportionation, giving the intermediate B. In the homocoupling reaction, KF might serve as an effective promoter in the formation of the intermediate B, and AgNO3 acts as an oxidizing agent to regenerate a Pd(II) catalyst from Pd(0) species.
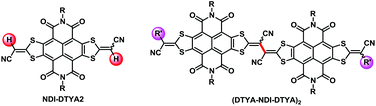 |
| | Fig. 1 Chemical structures of NDI–DTYA2 and (DTYA–NDI–DTYA)2 derivatives. | |
Herein, in order to endow (DTYA–NDI–DTYA)2 with fine-tunable molecular energy levels,13 the end-capped groups with different electron-accepting abilities were incorporated at one DTYA moiety of NDI–DTYA2 via the Suzuki–Miyaura cross coupling reaction between the monolateral bromide NDI–DTYA2 derivative and the corresponding aromatic boronic acid. Three largely π-conjugated scaffolds (4a–c, Scheme 1) based on NDI–DTYA2 units were achieved in 85–94% yields by using the vinyl C–H/C–H homocouplings of R′ monosubstituted NDI–DTYA2 derivatives (3a–c). Changing the end-capped groups of 4a–c (R′ = 4-(trifluoromethyl)phenyl, phenyl and 4-(diphenylamino)phenyl for 4a–c, respectively) makes it possible to fine-tune the molecular energy levels and optical properties of these materials. Compounds 4a–c exhibited broad and long-wavelength optical absorptions with narrow thin-film optical gaps <1.4 eV and low-lying LUMO energies <−4.2 eV. These three π-conjugated scaffolds, together with a dimeric compound (DTYM–NDI–DTYA)2 (R = 2-octyldodecyl, R′ = CN, Fig. 1) developed recently by us, can be regarded as NDI-fused vinylogous tetrathiafulvalene (TTF) derivatives.14
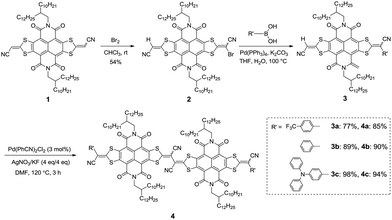 |
| | Scheme 1 Synthetic routes to largely π-conjugated scaffolds 4a–c. | |
Results and discussion
Synthetic procedures
The synthetic routes of 4a–c are shown in Scheme 1. The bromination of NDI–DTYA2 (1) with bromine in chloroform at room temperature afforded 2 in a moderate yield of 54%. Compound 2 then underwent Suzuki–Miyaura cross couplings with the corresponding boronic acids (4-(trifluoromethyl)phenyl boronic acid, phenyl boronic acid and 4-(diphenylamino)phenyl boronic acid) producing 3a–c in high yields of 77%, 89% and 98%, respectively. Under improved reaction conditions according to the literature,11 the homocouplings of 3a–c were performed with Pd(PhCN)2Cl2 as the catalyst and AgNO3/KF (add in one portion) as the oxidant/base at 120 °C for 3 h, giving the target dimeric NDI–DTYA2 derivatives (4a–c) as green solids with high yields of 85%, 90% and 94%, respectively. It should be noted that the homocoupling yields gradually improve with the increased electron-donating ability of the terminated R′ groups. A dimeric compound (DTYM–NDI–DTYA)2 (R = 2-octyldodecyl, R′ = CN, Fig. 1) reported recently by us could also be synthesized by using this C–H/C–H homocoupling strategy with a yield of 83%. This yield is much higher than that of the previous synthetic route (about 43%)14 from the same starting material DTYM–NDI–DTYA (Scheme S1†). These vinyl C–H/C–H homocouplings are uncommon for C–H activation, because most C–H activation processes occur at typical aromatic rings (such as thiophene and indole) and need a long reaction time (even more than 24 h).8b Notably, all the target compounds were obtained as the complex cis/trans-isomers, which is reflected by the 1H NMR with many overlapped signals. In addition, high performance liquid chromatography (HPLC) of the intermediate 2, (DTYM–NDI–DTYA)2, and 4a was performed to evaluate the composition of the mixed isomers. As shown in Fig. S1,† the isomerization of 2 with the ratio of anti/syn isomers close to 1/1 is almost the same with that of 1 reported by us previously,12 demonstrating that the isomerization results from the two different end-capped groups. The HPLC image (Fig. S2†) of the dimeric compound (DTYM–NDI–DTYA)2 exhibits two peaks with an integral area ratio of 3/2, implying that the isomerization was caused by the dicyano-substituted vinylogous tetrathiafulvalene unit. It is the different end-capped groups and the dicyano-substituted vinylogous tetrathiafulvalene unit that result in the complexity of isomerization and the infeasibility of separation for 4a–c. This deduction was partly confirmed by the broad and overlapped HPLC peaks of 4a (Fig. S3†).
The chemical structures of 4a–c were fully characterized by 1H and 13C NMR spectroscopy, high-resolution mass spectrometry and FT-IR spectrometry as well as elemental analysis. Compounds 4a–c, with the molecular weights (MWs) of about 2500–2800, are highly soluble in most common organic solvents such as CH2Cl2, CHCl3, THF, and toluene (>10 mg mL−1) at room temperature. The good solubility of 4a–c laid a foundation for the solution-processable fabrication of their optoelectronic devices.15
DFT calculations
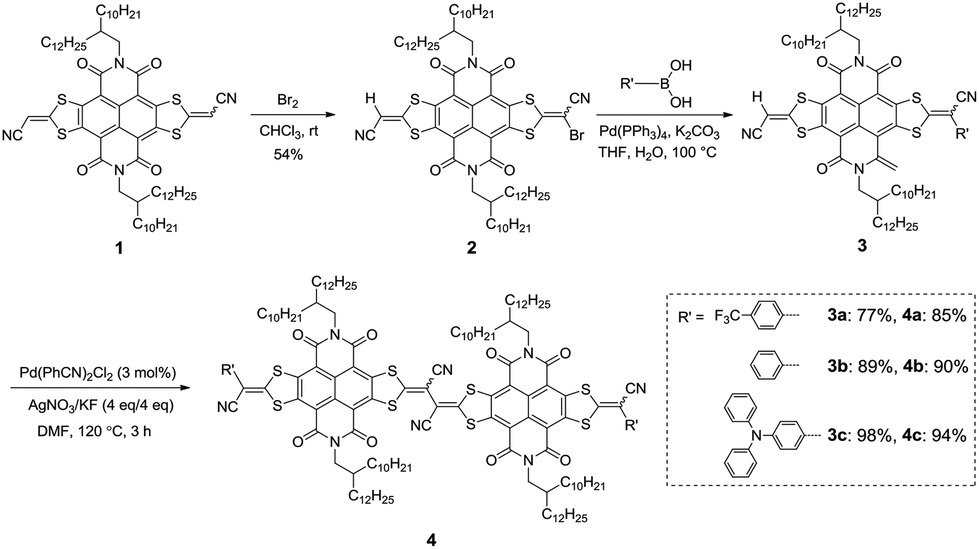
To evaluate the molecular electronic structures of 4a–c, Density Functional Theory (DFT) calculations were performed by using the Gaussian 09 program at the B3LYP/6-31G(d,p) level. Alkyl chains were replaced by methyl groups to reduce the calculation time. The optimized molecular geometries of 4a–c (Fig. 2) exhibited twisted molecular backbones with similar intramolecular dihedral angles of 117.72°, 117.14° and 115.52°, respectively, which could explain the good solubility of these largely π-extended building blocks in common organic solvents. As shown in Fig. 2, both the (highest occupied molecular orbital) HOMO and (lowest unoccupied molecular orbital) LUMO energy levels of 4a–c present a stepwise increase with the increased electron-donating ability of the end-capped groups, demonstrating that the incorporation of these aryl substituents can fine-tune the frontier molecular orbital (FMO) energy levels of these conjugated scaffolds. Compounds 4a and 4b exhibit similar HOMO/LUMO wave functions, the density distributions of the LUMO located on the two NDI segments and the density distributions of the HOMO distributed across the whole π-conjugated skeletons except the imide rings, and focus on the vinylogous TTF unit. The wave functions of FMOs of 4a and 4b indicate that there may be intramolecular charge transfers (ICTs). Notably, the HOMO density distribution of 4c is located at two terminated 4-(diphenylamino)phenyl moieties with no orbital density spreading over the central π-conjugated skeleton, whereas its LUMO density distribution on the NDI cores is almost complementary to the HOMO wave functions. The HOMO and LUMO energies of 4a shift downward versus those of 4b (HOMO: −5.94 eV; LUMO: −3.91 eV; HOMO–LUMO gap: 2.03 eV), giving relatively lower FMO energies (HOMO: −6.14 eV; LUMO: −4.06 eV) and a slightly broader HOMO–LUMO gap (2.08 eV). This downward-shift of FMO energies may be attributed to the electron-withdrawing ability of two 4-(trifluoromethyl)phenyl end groups. In comparison with the FMO energies of 4b, compound 4c presents a higher LUMO energy (LUMO: −3.80 eV) and an even higher HOMO energy (HOMO: −5.25 eV) induced by the electron-donating ability of two 4-(diphenylamino)phenyl end groups and thus a much narrowed HOMO–LUMO gap (1.45 eV).
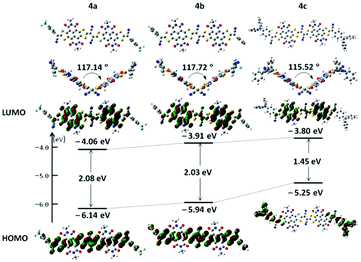 |
| | Fig. 2 Molecular geometries, frontier molecular orbitals and the energy level diagram of N,N′-bis(methyl)-substituted model molecules for 4a–c, obtained by DFT calculations. | |
Thermal properties
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out under a nitrogen atmosphere to evaluate the thermal properties of compounds 4a–c. The onset decomposition temperatures (Td) and the phase transition temperatures (Tpt) are listed in Table 1. TGA measurements demonstrate that 4a–c have excellent thermal stability with Td values of 426 °C, 414 °C and 412 °C, respectively (Fig. S4†). As shown in Fig. S5,†4a displays two couples of endothermic and exothermic peaks in the DSC cycles, the former peaks (61/48 °C) are relatively weak with low enthalpy, demonstrating a solid–solid phase transition, and the latter peaks (330/319 °C) including melting and crystallization are strong and sharp with high enthalpy. The second heating–cooling DSC scan cycle of 4b indicated that no endotherm or exotherm transitions were observed from rt to 350 °C. A couple of quasi-reversible endothermic and exothermic peaks (342/320 °C) were observed for 4c, indicating the melting/crystallization transition process. The melting points of 4a and 4c revealed by DSC analyses are 330 °C and 342 °C, respectively.
Table 1 Thermal, optical, electrochemical and DFT calculation data for 4a–c
| |
T
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (°C) a (°C) |
T
ptb (°C) heating/cooling |
λ
solmax (nm) (Eg/eV)c |
λ
filmmax (nm) (Eg/eV)d |
Δλsol→film (nm) |
E
onsetred1
(V) |
LUMOe (eV) |
HOMOf (eV) |
LUMOg (eV) |
HOMOg (eV) |
|
Onset decomposition temperature measured by TGA under a nitrogen flow at a temperature ramp of 10 °C min−1.
Phase transition temperature obtained from DSC scans of the second heating–cooling cycle under nitrogen at a scan rate of 10 °C min−1.
The maximum end absorption wavelength of the UV-vis-NIR spectrum in CHCl3, optical band gap (Eg) is estimated from the edge of end absorption.
The maximum end absorption wavelength of the UV-vis-NIR spectrum as drop-cast thin film on glass, optical band gap (Eg) is estimated from the edge of end absorption.
Measured in Bu4NPF6 (0.1 M) in CHCl3 (vs. SCE) at a scan rate of 50 mV s−1 and estimated from the equation LUMO = −(4.8 eV − EFc+/Fc + Eonsetred1).
Estimated from HOMO = LUMO − Eg.
Estimated from DFT calculations.
|
|
4a
|
426 |
61/48, 330/319 |
652 (1.69) |
709 (1.35) |
57 |
−0.19 |
−4.25 |
−5.94 |
−4.06 |
−6.14 |
|
4b
|
414 |
N.A. |
667 (1.71) |
721 (1.37) |
54 |
−0.22 |
−4.22 |
−5.93 |
−3.91 |
−5.94 |
|
4c
|
412 |
342/320 |
748 (1.36) |
821 (1.12) |
73 |
−0.23 |
−4.21 |
−5.57 |
−3.80 |
−5.25 |
Optical and electrochemical properties
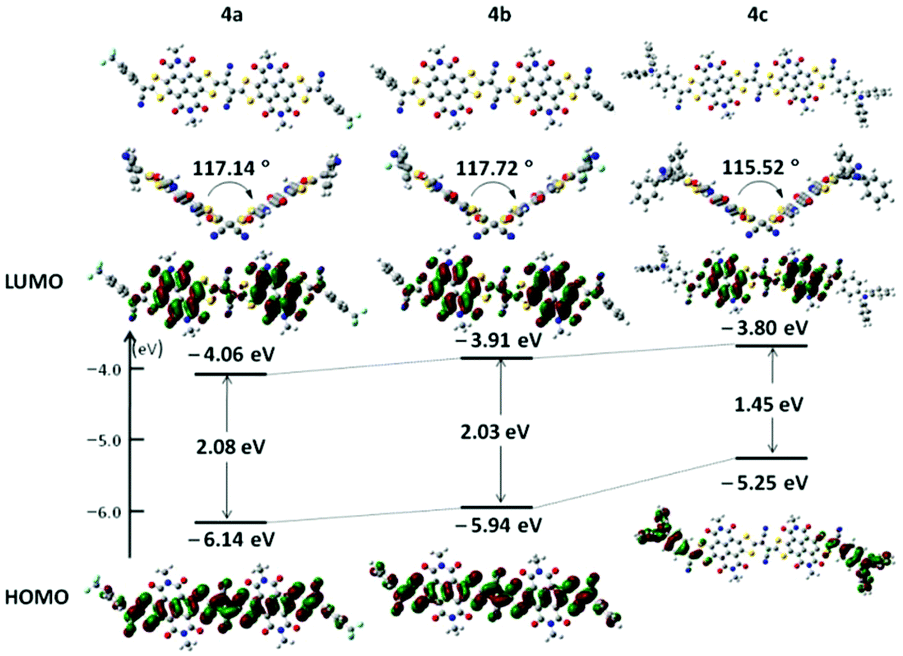
The absorption spectra of 4a–c in diluting solution and spin-coated film on quartz plates were measured by using ultraviolet-visible (UV-vis)-NIR spectroscopy. The data are collected in Table 1. As shown in Fig. 3a, 4a and 4b exhibited comparable broad and strong low-energy absorption bands in diluting solution (4a: λmax = 652 nm; 4b: λmax = 667 nm), indicating their similar electronic structure in the ground state.16 This is also in well accordance with the DFT calculations, since the electron distributions in the terminal 4-(trifluoromethyl)phenyl groups and phenyl groups are very limited. Changing the terminal aromatic ring with 4-(diphenylamino)phenyl moieties much red-shifted the absorption, as exhibited by compound 4c with a λmax of 748 nm. The large absorption red-shift of compound 4c relative to those of 4a and 4b should be ascribed to its strong ICT between the 4-(diphenylamino)phenyl moieties and NDI–DTYA2 moieties, as confirmed by the electron distributions in its HOMO and LUMO.17 All of the three compounds showed high molar absorptions (4a: ε = 8.98 × 104 M−1 cm−1, 4b: ε = 5.15 × 104 M−1 cm−1, and 4c: ε = 5.21 × 104 M−1 cm−1), possibly due to their large π-framework. It is worth noting that from solution to thin film, the absorption bands of compounds 4a–c showed considerable red-shifts (57, 54, and 73 nm for compounds 4a, 4b, and 4c, respectively), in spite of the high rigidity of NDI–DTYA2 (Fig. S6†). Because in the solution state the NDI–DTYA2 moieties in these three molecules are much twisted, the large absorption red-shifts of compounds 4a–c in the thin film state probably benefit from the much improved planarity due to the increased intermolecular interaction in the packing state.16
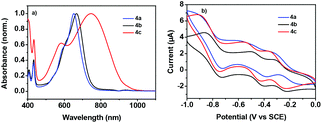 |
| | Fig. 3 (a) Optical absorption spectra of 4a–c in solution with a concentration of 2 × 10−5 M. (b) Cyclic voltammograms of 4a–c (10−3 mol L−1) with 0.1 M Bu4NPF6 in chloroform solution and a scan rate of 50 mV s−1 (vs. SCE). | |
The electrochemical properties of 4a, 4b, and 4c were examined by cyclic voltammetry (CV) (Fig. 3b and Table 1). All compounds display three quasi-reversible reduction processes, indicating that these dimers could successively accept and lose electrons reversibly.18,19 These three reduction waves might be assigned to three successive electron-accepting processes with three half-wave reduction potentials of E0/−11/2 (−0.24, −0.29, and −0.31 V for 4a, 4b, and 4c, respectively), E−1/−21/2 (−0.51, −0.46, and −0.45 V for 4a, 4b, and 4c, respectively), and E−2/−41/2 (−0.85, −0.80, and −0.83 V for 4a, 4b, and 4c, respectively).18,19 The first onset reductive potentials (Eonsetred1) are determined at −0.19, −0.22, and −0.23 V for 4a, 4b, and 4c, respectively, implicating that the incorporation of the end-capped aryl substituents with different electron-accepting abilities has a slight impact on their reductive processes. The LUMO energy levels of 4a–c, estimated from the onset reductive potentials, are positioned at −4.25, −4.22, and −4.21 eV, respectively, making them promising ambient stable semiconductors for organic optoelectronic devices.
Thin-film transistor device characterization
To study the charge transport properties of 4a–c, their organic thin film transistors (OTFTs) were fabricated by spin-coating methods, affording a bottom-gate top-contact device configuration (see the ESI† for details). As shown in Fig. S7, Tables S1 and S2,† OTFT devices based on 4a–c, when measured both under nitrogen and ambient atmosphere, exhibited comparable electron mobilities, indicating their ambient stable charge transport behaviors. The mobility of these three dimers showed no obvious performance change as a function of thin-film annealing temperatures. As shown in Fig. 4, the annealed thin films of 4b processed from toluene solution presented an average electron mobility of 0.02 cm2 V−1 s−1, which is much higher than that of 4a (0.003 cm2 V−1 s−1). It is very interesting to note that OTFTs based on 4c measured under ambient conditions exhibited obvious ambipolar charge transport behavior (Fig. S7c and d†), with hole and electron mobilities of up to 0.005 and 0.003 cm2 V−1 s−1, respectively. For OTFTs based on 4c, the oxygen doping in the thin film20 and the relatively higher HOMO energy level of 4c might lead to its ambipolar charge transport behavior. The ambient stable electron-transporting properties of 4a–c together with their deep LUMO energies and narrow band gaps make them promising candidates for organic optoelectronic devices.
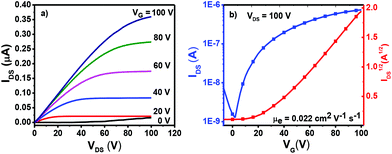 |
| | Fig. 4 Output (a) and transfer (b) characteristics of OTFTs based on 4b annealed at 120 °C. | |
Thin-film morphologies
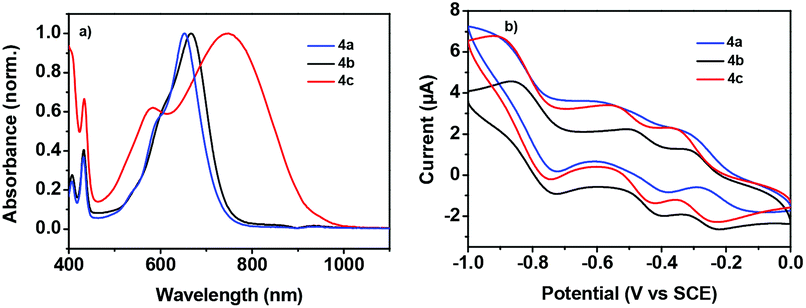
To understand the charge transport properties of these semiconductors, we investigated their thin film microstructures and morphologies by X-ray diffraction (XRD) and atomic force microscopy (AFM). Fig. S8† shows the XRD patterns of the thermally annealed thin films of 4a–c (annealed at 120 °C). XRD plots of the thin films of 4a and 4b showed the first order reflections, while the thin film of 4c exhibited higher order reflection, indicating the better long-range order of the thin films of 4c relative to 4a and 4b.21 As shown in Fig. 5, compounds 4a–c all showed a connected film morphology, but with obviously different morphology features in terms of grain sizes and the density of grain boundaries. It should be noted that the thin films of 4b showed well aligned micronano-sized fibres, which can form interconnected network-like patterns and might contribute to charge transport.22 Therefore, the better film morphology should be responsible for the higher electron mobility of compound 4b.
 |
| | Fig. 5 AFM images of spin-coated thin films of 4a (a), 4b (b), and 4c (c) annealed at 120 °C. | |
Conclusions
In conclusion, three new largely π-extended systems (4a–c) based on NDI–DTYA2 were synthesized by using C–H/C–H homocoupling reactions in high yields (85–94%). This protocol would be a useful and operationally simple access to largely π-conjugated structures for organic electronic materials. The largely π-extended scaffolds (4a–c) with different end-capped aromatic groups showed good solubility in most common solvents and fine-tuned LUMO energies (−4.21 to −4.25 eV) as well as narrow band gaps (<1.4 eV in thin film); they would be promising candidates for solution-processed and ambient stable organic optoelectronic materials. Syntheses of largely π-extended oligomers and polymers based on NDI-fused vinylogous TTFs via C–H activation together with their applications in organic optoelectronic devices are currently underway in our lab.
Acknowledgements
This research was financially supported by the National Natural Science Foundation (21522209 and 20902105), the “Strategic Priority Research Program” (XDB12010100) and the Shanghai Science and Technology Committee (16JC1400603).
Notes and references
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
-
(a) L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed;
(b) R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
-
(a) Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed;
(b) X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947–968 CrossRef CAS.
- X. Gong, M. Tong, Y. Xia, W. Cai, J. S. Moon, Y. Cao, G. Yu, C.-L. Shieh, B. Nilsson and A. J. Heeger, Science, 2009, 325, 1665–1667 CrossRef CAS PubMed.
-
(a) X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed;
(b) M. A. Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef CAS PubMed;
(c) M. Stolte, M. Gsanger, R. Hofmockel, S. L. Suraru and F. Würthner, Phys. Chem. Chem. Phys., 2012, 14, 14181–14185 RSC.
-
(a) G. Marzano, C. V. Ciasca, F. Babudri, G. Bianchi, A. Pellegrino, R. Po and G. M. Farinola, Eur. J. Org. Chem., 2014, 6583–6614 CrossRef CAS;
(b) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 CAS.
-
(a) D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172–1175 CrossRef CAS PubMed;
(b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed.
-
(a) W. Wu, H. Xin, C. Ge and X. Gao, Tetrahedron Lett., 2017, 58, 175–184 CrossRef CAS;
(b) J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- S.-L. Suraru, J. A. Lee and C. K. Luscombe, ACS Macro Lett., 2016, 5, 724–729 CrossRef CAS.
- M. Takahashi, K. Masui, H. Sekiguchi, N. Kobayashi, A. Mori, M. Funahashi and N. Tamaoki, J. Am. Chem. Soc., 2006, 128, 10930–10933 CrossRef CAS PubMed.
- Z. Zhao, F. Zhang, Y. Hu, Z. Wang, B. Leng, X. Gao, C.-a. Di and D. Zhu, ACS Macro Lett., 2014, 3, 1174–1177 CrossRef CAS.
- A. Nowak-Król, R. Wagener, F. Kraus, A. Mishra, P. Bäuerle and F. Würthner, Org. Chem. Front., 2016, 3, 545–555 RSC.
- Y. Hu, Z. Wang, X. Zhang, X. Yang, C. Ge, L. Fu and X. Gao, Org. Lett., 2017, 19, 468–471 CrossRef CAS PubMed.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- A. Salleo, R. J. Kline, D. M. DeLongchamp and M. L. Chabinyc, Adv. Mater., 2010, 22, 3812–3838 CrossRef CAS PubMed.
-
(a) Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed;
(b) H. Shang, H. Fan, Y. Liu, W. Hu, Y. Li and X. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed.
- B. Leng, D. Lu, X. Jia, X. Yang and X. Gao, Org. Chem. Front., 2015, 2, 372–377 RSC.
- L. E. Polander, S. P. Tiwari, L. Pandey, B. M. Seifried, Q. Zhang, S. Barlow, C. Risko, J.-L. Brédas, B. Kippelen and S. R. Marder, Chem. Mater., 2011, 23, 3408–3410 CrossRef CAS.
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
- W. Cui, M. Yao, S. Liu, F. Ma, Q. Li, R. Liu, B. Liu, B. Zou, T. Cui and B. Liu, Adv. Mater., 2014, 26, 7257–7263 CrossRef CAS PubMed.
-
(a) S. Wang, W. Pisula and K. Müllen, J. Mater. Chem., 2012, 22, 24827 RSC;
(b) J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 124, 3651–3655 CrossRef;
(c) Z. Zhao, Z. Wang, C. Ge, X. Zhang, X. Yang and X. Gao, Polym. Chem., 2016, 7, 573–579 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00061h |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.