DOI:
10.1039/C6QM00380J
(Research Article)
Mater. Chem. Front., 2017,
1, 1200-1206
The photo-controlled polymerization-induced self-assembly and reorganization process for fabrication of polymeric nanomaterials†
Received
30th December 2016
, Accepted 23rd January 2017
First published on 24th January 2017
Abstract
A visible light mediated reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization of benzyl methacrylate (BzMA) is conducted in ethanol using poly(2-dimethylaminoethyl methacrylate) (PDMAEMA) as a macro-chain transfer agent (macro-CTA), affording polymeric nanomaterials with various morphologies. Unlike the polymerization-induced self-assembly and reorganization (PISR) process initiated by thermal stimuli, the photo-controlled PISR can be performed under more mild conditions, such as visible light and room temperature. Photoredox catalysis is employed to regulate the polymerization via a photoinduced electron transfer (PET) process. It is observed that the polymerization can be affected by catalyst concentration and light intensity, and “ON/OFF” control over polymerization is also demonstrated.
Introduction
Polymeric nanomaterials with diverse morphologies have attracted great attention due to their promising applications in many fields, such as diagnostic, imaging, nanoreactors and drug delivery.1–3 Generally, the fabrication strategy of such polymeric nanomaterials includes traditional self-assembly in a selective solvent and polymerization-induced self-assembly and reorganization (PISR).4,5 The former strategy has been studied for several decades, fabricating a broad range of nanomaterials including micelles, nanowires, vesicles and compound vesicles.6–8 However, the multi-step synthesis and low concentration of prepared nanomaterials (<1 wt%) in the traditional self-assembly process limit its further applications. A PISR process was developed several years ago and has become popular for its convenience and productiveness compared to traditional self-assembly. The strategy of PISR can realize the formation of diblock copolymers, self-assembly and morphology transition of nano-objects in one-pot, and diverse nanomaterials can be efficiently prepared on a large scale. The polymerizations performed in water, alcohol or other solvents have been carefully studied by Armes, our group and other researchers.9–20 Among the previous research on PISR, RAFT polymerization is the most popular technique to generate insoluble blocks, and thermal initiators, such as azobis(isobutyronitrile) (AIBN) and azobis(4-cyanopentanoic acid) (ACVA), are usually employed as the radical source to initiate the polymerization. Thus the fabrication of nanomaterials usually requires relatively high reaction temperature (above 60 °C) in the thermal-initiated PISR system, which is unfavorable for the synthesis of bio-related or environmentally responsive nanomaterials. However, the PISR process performed at low temperature has rarely been reported.21,22
Photo-controlled living radical polymerization (photo-CRP) has attracted wide interest due to its unique abilities to control polymerizations in both space and time. Besides, the photo-CRP approach enables the polymerization to be carried out at room temperature.23–29 Well-defined polymers, such as block, graft and star polymers, have been prepared by employing this technique.30–36 Recently, efforts have been made to prepare polymeric nanomaterials using the photo-CRP method.21,22,37–41 Chen and co-workers42 reported UV light-initiated RAFT polymerization induced self-assembly (PISA) using AIBN as a photoinitiator. The polymerization can be carried out under mild conditions to prepare polymeric micelles. Cai and co-workers43 fabricated vesicles and nanotubes at 25 °C through visible-light-mediated PISA by employing a water-soluble photoinitiator sodium phenyl-2,4,6-trimethylbenzoylphosphinate (SPTP). Boyer et al.44 developed a blue light-mediated RAFT polymerization in a PISA dispersion process. The ruthenium-based photoredox catalyst, Ru(bpy)3Cl2, was employed to activate the polymerization via a photoinduced electron transfer (PET) process. The formation of different nanomaterial morphologies was demonstrated. However, the heavy metal catalyst, which is usually difficult to get rid of, might limit the practicality of this system.
Herein, we report visible light mediated RAFT dispersion polymerization for the preparation of various morphological nanomaterials. In this PISR approach, an organic compound, 10-phenylphenothiazine (PTH), was utilized as a photoredox catalyst to regulate the dispersion polymerization of BzMA via a PET process using a PDMAEMA macro-CTA. Spheres, nanowires and vesicles were successfully produced under visible light irradiation at ambient temperature. The control over polymerization was also demonstrated by adjusting the light “ON/OFF” and light intensity. This study provides a mild and easily manipulated strategy to prepare polymeric nanomaterials via a visible light-mediated PISR process, which is valuable for the extension and development of the PISR system.
Materials
The 4-cyanopentanoic acid dithiobenzoate (CPADB) was synthesized as reported.45 2-Dimethylaminoethyl methacrylate (DMAEMA, Aladdin, 99%) and BzMA (Aladdin, 99%) were purified by passing through an alumina column for the removal of inhibitor, followed by vacuum distillation. Toluene and tetrahydrofuran (THF, Sinopharm Chemical Reagent Co., 99%) were refluxed over sodium for 24 h, and then distilled prior to use. AIBN (Aldrich) was recrystallized from ethanol. Phenothiazine (Aladdin, 98%), iodobenzene (Aladdin, 99%), tri(dibenzylideneacetone)dipalladium (Pd2(dba)3, Aladdin, 97%), 1,1-bis(diphenylphosphino)ferrocene (DPPF, Aladdin, 97%) and sodium tert-butoxide (NaOtBu, Aladdin, 98%) were all used as received.
Synthesis of 10-phenylphenothiazine (PTH)
10-Phenylphenothiazine was synthesized as shown in Scheme S1 (ESI†). Phenothiazine (500 mg, 2.50 mmol), Pd2(dba)3 (46 mg, 0.05 mmol), DPPF (55 mg, 0.1 mmol) and NaOtBu (360 mg, 3.75 mmol) were added into a reaction tube with a magnetic stir bar. The tube was evacuated and backfilled with nitrogen three times, then dry toluene (5 mL) and iodobenzene (245 μL, 2.2 mmol) were added under the protection of nitrogen. The reaction tube was placed in an oil bath at 160 °C for 16 h. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (CH2Cl2, 50 mL), and washed with water three times and saturated NaCl solution once. The organic layer was dried with anhydrous Na2SO4 and rotary evaporated. The crude product was purified using silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 20/1, v/v) to afford 10-phenylphenothiazine. The product was dried under reduced pressure to obtain a pale yellow solid (75% yield). 1H NMR (300 MHz, CDCl3) δ: 7.60 (t, J = 7.5 Hz, 2H), 7.47 (t, J = 7.4 Hz, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.02 (d, J = 7.0 Hz, 2H), 6.87–6.77 (m, 4H), 6.20 (d, J = 7.7 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ: 144.3, 141.1, 130.8, 128.8, 128.1, 127.2, 126.8, 122.5, 120.3, 116.1 ppm. GC-MS C18H13NS found 275.15, calc. 275.37.
Synthesis of CPADB-terminated poly(2-dimethylaminoethyl methacrylate) (PDMAEMA-CPADB)
The general synthetic procedure of PDMAEMA-CPADB is as follows: CPADB (282 mg, 1.02 mmol), DMAEMA (8.0 g, 51 mmol), AIBN (16.7 mg, 0.102 mmol), and THF (8 mL) were added into a 50 mL polymerization tube with a magnetic bar. The polymerization tube was sealed after three freeze–pump–thaw cycles, and then placed in a thermostated oil bath at 70 °C for 8 h. The polymerization was stopped by opening the polymerization tube to air. The polymer solution was precipitated into an excess of petroleum ether three times. The obtained product was dried under vacuum at room temperature overnight. 1H NMR and GPC were used to characterize the resultant polymer, and the results are shown in Fig. S1 and S2 (ESI†), respectively.
Synthesis of thiol-terminated PDMAEMA (PDMAEMA-SH)
PDMAEMA-CPADB (500 mg) and ethanol (15 mL) were added into a two-neck round-bottom flask with a magnetic bar. After the PDMAEMA-CPADB had dissolved, hydrazine (1.0 mL, 50 wt%) was injected into the solution with a syringe under nitrogen. The pink color of the solution faded quickly. After hydrazinolysis for 4 h at room temperature, the solvent was removed by rotary evaporation under reduced pressure, and the obtained crude product was dissolved in dichloromethane (20 mL) and then washed with alkaline water three times. The organic phase was collected and concentrated by rotary evaporation and poured into petroleum ether to get the polymer. The obtained product was dried under vacuum at room temperature overnight. 1H NMR was used to characterize the resultant polymer, and the result is shown in Fig. S3 (ESI†).
Synthesis of PDMAEMA-b-PBzMA block copolymers by visible light mediated RAFT dispersion polymerization
PDMAEMA-b-PBzMA block copolymers were synthesized by visible light mediated RAFT dispersion polymerization in ethanol. A typical experiment was set up as follows: PDMAEMA-CPADB (160 mg, 2.5 × 10−5 mol), BzMA (0.88 g, 5.0 × 10−3 mol), PTH (1.375 mg, 5.0 × 10−6 mol) and ethanol (5.895 g) were successively added into a 10 mL glass tube with a magnetic bar, and the system was degassed by three freeze–pump–thaw cycles. The tube was sealed under vacuum, and the polymerization mixture was irradiated with LED light in a circular reactor (24 W) at room temperature while stirring. After 24 h, the polymerization was quenched by opening the tube to air. 100 μL of the reaction mixture was immediately extracted for 1H NMR analysis to determine the monomer conversion. A portion of the mixture was diluted with ethanol to prepare the samples for TEM observation. For measuring the GPC of PDMAEMA-b-PBzMA, the mixture was precipitated by pouring the solution into excess petroleum ether while stirring. The obtained solid was collected and then dried in a vacuum oven at room temperature.
Characterization
Nuclear magnetic resonance (NMR) measurements were performed on Bruker DMX300 and DMX400 spectrometers using CDCl3 as the solvent. The molecular weight and molecular weight distribution were determined on a Waters 150C gel permeation chromatograph (GPC) equipped with microstyragel columns (500, 103 and 104 Å) and a RI 2414 detector at 30 °C; monodispersed polystyrene standards were used in the calibration of Mn, Mw and Mw/Mn, and THF was used as an eluent at a flow rate of 1.0 mL min−1. Transmission electron microscopy (TEM) observations were performed on a Hitachi H-800 TEM at an accelerating voltage of 100 kV. The samples for TEM observations were prepared by depositing a drop of dispersion of nano-aggregates in ethanol on copper grids. UV/Vis measurement was performed on a Unico UV/vis 2802PCS spectrophotometer. Fluorescence spectra were recorded on a RF-5301PC fluorescence spectrometer. Gas chromatography-mass spectrometry (GC-MS) analysis was performed on an Agilent Technologies 7890A GC system and an Agilent Technologies 5975C MS instrument with a triple-axis detector.
Results and discussion
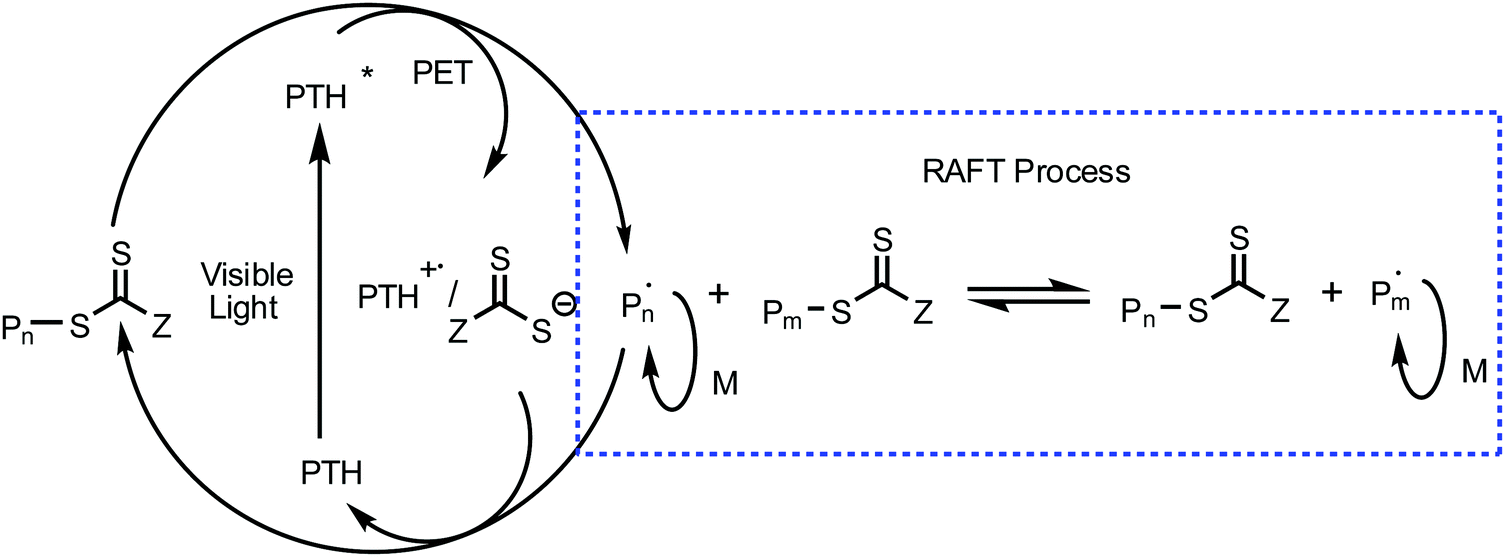
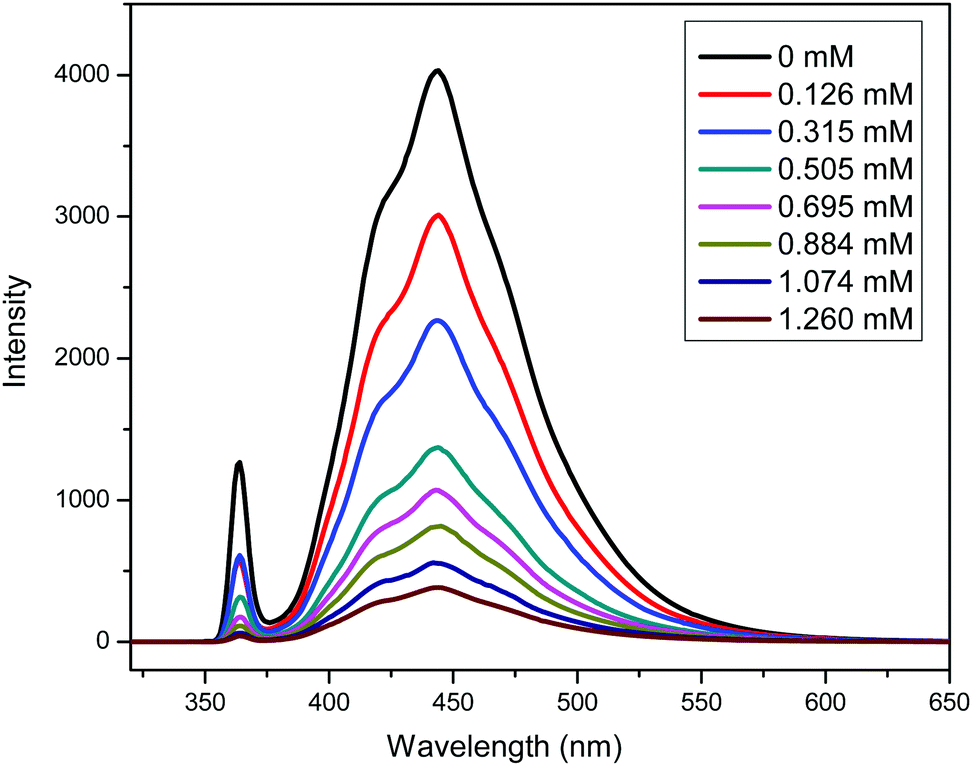
A PDMAEMA-CPADB homopolymer used as macro-chain transfer agent was synthesized via homogeneous RAFT polymerization of DMAEMA using CPADB as a RAFT agent and AIBN as an initiator. The 1H NMR spectrum of PDMAEMA-CPADB is shown in Fig. S1 (ESI†); all characteristic peaks have been marked, and the DP of PDMAEMA-CPADB is calculated to be 38 based on the integral ratio of the peaks at 4.10 and 7.30–7.90 ppm, which are attributed to the ester methylene protons of the DMAEMA units and the phenyl protons in CPADB, respectively. The GPC analysis (Fig. S2, ESI†) indicates that the number-average molecular weight (Mn) and polydispersity (Mw/Mn) are 1900 g mol−1 and 1.28, respectively. The PDMAEMA macro-CTA was then chain extended with BzMA through the photoinduced electron transfer RAFT polymerization in ethanol. PTH was selected as the organic photoredox catalyst, and the proposed mechanism for this photoinduced polymerization is described in Scheme 1.32 The redox potential of CPADB is −0.4 V (potential versus the saturated calomel electrode (SCE)),31 which is greater than that of PTH+/PTH* (−2.1 V),46 indicating that PTH* would be able to reduce CPADB via the PET process. Fluorescence quenching studies were conducted to investigate the electron transfer between the chain transfer agent and PTH. The fluorescence spectra of solutions of PTH (0.669 mM) in ethanol with varying concentrations of PDMAEMA-CPADB under irradiation at λex = 360 nm are shown in Fig. 1. It was observed that the intensity of the fluorescence emission of PTH at 444 nm decreased obviously, when the concentration of the PDMAEMA-CPADB in the solution of PTH increased, which confirmed the energy transfer between PDMAEMA-CPADB and PTH. Subsequently, thiol-terminated PDMAEMA (PDMAEMA-SH) was prepared by hydrazinolysis of PDMAEMA-CPADB. The 1H NMR spectrum of PDMAEMA-SH is shown in Fig. S3 (ESI†), and the disappearance of the peaks at 7.30–7.90 ppm, which are assigned to the phenyl protons in CPADB, confirmed the rupture of dithiocarbonate in PDMAEMA-CPADB. The obtained PDMAEMA-SH was added into the solution of PTH in ethanol, and the fluorescence property of PTH was characterized as shown in Fig. S4 (ESI†). With an increasing concentration of PDMAEMA-SH in the solution, no fluorescence quenching of PTH was observed. This result indicated that there was no energy transfer between PDMAEMA-SH and PTH. Considering the fluorescence quenching of PTH in the presence of PDMAEMA-CPADB in solution, it can be inferred that energy transfer occurs between PTH and the CPADB terminal of PDMAEMA-CPADB.
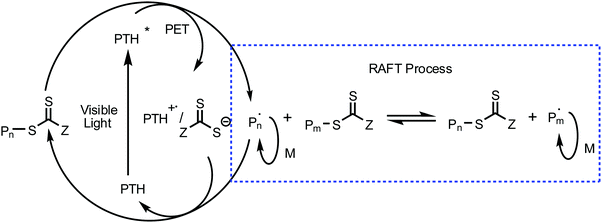 |
| | Scheme 1 Proposed PET-RAFT polymerization mechanism with PTH as the catalyst. | |
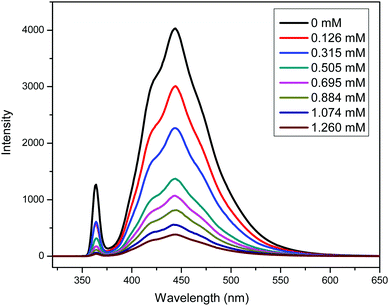 |
| | Fig. 1 Fluorescence quenching study of solutions of PTH (0.669 mM) in ethanol with varying concentrations of PDMAEMA-CPADB. | |
The PDMAEMA-CPADB macro-CTA mediated RAFT dispersion polymerization of BzMA in ethanol was investigated using different catalyst concentrations with [BzMA]/[PDMAEMA] = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The reactions were irradiated with visible light for 24 h, and the monomer conversions, number-average molecular weights and polydispersities of the resultant polymers are shown in Table 1. The monomer conversions, which were calculated based on eqn (S1) (ESI†), gradually increased with PTH concentration. GPC analysis indicated that the molecular weights of the polymers were narrowly distributed (Mw/Mn < 1.20), suggesting good control of the molecular weight during the polymerization. In the 1H NMR spectrum of the diblock copolymer (Fig. S6, ESI†), characteristic peaks at 4.07 ppm and 4.87 ppm are assigned to the ester methylene protons for PDMAEMA and PBzMA, respectively, confirming the successful chain extension.
:1. The reactions were irradiated with visible light for 24 h, and the monomer conversions, number-average molecular weights and polydispersities of the resultant polymers are shown in Table 1. The monomer conversions, which were calculated based on eqn (S1) (ESI†), gradually increased with PTH concentration. GPC analysis indicated that the molecular weights of the polymers were narrowly distributed (Mw/Mn < 1.20), suggesting good control of the molecular weight during the polymerization. In the 1H NMR spectrum of the diblock copolymer (Fig. S6, ESI†), characteristic peaks at 4.07 ppm and 4.87 ppm are assigned to the ester methylene protons for PDMAEMA and PBzMA, respectively, confirming the successful chain extension.
Table 1 Characterization of PDMAEMA-b-PBzMA diblock copolymers synthesized using different molar ratios of [PTH]/[PDMAEMA]
| Sample |
[PTH]/[PDMAEMA] |
Conv.a (%) |
M
n
|
M
w/Mnb |
|
The results were calculated based on 1H NMR spectra in CDCl3.
The molecular weights and molecular weight distributions of di-block copolymers were determined by GPC measurements. The polymerization was conducted in ethanol with a molar ratio of [BzMA]/[PDMAEMA] = 200/1 at 15% solids concentration for 24 h.
|
| 1 |
1/200 |
62 |
23300 |
1.12 |
| 2 |
1/50 |
76 |
26600 |
1.06 |
| 3 |
1/20 |
77 |
28200 |
1.08 |
| 4 |
1/5 |
85 |
31000 |
1.08 |
| 5 |
1/2 |
97 |
33000 |
1.09 |
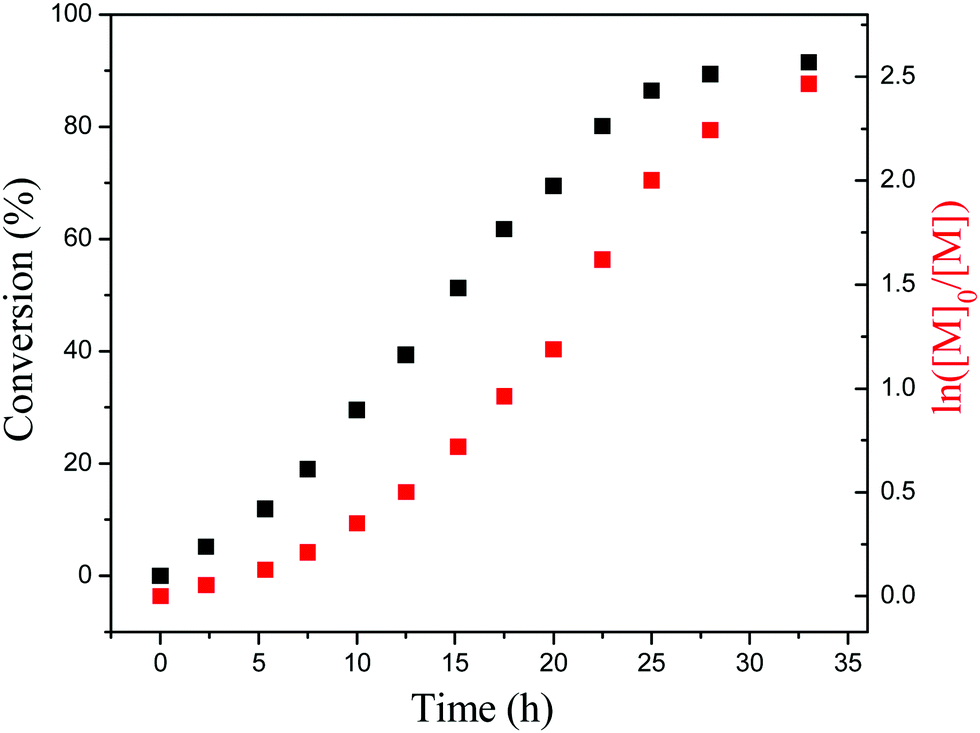
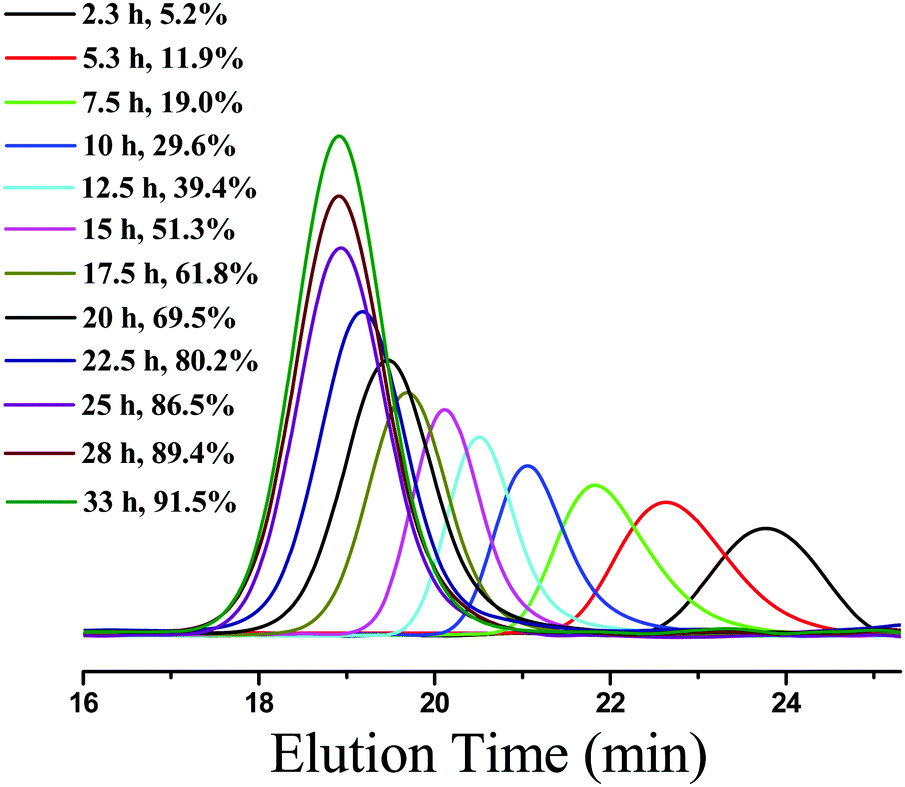
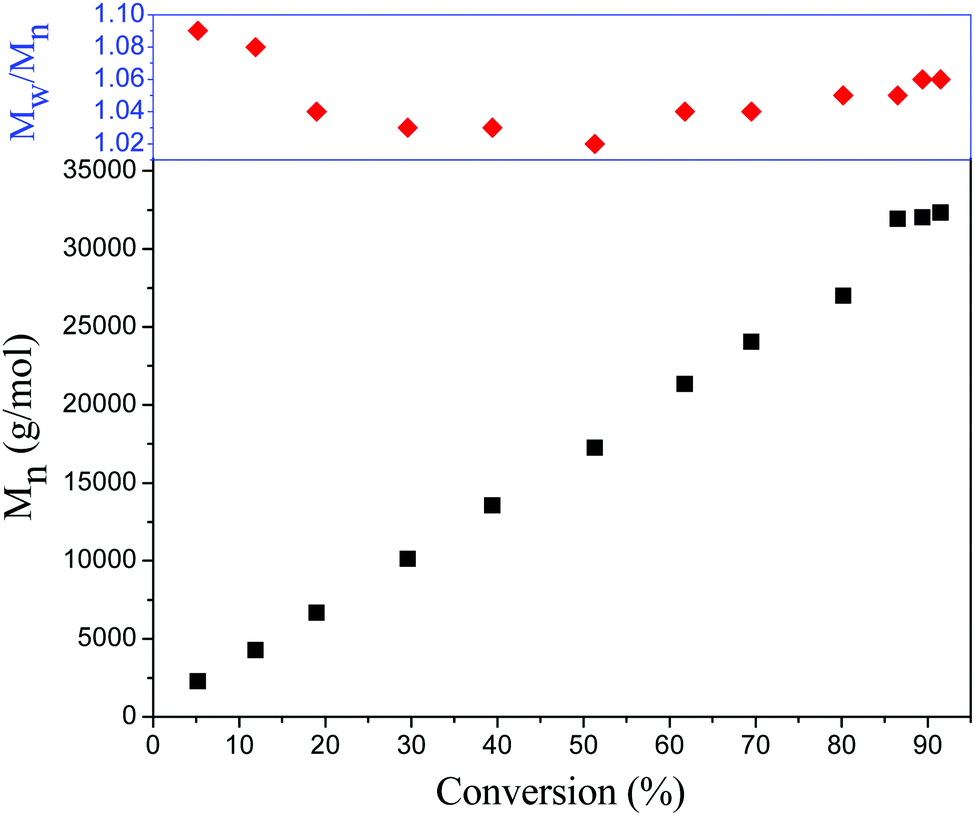
To further investigate the PET-RAFT dispersion polymerization, a kinetics study was conducted. The polymerization was performed in ethanol at 15% solids concentration with a molar ratio of [BzMA]/[PDMAEMA]/[PTH] = 1000/5/1. The polymerization process was monitored by 1H NMR analysis. The plot of conversion vs. increasing exposure time and the evolution of ln([M]0/[M]) vs. exposure time are shown in Fig. 2. It is indicated that the conversion of BzMA could achieve above 85% after polymerization for 24 h. There are two stages involved in the PET-RAFT dispersion polymerization process, which is similar to previously reported observations for thermal-initiated RAFT dispersion polymerization.47 In the initial stage of polymerization, the monomer conversion increased slowly. After about 10 h, the rate of polymerization discernibly accelerated. This time period corresponded to the onset of micellar nucleation, which meant that the polymerization changed from homogeneous RAFT polymerization into a heterogeneous one. The enhanced rate of polymerization was generally because of the solubilization of BzMA within the growing nanoparticles and the radical compartmentalization effect in heterogeneous RAFT polymerization.48 PDMAEMA-b-PBzMA diblock polymers synthesized at different polymerization times were characterized by GPC analysis. The GPC traces are shown in Fig. 3, and the results of the number-average molecular weights and polydispersities are summarized in Fig. 4 and Table S1 (ESI†). The GPC traces are all unimodal and clearly shift to the high molecular weight region with the increasing monomer conversion. The plot of Mnvs. conversion shows a linear increase, and the molecular weights of the PDMAEMA-b-PBzMA diblock polymers were narrowly distributed (Mw/Mn < 1.10), suggesting good control over the molecular weight during the RAFT dispersion copolymerization. At different polymerization times, the morphologies of the PDMAEMA-b-PBzMA di-block polymer nano-objects were checked by TEM, and the results are shown in Fig. 5. Micelles (Fig. 5a), nanowires (Fig. 5b) and vesicles (Fig. 5c and d) successively formed with the prolonging of polymerization time. Moreover, different rheological behavior was observed during the polymerization. At the beginning of polymerization, the reaction solution was clear, because all reactants were soluble in the solvent. After micellar nucleation, the polymerization system turned from transparent to a lightly blue opalescence, and TEM examination revealed the spherical morphology (Fig. 5a) of the polymer nano-objects at that moment. Subsequently, the polymerization system underwent a gradual increase in viscosity and acquired a gelatinous modality. The formation of worm-like nano-objects was confirmed by TEM characterization (Fig. 5b). The macroscopic gelation behavior may be due to strong inter-worm entanglements as reported previously.49 When further prolonging the irradiation time, the polymerization system showed a decrease in viscosity and transformed to a free-flowing milky solution. TEM indicated that vesicles were obtained as shown in Fig. 5c and d. This phenomenon shown in the process of polymerization, which can be detected by the naked eye, provides a convenient method to judge the morphological evolution and to get a pure phase of the polymer nano-objects that we wanted.
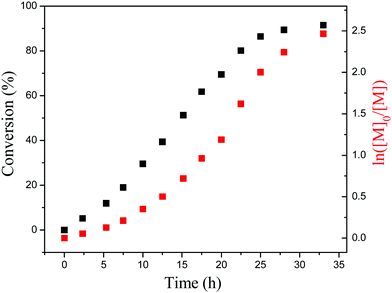 |
| | Fig. 2 Total monomer conversion vs. time plot and the corresponding semilogarithmic plot. | |
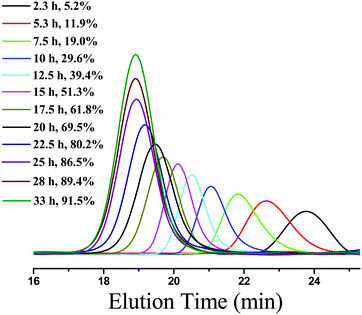 |
| | Fig. 3 GPC traces of PDMAEMA-b-PBzMA diblock copolymers synthesized under different irradiation times. | |
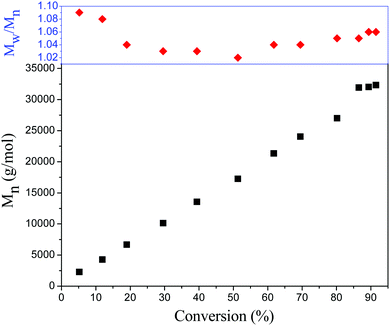 |
| | Fig. 4 Evolution of the number-average molecular weights and polydispersities (Mw/Mn) of PDMAEMA-b-PBzMA diblock copolymers as a function of monomer conversion. | |
 |
| | Fig. 5 TEM images of PDMAEMA-b-PBzMA nano-objects fabricated by PET-RAFT dispersion polymerization at different polymerization times. | |
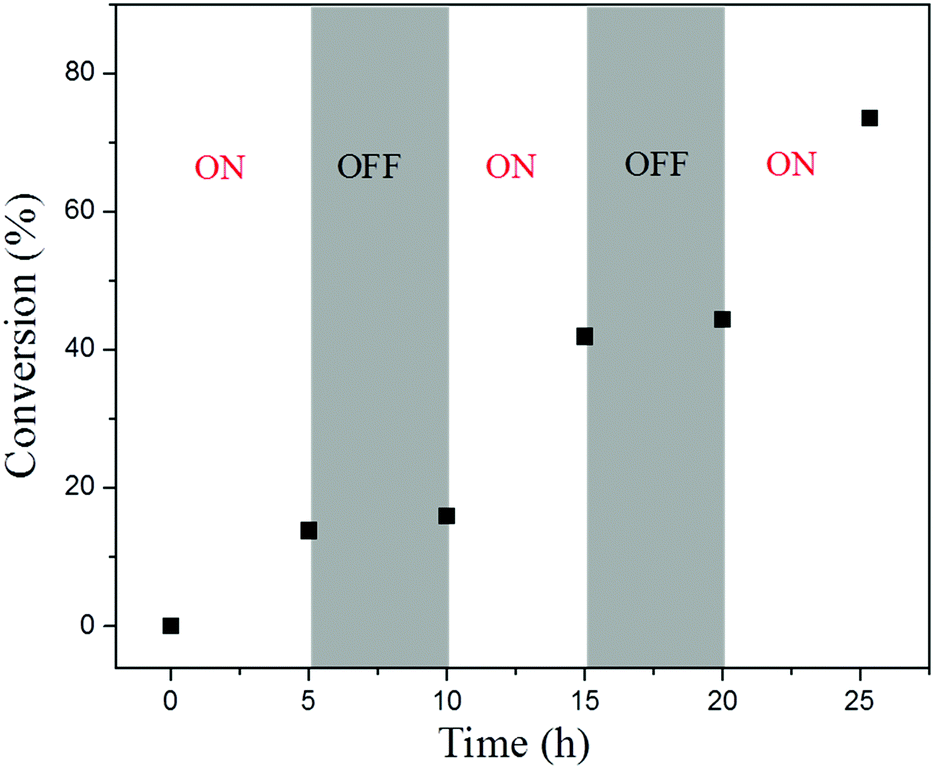
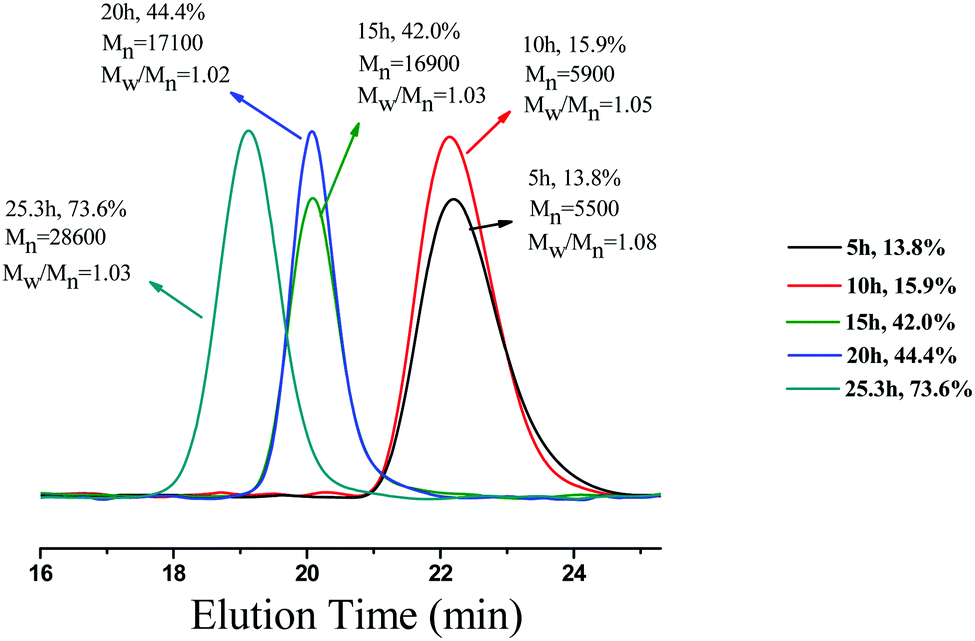
To examine the capability of activating or deactivating polymerization by light in this PET-RAFT PISR system, the polymerization was exposed to alternating “ON/OFF” periods of external light stimulation. The experiment was conducted at 15% solids concentration with a molar ratio of [BzMA]/[PDMAEMA]/[PTH] = 1000/5/1, and the light source was alternated between “ON/OFF” every 5 h. The conversion of BzMA and the molecular weights of the PDMAEMA-b-PBzMA diblock polymers were monitored by 1H NMR and GPC analysis. As shown in Fig. 6, the monomer consumption occurred during the periods of light exposure. When the light source was OFF, the system turned into a dormant state, and monomer consumption was halted. While switching the light back ON, the polymerization could be reactivated, and monomer consumption was resumed. The GPC traces of the PDMAEMA-b-PBzMA diblock polymers (Fig. 7) in the ON/OFF experiment showed that the molecular weight obviously increased during light exposure, but the value of the molecular weight changed little during periods of light OFF, which was consistent with the monomer consumption. The results indicate that the PET-RAFT PISR system can be temporally controlled by simply regulating the light source ON or OFF.
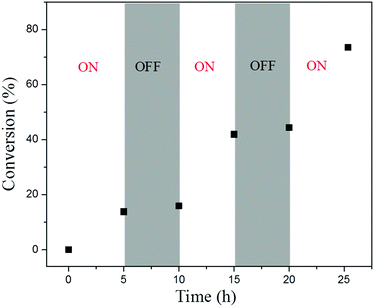 |
| | Fig. 6 ON/OFF experiment of PET-RAFT dispersion polymerization with the PTH photoredox catalyst. | |
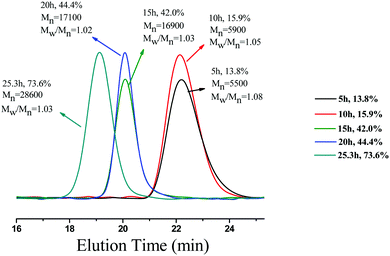 |
| | Fig. 7 The GPC traces of PDMAEMA-b-PBzMA diblock copolymers in ON/OFF experiments. | |
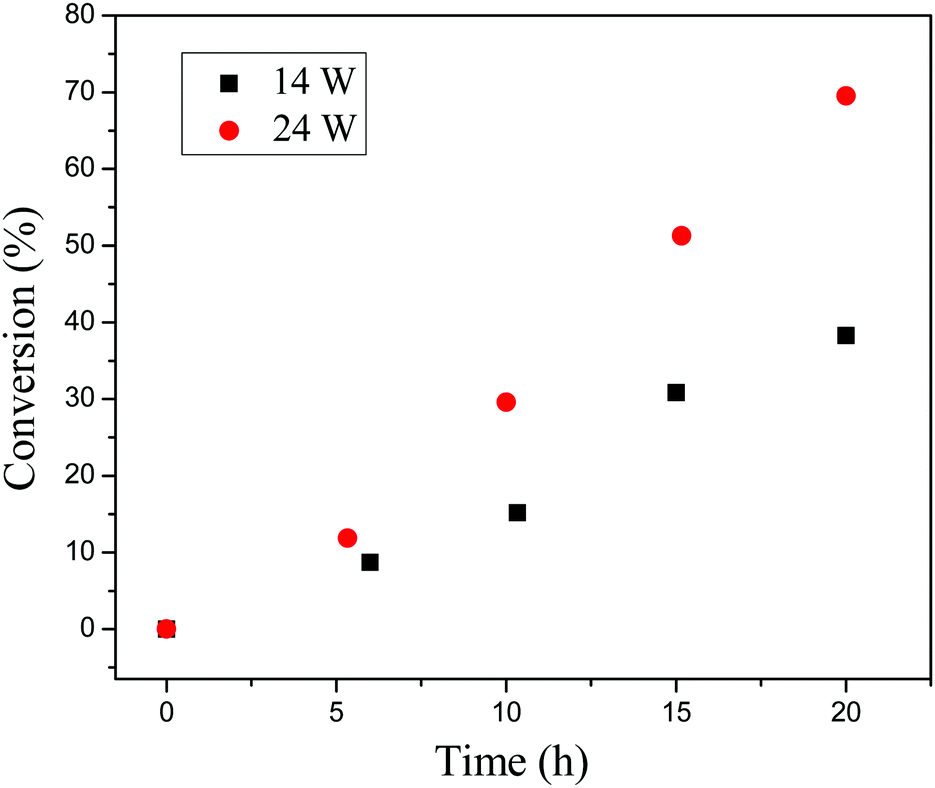
Regulating the rate of polymerization by changing the light intensity has been investigated. The polymerizations were conducted under different light sources (14 W and 24 W) with the same molar ratio of [BzMA]/[PDMAEMA]/[PTH] = 1000/5/1. When the intensity of the light source was 24 W, the monomer conversion could reach 70% after irradiation for 20 h as shown in Fig. 8. When changing the light source to 14 W, the rate of polymerization decreased, and only 38% monomer was consumed after polymerization for 20 h. This indicates that the rate of polymerization is obviously dependent on the light intensity. In other words, we could manipulate the polymerization conveniently by tuning the light intensity.
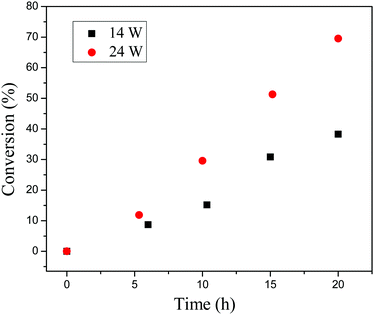 |
| | Fig. 8 Monomer conversion vs. exposure time under light intensity of 14 W (black squares) and 24 W (red circles). | |
Conclusions
Visible light-mediated PET-RAFT polymerization using an organic photoredox catalyst was applied in a PISR system. The PET-RAFT process enabled the fabrication of nano-objects with different morphologies, such as micelles, nanowires and vesicles at ambient temperature. Based on the rheological behavior during the polymerization, the morphological evolution can be conveniently judged to obtain a pure phase of the polymer nano-objects that we wanted. The ability to temporally control the polymerization in the PET-RAFT process by modulating the light source has been demonstrated, and the rate of polymerization can also be regulated by tuning the light intensity. We expect that this research will be useful for the fabrication of various nano-objects at ambient temperature and valuable for the further study and development of PISR system.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21525420 and 21374107) and the Fundamental Research Funds for the Central Universities (WK 2060200012).
References
- Y. M. Li, Y. F. Qian, T. Liu, G. Y. Zhang and S. Y. Liu, Biomacromolecules, 2012, 13, 3877–3886 CrossRef CAS PubMed.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS PubMed.
- C. Hu, T. Xia, Y. Gong, X. Wang, R. Q. Liu, Q. Y. Zhang, C. F. Yi, Z. S. Xu and D. Z. Guo, Chin. J. Polym. Sci., 2016, 34, 135–146 CrossRef CAS.
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. T. Sun, C. Y. Hong and C. Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- F. Schacher, A. Walther, M. Ruppel, M. Drechsler and A. H. E. Müller, Macromolecules, 2009, 42, 3540–3548 CrossRef CAS.
- Y. F. Zhou and D. Y. Yan, Angew. Chem., Int. Ed., 2005, 44, 3223–3226 CrossRef CAS PubMed.
- K. J. Gao, G. T. Li, X. P. Lu, Y. G. Wu, B. Q. Xu and J. H. Fuhrhop, Chem. Commun., 2008, 1449–1451 RSC.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromolecules, 2014, 47, 1664–1671 CrossRef CAS.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2015, 36, 1428–1436 CrossRef CAS PubMed.
- J. M. Zhou, W. J. Zhang, C. Y. Hong and C. Y. Pan, Polym. Chem., 2016, 7, 3259–3267 RSC.
- W. M. Cai, W. M. Wan, C. Y. Hong, C. Q. Huang and C. Y. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- W. Zhou, W. J. Yu and Z. S. An, Polym. Chem., 2013, 4, 1921–1931 RSC.
- X. W. Zhang, S. Boisse, W. J. Zhang, P. Beaunier, F. D. Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- S. T. Li, X. He, Q. L. Li, P. F. Shi and W. Q. Zhang, ACS Macro Lett., 2014, 3, 916–921 CrossRef CAS.
- P. F. Shi, C. Q. Gao, X. He, P. C. Sun and W. Q. Zhang, Macromolecules, 2015, 48, 1380–1389 CrossRef CAS.
- E. Groison, S. Brusseau, F. D'Agosto, S. Magnet, R. Inoubli, L. Couvreur and B. Charleux, ACS Macro Lett., 2012, 1, 47–51 CrossRef CAS.
- Y. Y. Jiang, N. Xu, J. Han, Q. P. Yu, L. Guo, P. Gao, X. H. Lu and Y. L. Cai, Polym. Chem., 2015, 6, 4955–4965 RSC.
- J. B. Tan, H. Sun, M. G. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS PubMed.
- S. Shanmugam, J. T. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC.
- H. X. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- J. T. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- G. Liu, H. Shi, Y. Cui, J. Tong, Y. Zhao, D. Wang and Y. Cai, Polym. Chem., 2013, 4, 1176–1182 RSC.
- J. T. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- M. Chen, M. J. Macleod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 CrossRef CAS.
- J. T. Xu, S. Shanmugam and C. Boyer, ACS Macro Lett., 2015, 4, 926–932 CrossRef CAS.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- S. Park, H. Y. Cho, K. B. Wegner, J. Burdynska, A. J. D. Magenau, H.-j. Paik, S. Jurga and K. Matyjaszewski, Macromolecules, 2013, 46, 5856–5860 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen, W. Li, K. Matyjaszewski and Y. Yagci, Macromolecules, 2013, 46, 9537–9543 CrossRef CAS.
- K. Jung, J. T. Xu, P. B. Zetterlund and C. Boyer, ACS Macro Lett., 2015, 4, 1139–1143 CrossRef CAS.
- Q. P. Yu, Y. Ding, H. Cao, X. H. Lu and Y. L. Cai, ACS Macro Lett., 2015, 4, 1293–1296 CrossRef CAS.
- J. B. Tan, X. Rao, J. W. Yang and Z. H. Zeng, Macromolecules, 2013, 46, 8441–8448 CrossRef CAS.
- X. Wang, Q. P. Lu, X. F. Wang, J. Joo, M. Dahl, B. Liu, C. B. Gao and Y. D. Yin, ACS Appl. Mater. Interfaces, 2016, 8, 538–546 CAS.
- J. B. Tan, M. G. Yu, X. Rao, J. W. Yang and Z. H. Zeng, Polym. Chem., 2015, 6, 6698–6708 RSC.
- Z. Z. Liu, G. J. Zhang, W. Lu, Y. J. Huang, J. W. Zhang and T. Chen, Polym. Chem., 2015, 6, 6129–6132 RSC.
- P. Gao, H. Cao, Y. Ding, M. Cai, Z. G. Cui, X. H. Lu and Y. L. Cai, ACS Macro Lett., 2016, 5, 1327–1331 CrossRef CAS.
- J. Yeow, J. T. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. D. Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CrossRef CAS.
- M. J. Monteiro and J. D. Barbeyrac, Macromolecules, 2001, 34, 4416–4423 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00380j |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.