
pH-Responsive polymers
G.
Kocak
 ,
C.
Tuncer
and
V.
Bütün
*
,
C.
Tuncer
and
V.
Bütün
*
Department of Chemistry, Faculty of Arts and Science, Eskisehir Osmangazi University, Eskisehir, 26480, Turkey. E-mail: gkocak@ogu.edu.tr; ctasagir@ogu.edu.tr; vbutun@ogu.edu.tr; Fax: +90 222 2393578; Tel: +90 222 2393750/2751
First published on 14th November 2016
Abstract
In this review, we provide an analysis of some of the recent literature reports on the synthesis and applications of pH-responsive polymers. Depending on the solution pH, such copolymers can self-assemble and form various nanosized structures including core–shell micellar structures, micelles/reverse micelles, hollow spheres, vesicle structures, adsorbed species at the water–air interface, and more complex architectures. Their self-assembly behaviors open the door for the production of various novel nanostructures including shell/core cross-linked micelles, hollow spheres, hydrogels, microgels, layer-by-layer (LbL) nanofilms, controlled releasing systems, drug carrier systems, etc. The review consists of various major parts including types of pH-responsive polymers, synthetic methods for their synthesis and their solution behaviors, their nanostructures in aqueous media, applications as LbL nanofilms, delivery devices, controlled release systems, sensors, stabilizers, solubilizers, etc. In the last two decades, there have been great developments in synthetic methods and strategies for the preparation of novel pH-responsive polymers or polymeric materials providing possible materials for various applications including biotechnology, nanotechnology, colloid and surface science, materials science, etc.
 G. Kocak | Gökhan Kocak obtained his BSc and MSc degrees in chemistry from the University of Gaziosmanpasa and Adiyaman University in 2007 and 2011, respectively. He is currently a PhD student in Prof. Butun's research group at Eskisehir Osmangazi University. His current research interests include block copolymers, post-polymerization modifications, cross-linked micelles, water-soluble polymers, hydrogels, stimulus-responsive polymers and their applications. He has good experience in RAFT, ATRP, and free radical polymerizations. |
 C. Tuncer | Dr Cansel Tuncer obtained her BSc and MSc degrees in chemistry from Hacettepe University in 2006 and 2009, respectively. She has been working as a research assistant at the same university since 2009. She received her PhD from Eskisehir Osmangazi University in 2015. Her research interests include the synthesis, evaluations and derivatizations of novel polymers, hydrogels, microgels, and metal nanoparticle dispersions. She is an expert in GTP, ATRP, free radical polymerization, and heterogeneous polymerization techniques. |
 V. Bütün | Prof. Dr Vural Bütün received his PhD at the University of Sussex-UK in 1999. He has been working at the Chemistry Department of Eskisehir Osmangazi University (ESOGU-Turkey) since 1999. His research interests mainly focus on the synthesis and characterization of water soluble polymers and investigation of their self-assembly behaviors. He has published 60 papers with 2850 citations. He is the recipient of various scientific awards including the TWAS-Science Encouragement Award, TUBA-Outstanding Young Scientist Award, and TUBITAK-Science Encouragement Award. Currently, he is the Director of the Central Research Laboratory and Application Center and the Dean of the Faculty of Science at ESOGU. |
Introduction
Stimuli responsive polymers are a class of polymers that can self-assemble or undergo phase transitions or morphology changes via physical or chemical changes in response to small external changes in the environment. They have also been named intelligent, smart, or environmentally-responsive polymers. These polymers can be responsive to a number of factors such as temperature, pH, biomaterials, solvent, ionic strength, chemical agents, light, electrical field, and magnetic field.1–3 The field of pH-responsive polymers has gained great academic and commercial interest in the last two decades parallel to not only developments in the synthetic methodologies to make these materials but also their wide-span application potentials.pH-Responsive polymers are a group of stimuli-responsive polymers that can respond to solution pH by undergoing structural and property changes such as surface activity, chain conformation, solubility, and configuration. The term “pH-responsive polymers” is commonly used to describe polymers having ionisable acidic or basic residues whose ionization depends on solution pH. The subject of pH-responsive polymers has become very popular in recent years and new studies have been added year after year. These unique properties of pH-responsive polymer systems consequently make them very useful in various applications such as drug delivery, gene delivery, sensors, surfaces, membranes, and chromatography.4–6
pH-Responsive polymers can have linear, branched or network structures. They may show different responses to solution conditions and different self-assembly behaviors depending on their structures. For example, a pH change may cause (de)protonation of functional groups in polymer chains. In some cases, it may cause flocculation, chain collapse-extension, and precipitation for homopolymers. It may also cause self-assembly such as formation of micelles, unimers, gels, vesicles, swelling, deswelling, etc. Block (co)polymers, branched (co)polymers, and star (co)polymers having pH-responsive block(s) show surface active behaviors by pH change. Additionally, hydrogel and dendrimer like structures show (de)swelling behavior by pH change. Surfaces modified with polymers give a chance to obtain ionic surfaces and thin/thick layer formation by pH change. The changes observed in polymers of different architectures by pH change are shown in Fig. 1.
 | ||
| Fig. 1 pH-Responsive polymers of different architectures: (a) unimer–micelle, (b) micelle–reverse micelle, (c) nanogels or microgels, (d) hollow–reverse hollow, (e) dendrimer, (f) hyper-branched, (g) micelle morphology changes (from worm-like to hollow), and (h) polymer brushes. | ||
pH-Responsive polymers can be defined as polyelectrolytes that include in their structure weak acidic or basic groups that either accept or release protons in response to a change in the environmental pH. Polymers having acidic or basic groups like carboxyl, pyridine, sulfonic, phosphate, and tertiary amines are typically described as pH-responsive polymers because the ionization of the groups with pH change results in a change in the structure. In addition to their biotechnological applications, their pH-responsiveness or ionization allows us to tune their self-assembly behavior, hydrophilicity phase separation, polyelectrolyte nature, etc. It is possible to prepare a polymer having a pKa between pH 1 and 14. In general, pH-responsive polymers of basic monomers behave as cationic polymers under acidic conditions and polymers with acidic monomers behave as anionic polymers under basic conditions. Depending on the application, it is necessary to choose one of these two types or a combination of them with the right composition. Apart from synthetic polymers, natural polymers have often been studied. The use of natural polymers is the most common focus of interest because of their abundance in nature, degradability, biocompatibility and ability to be modified. Synthetically, pH-responsive polymers can be produced from polypeptides such as poly(L-glutamic acid) (PLGA), poly(histidine) (PHIS), and poly(aspartic acid) (PASA). These polymers are biocompatible and degradable just like natural polymers. These natural polymers have great importance among pH-responsive polymers.5,7,8 In this review, the type of pH-responsive polymer, synthesis methods, architectures, and their applications will be summarized in detail.
Classification of pH-responsive polymers
A large number of pH-responsive polymers can be designed using various electrolyte groups. There are two kinds of pH-responsive polymers. One is polymers with acidic groups and the other one is polymers with basic groups. Weak polyacids such as poly(methacrylic acid) (PMAAc) accept protons at low pH and release protons at neutral and high pH. Weak polybases such as poly[(2-dimethylamino)ethyl methacrylate] (PDMA) accept protons at low pH and form a positively charged polymer chain. In the following section, acidic, basic, and neutral pH-responsive polymers reported in the literature will be summarized.pH-Responsive acidic polymers
Polymers having weak acidic or basic residues are utilized as pH-responsive polymers (Fig. 2). Such weak acidic/basic pendant groups accept protons at low pH and release them at high pH. Therefore, they gain polyelectrolyte nature at acidic or basic pH values depending on their pKa values. Such an ionic/non-ionic transition allows us to tune their hydrophilicity in the aqueous phase. This tuning results in precipitation/solubilization of polymer chains, swelling/deswelling of their hydrogels, or hydrophobic/hydrophilic characteristic changes of such polymeric surfaces and their particles.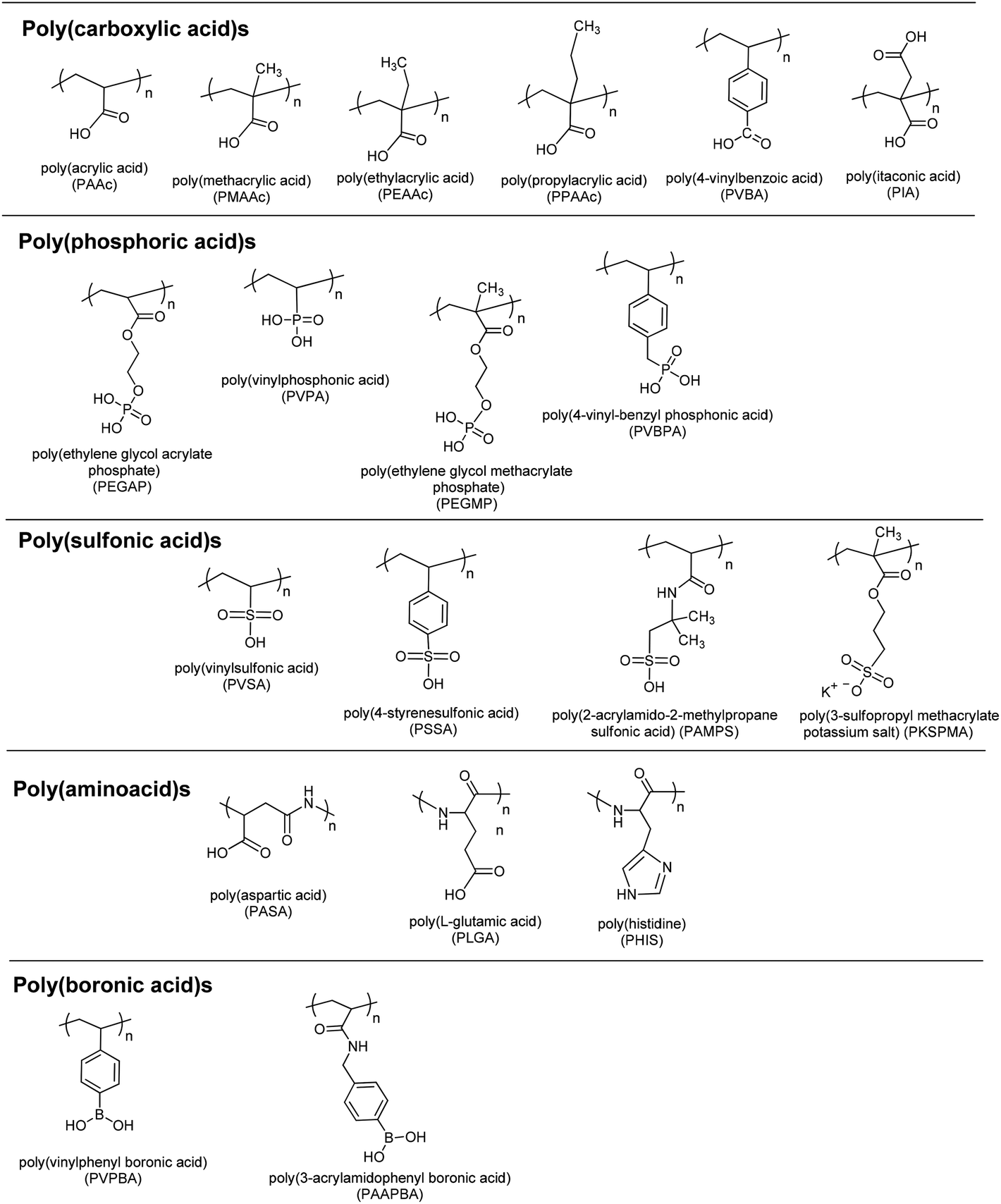 | ||
| Fig. 2 Chemical structures of pH-responsive acidic polymers. | ||
pH-Responsive basic polymers
Weak polybases, which undergo ionization/deionization transitions from pH ∼7–11, are utilized as pH-responsive polymers. The amine groups are located in their side chains. These groups accept protons at low pH values by forming polyelectrolytes and releasing them under basic conditions. The (meth)acrylate, (meth)acrylamide, and vinylic polymers containing tertiary amine, morpholino, pyrrolidine, imidazole, piperazine, and pyridine groups are shown in Fig. 3.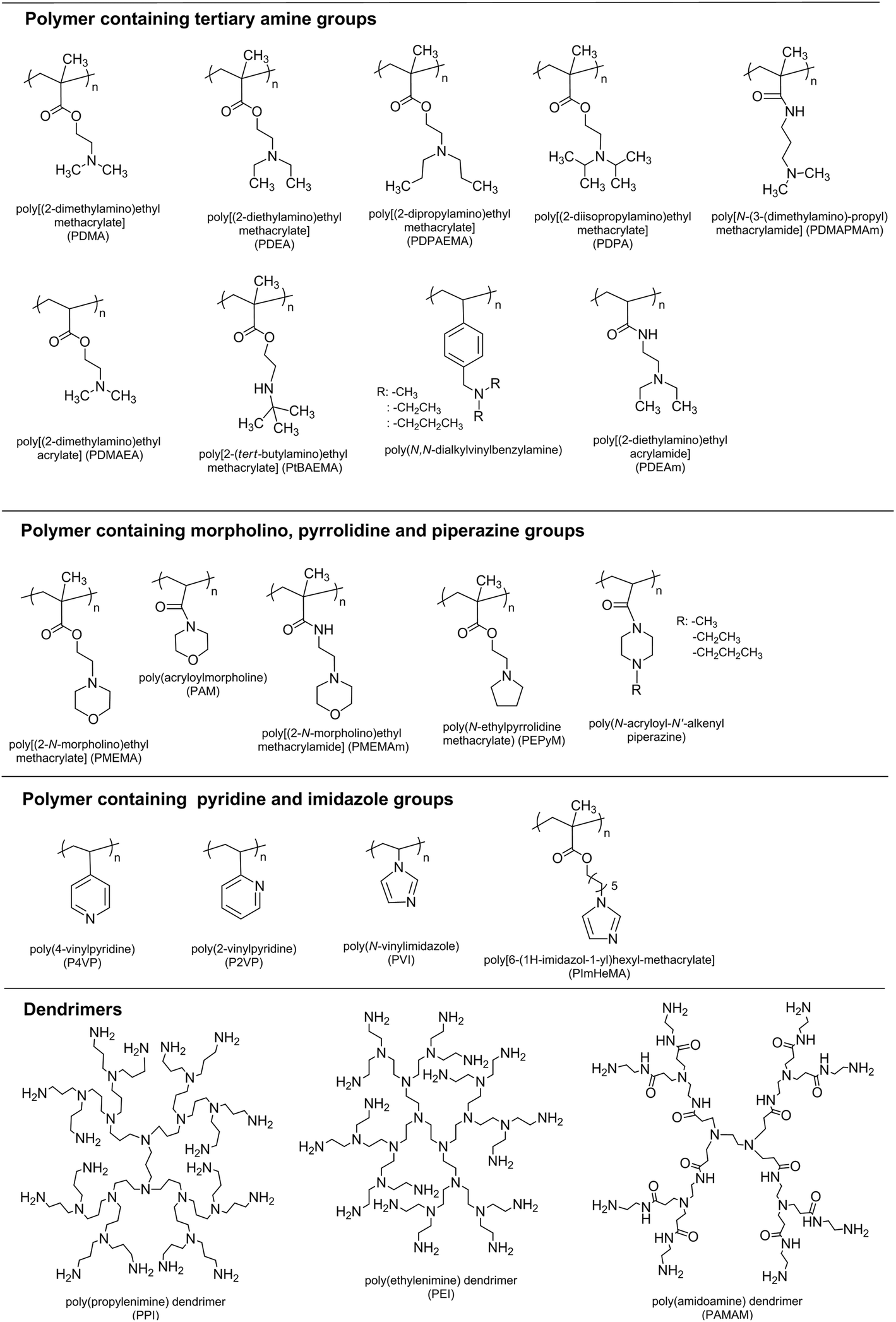 | ||
| Fig. 3 Chemical structures of pH-responsive basic polymers. | ||
Vinyl, (meth)acrylamide, and (meth)acrylate polymers containing tertiary amine groups have also received great attention.4,20–24 Tertiary amine methacrylate based polymers such as PDMA, poly[(2-diethylamino)ethyl methacrylate] (PDEA), and poly[(2-diisopropylamino)ethyl methacrylate] (PDPA) are the most preferred species among the basic polymers. Particularly, PDMA is the most popular weak basic polymer having not only pH-responsive nature but also thermo-responsive nature. Their polymers obtained from various techniques are commercially readily available. Poly(vinyl pyridine) based polymers are also widely used polymers which include poly(4-vinylpyridine) (P4VP) and poly(2-vinylpyridine) (P2VP). These polymers undergo a phase transition above pH 5 owing to deprotonation of pyridine groups.25–27
Other pH-responsive polymers are the polymers containing functional groups, such as imidazole,28–32 piperazine,33,34 pyrrolidine,35 and morpholino.31,36–38 Poly[(2-N-morpholino)ethyl methacrylate] (PMEMA) is an important polymer having morpholino groups and a response to pH, temperature, and ionic strength of the medium. Both Armes and Butun's groups have reported the synthesis of various PMEMA based polymers and investigation of their solution behaviors.36,39–41 It is also necessary to mention that dendrimers such as poly(ethylene imine) (PEI), poly(propylene imine) (PPI), and poly(amidoamine) (PAMAM) can be classified as pH-responsive polymers. They can be modified with different functionalities and can be grafted with various polymers.42–44 Some basic polymers are shown in Fig. 3.
pH-Responsive natural polymers
During the last decade, there have been great scientific developments in synthetic biodegradable polymers for various biomedical applications. Additionally, the use of natural biodegradable polymers has also gained great attention owing to their good biocompatibility and easy modification by simple chemistry. Some natural polymers are also used in the pH-responsive self-healing gel system.5,7,45 Dextran, hyaluronic acid, alginic acid, chitosan, and gelatine are among the most widely used natural polymers (Fig. 4). On suitable chemical modification, these polymers can provide better materials for drug delivery systems. Another popular preparation strategy is the grafting of pH-responsive polymers onto a polysaccharide backbone. Hydrogels of these polymers in many studies using cross-linking agents have been produced. Chitosan has been the most studied species in such polymers.46–51 Some of the natural and semi-natural polymers are given in Table 1.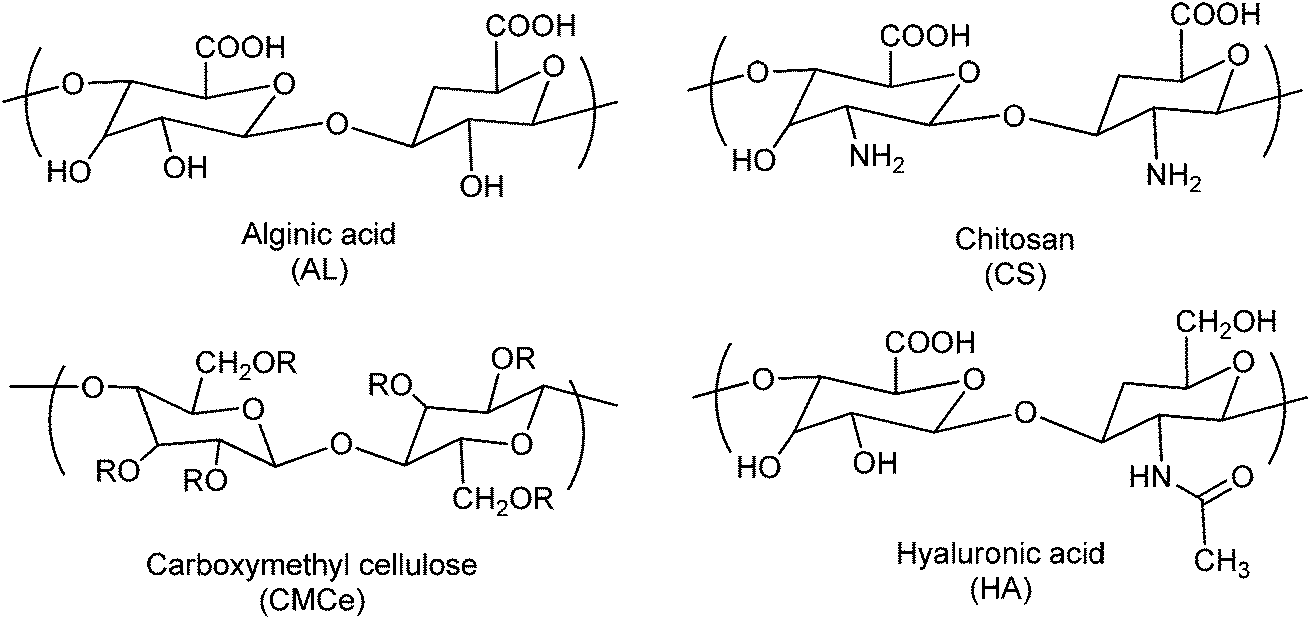 | ||
| Fig. 4 Chemical structures of representative pH-responsive natural polymers. | ||
| Natural polymers | Functional groups | Swelling pH | Ref. |
|---|---|---|---|
| Chitosan (CS) | –NH2, –OH | Acidic | 46–51 |
| Guar gum (semi-natural) | –COOH | Basic | 52 and 53 |
| Alginic acid | –COOH, –OH | Basic | 54–56 |
| Hyaluronic acid (HA) | –COOH, –OH | Basic | 57 |
| Carboxymethyl dextran (CM-Dex) | –COOH, –OH | Basic | 58 |
| Carboxymethyl cellulose | –COOH, –OH | Basic | 59–61 |
| Gelatine A and B | –NH2, –OH, –COOH | Acidic and basic | 45 and 57 |
| Tertiary amine starch ether | –N<, –OH | Acidic | 62 |
Multi-responsive polymers
Multi-responsive polymers have recently become very popular. They give better results in various applications than other alternatives. Responsive polymers can have responses to a number of factors, such as temperature, pH, biomaterials, redox, light, electrical field, magnetic field, etc.63,64 These polymers can be prepared by selecting two or more external impact sensitive specific monomers or polymers.63–66 For example, PDMA and PMEMA polymers are both temperature and pH-responsive.4,65 By combining two or more different monomers having different stimuli responsive behaviors, a number of multi-responsive copolymers can be prepared. Temperature and pH-responsive polymers are probably the most studied among multi-responsive polymers. Such copolymers can be fabricated from polymers having one block that is temperature responsive and the other block having pH-responsiveness. In general, PNIPAm is chosen as a thermo-responsive block due to its lower critical solution temperature (32 °C) in water, which is close to body temperature.63 Some of the multi-responsive polymers are given in Table 2.| Responsive to | Polymersa | Type | Ref. |
|---|---|---|---|
| a See abbreviations for the definitions of polymers and reagents. | |||
| Glucose, pH, and thermo | PDMA-co-PAAPBA | Macrogel | 67 |
| Light, pH, and thermo | PDMA-co-PSP | Micelle | 68 and 69 |
| Glucose and pH | PVPBA-co-PDMAEA | Nanogel | 70 |
| Glucose and light | MePEGA-b-(PNBA-co-PAAPBA) | Micelle | 71 |
| pH and magnetic | PMAAc | Microgel | 72 |
| pH and electric | PAAc-co-PVSA | Macrogel | 73 |
| pH and reduction | PEO-b-(PMAAc-g-Hyd) | Micelle | 74 |
| RPHA-g-coumarin | Micelle | 75 | |
| Thermo and enzyme | PPDPMA-co-PTEGMA | Micelle | 76 |
| pH and thermo | PVI-co-PIMMA | Copolymer | 30 |
| PNIPAm-co-PDMA | Nanogel | 77 | |
| PEPyM | Macrogel | 35 | |
| PNIPAm-b-PAAc | Micelle | 78 | |
| PEG-b-P4VP-b-PNIPAm | Micelle | 79 | |
| PNIPAm-b-PDEA | Micelle | 66 | |
| PDPA-b-PDMA-b-PDPA | Micelle | 80 | |
| PDMA-b-PAAc | Micelle | 20 | |
| PDMA-b-PDEA | Micelle | 81 | |
| PDMA-b-PDPA | Micelle | 36 | |
| [PMEO2MA-b-(PDEA-co-P TPHMA)] | Micelle | 82 | |
| PEO-b-[PGMA-g-(PDEA)(PMEO2MA)] | Micelle | 83 | |
| PMAAc-co-PNIPAm | Yolk/shell | 84 | |
| PDMA-b-PMPS | Micelle | 85 | |
| PNIPAm-b-PLGA | Micelle | 86 | |
| pH, thermo, and salt | PMEMA | Microgel | 65 |
| PDEA-b-PMEMA | Micelle | 87 | |
| PQDMA-b-PMEMA | Micelle | 88 | |
| PβDMA-b-PMEMA | Micelle | 89 | |
Most commonly used methods for the synthesis of pH-responsive polymers
Vinyl-based pH-responsive polymers can be synthesized with different polymerization methods such as free radical polymerization, cationic polymerization, anionic polymerization, group transfer polymerization (GTP), atom transfer radical polymerization (ATRP), nitroxide-mediated radical polymerization (NMP), and reversible addition–fragmentation chain transfer (RAFT) polymerization. Polymers with various molecular structures or shapes including random, gradient, block, star, branched, brushes and graft (co)polymers can be synthesized with these methods. Among them, free radical polymerization is the simplest one. But it is not suitable for the synthesis of polymers with controlled molecular weight, narrow molecular weight distributions, and well defined structures.For well-defined polymers with narrow molecular weight distributions, controlled living polymerization techniques are developed. These methods include either ionic or radical chemistry. Webster et al. developed an anionic living polymerization chemistry, namely group transfer polymerization (GTP), in 1983.90 GTP allows the living polymerization of (meth)acrylates at room temperature. Due to the strict reaction conditions and being not suitable for all monomers, the application of this technique is limited. In spite of difficult reaction conditions, pH-responsive polymers consisting of tertiary amine methacrylates such as DMA,91–93 DEA,81,92,94 DPA36,39,80 and MEMA36,39,94,95 have been prepared using GTP in a variety of architectures such as block,36,91,93 star96, branched,92etc. Functional monomers such as methacrylic acid cannot be used in GTP since their labile protons terminate the polymerization. In order to polymerize such monomers, the functional groups need to be masked using protecting groups that are readily converted back to the functional species after the polymerization.91,97
Using controlled/living radical polymerization (CRP or CLP) methods, well-defined polymers can be synthesized under mild conditions with predictable molecular weights and content, narrow molecular weight, end-functional, and different polymer architectures.98,99 CRP is a chain polymerization that proceeds in the absence of chain transfer and termination reactions.98 Among CRP methods, ATRP, NMP and RAFT are prominent in the preparation of pH-responsive polymers. Some examples of pH-responsive polymers with various architectures reported with the NMP method are P2VP-b-PNIPAAm,100 PPO-b-P(DMA-stat-2VP),101 PDEVBP-b-P2VP,102 PDEVBP-b-P(NIPAm-stat-DMAAm),102 PDMA-ra-PVBK,103 and PAAc-grad-PS.104
Another CRP type ATRP chemistry was independently discovered by Matyjaszewski et al.105 and Sawamoto et al.106 in 1995. In recent years, it became the most popular CRP method which is suitable for a wide range of monomers, including (functionalized) styrenes,107 (meth)acrylates,107,108 and some (meth)acrylamides.109–113 But it has some limitations on the polymerization of acrylamide and its derivatives. Unlike ionic polymerization, ATRP exhibits a tolerance of trace impurities, e.g. no moisture sensitivity and functional group tolerance. A wide variety of mono- and multi-functionalized ATRP initiators can easily be prepared or obtained commercially. Additionally, the polymers synthesized via ATRP can be used as a macro-initiator for further steps, which is another important advantage of ATRP. Due to easy production of pH-responsive polymers with various architectures such as block,108 star,114–116 gradient,108,117 brushes,118,119 and branched120 (co)polymers, this method has been frequently used in the synthesis of pH-responsive polymers.
The first reports of the direct use of addition–fragmentation transfer agents to control radical polymerization appeared in the 1980s.121–123 Reversible addition–fragmentation chain transfer (RAFT) polymerization is one of the most widely used processes for synthesizing well-defined blocks,22,124 stars,125–128 branches128,129 and brushes85,130–132 in recent years. The difference of this method from free radical polymerization is the addition of the transfer agent (RAFT-CTA) to the polymerization medium. RAFT polymerization is appropriate for (meth)acrylates, styrenics, and (meth)acrylamides, whereas RAFT reagents are not tolerant to primary and secondary amines.133 Just as in the ATRP method, the polymer obtained by the RAFT method can be used as a macro-RAFT agent and can be easily obtained commercially. In addition, the polymers obtained by ATRP and RAFT methods have mono- or multi-functional end groups. These groups on polymers can also be modified to obtain different polymeric materials. As an example, Armes’ group has prepared non-ionic PG2MA-b-PHPMA diblock copolymers which can exhibit pH-responsive behavior. This behavior is due to the fact that the carboxylic acid-functional RAFT agent is used in the synthesis of polymers.134 Due to easy modification of silicon and gold surfaces with an ATRP initiator and/or RAFT agent, surface initiated-atom transfer radical polymerization (SI-ATRP) and surface initiated-reversible addition–fragmentation chain transfer (SI-RAFT) methods are used frequently in the synthesis of pH-responsive polymeric brushes.
Different sizes of pH-responsive crosslinked hydrogels (micro- and nano-) by emulsion polymerization techniques can be prepared.135–138 Thanks to the developed techniques, the production of hydrogels with narrow size distribution and smaller size has provided more effective use. The type of monomer or monomers used in the synthesis of hydrogels is the most important factor that affects their swelling–deswelling behavior at pH changes. pH-Responsive degradable cross-linking agents which can be degraded by pH change have also been reported.139–142
Different architectures of pH-responsive polymers
The most common pH-responsive polymer structures described in the literature are linear homopolymers, amphiphilic, and double hydrophilic block copolymers which form micelles or vesicles, star, branched, and hyper-branched polymers, polymer brushes, dendrimer polymers, nanogels, microgels, and hydrogels (macrogels). The unique self-assembly properties of block, star, and branched (co)polymers make them a promising field of research for use in applications such as drug delivery. The diameter of the prepared pH-responsive polymers and their self-assembled structures are important factors in determining their applications. The polymeric materials used in biological applications are especially required to be biocompatible and less than 100 nm in diameter. These structures and their sizes are schematically depicted in Fig. 5. pH-Responsive polymer structures attracting attraction for different applications are underlined in this section.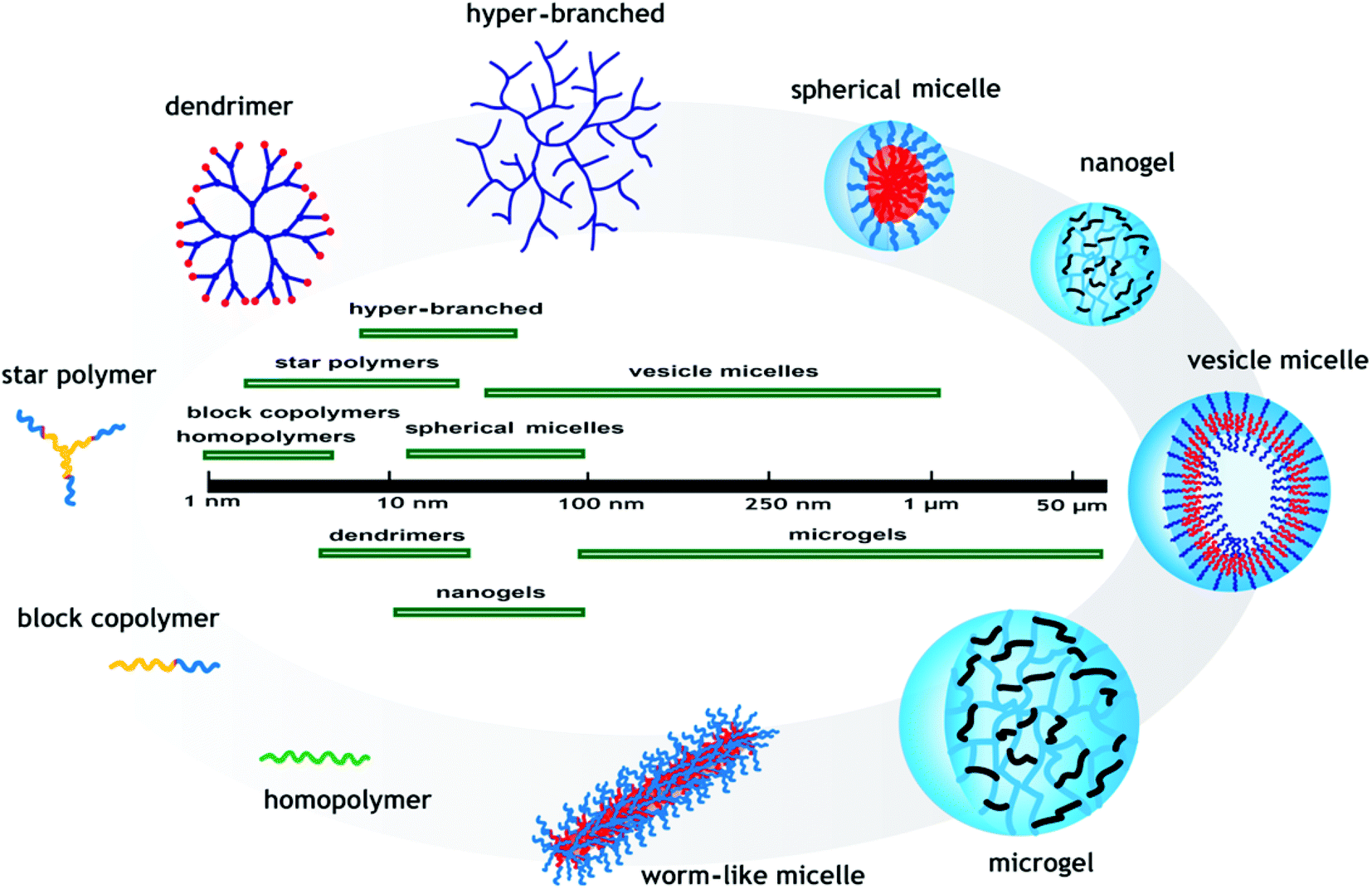 | ||
| Fig. 5 Average sizes of pH-responsive polymers with different structures. | ||
pH-Responsive linear block copolymer
pH-Responsive linear polymer structures are generally reported to be in the form of homopolymers, random copolymers, amphiphilic, and double hydrophilic block copolymers (Table 3). pH-Responsive homo- and random polymers are not preferred in a lot of applications due to their only crash-dissolution behavior by pH changes. As is known, block copolymers consist of two or more different polymer segments. If one block is soluble in a solvent but the second block is not soluble, diblock copolymers tend to self-assemble into micellar structures by the insoluble block forming the core of the micelle and the solvated soluble block forming the corona. This self-assembly behavior provides a great possibility to scientists to prepare various micellar structures, nanoparticles and different materials.| pH-responsive diblock copolymersa | |
|---|---|
| a See abbreviations for definitions of the terms used. | |
| Double hydrophilic diblock copolymers (DHBCs) (hydrophilic–hydrophilic) | Acidic-b-neutral block copolymers: PEO-b-PMAAc,147 PAAc-b-PNIPAm78 |
| Basic-b-neutral block copolymers: P2VP-b-PEG,144 PNIPAm-b-PDEA,66 PImHeMA-b-PG2MA32 | |
| Basic-b-acidic block copolymers: PVBA-b-PMEMA,148 PDMA-b-PAAc,149 PMAAc-b-PDEA,40 PAAPBA-b-PAEAm150 | |
| Basic-b-basic block copolymers: PMEMA-b-PDEA151 | |
| Acidic-b-asidic block copolymers: PNaAMPS-b-PAaH,152 PNaStS-b-PNaVBA153 | |
| Acidic-b-zwitterionic block copolymers: PNaSS-b-PSVBP154 (no micelle by pH changes) | |
| Basic-b-zwitterionic block copolymers: PMPC-b-PDPA,155 PMPC-b-PDEA,156 PDPA-b-PβDMA,89 PMEMA-b-PβDMA89 | |
| Amphiphilic diblock copolymers (hydrophilic–hydrophobic) | Acidic-b-neutral block copolymers: PS-b-PAAc,157 PCL-b-PAAc,158 PVPBA-b-PS159 |
| Basic-b-neutral block copolymers: PDMA-b-PS107, PDMA-b-PMPS,85 PPO-b-PDEA160 | |
Block copolymers which consist of both hydrophobic and hydrophilic blocks can behave as a surfactant. AB-type amphiphilic block polymers form micelles in aqueous solution by hydrophobic blocks moving to the interior to minimize their contact with water and hydrophilic blocks remaining on the outer surface of the micelles to maximize their contact with water (see Fig. 6). Various self-assembly structures such as spherical, flower, worm-like, and vesicle (hollow) micelles are formed by amphiphilic and double hydrophilic block copolymers by pH changes (Table 4).4,80,143–145 The factors which determine the self-assembly and/or micelle morphology are temperature, pH, salt, polymer concentration, solvent type, and the structure/length of blocks.146
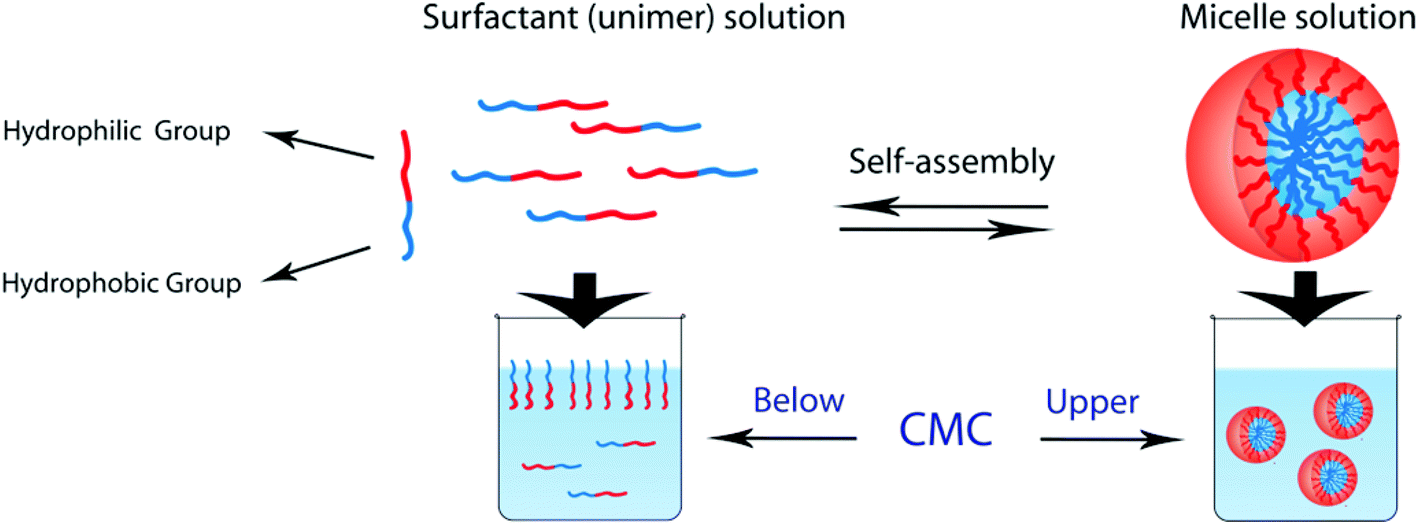 | ||
| Fig. 6 Schematic representation of unimer–micelle equilibrium in water. | ||
| Polymersa | Micelle type | Diameter | Ref. |
|---|---|---|---|
| a See abbreviations for definitions of the terms used. | |||
| Diblock copolymers (AB-type) | |||
| P2VP-b-PEG | Spherical | 100–170 nm | 144 |
| PPO-b-PDEA | Spherical | 40–80 nm | 160 |
| PβDMA-b-PDPA | Spherical | ∼20 nm | 167 |
| PCL-b-PAAc | Spherical | 100–200 nm | 158 |
| PLGA-b-PLL | Vesicle | 120–180 nm | 145 |
| PB-b-PLGA | Vesicle | — | 168 |
| PEG-b-P(DEA-co-TMSPMA) | Vesicle | 0.6–1.6 μm | 169 |
| Triblock copolymers (ABC- and ABA-type) | |||
| PDPA-b-PDMA-b-PDPA | Flower | 25–30 nm | 80 |
| PAAc-b-PS-b-P4VP | Vesicle | 100–150 nm | 170 |
| PDMA-b-PMMA-b-PMAAc | Spherical | ∼11 nm | 97 |
| PDMA-b-PPO-b-PDMA | Worm-like | 212 nm | 143 |
In addition to amphiphilic block copolymers, double hydrophilic block copolymers (DHBCs) can also form micelles due to their blocks with different stimuli responsiveness. These DHBCs can be responsive to pH in either block or in only one block. When the pH value is changed, these groups can accept or donate protons in aqueous solution and change the hydrophilicity of the related block. If there is a difference in pH-responsiveness of both blocks, pH changes may cause a self-assembly due to the dehydration of one block when the other one remains hydrophilic under related conditions. In contrast to the permanently amphiphilic block copolymers, so-called pH-responsive double hydrophilic block copolymers offer an assembly strategy without the usage of a cosolvent.4,161,162 Such amphiphilic pH-responsive block copolymer examples are given in Table 3.
There are different architectures of linear block copolymers, namely AB-type diblocks, ABA-type triblocks, ABC-type triblocks, and other multi-blocks (three or more).163–165 In general, the ABA-type triblock copolymer can form flower type micelles at low concentrations and gels at high concentrations by pH changes. pH-Responsive ABA-type PDPA-b-PDMA-b-PDPA80 having basic segments and PMAAc-b-PEG-b-PMAAc166 having acidic segments triblock copolymers have also been reported as such systems. As an amphoteric triblock copolymer, the pH-responsive ABC-type PDMA-b-PMMA-b-PMAAc triblock copolymer has been reported by Patrickios et al. by examining its solution behavior.97 In the following years, ABC, ACB, and BAC type triblock copolymers have also been demonstrated using hydrophobic MMA, basic DMA, and acidic MAAc monomers.91 Some of the pH-responsive AB, ABC and ABA type block polymers are presented in Table 4.
The micelles can form or undergo a change of micellar morphology in an aqueous solution of block copolymers by pH changes. An example of the change of micelle morphology obtained by using schizophrenic polymers is micelles–reverse micelles.161 In 1998, Armes’ group reported a new type of pH- and salt-responsive block copolymer called “schizophrenic” AB diblock copolymers (PMEMA-b-PDEA, see Fig. 7). PMEMA-core micelles with cationic PDEA shells at pH 6.5 in the presence of Na2SO4 and PDEA-core micelles with neutral PMEMA shells at alkali pH have been successfully obtained in aqueous solution.151 It is the first example of a pH and electrolyte responsive schizophrenic copolymer formed by a double hydrophilic block copolymer comprising weak polybase blocks. Later, the PDEA-b-PVBA block copolymer has been reported to be the only pH-responsive schizophrenic block copolymer formed by PVBA-core micelles with cationic PDEA shells at pH 2 and PDEA-core micelles with anionic PVBA shells in aqueous solution at pH 10.171 Many pH-responsive schizophrenic polymers such as PVBA-b-PMEMA, PLGA-b-PLL, PPO-b-PDEA AB-type diblock copolymers and PDEA-b-PDMA-b-PMEMA and PAAc-b-PS-b-P4VP ABC-type triblock copolymers have been reported after these pioneering studies.78,94,145,160,161,170,172 In later years, Lecommandoux and co-workers have succeeded in preparing schizophrenic vesicles using the PLGA-b-PLL diblock copolymer.145
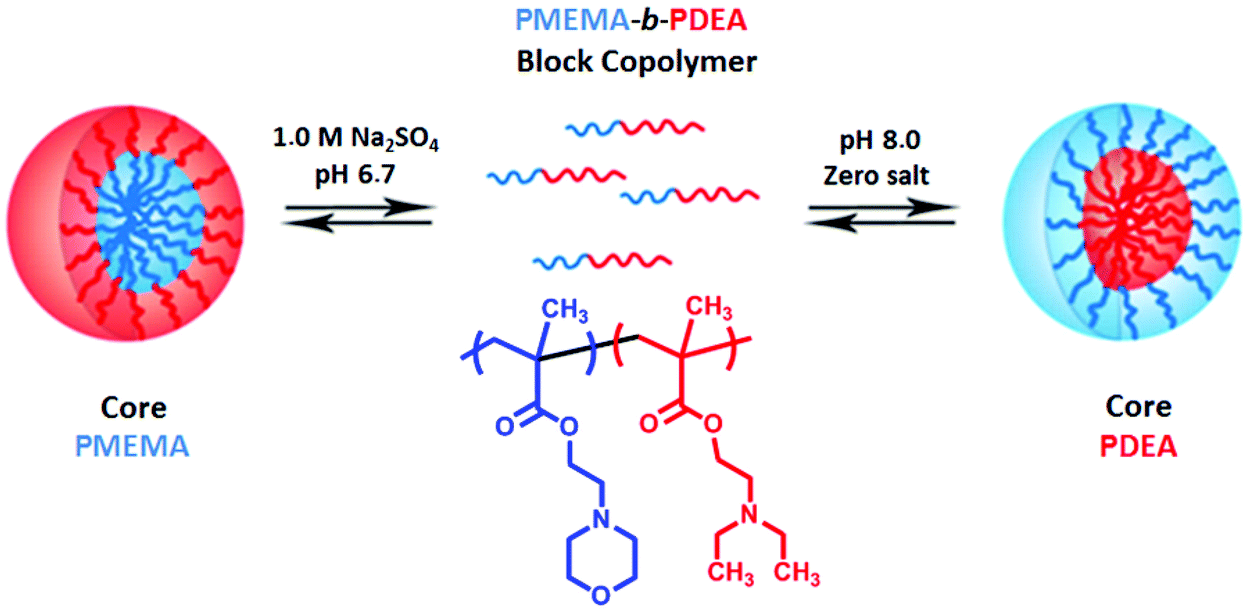 | ||
| Fig. 7 Schematic representation of the formation of micelles and reverse micelles for a schizophrenic PMEMA-b-PDEA diblock copolymer in aqueous solution.151 | ||
In 2015, Armes’ group achieved non-ionic PG2MA-b-PHPMA diblock copolymers which can exhibit pH-responsive behavior. This behavior is due to the fact that the carboxylic acid-functional RAFT agent is used in the synthesis of the PG2MA-b-PHPMA block copolymers. This study is a good report on the reversible transition from worm-like micelles to spherical micelles depending on pH change (Fig. 8).134
 | ||
| Fig. 8 TEM images obtained on addition of NaOH followed by dilution of a 10% w/w aqueous dispersion of a PG2MA56-b-PHPMA155 diblock copolymer prepared using the carboxylic acid functionalized PETTC RAFT agent: (a) pH 3.5 (initial worms); and (b) pH 6.0 (spheres) (Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, modified from ref. 134). | ||
The practical applications of micelles are limited due to their structural instability since the micellar structure can hardly remain stable upon dilution or changes of external conditions such as changes in pH, ionic strength, type of solvent, and temperature.173 In order to enhance the stability, cross-linking of the micelle core or corona by reacting functional groups of polymer chains with a bifunctional cross-linker was reported to be a useful approach. Based on cross-linking chemistry, core cross-linked micelles (CCL),174,175 shell cross-linked micelles (SCL),173–175 and intermediary layer cross-linked micelles (ILCL)41,174 have been developed (see Fig. 9). In 1996, Wooley and co-workers reported the first example of covalently-stabilized block copolymer micelles. In this first example, polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) block copolymers quaternized with 4-(chloromethyl)styrene have been used as building blocks to prepare conventional core–shell micelles using a THF/water cosolvent approach.176 Cross-linked micelles (CLMs) are potentially useful as nanosized vehicles for the delivery of various actives.177 The CLMs also show swelling–deswelling behavior with pH change. These CLMs are also called nanogels in the literature.178,179 pH-Responsive cross-linked spherical micelles are the most studied species. Although limited in number, they have been made during the studies of the vesicle169,180,181 and worm-like143,181 micelles. CLMs have been obtained from various functional monomers such as TMSPMA, MPMA, and CMA169,179,182 with various cross-linking agents such as BIEE, DVS, dicarboxylic acid, etc.74,173–175,183 Degradable CLMs which are especially important for biomedical applications can be degraded by photo-, pH, biomaterials, and other chemicals.75,173–175,184 Some of them are given in Table 5.
 | ||
| Fig. 9 Types of cross-linked micelles: (a) SCL micelles, (b) ILCL micelles, and (c) CCL micelles. | ||
| Block copolymers | Cross-linking moieties | Cross-linking agent | Core of micelle | Response | Ref. |
|---|---|---|---|---|---|
| a Drug delivery. b Nanoreactors, (↑) represents the swelling (see abbreviations for the definitions of polymers and reagents). “A” represents the first block, “B” represents the second block and “C” represent the third block. | |||||
| Spherical CCL micelles | |||||
| PMEO2MA-b-P(DEA-co-TPHMA) | TPHMA | RuCl3 | B/C | Low pH↑a | 82 |
| MPEO-b-PG2MA-b-PDPA | PG2MA | DVS | B | Low pH↑ | 185 |
| PS-b-PAAc | PAAc | BIEE | A | — | 186 |
| P(QDMA-co-DMA)-b-PMEMA | DMA | BIEE | C | Low pH↑ | 187 |
| P(MMA-co-MPMA)-b-PNIPAm | MPMA | Si–O–Si | A/B | Low pH↑ | 182 |
| P(MMA-co-MPMA)-b-PDEA | MPMA | Si–O–Si | A/B | Low pH↑ | |
| PAAPBA-b-PAEAm | AEAm | Dicarboxylic acid | A | Anionic/cationic | 150 |
| MPEO-b-P(DEA-co-CMA) | CMA | UV light | B/C | Low pH↑b | 179 |
| PEO-b-(PMAAc-Hyd) | MAAc-Hyd | DTE | B | High pH↑a | 74 |
| PVAm-b-PNIPAm | PVAm | Anthracene | A | pH degradable | 184 |
| Spherical ILCL micelles | |||||
| PDEA-b-PDMA-b-PMEMA | DMA | BIEE | A or C | Low pH↑ | 94 |
| PDPA-b-PDMA-b-PMEMA | DMA | BIEE | A or C | Low pH↑ | 39 |
| MPEO-b-PG2MA-b-PDEA | G2MA | DVS | C | Low pH↑b | 188 |
| MPEO-b-PDMA-b-PMEMA | DMA | BIEE | C | Low pH↑ | 41 |
| PEG-b-P(CGMA-co-G2MA)-b-PDEA | CGMA | UV light | D | Low pH↑ | 189 |
| MPEO-b-PG2MA-b-PDPA | G2MA | DVS | C | Low pH↑ | 185 |
| MPEO-b-PAPMAm-b-PNIPAm | APMAm | TDA | C | pH degradablea | 190 |
| MPEG-b-P(LGA-co-CELG) | LGA | DTbDEA | B/C | High pH↑a | 183 |
| Spherical SCL micelles | |||||
| PDMA-b-P(MMA-co-CMA) | CMA | UV light | A | Desolve/low pH | 191 |
| PAAc-b-PDMA | DMA | BIEE | A | High pH↑ | 149 |
| PSPMA-b-P(DEGMMA-co-CMA) | CMA | UV light | Both | High pH↑ | 178 |
| PDMA-b-PMAAc | Both | BIEE | Both | — | 40 |
| (CNPBA-Dex)-b-PLA | CNPBA | CNPBA | B | pH degradablea | 192 |
| MPEO-b-PDMA-b-PMEMA | DMA | BIEE | C | — | 41 |
| Other micelle types | |||||
| MPEO-b-P(DEA-co-TMSPMA) | TMSPMA | Si–O–Si bond | B/C | Low pH↑b | 169 |
| PDMA-b-PPO-b-PDMA | DMA | BIEE | B | — | 143 |
| MPEO-b-P(DEA-co-GMA) | GMA | EN | B | Low pH↑ | 180 |
Spherical micelles and their SCL micelles were first prepared from PCL-b-PAAc diblock copolymers by Wooley and co-workers by first cross-linking of the PAAc shell. Then, the polyester core was degraded hydrolytically to obtain hollow spheres.158 Similarly, Fustin and co-workers have reported hollow spheres by using PtBA-hv-PDMA block copolymers by cross-linking the PDMA shell followed by UV light irradiation of the PtBMA-core.193 A CCL mixed micelle with dual responsive shells has also been constructed from two amphiphilic block copolymers P(MMA-co-MPMA)-b-PNIPAm and P(MMA-co-MPMA)-b-PDEA via a two-step process by Zhang and co-workers. PMPMA behaves as a cross-linker depending on solution conditions. Both block copolymers form core–shell mixed micelles in acidic aqueous solution at room temperature followed by using the inorganic “silica-based” cross-linking strategy without an additional cross-linker.182 ILCL micelles from the MPEO-b-PAPMAm-b-PNIPAm triblock copolymer have been reported by McCormick and co-workers by using terephthaldicarboxaldehyde as a cross-linker. ILCL micelles can be reversibly cleaved by simply adjusting the solution pH.190 Such polymers are used in drug release studies.
pH-Responsive star polymers
Star polymers, containing several linear polymer chains connected to one central core, are another class of macromolecules with a precisely controlled architecture. Most commonly used synthetic approaches for the preparation of star polymers using different polymerization techniques are “core-first”, “arm-first”, and “coupling-onto” approaches.194,195 There are different star-shaped segmented (co)polymers with a diameter between 7 and 30 nm.196–199 Particularly, the so-called pH-responsive star polymers have attracted great interest since the stars bearing block/arms undergo sharp phase transitions upon responding to pH changes. These polymers can form micellar self-assembly or undergo a change of micelle morphology by pH changes. In vesicle, rod, spherical, and flower micelles morphology is obtained using star polymers.96,114,115,200 For star polymers having very different segments, it is quite difficult to show the morphology of the micelles via self-assembly.198 In addition, some of them may form gels by pH changes.201 Some pH-responsive star polymers and their behaviors exhibited by pH changes are given in Table 6.| Star polymersa | Micelle type | Core block of micellesa | Diameter of micelles | Ref. |
|---|---|---|---|---|
| a See abbreviations for definitions of the terms used. b Drug delivery. c Enzyme carrier. | ||||
| PMPC-b-P(DMA-co-DPA) | Gel formation | — | — | 201 |
| star-P(DEA-b-MMA-b-PEGMA) | Multi-compartment spherical micelle | PDEA/PMMA | 60–110 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
114 |
| PS(PNIPAm-b-P4VP)2 | Spherical and rod | — | 100–200 nm | 115 |
| SPCL-b-PCEMA-b-PDMA | Spherical-ILCL | SPCL | 87–230 nm | 127 |
| PSn(P2VP-b-PAAc)n | Worm-like, spherical multicore | — | 30–500 nm | 202 |
| PDMA-star-PDEA | Spherical | PDEA | 25–50 nm | 95 |
| PMMA-b-P(NIPAm-co-DMA)3 | Spherical | PMMA | 85–110 nmb |
77 |
| 4AS-PCL-b-PDEA-b-PPEGMA | Spherical | PCL | 60–220 nmb |
116 |
| MPEG-b-PtBA-b-PCL | Spherical | PCL | 120–237 nmb |
203 |
| (PEO-b-PDEA)4 | Spherical | PDEA | 21–56 nm | 196 |
| PEG-b-PMAAc-b-PDEA | Spherical | Both | 57–94 nm | 197 |
| PEO-b-PDMA-b-PHEA | Spherical | PHEA | —c | 204 |
| PLL-b-(PLGA)2 | Spherical | PLL or PLGA | 77–110 nm | 205 |
| μ-(PtBA)(PCEMA)(PEO) | Vesicle | — | 188 nm | 200 |
| PDEA-g-CD-g-PNIPAm | Vesicle | — | 30–85 nm | 199 |
| P2VP-b-PMMA-b-PAAc | Vesicle | PMMA | 0.1–1.5 μm | 206 |
| MPEG-b-PHIS | Vesicle | PHIS | ∼70 nm | 207 |
pH-Responsive star block copolymers are also good polymers for the preparation of hydrogels as reported by Armes and co-workers. They can be synthesized using 2-(methacryloyloxy)ethyl phosphorylcholine (MPC) as the inner block and followed by sequential monomer addition of various tertiary amine methacrylates or mixtures thereof such as DMA, DMA/DEA, and DMA/DPA. Star diblock copolymer gel agents having both thermo- and pH-responsive nature have also been reported by copolymerization of DMA with DPA.201 Whittaker and co-workers have reported the successful synthesis of PPEGMA-b-P(TFEMA-co-DMA) degradable core cross-linked star (CCS) polymers through the arm-first approach. The related polymer could form nanoparticles in aqueous solution and the particle size is dependent on solution pH.208 The star-P(DEA-b-MMA-b-PEGMA)6 triblock copolymer has self-assembly behavior depending on pH change. The star copolymer can self-assemble into multi-compartment micelles at pH 10.5, vesicles at pH 7.4 and micelles at pH 2.0.114 Multiarm star-shaped terpolymers based on PSn−core-(P2VP-b-PAAc)n are known to be pH-responsive in dilute aqueous solutions (Fig. 10). A variety of amphoteric self-assemblies such as unimolecular micelles, multicore micelles, and worm-like micelles have been observed by pH changes.202
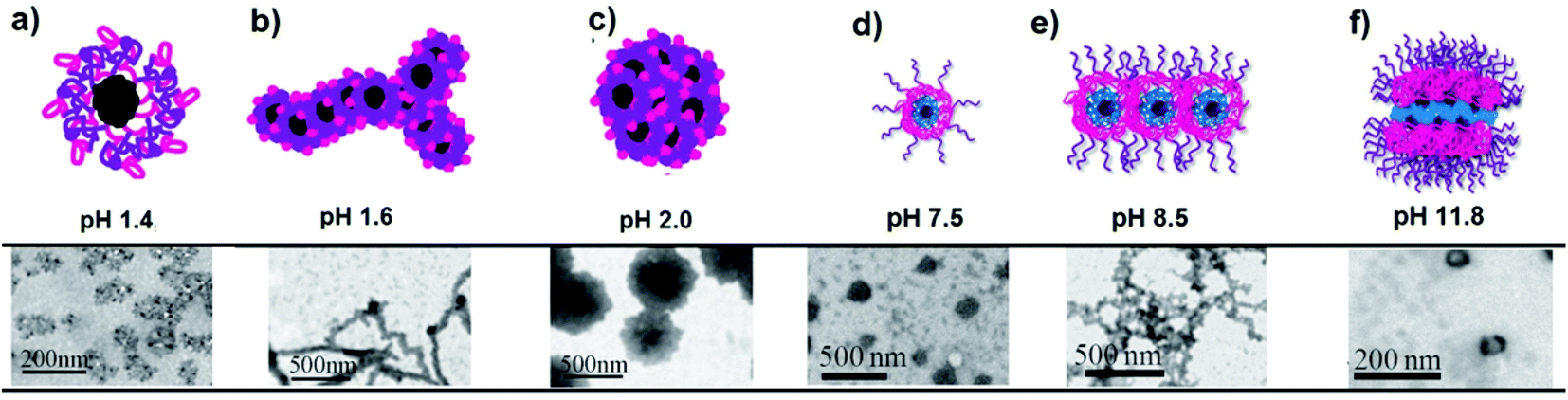 | ||
| Fig. 10 TEM images of the PSn−core-(P2VP-b-PAAc)n star conformation and the corresponding self-assemblies aqueous solution at: (a) pH 1.4 unimolecular micelle, (b) pH 1.6 worm-like micelle, (c) pH 2.0 multicore micelle, (d) pH 7.5 unimolecular micelle, (e) pH 8.5 unimolecular micelle association towards network-like assemblies, and (f) pH 11.8 multi-compartment multi-molecular micelle (Copyright 2011, The Royal Society of Chemistry, modified from ref. 202). | ||
The miktoarm copolymer μ-(PtBA)(PCEMA)(PEO)1.14 has been used for the preparation of various vesicles via a double assembly strategy. PCEMA units have been used as photo cross-linkers and then hydrolysed PtBA units. These cross-linked vesicles exhibited pH-responsive reagent release in aqueous media.200 Similarly, Lang and co-workers have also reported cinnamate-functionalized star amphiphilic triblock copolymers SPCL-b-PCEMA-b-PDMA which can self-assemble into core–shell-corona micelles in aqueous solution. The micelle stability has been improved by photo cross-linking by PCEMA units. pH changes provided swelling–deswelling on the cross-linked micelles.127 The Y-shaped miktoarm star polypeptide PLL-b-(PLGA)2 copolymer has also been reported to be an interesting polymer which can form both PLGA-core micelles at acidic pH and PLL-core micelles under alkaline pH conditions (Fig. 11). Thus, a pH-responsive self-association “schizophrenic” behavior of this segmented star polypeptide has been revealed in aqueous media.205
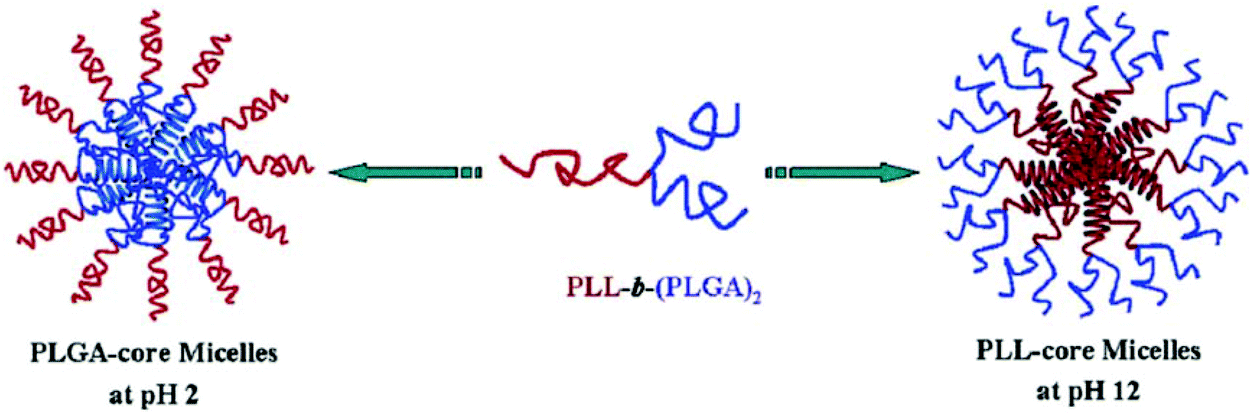 | ||
| Fig. 11 pH-Induced micellisation of PLL-b-(PLGA)2 associated with coil-to-helix transitions (Copyright 2008, The American Chemical Society, reprinted from ref. 205). | ||
pH-Responsive branched and hyper-branched polymers
Among various polymer architectures, hyper-branched polymers are interesting materials since they have unique properties compared to their respective linear analogues. They have better solubility in a wide range of solvents due to their decreased chain entanglement, low melt and solution viscosities, reduced hydrodynamic volume, and critical phase transition behaviors. They also provide various advantages for further derivative materials due to the presence of a huge number of chain end functionalities.209 In 1990, the first branched polymer using the Suzuki-type coupling of dibromoboronic acid monomers was reported.210 In recent years, various branched and hyper-branched polymers have been reported with various application possibilities. Branched polymers have rather broad molecular weight distributions due to uncontrolled polymerization chemistry. However, novel approaches have recently been applied with controlled polymerization techniques which allow one to control the primer chain-length. Their synthesis via various polymerization methods such as GTP, RAFT, NMP, and ATRP became very easy.92,209 These polymers can self-assemble and form micelles211–213 and/or (de)swellable-particles by pH changes.214,215 Worm-like, vesicles, and spherical micelles are obtained using pH-responsive branched and hyper-branched polymers. pH-Responsive branched and hyper-branched polymers and their solution behaviors are given in Table 7.| Polymer containsa | Structure formed by pH change | Applications | Ref. |
|---|---|---|---|
| a See abbreviations for definitions of the terms used. | |||
| Hyper-branched polymers | |||
| PEGMA-co-PDEA-co-PtBAEMA-co-EGDMA | (De)swelling | Drug delivery | 214 |
| P(Boc-VaI-HEA-star-MEO2MA/PEGMA) | Multi-micelle | — | 128 |
| PEGMA/PDEA-PEG | Spherical micelle | — | 216 |
| P(BAC-AMPD)-PEG | (De)swelling | Drug delivery | 215 |
| BP(DMA-co-MAEBA-co-DTDMA)(PMAGP)n | Spherical micelle | Drug delivery | 126 |
| HBPE-PDMA | Vesicle micelle | — | 217 |
| PAAc-b-PMEA-b-PNIPAm-b-MPEG | Vesicle micelle | Drug delivery | 213 |
| Branched polymers | |||
| PBIEM-g-PDMA | Worm-like micelle | — | 212 |
| PNIPAm-b-P(EA-g-DEA) | Schizophrenic micelle | — | 211 |
| PEO-b-[PGMA-g-(PDEA)(PMEO2MA)] | Schizophrenic micelle | — | 83 |
| PDEA-b-P(DMA-co-EGDMA) | Spherical micelle | — | 92 |
The hyper-branched polyethylene with thiocarbonyl thio moiety ends (HBPE-BSPA) can be used as a macro-RAFT agent for the synthesis of hyper-branched polyethylene amphiphiles, HBPE-PDMA, by RAFT polymerization of DMA. The resultant HBPE-PDMA can self-assemble to form supra-molecular polymer vesicles in aqueous solution. A preliminary investigation on the thermo- and pH-responsive behaviors of the polymer is also reported (see Fig. 12).217
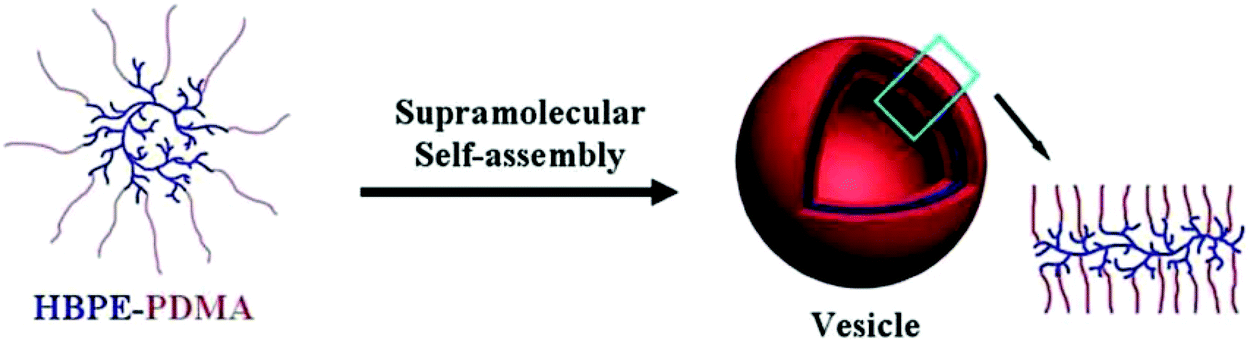 | ||
| Fig. 12 A possible self-assembly mechanism for polymer vesicles (Copyright 2012, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, modified from ref. 217). | ||
The PEO-b-[PGMA-g-(PDEA)-(PMEO2MA)] coil–rod diblock copolymer has been synthesized via a combination of ATRP and click reaction. This coil–rod diblock copolymer exhibits pH- and thermo-responsive supra-molecular aggregation behavior in aqueous solution. It behaves as schizophrenic and shows micelles–reverse micelles self-assembly at solution pH and temperatures (Fig. 13).83
 | ||
| Fig. 13 Schematic illustration of the synthesis and multi-responsive supra-molecular self-assembly of a coil–rod double hydrophilic diblock copolymer: PEO-b-[PGMA-g-(PDEA)(PMEO2MA) (Copyright 2009, The American Chemical Society, reprinted from ref. 83). | ||
A series of novel pH-responsive, amphiphilic branched copolymers based on PEGMA, DEA, and tBAEMA have also been prepared. Dynamic light scattering data indicated micelle formation with a diameter of about 16 nm by the formation of the hydrophobic PtBAEMA and PDEA core and the hydrophilic PEGMA corona above pH 8. With the decrease of pH from 8 to 6, a dramatic increase in the hydrodynamic radius of polymer particles from 16 nm to 130 nm has been observed, resulting from the protonation of the PDEA segment.214
pH-Responsive dendrimer polymers
Dendrimers are an important member of the class of polymers. The first report on dendrimer synthesis is attributed to Vögtle and co-workers218 in 1978, followed by the work of Tomalia and co-workers in the early 1980s.219 In later years, many dendrimers were reported with several application studies. They have great control over polydispersity, molecular weight and architecture with a high surface functionality. Their water solubility is an important property that makes them attractive for drug delivery and biomedical applications.219–222 Among them, pH-responsive dendrimers have attracted great interest in recent years. Some dendritic molecules such as PEI, PPI, and PAMAM already show pH-responsive behavior without requiring further modification. Depending on requirements, some dendrimers have to be modified to provide a different functionality. Despite the superior properties of dendrimers, their diameters being less than 15 nm cause some difficulties in some application processes. To overcome this problem, various polymers have been grafted onto dendrimers (Fig. 14 and Table 8).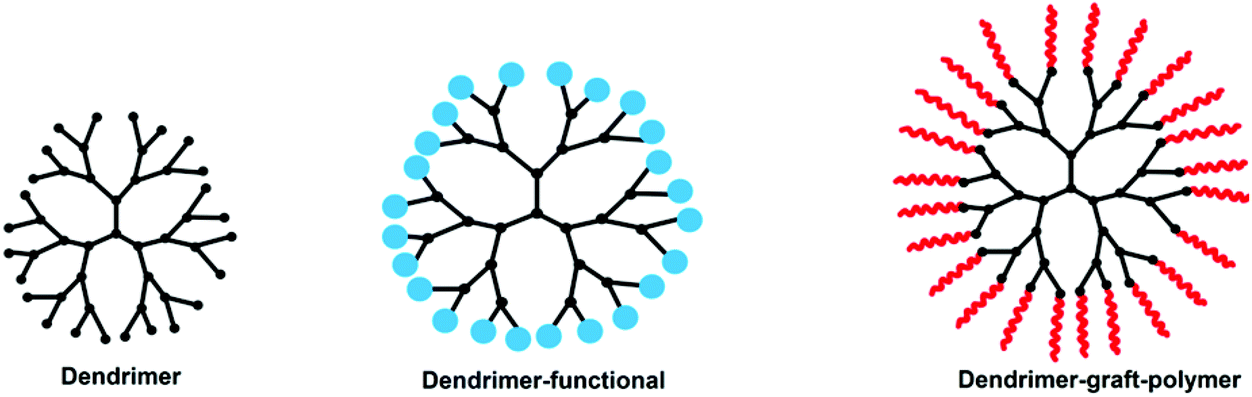 | ||
| Fig. 14 Chemical structures of a dendrimer, a functional dendrimer and a dendrimer-graft-polymer. | ||
| Dendrimer containsa | Applications | Ref. |
|---|---|---|
| a See abbreviations for definitions of the terms used. | ||
| Dendrimer | ||
| PAMAM | Drug delivery | 223 |
| PEI | — | 224 |
| PPI | Carrier | 225 |
| Funtional dendrimer | ||
| PEI-sulfopropylated | — | 226 |
| PEI-phosphonated terminated | — | 227 |
| PPI-carboxylic acid terminated | — | 228 |
| HBPO-carboxylic acid terminated | — | 10 |
| Dendrimer-graft-polymers | ||
| H40-PCL-b-PAAc-b-MPEG/PEG-FA | Drug delivery | 229 |
| PEI-g-(PLG-b-PEG) | Drug delivery | 230 |
| PEI-PLL-b-PEG | Protein carrier | 231 |
| PAMAM-g-PDMA | Drug delivery | 232 |
| PAMAM-g-PDMAPS | Drug delivery | 233 |
| PAMAM-g-PEG (vesicle micelle) | — | 234 |
pH-Responsive brush and comb polymers
Polymer brushes are one kind of ultrathin polymer layers linked with one chain end to the surface of the solid substrate. As shown in Fig. 15, there are two common methods for the synthesis of such brushes on an inorganic surface. They are “grafting to” (attachment of the side chains to the backbone) and “grafting from” (grafting the side chains from the backbone).235,236 In a “grafting to” method, pre-synthesized polymer chains are grafted to a backbone through the physicochemical adsorption or chemical reactions. On the other hand, during the last decade, the development of surface-initiated controlled/living radical polymerization (SI-CRP) provides a “grafting from” approach allowing one to tune the brush thickness, composition and architecture of brush polymers. Well-defined polymer brushes based on the “grafting from” strategy can be initiated directly from initiator-functionalized surfaces. The most commonly used polymerization techniques which have been applied in the “grafting from” approach via SI-CRP methods are NMP, RAFT, and ATRP.235,236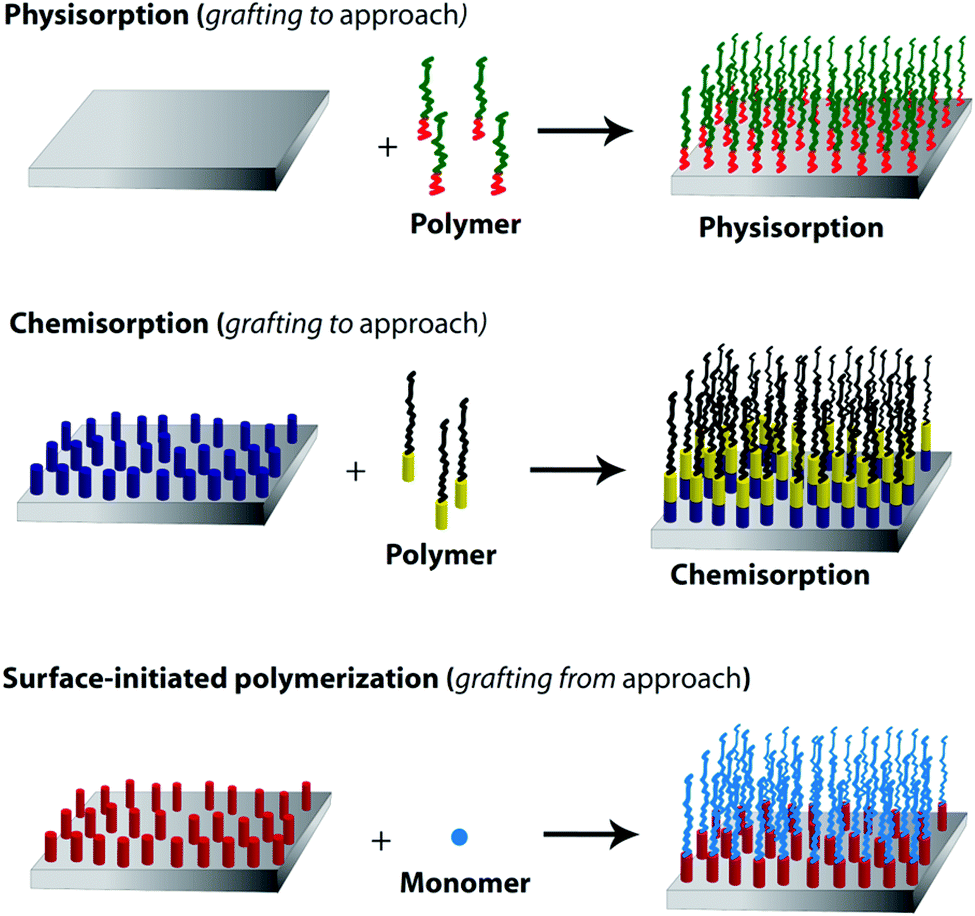 | ||
| Fig. 15 Synthetic strategies for the preparation of polymer brushes: (a) physisorption of diblock copolymers (grafting to), (b) chemisorption (grafting to) and (c) polymer brushes grown via surface-initiated polymerization (grafting from). | ||
If any segment of related brushes has a response to external stimuli, it is possible to control the conformation, surface energy, and phase transition of a polymer brush by tuning the composition, density and length of the brushes. There are a large number of reports in the literature that describe their pH-responsive nature.235–237 These brushes contain ionisable pendant groups that can accept or donate protons in response to an environmental change in pH that leads to tuning of surface wettability. pH-Responsive brush polymers are used in many applications such as drug delivery or carriers,238 non-biofouling,239 membranes,240 cell adhesive surfaces239 and protein adsorption–desorption241 (Table 9).
| Polymersa | Method for synthesis | Applications | Surfaces | Ref. |
|---|---|---|---|---|
| a See abbreviations for definitions of the terms used. | ||||
| Basic brush polymers | ||||
| PDMA | Grafting from | Protein ads. | Au coated quartz | 242 |
| PDMA | Grafting from | Wettability | Silicon substrates | 243 |
| PDEA and PDPA | Grafting from | Wettability | Silicon substrates | 244 |
| PNIPAm-co-PVI | Grafting from | — | Au coated silicon | 119 |
| PSPEA-co-PDMA | Grafting from | Catalyst surface | Silicon nanospheres | 245 |
| Acidic brush polymers | ||||
| PMAAc | Grafting from | Membrane | Silicon nitride | 240 |
| P(MAAc-co-DVB)-g-PNIPAm | Grafting from | Drug release | Silicon nanospheres | 238 |
| PAAPBA | Grafting from | Cell capture and release | Silicon nanowire | 246 |
| PEGMP | Grafting from | membrane | Silica film | 247 |
| PTMA-co-PCAA | Grafting from | Non-fouling and adhesive surface | Au chips and Au coated silicon | 239 |
| PKSPMA | Grafting from | — | Au coated silicon | 248 |
| Acidic–basic brush polymers | ||||
| PDMA-b-PMAAc | Grafting from | Protein ads. | Silicon wafer | 241 |
| PAAc-b-P2VP and PAAc-b-P4VP | Grafting to | — | Silicon wafer | 249 |
| Mixed PAAc-P2VP | Grafting to | Drug delivery | ITO glass and silicon | 250 |
| Mixed PAAc-P2VP | Grafting to/grafting from | — | Silica particles | 251 |
Similar to brush type polymer synthesis, there are various reports based on the synthesis of pH-responsive comb type polymers using both the “grafting from” and “grafting to” approaches (see Table 10). Additionally, the “grafting through” method with the usage of macromolecular monomers is also reported to be useful for the same purpose similarly to the “grafting from” approach.252,253 The related reports are mainly based on PAAc, CS and PDMA type polymers. ATRP is the mostly preferred chemistry for the “grafting from” approach. In the “grafting to” pathway, chain ends of polymer molecules are commonly functionalized with alkyl-azide, thiol–ene and succinimide chemistries. As an example, the P(NVK-co-VBC)-g-P(DMA-co-AAc) comb polymer has been reported to be pH-responsive and has potential in drug delivery applications since it can self-assemble with pH change and form hollow micelles with different diameters at different temperatures.254
| Polymersa | Method for synthesis | Polymerization method | Ref. |
|---|---|---|---|
| a See abbreviations for definitions of the terms used. | |||
| P(MMA-co-HEMA)-g-PDMA | Grafting from | ATRP | 255 |
| Pluronic-g-PAAc | Grafting from | — | 256 |
| P(NVK-co-VBC)-g-P(DMA-co-AAc) | Grafting from | ATRP | 254 |
| Chitosan-g-PNIPAm | Grafting from | ATRP | 257 |
| PAMA-g-PLGA | Grafting from | ROP | 258 |
| PEGMA-g-PMAAc | Grafting through/from | ATRP | 252 |
| PEGMA-g-PDEA | Grafting through/from | ATRP | 253 |
| PNBC-g-PAAc | Grafting to | — | 259 |
| PNBC-g-PDMA | Grafting to | — | 259 |
| CS-g-PNIPAm | Grafting to | — | 260 |
| CS-g-PNIPAm | Grafting to | — | 261 |
| CS-g-PDMA | Grafting to | — | 262 |
| Alginate-g-PNIPAm | Grafting to | — | 263 |
pH-Responsive hydrogels (macrogels), microgels and nanogels
It is also worth mentioning that pH-responsive hydrogels are another type of pH-responsive polymer family. Hydrogels are network polymeric materials that have chemical or physical cross-linking among their macromolecular chains and remain insoluble in aqueous media.264 Cross-linking of polymers can be carried out by electrostatic or molecular interactions between polymer chains (physical gels) or by chemically linking with cross-linking agents of polymer chains (chemical gels). Hydrogels are hydrophilic polymer networks and may absorb water up to thousands of times their dry weight. Hydrogel compositions can be divided into three classes: natural polymer hydrogels, synthetic polymer hydrogels and a combination of the two classes.7Hydrogels can exhibit a phase transition with a change in external conditions such as pH, temperature, electric field, magnetic field, light, ionic strength, solvent, etc. and are known as ‘stimuli-responsive’ or ‘smart’ gels.265 pH-Responsive hydrogels can be synthesized from pH-responsive polymers possessing ionisable functional groups which either accept or release protons in response to changes in environmental pH. As seen in Fig. 16, acidic hydrogels prepared with acidic monomers become negatively charged by releasing protons at high pH and swell. On the contrary, basic hydrogels prepared with basic monomers become positively charged by accepting protons at low pH and swell. Such ionizations cause their swelling due to an increase in hydrophilicity of related groups, changes in osmolarity and ionic interactions within the gel.266 Swelling of a pH-responsive hydrogel is slow and takes hundreds of hours until it reaches equilibrium, but its deswelling is very fast (minutes). Amphoteric hydrogels such as PDMA-co-PMAAc contain both basic and acidic monomers, swell at low or high pH but deswell at isoelectric point.267 Such hydrogels swell in a wider pH range compared to other hydrogels.70,267–269
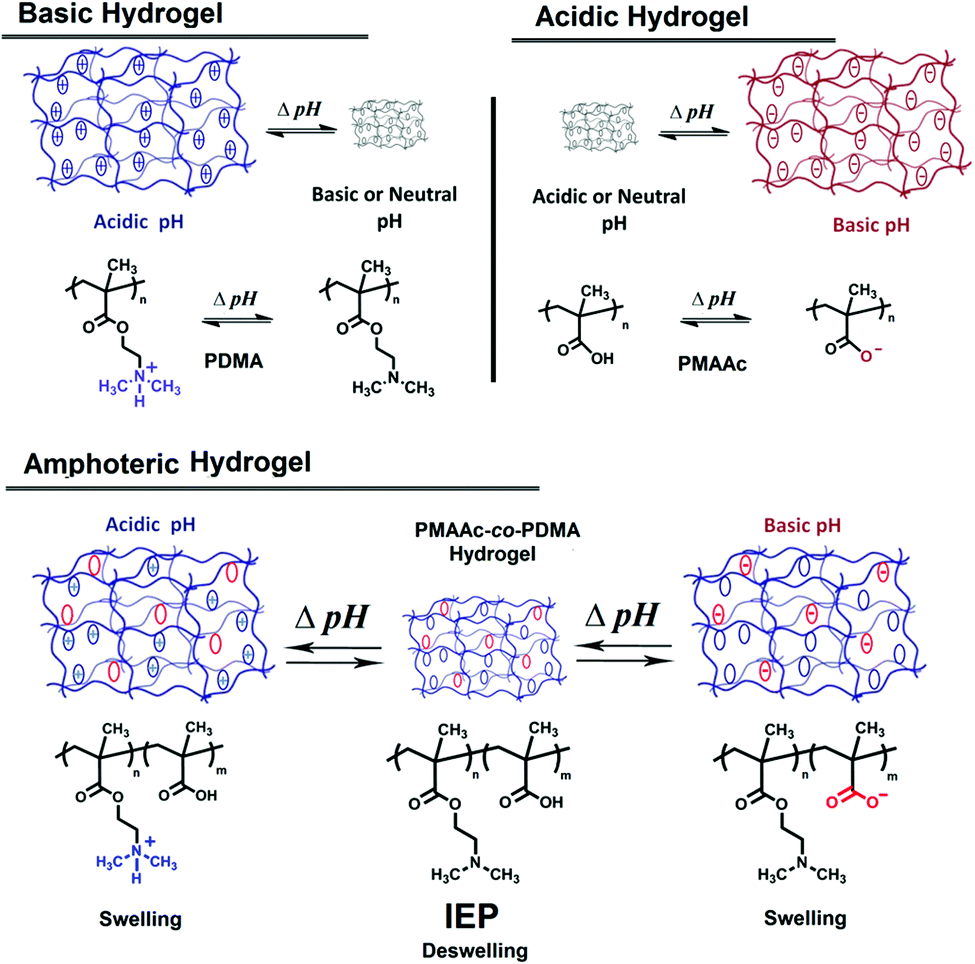 | ||
| Fig. 16 Basic, acidic, and amphoteric hydrogels and their swelling–deswelling behaviors. | ||
Hydrogels are also classified according to their size: macrogels (≥100 μm), microgels (0.1–100 μm) and nanogels (1–100 nm).264 Various macro-, micro-, and nanogels have been synthesized containing different hydrophilic polymers. Nanosized PMMA-co-PAAc hydrogels were first prepared in 1991.270 In 1996, a pH-responsive nanogel was developed for biomedical application as a drug delivery carrier by Gurny et al.271 Such smart polymers are very promising polymers in drug delivery applications as a carrier and/or release system because of their high loading capacity, high stability and responsiveness to pH of the medium.272 In the synthesis of pH-responsive hydrogels, synthetic polymers such as PDMA,273,274 PAAc and PMAAc84,142 and the natural polymer chitosan appear to be frequently used.50,275 A few examples of pH-responsive hydrogels in natural, synthetic or combinations of the two classes with a diameter between 50 and 1000 nm are given in Table 11.
| Polymersa | Types | Response | Cross-linking agentsa | Ref. |
|---|---|---|---|---|
| Arrows (↑) represent the swelling.a See abbreviations for definitions of the terms used. | ||||
| Mussel adhesive proteins | Macro | pH degradable | Fe3+ | 276 |
| CS-PVP | Macro | Low pH↑ | GA | 48 |
| PAAc-PA-PBuAA-PMAGGONp | Macro | Low pH↑ | ACDAAB | 140 and 277 |
| PHPMAm-co-PAAc-co-PBuAm | Macro | Low and high pH↑ | AB | 278 |
| PAM-co-PAAc-co-PBuAm | ||||
| PMA-co-PAAc-co-PBuAm | ||||
| PDMA-co-PAAc-co-PBuAm | ||||
| PAAPBA-co-PVDT-co-POEGMA | Macro | High & low pH↑ | PBAC | 279 |
| Dextrin/PAAc | Macro | High pH↑ | MBA | 141 |
| PGH-PAAc, PGH-PMAAc | Macro | High pH↑ | Modified PLGA | 142 |
| PGH-PEAAc, PGH-PPAAc | ||||
| 4-Arm PEG catechol/BDBA | Macro | pH degradable | BDBA | 280 |
| PEGMP-PHEMA | Macro | High pH↑ | — | 14 |
| PASA | Macro | High pH↑ | — | 281 and 282 |
| PVP-co-PIA | Macro | Low pH↑ | EGDMA | 283 |
| P2VP | Micro | Low pH↑ | DVB | 135 |
| PEA-co-PMAAc | Micro | High pH↑ | BDDA | 284 |
| PMMA-PMAAc | Micro | High pH↑ | EGDMA | 136 |
| Chitosan/gelatin | Micro | Low pH↑ | GA | 285 |
| PMAAc/PAAc-g-PPEGMA | Micro | High pH↑ | TEG | 286 |
| PEG-chitosan | Micro | Low pH↑ | PEG | 50 |
| PMEMA | Micro | Low pH↑ | EGDMA | 65 |
| PMAAc-co-PNIPAm (yolk/shell) | Micro | High pH↑ | EGDMA | 84 |
| PS-co-PtBAEMA | Micro | Low pH↑ | DVB | 287 |
| PDMA | Nano | Low pH↑ | EGDMA | 274 |
| PMMA-co-PAAc | Nano | High pH↑ | EGDMA | 270 |
| MPEG-g-PDEA | Nano | Low pH↑ | EGDMA | 288 |
| PNIPAm-co-PDMA | Nano | Low pH↑ | MBA | 273 |
| MPEG-b-P(LGA-co-CLG) | Nano | Low pH↑ | CLG | 289 |
| PMAAc-g-PPEGMA | Nano | High pH↑ | TEG | 290 |
| PDEA-co-PTBAEMA-g-PPEGMA | Nano | Low pH↑ | TEG | 138 |
| P2VP | Nano | Low pH↑ | DVB | 137 |
| Lipoic acid modified PLL | Nano | Low pH↑ | Lipoic acid | 291 |
Recently, pH-responsive degradable hydrogels which can be degraded by biomaterial or physiological conditions,139–142,277,278 oxidative processes,183,279 and pH changes276,279,280,292 have been reported. This behavior occurs by degradation of the polymer skeleton or cross-linking agents. The synthesis of biodegradable hydrogels which are used in medical and pharmaceutical applications has gained great importance in recent years.
As shown in Fig. 17, a successful synthesis of a boron containing hydrogel based on 4-arm PEG catechol and 1,3-benzenediboronic acid (BDBA) has been reported as a novel pH-responsive hydrogel. This hydrogel has been produced by complexation of a multifunctional catechol polymer with a bifunctional borate compound. The dynamic nature of the boronate ester linkages gives rise to self-healing hydrogels exhibiting high stability at alkaline pH (∼9.0) and low stability at acidic pH (∼3.0).280
 | ||
| Fig. 17 Schematic illustration of pH-responsive hydrogels based on 4-arm PEG catechol and BDBA in aqueous solution at 20 °C (Copyright 2011, The Royal Society of Chemistry, modified from ref. 280). | ||
As shown in Fig. 18, pH-responsive hollow P(MBA-co-MAAc) microgels have been successfully synthesized and used in drug delivery studies.293 In another study, pH-responsive hydrogel fibers by the electrospin method have been reported by using PNIPAm and PAAc monomers.294 In another study, enzymatically digested pig hair keratin has been modified by grafting copolymerization with a functional monomer, MAAc, which afforded an enzymatically digested pig hair keratin-based biopolymer hydrogel, a type of novel pH-responsive hydrogel. The hydrogel has been examined as a drug carrier, and its swelling and drug release properties have been defined.295
 | ||
| Fig. 18 Preparation of pH-responsive P(MBA-co-MAAc) hollow microspheres and TEM micrographs of polymer microspheres: (a) PMAAc seeds, (b) PMAAc/P(MBA-co-MAAc) core–shell microspheres, (c) hollow P(MBA-co-MAAc) microspheres with a cross-linking degree of 40 wt% (Copyright 2009, Elsevier Ltd, modified from ref. 293). | ||
Application areas of pH-responsive polymers
pH-Responsive polymers are intelligent polymers that undergo changes in structures and properties such as volume, chain conformation, solubility, configuration and so on in response to changes in pH. pH-Responsive polymers have various potential application areas in biotechnology, nanotechnology, chromatography, membranes, coatings, etc. They can be used as gene delivery systems, controlled drug release systems, drug carriers, sensors, stabilizers, viscosity modifiers, etc.4–6,9,296,297 The subject of pH-responsive polymers has become very popular in recent years and new studies have been added year after year.One successful application of pH-responsive copolymers is their usage as stabilizers in dispersion and emulsion polymerizations.297 Tertiary amine methacrylate based block copolymers can be used as stabilizers in heterogeneous polymerizations. For example, the PDPA-b-PMEMA diblock copolymer has been reported to be a good stabilizer in the production of monodisperse PMEMA microgels via emulsion polymerization.298 As an example, MPEO-b-PDEA-b-PMPC is a successful stabilizer for such a purpose. The MPEO-b-PDEA-b-PMPC triblock copolymer can form micelles at pH >7.5 by the middle block forming micelle core and the outer blocks forming hydrated coronas. Nitrogen atoms of PDEA blocks are in the deprotonated form at pH >7.5 and PDEA blocks become insoluble due to dehydration. But both outer blocks show hydrophilic character over the entire pH range. Thus, this block copolymer provides successful stabilization for multi-responsive PMEMA microgels in aqueous media.65 Another example, DMA and MMA monomers, can be used to prepare different types of polymers such as AB diblock, ABA and BAB triblock copolymers. With these copolymers, polystyrene latexes can be synthesized and these copolymers stabilize PS latexes well. At low pH, PS latexes have exhibited no surface activity, but showed surface activity at the air–water interface at high pH.93
Stabilization of inorganic particles which are metal oxides and metals is one of the most important problems in modern colloid chemistry and technology known nowadays as nanoscience and nanotechnology. The synthesis of nanoparticles with small size, narrow size distribution and long term stability requires the use of a stabilizer system that consists of small molecules and/or macromolecules. In the synthesis of inorganic particles, the use of polymers as a stabilizing agent is very common. Hydrogels, microspheres, nanospheres, homopolymers, dendrimers, brushes and block copolymers are widely used as stabilizer systems.299–310 Among them, amphiphilic and double-hydrophilic block copolymers have attracted great attention since they allow inorganic particle formation within the core of their micelles. For such micellar systems, the block copolymer requires one block forming the micelle core by coordinating with metal containing ions and the second block forming the corona, which provides good stability in the solvent medium.297,311,312
Drug release by pH changes can occur by two different strategies. In the first strategy, polymeric materials containing pH-responsive groups release drug molecules due to swelling/deswelling behavior or degradation of the micelle structure with pH change. In the second strategy, polymeric materials release drugs with cleavage of covalent bonds between the drug and polymer by pH changes. pH-Labile bonds (Fig. 19), used in polymer structures, are hydrazone,316–321 acetal/ketal,322–325cis-acotinyl,326–330 imine,184,331,332 substituted trityl,333–335 orthoester,336,337 and others.338,339
 | ||
| Fig. 19 Structures of pH-labile bonds and their degradation products. | ||
Storage and release of biomaterials, such as proteins, gene, enzymes and particularly drugs, can be carried out via the usage of pH-responsive micelles and hydrogels.293,296,313,340 Owing to pH-responsive groups in the structure, the release of various compounds is possible. Particularly smart polymers are very promising as delivery and carrier systems because of their high loading capacity, high stability, and responsiveness to change in environmental factors.272 It is well known that micelles can solubilize some compounds which are not soluble in the aqueous phase.296 Self-assembled pH-responsive vesicle structures are mostly used for drug transport and release.341 Also cross-linked micelles have been used in the drug release study. These cross-linked micelles like nanogels can swell or deswell by pH changes and might be useful in drug release studies as releasing systems. There have been various reports on pH-responsive degradable cross-linked micelles and hydrogels in recent years.139–142,178,190,192
pH-Responsive polymeric micelles have also been used as controlled delivery systems for anticancer drugs in tumor-targeted studies. The micelles of poly(2-ethyl-2-oxazoline)-poly(D,L-lactide) have been used in the encapsulation of both the D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS1000) and doxorubicin (DOX) drugs. The micelles had a high amount of drug encapsulation capability within the small size. They are reported to be perform well in drug release studies at low pH and in tumor targeting applications.342
Folic acid (FA)-functionalized well-defined PMPC-b-PDPA diblock copolymer (FA-PMPC-b-PDPA) micelles are reported to be useful to encapsulate the anti-cancer drugs tamoxifen and paclitaxel. In vitro cell viability studies demonstrated that both tamoxifen- and paclitaxel-loaded FA-MPC-b-DPA copolymer micelles are more toxic toward tumor cells in an acid environment. Such micelles dissociate at pH 5.5, thereby triggering drug release in the acid media. In contrast, minimal cytotoxicity is observed at body pH, because the drug-loaded micelles remain intact at this solution pH. Cell viability studies have been carried out on human chronic myelogenous leukemia (K-562) and colon carcinoma cell lines (Caco-2) in order to demonstrate that drug-loaded FA-PMPC-b-PDPA micelles exhibited higher cytotoxicities toward cancer cells than unfunctionalized PMPC-b-PDPA micelles. Uptake studies confirmed that folate conjugated micelles led to increased drug uptake within cancer cells, demonstrating the expected selectivity toward these tumor cells.343 In another study, it has been reported that the PMPC-b-PDPA diblock copolymer is soluble molecularly in dilute acid solution, since the pH-responsive PDPA block is protonated and hence becomes hydrophilic under these conditions. On adjusting the copolymer solution to around pH 5–7, the PDPA blocks become deprotonated and hence hydrophobic, leading to the formation of micelles with dehydrated PDPA cores and hydrated PMPC coronas. These pH-responsive micelles have been used for encapsulation and controlled release of the dipyridamole drug.344 Similarly, the controlled release of the same drug can be carried out by using amphiphilic MPEG-b-PDMA-b-PDEA triblock copolymers as well.345 Tumoral acidic pH targeting of pH-responsive MPEG-b-PAE micelles has been examined for cancer targeting therapy. The anticancer drug, camptothecin (CPT), has been simply encapsulated into the pH-responsive micelles with higher loading efficiency and the CPT encapsulated pH-responsive MPEG-b-PAE micelles showed distinguished pH-responsive drug release characteristics under weakly acid conditions.315
A series of novel pH-responsive ABA triblock copolymers has been reported as a novel polymeric gelling agent. PDPA has been chosen as the A block while PDMA has been chosen as the B block. While the PDPA-b-PDMA-b-PDPA triblock copolymer is molecularly soluble in acidic aqueous media due to protonation of all tertiary amine residues, they formed either gels by the chain-end hydrophobic interactions with a relatively high polymer concentration (10 wt%) or near-monodisperse “flower” micelles with a low polymer concentration in neutral and basic aqueous solutions. A model hydrophobic drug release study has been carried out in a sustained manner from the gels at pH 7.4 by varying the polymer concentration, the polymer molecular weight, and the temperature of the medium. Studies indicate that both slow, sustained release and fast, triggered release of a model hydrophobic drug, dipyridamole, can be achieved by tuning the solution pH, polymer concentration, polymer molecular weight and temperature of the gel. In addition to pH, the thermo-responsive nature of the middle PDMA block has an important effect on the dipyridamole releases from the hydrogel.80 PEG-b-PDEA-b-PCL amphiphilic triblock copolymers have been determined to be a good source for three layer micelle preparation in aqueous solution. The pH-responsive PDEA middle layer is hydrophobic and collapses on the micellar core at physiological pH (7.4) to prevent the premature burst drug-release. But it becomes positively charged below pH 6.5 and protrudes out at the solid-tumor interstitial pH. Its positive nature causes adsorption of nanoparticle onto the negatively-charged cell membrane and subsequently induces adsorptive endocytosis of the nanoparticle. After its transfer to a lysosome, PDEA layer is further charged, disrupting the lysosomal membrane to release the nanoparticles into the cytosol.346 The pH-responsive glycol chitosan-g-3-diethylaminopropyl (GCS-g-DEAP) nanogel having both hydrophobic DEAP residues and hydrophilic GCS residues has been prepared and loaded with DOX. The release of DOX increased at pH 6.8 compared to that at normal body pH 7.4. This study showed that these self-assembled nanogels are appropriate for transport to the target cells and release of anticancer drugs.347
Duan et al. synthesized CS-g-PNIPAm based pH-responsive and biocompatible nanogels via free radical copolymerization. These nanogels have been evaluated as drug delivery systems. Nanogels have been loaded with oridonin (ORI) with a self-assembly method. ORI-loaded gels showed pH-responsive release behavior. Release of drugs from ORI-loaded gels is very slow at pH 7.4 but faster at pH 6.0–6.5.348 Injectable hydrogels that have responses to pH and glucose have also been prepared with oxidized dextran and biocompatible phenylboronate ester. Imine residues behave as the pH-responsive part and phenylboronate ester behaves as the glucose responsive part of the polymer. The anticancer drug (DOX) can also be loaded successfully to the hydrogels.349
PHIS and PHEMA based synthetic polymers have been synthesized via ring opening polymerization and ATRP. PHEMA-b-PHIS polymers are both membranolytic and biocompatible. These pH-responsive polymers have been used as a drug carrier for tumor targets. Its nanosized micelles can be formed by self-assembly with changing solution pH and DOX which is encapsulated in the micellar system. Release of DOX has been studied under different pH conditions. PHEMA-b-PHIS micelles release DOX and do not lose their biological activity. At acidic pH, cancer cells have been killed by the DOX drug. The PHEMA-b-PHIS systems are suitable carriers of drug molecules for tumor targeting and productively encapsulating the chemo-therapeutic drug DOX.350
For a similar purpose, poly(2-(diethylamino)ethyl methacrylate)-b-poly(oligo(ethylene glycol) methacrylate)-coated silica nanotubes (SNT-PDEA-b-POEGMA) have been prepared via surface RAFT polymerization. DOX has been loaded into this polymer and controlled release of DOX has been monitored by changing the pH (buffer solution pH = 4.0, 5.0, and 7.4). 13% of drug release has been observed at pH 7.4 whereas 77% of drug release has been observed at pH 4.0 within 24 h. PDEA chains being protonated and stretched in the open state provide easier release of DOX under acidic conditions.351 Mesoporous silica-coated magnetic graphene oxide is another interesting material as a multifunctional drug nanocarrier. After attaching polyglycerol-g-polycaprolactone on the nanocarrier DOX is loaded to the system via electrostatic interaction between the DOX molecule and mesopores and adsorbed on graphene. Release of DOX has been examined at pH 7.4 (blood pH) and 5.5 (endosomal pH) at body temperature. The amount of drug release has been determined to be 86% at the endosomal pH for 48 h, but it is 61% at normal blood pH for the same period. This indicates faster release of the DOX at the acidic pH.352
In the last decade, another important application of such micellar systems was found via the layer-by-layer assembly method, which has also been widely applied in drug and different compound carrier systems using block copolymer micelles.353 Tertiary amine methacrylate based copolymers have been determined to be good adsorbents on silica and glass surfaces, which results in the formation of nanofilms on the surface.354–356 This nanofilm formation has opened the door for the preparation of LbL nanofilms on such surfaces as well.357,358 Multi-layer films are prepared from block copolymer micelles such as PDMA-b-PDEA,354,358–360 PMEMA-b-PDPA,361 PβDMA-b-PDPA,167,362–364 PDMA-b-PDEGMA,365 PAMPS-b-PAAL366 and PDEA-b-PNIPAm367 loaded with various compounds by the layer-by-layer method with the release of these compounds by pH changes.
A novel pH-responsive nanogel with a pH-dependent charge conversion feature has also been developed for use as a possible anticancer drug delivery agent. The nanogel can be transformed from a negatively-charged to a positively-charged form in a slightly acidic tumor extracellular environment, which enhances the cellular uptake of the nanogel and promotes the efficiency of drug release in killing cancer cells. Positively charged DOX hydrochloride has been used to investigate the drug-loading capacity of the PAMA-co-DMMA nanogel (see Fig. 20).368
 | ||
| Fig. 20 Schematic illustration of the performance of the drug-loaded pH-responsive charge conversional PAMA-co-DMMA nanogel. In the acidic tumor extracellular environment, the PAMA-co-DMMA nanogel is activated to be positively charged and is thus readily internalized by tumor cells with subsequent intracellular drug release (Copyright 2010, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, reprinted from ref. 368). | ||
pH-Responsive polymer systems can behave as the host for the production of metal nanoparticles. For example, PEG-co-PMAAc gels can be used for Ni–Ag bimetallic NP production by the resulting core–shell hybrid system. These pH-responsive hybrid systems have also magnetic susceptibility with fluorescent pH sensing over the physiologically important pH range of 5.0–7.4. The hybrid nanogels could be used in pH-responsive delivery of the anticancer drug 5-fluorouracil (5-FU).369
Silica nanoparticles (SiNPs) are the most common materials for surface modification such as pH-responsive guanidine containing polymers (SiNP@PMCGH). Polymer coated silica particles do not form agglomerates after modification. The drug loading behavior of doxorubicin (+) and sulfasalazine (−) has been examined in a wide range of pH and drug loading and release has been determined to be dependent on the type of grafted polymer and its content on silica nanoparticles. The rate and extent of drug release can be controlled by varying the values of pH. DOX solubility is low at low pH values. It can be released faster at pH 5 with the effect of solubility and electrostatic repulsion. Sulfasalazine is not a hydrophilic drug. Therefore, compared with DOX, sulfasalazine has a slower release profile.370
The hairy polymeric nanocapsules with a pH-responsive PMAAc-co-PDVB inner shell and temperature-responsive PNIPAm brushes can be successfully prepared as seen in Fig. 21. The hairy P(MAAc-co-DVB)-g-PNIPAm nanocapsules as a drug carrier show pH/temperature dual-responsive drug release behavior. Changing the external pH and temperature could effectively control the shell permeability for loaded drug molecules passing through the PMAAc-co-PDVB inner shell and the PNIPAm brush layer.238
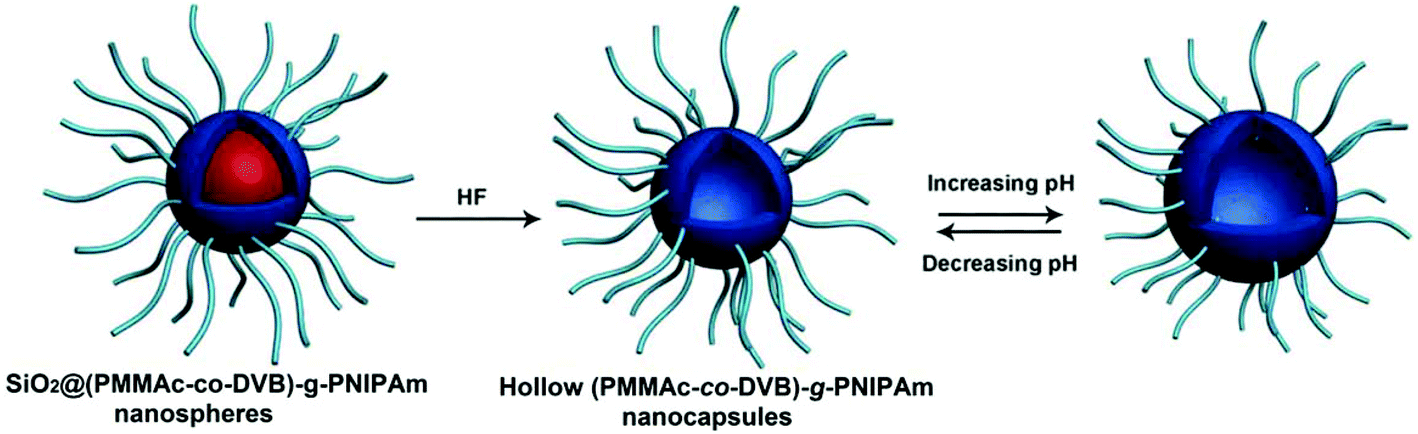 | ||
| Fig. 21 Preparation of pH-responsive hairy P(MAAc-co-DVB)-g-PNIPAm nanocapsules (Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, modified from ref. 238). | ||
The layer-by-layer (LbL) self-assembly technique has been successfully used to design superparamagnetic pH-responsive hybrid hollow microspheres [(CS-g-PEG/Fe3O4-CA)4/CS-g-PEG]. DOX has been chosen as a model hydrophobic drug. The controlled release behavior has been investigated in in vitro studies by differing pH values. The release of DOX has been determined to be slower at pH 7.4 or pH 6.5 than at pH 5.0.371 In another study, an acrylamide modified chitin based microsphere has been prepared and loaded with the vancomycin hydrochloride drug. Release behavior of vancomycin has been investigated under different pH conditions. In contrast to the previous work, in this study, release of the drug has been determined to be faster at pH 7.4 than under low pH conditions.372
Different sized chitosan coated magnetic nanoparticles have been synthesized and efficiently loaded with DOX for the in vitro targeted delivery applications on MCF (Michigan Cancer Foundation) breast cancer cells. The optimal loading efficiency, stability, and release profiles of DOX loaded nanoparticles have been determined. The DOX release profiles showed a pH dependent and slow release pattern. As the pH decreased, the swelling ratio of chitosan increased, therefore the drug release increased. The chitosan coated magnetic nanoparticles released most of the DOX at pH 4.2, while the nanoparticles are quite stable at pH 7.4.373
Biodegradable microgel systems based on glycerol-1,3-diglycidyl ether cross-linked TEMPO-oxidized potato starch polymers are capable of absorbing a large amount of lysozyme. The results provide insight into the factors that control the uptake and release of lysozyme by oxidized starch microgels. The protein uptake at saturation (Γsat) is the highest at high pH and low ionic strength. It is found that Γsat increases with increasing pH. This is due to the protein binding capacity which is mainly determined by charge compensation: with increasing pH, the positive charge on lysozyme decreases, while the negative charge on the microgel particle increases. Therefore, more protein molecules are required to neutralize for the charge on the gel and the binding capacity increases. The decreased Γsat with increased ionic strength is mainly due to the shielding effect on the electrostatic interaction between the gel and proteins caused by a high salt concentration. Protein release has been triggered by decreasing the pH and/or increasing the ionic strength, since the binding strength is the lowest at low pH and high ionic strength. The results suggest that oxidized microgels can be potentially applied in the controlled uptake and release of proteins.374
DMA and AAc copolymerized using free radical aqueous copolymerization and a series of pH-responsive hydrogels have been obtained. The reported formulations, being devoid of any chemical cross-linkers, remained dimensionally stable in buffer solutions of pH 1.2–7.4 with inter-locked nanogels being identified as the building blocks of the network structures. Swelling behavior and release kinetics of bovine serum albumin have been investigated for PAAc-co-PDMA in various buffer solutions that mimic the pH-metric hierarchy in the gastrointestinal tract. The PAAc-co-PDMA hydrogel has been prepared with different compositions and investigated for their possibile drug release behaviours.375
pH-Responsive polymers are also used in insulin transport systems. It is important that the right amount of insulin is used at the right time. Therefore the transport of insulin is very important. The oxidation of glucose to gluconic acid catalysed by glucose oxidase can lower the pH to nearly 5.8 when there is a rich glucose environment. This enzyme is generally used in glucose sensing and makes possible the use of different types of pH-responsive hydrogels for modulated insulin delivery.376 Some of the polymers used in insulin release studies are PMAAc-co-PDMA microgels,267 PVPBA-co-PDMAEA nanogel,70 PLGA-g-PHEMA,142 and PAAc-g-PEG and PMAAc-g-PEG.286
The pH-responsive polyacrylamide-modified hydroxypropyl methyl cellulose [g-HPMC (M)] graft copolymer has been synthesized via a microwave-assisted grafting method. This polymer has been examined as a drug release system for ornidazole. The drug is delivered to the colonic region with a copolymer at pH 7.4.377 Poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-b-PCL) and folate-PDMA-b-PCL (Fol-PDMA-b-PCL) diblock copolymers form complex micelles in acidic water. When pH has been adjusted to above the pKa of PDMA (pH 7.4), core–shell-corona (hydrophobic PCL-collapsed PDMA-soluble PEG) micelles have been obtained. After loading DOX into the micelle-core (PCL), the DOX release profile indicated that complex micelles with a core–shell structure showed a faster drug release rate at pH 5.5.378 pH-Responsive NaAlg-graft-poly(N-vinyl-2-pyrrolidone) copolymer beads are another polymer which has been prepared with microwave-assisted synthesis in order to examine the release profile of the ibuprofen (IB) model drug. The release of IB has been determined to be much slower at pH 1.2 than pH 7.4 release.379
PDMA with a high molecular weight has been very productive for condensed DNA.4 When PDMA is copolymerized with any monomer, its cytotoxicity and transfection efficiency could be changed.380–382 Various hyper-branched poly(ester amine) polymers have been synthesized and used as gene carriers. Such hyper-branched poly(ester amine)s, due to their highest transfection efficiency and low cytotoxicity, have been determined to be safe and productive gene carriers. In another work, PNIPAm-co-PDMA-co-PBMA copolymers have been synthesized and used as a gene carrier. Gene-transfection efficiency has been studied at different temperatures. When the temperature is decreased, the transfection efficiency is increased. This case demonstrated the effect of temperature changes during formation and dissociation control of the complexes.383,384 Similarly, PDEA containing polymers such as PEO-b-PDEA and plasmid DNA binding interactions have been determined to be due to enthalpy change. A pH change causes a change in the DNA compaction. In this case, the binding force of DNA and polymers depends on the stoichiometric balance between the molar ratio of DNA nucleoids and amine groups on PDEA.
Polycations consisting of poly(L-lysine) (PLL) side chains and a PDEA backbone have been synthesized for use as a new pH-responsive DNA carrier. Polycations attached with several ligands, such as insulin, transferrin and antibody molecules, have been examined for the efficient internalization of DNA–ligand complexes. The copolymer has shown proton dissociation and dual ionic character. The copolymer has shown no significant turbidity even at pH 10, whereas the PDEA homopolymer precipitated at pH greater than 7.5 owing to the deprotonation of amino groups. The discontinuous turbidity change of DNA-PDEA-g-PLL solution at pH 7.5 suggested that the solubility of the complex varied with pH.385
Functionalized membranes with three different acidic polymers, PAAc, PEGMP, and PAMPS, have been prepared by UV initiated grafting of functional monomers in the pores of the poly(propylene) host membrane. The adsorptions of different ions in the functionalized membranes have been studied.13 The design of hybrid interfaces based on the use of mesoporous thin films incorporating polymer brushes as versatile functional units offers major opportunities for controlling molecular transport through interfaces. They conducted charge-selective ionic transport via pH changes. A PEGMP brush was designed via the “growth from” approach after initiator-functionalization of the mesoporous silica surface.247 Increasing pH from 4 to 8 led to a significant increase in (anion) permselectivity and (cation) preconcentration, thus reflecting the ability of the PEGMP brush-modified mesoporous silica thin film to act as a selective “electrostatic nanovalve” precluding and boosting the anionic and cationic transport, respectively (Fig. 22). On the other hand, the complexation/chelation of phosphate groups with Ca2+ ions also allowed the generation of gate-like hybrid ensembles.247
 | ||
| Fig. 22 Schematic depiction of the ionic transport processes taking place in the hybrid polymer–inorganic interfacial assembly at different pH values: (a) pH < 5, ionic mesochannel (no exclusion of ionic species) and (b) pH > 5, permselective transport of cations (Copyright 2012, The American Chemical Society, reprinted from ref. 247). | ||
As an example, the pH sensitivity of PEGMP brushes bearing orthophosphoric acid with two ionization states for switching surface wettability (pKa1 in the range of pH 1–2 and pKa2 in the range of pH 6–7) has been well documented (Fig. 23). The phosphate groups are completely protonated (diacid) at pH 1, partly protonated when the pH is close to 7 and are in completely dibasic form when pH >7. The charges on the brush, the concentration of free counter ions and the degree of swelling can therefore all be tuned via the adjustment of the pH.392
 | ||
| Fig. 23 Procedure for patterned polymer brush formation on gold substrates. Polymer brush growth from initiator modified areas (Copyright 2005, The Royal Society of Chemistry, modified from ref. 392). | ||
A zwitterionic polymeric surface which has a tunable mixed-charge copolymer containing both positive quaternary amines and negative carboxylic acid residues has been determined to be useful for bacteria adhesive/resistance transition. The non-fouling properties of such a polymeric surface depend on the pH of the medium (Fig. 24). The surface has charge neutrality under neutral and basic conditions. It is positively charged under acidic conditions due to the protonation of the carboxylic acid and quaternary amine groups. This surface charge transition with respect to pH allows the surface to be switched from bacteria-adhesive to bacteria-resistant.239
 | ||
| Fig. 24 A surface switching from fouling to non-fouling in response to pH change: (a) in low pH solutions where the surface bears a moderately positive charge, favoring the attachment of bacteria cells, and (b) in neutral or higher pH solutions where the surface becomes non-fouling, releasing the bacteria cells (Copyright 2010, Elsevier Ltd, reprinted from ref. 239). | ||
Closing remarks
The fascinating properties of pH-responsive polymers are likely to continue to generate more activities in scientific pathways. They are good candidates for further material and nanostructure developments. They can be used as stabilizers in heterogeneous polymerizations, hosts for nanometal dispersion preparation, and precursors of cross-linked micelles, hallow spheres, controlled drug-releasing systems, etc. In this review, we have highlighted some recently reported examples of pH-responsive monomers, polymers and their derivative nano- and micro-structures including copolymers, nanogels, microgels, hydrogels, cross-linked structures, LbL nanofilms, etc. We have also focused on their classifications, synthetic pathways for their synthesis, self-assembly behaviors depending on solution pH, nano-aggregates and further chemistries for their derivative nanostructures or materials, application possibilities in various fields, etc. Given the number of synthetic procedures and the variety of biocompatible monomers now available, it may be anticipated that biodegradable pH-responsive polymers will be the most prefered polymers in biotechnological applications owing to their response to biological conditions and favorable interactions with related biological environments.Abbreviations
| AAc | Acrylic acid |
| ACDAAB | N,N-(ω-Aminocaproyl)-4,4′-diaminoazobenzene |
| AMPD | 4-(Aminomethyl)piperidine |
| AMPD | 4-(Aminomethyl)piperidine |
| ATRP | Atom transfer radical polymerization |
| BAC | N,N-Cystaminebis(acrylamide) |
| BDBA | 1,3-Benzenediboronic acid |
| BDDA | 1,4-Butanediol diacrylate |
| BIEE | 1,2-Bis-(2-iodoethoxy) ethane |
| Boc-VaI-HEA | tert-Butyl carbamate-L-valine-acryloyloxyethyl ester |
| BSPA | 3-Benzylsulfanylthiocarbonyl sulfanylpropionic acid |
| CCL | Core cross-linked |
| CCS | Core cross-linked star |
| CELG | γ-2-Chloroethyl-L-glutamate |
| CLMs | Cross-linked micelles |
| CMA | 4-Methyl-(7-(methacryloyl) oxyethyloxy)coumarin |
| CNPBA | 3-Carboxy-5-nitrophenylboronic acid |
| CRP | Controlled radical polymerization |
| Dex | Dextran |
| DHBCs | Double hydrophilic block copolymers |
| DTbDEA | 2,2′-Dithiobis(N,N-dimethylethylamine) |
| DTDMA | 2,2′-Dithiodiethoxyl dimethacrylate |
| DVB | Divinylbenzene |
| DVS | Divinyl sulfone |
| EGDMA | Ethylene glycol dimethacrylate |
| GA | Glutaraldehyde |
| GTP | Group transfer polymerization |
| H40 | Hyper-branched polyester |
| HA | Hyaluronic acid |
| HBPE | Hyper-branched polyethylene |
| HBPO | Poly[3-ethyl-3-(hydroxymethyl)oxetane] |
| Hyd | Hydrazine functionalized |
| ILCL | Intermediary layer cross-linked |
| ITO | Indium tin oxide |
| MAAc | Methacrylic acid |
| MAEBA | p-(Methacryloxyethoxy)benzaldehyde |
| MBA | N,N′-Methylene-bis-acrylamide |
| MePEGA | Poly(methoxypolyethylene glycol acrylamide) |
| MPEG | Methoxypolyethylene glycol |
| NMP | Nitroxide-mediated radical polymerization |
| P2VP | Poly(2-vinylpyridine) |
| P4VP | Poly(4-vinylpyridine) |
| PAAc | Poly(acrylic acid) |
| PAaH | Sodium 6-acrylamidohexanoate |
| PAAPBA | Poly(3-acrylamidophenylboronic acid) |
| PAEAm | Poly(acrylamidoethylamine) |
| PAMA | Alkyne-poly(2-aminoethyl methacrylate) |
| PAM | Poly(acryloylmorpholine) |
| PAMAM | Poly(amidoamine) |
| PAMPS | Poly(2-acrylamido-2-methylpropane sulfonic acid) |
| PANMP | Poly(N-acryloyl-N′-methylpiperazine) |
| PAPMAm | Poly[N-(3-aminopropyl)methacrylamide] |
| PASA | Poly(aspartic acid) |
| PBAC | N,N′-Bis(acryloyl)cystamine) |
| PBIEM | Poly[2-(2-bromoisobutyryloxy)ethyl methacrylate] |
| PBuAm | Poly(N-tert-butylacrylamide) |
| PCAA | 2-Carboxy ethyl acrylate |
| PCEMA | Poly(2-cinnamoyloxyethyl methacrylate) |
| PCGMA | Poly(3-cinnamoyl glycerol monomethacrylate) |
| PCL | Poly(ε-caprolactone) |
| PDEA | Poly[(2-diethylamino)ethyl methacrylate] |
| PDEAm | Poly[(2-diethylamino)ethyl acrylamide] |
| DEGMMA | Methoxydi(ethylene glycol) methacrylate |
| PDEVBP | Poly(diethyl-4-vinyl-benzyl phosphonate) |
| PDMA | Poly[(2-dimethylaminoethyl) methacrylate] |
| PDMAEA | Poly[(2-dimethylaminoethyl) ethacrylate] |
| PDMAPAm | Poly[(3-dimethylamino)propyl acrylamide] |
| PDMAPMAm | Poly[N-(3-(dimethylamino)-propyl)methacrylamide] |
| PDPA | Poly(2-diisoprophylamino)ethyl methacrylate |
| PEAAc | Poly(ethyl acrylic acid) |
| PEG | Polyethylene glycol |
| PEGAP | Poly(ethylene glycol acrylate phosphate) |
| PEG-FA | Poly(ethylene glycol)-folate |
| PEGMA | Poly(ethylene glycol) methacrylate |
| PEGMP | Poly(ethylene glycol methacrylate phosphate) |
| PEI | Poly(ethylene imine) |
| PEO | Polyethylene oxide |
| PEPyM | Poly(N-ethylpyrrolidine methacrylate) |
| PG2MA | Poly(glycerol monomethacrylate) |
| PGMA | Poly(glycidyl methacrylate) |
| PHEA | Poly(2-hydroxyethylacrylate) |
| PHEMA | Poly(2-hydroxyethyl methacrylate) |
| PHIS | Poly(histidine) |
| PHPMA | Poly[N-(2-hydroxypropyl)methacrylate] |
| PHPMAm | Poly[N-(2-hydroxypropyl)methacrylamide] |
| PImHeMA | Poly[6-(1H-imidazol-1-yl)hexyl-methacrylate] |
| PIMMA | Poly[2-(isobutyramido)-3-methylbutyl methacrylate] |
| PKSPMA | Poly(3-sulfopropylmethacrylate potassium) |
| PLA | Polylactide |
| PLGA | Poly(L-glutamic acid) |
| PLL | Poly(L-lysine) |
| PMAAc | Poly(methacrylic acid) |
| PMAGGONp | Poly(N-methacryloylglycylglycine p-nitrophenyl ester) |
| PMAGP | Poly(6-O-methacryloyl-D-galactopyranose) |
| PMEA | Poly(2-methacryloylethylacrylate) |
| PMEMA | Poly(2-N-morpholinoethyl)methacrylate |
| PMEO2MA | Poly[2-(2-methoxyethoxy)ethyl methacrylate] |
| PMMA | Poly(methyl methacrylate) |
| PMPC | Poly(2-methacryloyloxyethyl phosphorylcholine) |
| PMPMA | Poly(γ-methacryloxypropyltrimethoxysilane) |
| PMPS | Poly[3-(trimethoxysilyl)propyl methacrylate] |
| PNaAMPS | Poly[sodium 2-(acrylamido)-2-methylpropanesulfonate] |
| PNaSS | Poly(sodium 4-styrenesulfonate) |
| PNBC | Poly(S-(o-nitrobenzyl)-L-cysteine) |
| PNIPAm | Poly(N-isopropylacrylamide) |
| POEGMA | Poly[oligo(ethylene glycol) monomethyl ether methacrylate] |
| PPAAc | Poly(propylacrylic acid) |
| PPDPMA | Poly[N-2(3-pentadecylphenoxy)ethyl methacrylamide] |
| PPEGMA | Poly[(poly(ethylene glycol) methyl ether methacrylate) |
| PPI | Poly(propylene imine) |
| PPO | Poly(propylene oxide) |
| PS | Polystrene |
| PSP | Poly(spiropyan-functionalized) |
| PSPEA | 1′-(2-Acryloxyethyl)-3′,3′-dimethyl-6-nitrospiro-(2H-1-benzopyran-2,2′-indoline) |
| PSPM | Poly(3-sulfopropyl methacrylate) |
| PSPMA | Poly[1-3-[(2-methyl-1-oxo-2-propen-1-yl)oxypropyl] ester-b-poly(methoxydi(ethylene glycol) methacrylate)] |
| PSSA | Poly(4-styrenesulfonic acid) |
| PSVBP | Poly[4-(2-sulfoethyl)-1-(4-vinyl-benzyl)pyridinium betain] |
| PtBA | Poly(tert-butyl acrylate) |
| PtBAEMA | Poly[(2-(tert-butylamino)ethyl methacrylate)] |
| PTEGMA | Poly[2-(2-(2-methoxyethoxy)ethoxy)ethyl methacrylate] |
| PTFEMA | Poly(2,2,2-trifluoroethyl methacrylate) |
| PTMA | Poly[2-(acryloyloxy)ethyl] trimethyl ammonium chloride |
| PTMSPMA | Poly[3-(trimethoxysilyl)propyl methacrylate] |
| PTPHMA | Poly[4′-(6-methacryloxyhexyloxy)-2,2′:6′,2′′-terpyridine] |
| PVBA | Poly(4-vinylbenzoic acid) |
| PVBK | Poly[9-(4-vinylbenzyl)-9H-carbazole)] |
| PVDT | Poly(2-vinyl-4,6-diamino-1,3,5-triazine) |
| PVAm | Poly(N-vinyl amine) |
| PVI | Poly(N-vinylimidazole) |
| PVP | Polyvinylpyrrolidone |
| PVPBA | Poly(vinylphenyl boronic acid) |
| PVSA | Poly(vinylsulfonic acid) |
| QDMA | Quaternized DMA |
| RAFT | Reversible addition–fragmentation chain transfer |
| ROP | Ring opening polymerization |
| RPHA | Reducible poly(β-hydroxy amine)s |
| SCL | Shell cross-linked |
| SI-ATRP | Surface initiated-atom transfer radical polymerization |
| SI-CRP | Surface initiated-controlled/living radical polymerization |
| SPCL | Star poly(ε-caprolactone) |
| TDA | Terephthaldicarboxaldehyde |
| TEMPO | 2,2,6,6-Tetramethyl-1-piperidinyloxy |
| βDMA | Sulfobetaine DMA |
Notes and references
- A. S. Hoffman, P. S. Stayton, V. Bulmus, G. H. Chen, J. P. Chen, C. Cheung, A. Chilkoti, Z. L. Ding, L. C. Dong, R. Fong, C. A. Lackey, C. J. Long, M. Miura, J. E. Morris, N. Murthy, Y. Nabeshima, T. G. Park, O. W. Press, T. Shimoboji, S. Shoemaker, H. J. Yang, N. Monji, R. C. Nowinski, C. A. Cole, J. H. Priest, J. M. Harris, K. Nakamae, T. Nishino and T. Miyata, J. Biomed. Mater. Res., 2000, 52, 577–586 CrossRef CAS PubMed.
- C. De Las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- A. E. Smith, X. W. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- J. Hu, G. Zhang, Z. Ge and S. Liu, Prog. Polym. Sci., 2014, 39, 1096–1143 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- C. Alvarez-Lorenzo, B. Blanco-Fernandez, A. M. Puga and A. Concheiro, Adv. Drug Delivery Rev., 2013, 65, 1148–1171 CrossRef CAS PubMed.
- T. Y. Jiang, Z. Y. Wang, L. X. Tang, F. K. Mo and C. Chen, J. Appl. Polym. Sci., 2006, 99, 2702–2709 CrossRef CAS.
- A. E. Felber, M.-H. Dufresne and J.-C. Leroux, Adv. Drug Delivery Rev., 2012, 64, 979–992 CrossRef CAS PubMed.
- W. Dong, Y. Zhou, D. Yan, H. Li and Y. Liu, Phys. Chem. Chem. Phys., 2007, 9, 1255–1262 RSC.
- L. I. Gabaston, S. A. Furlong, R. A. Jackson and S. P. Armes, Polymer, 1999, 40, 4505–4514 CrossRef CAS.
- S. Monge, B. Canniccioni, G. David and J.-J. Robin, in Phosphorus-Based Polymers: From Synthesis to Applications, The Royal Society of Chemistry, 2014, pp. 1–18 Search PubMed.
- T. Vasudevan, S. Das, S. Sodaye, A. K. Pandey and A. V. R. Reddy, Talanta, 2009, 78, 171–177 CrossRef CAS PubMed.
- T. Miyata, K. Onakamae, A. S. Hoffman and Y. Kanzaki, Macromol. Chem. Phys., 1994, 195, 1111–1120 CrossRef CAS.
- B. Bingöl, C. Strandberg, A. Szabo and G. Wegner, Macromolecules, 2008, 41, 2785–2790 CrossRef.
- K. Nakamae, T. Nishino, K. Kato, T. Miyata and A. S. Hoffman, J. Biomater. Sci., Polym. Ed., 2004, 15, 1435–1446 CrossRef PubMed.
- H. M. Kang, Y. L. Cai, J. J. Deng, H. J. Zhang, Y. F. Tang and P. S. Liu, Eur. Polym. J., 2006, 42, 2678–2685 CrossRef CAS.
- A. Popa, C. M. Davidescu, P. Negrea, G. Ilia, A. Katsaros and K. D. Demadis, Ind. Eng. Chem. Res., 2008, 47, 2010–2017 CrossRef CAS.
- Y. Guan and Y. J. Zhang, Chem. Soc. Rev., 2013, 42, 8106–8121 RSC.
- X. Han, X. X. Zhang, H. F. Zhu, Q. Y. Yin, H. L. Liu and Y. Hu, Langmuir, 2013, 29, 1024–1034 CrossRef CAS PubMed.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS PubMed.
- Y. Mitsukami, A. Hashidzume, S. Yusa, Y. Morishima, A. B. Lowe and C. L. McCormick, Polymer, 2006, 47, 4333–4340 CrossRef CAS.
- Z. F. Song, K. Wang, C. Q. Gao, S. Wang and W. Q. Zhang, Macromolecules, 2016, 49, 162–171 CrossRef CAS.
- M. H. Dan, Y. Su, X. Xiao, S. T. Li and W. Q. Zhang, Macromolecules, 2013, 46, 3137–3146 CrossRef CAS.
- J. Seidel, V. T. Pinkrah, J. C. Mitchell, B. Z. Chowdhry and M. J. Snowden, Thermochim. Acta, 2004, 414, 47–52 CrossRef CAS.
- V. T. Pinkrah, M. J. Snowden, J. C. Mitchell, J. Seidel, B. Z. Chowdhry and G. R. Fern, Langmuir, 2003, 19, 585–590 CrossRef CAS.
- S. Wohlrab and D. Kuckling, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3797–3804 CrossRef CAS.
- N. Sahiner and O. Ozay, React. Funct. Polym., 2011, 71, 607–615 CrossRef CAS.
- R. C. Sutton, L. Thai, J. M. Hewitt, C. L. Voycheck and J. S. Tan, Macromolecules, 1988, 21, 2432–2439 CrossRef CAS.
- B. Wang, H. J. Liu, T. T. Jiang, Q. H. Li and Y. Chen, Polymer, 2014, 55, 6036–6043 CrossRef CAS.
- M. M. Fares and A. M. Al-Shboul, J. Biomed. Mater. Res., Part A, 2012, 100, 863–871 CrossRef PubMed.
- T. Matini, N. Francini, A. Battocchio, S. G. Spain, G. Mantovani, M. J. Vicent, J. Sanchis, E. Gallon, F. Mastrotto, S. Salmaso, P. Caliceti and C. Alexander, Polym. Chem., 2014, 5, 1626–1636 RSC.
- L. H. Gan, G. R. Deen, X. J. Loh and Y. Y. Gan, Polymer, 2001, 42, 65–69 CrossRef CAS.
- L. H. Gan, Y. Y. Gan and G. R. Doon, Macromolecules, 2000, 33, 7893–7897 CrossRef CAS.
- N. González, C. Elvira and J. S. Román, Macromolecules, 2005, 38, 9298–9303 CrossRef.
- V. Butun, S. P. Armes and N. C. Billingham, Polymer, 2001, 42, 5993–6008 CrossRef CAS.
- D. Velasco, C. B. Danoux, J. A. Redondo, C. Elvira, J. San Roman, P. S. Wray and S. G. Kazarian, J. Controlled Release, 2011, 149, 140–145 CrossRef CAS PubMed.
- Y. S. Jo, A. J. van der Vlies, J. Gantz, S. Antonijevic, D. Demurtas, D. Velluto and J. A. Hubbell, Macromolecules, 2008, 41, 1140–1150 CrossRef CAS.
- V. Butun, F. F. Taktak and C. Tuncer, Macromol. Chem. Phys., 2011, 212, 1115–1128 CrossRef.
- V. Butun, A. B. Lowe, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1999, 121, 4288–4289 CrossRef.
- V. Butun, X. S. Wang, M. V. D. Banez, K. L. Robinson, N. C. Billingham, S. P. Armes and Z. Tuzar, Macromolecules, 2000, 33, 1–3 CrossRef.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS PubMed.
- M. X. Tang, C. T. Redemann and F. C. Szoka, Bioconjugate Chem., 1996, 7, 703–714 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS PubMed.
- S. K. Samal, M. Dash, S. Van Vlierberghe, D. L. Kaplan, E. Chiellini, C. van Blitterswijk, L. Moroni and P. Dubruel, Chem. Soc. Rev., 2012, 41, 7147–7194 RSC.
- J. R. Santos, N. M. Alves and J. F. Mano, J. Bioact. Compat. Polym., 2010, 25, 169–184 CrossRef CAS.
- A. Basu, K. R. Kunduru, E. Abtew and A. J. Domb, Bioconjugate Chem., 2015, 26, 1396–1412 CrossRef CAS PubMed.
- M. V. Risbud, A. A. Hardikar, S. V. Bhat and R. R. Bhonde, J. Controlled Release, 2000, 68, 23–30 CrossRef CAS PubMed.
- K. Kaminski, K. Zazakowny, K. Szczubialka and M. Nowakowska, Biomacromolecules, 2008, 9, 3127–3132 CrossRef CAS PubMed.
- T. Zhou, C. F. Xiao, J. Fan, S. M. Chen, J. Shen, W. T. Wu and S. Q. Zhou, Acta Biomater., 2013, 9, 4546–4557 CrossRef CAS PubMed.
- N. Cao, X. Xie, Y. Zhang, Y. B. Zhao, L. Q. Cao and L. Sun, J. Ind. Eng. Chem., 2016, 34, 9–13 CrossRef CAS.
- S. Sharma, J. Kaur, G. Sharma, K. K. Thakur, G. S. Chauhan and K. Chauhan, Int. J. Biol. Macromol., 2013, 62, 636–641 CrossRef CAS PubMed.
- K. S. Soppimath, A. R. Kulkarni and T. M. Aminabhavi, J. Controlled Release, 2001, 75, 331–345 CrossRef CAS PubMed.
- Y. H. Lin, H. F. Liang, C. K. Chung, M. C. Chen and H. W. Sung, Biomaterials, 2005, 26, 2105–2113 CrossRef CAS PubMed.
- R. Guo, L. Y. Zhang, Z. P. Jiang, Y. Cao, Y. Ding and X. Q. Jiang, Biomacromolecules, 2007, 8, 843–850 CrossRef CAS PubMed.
- H. K. Ju, S. Y. Kim, S. J. Kim and Y. M. Lee, J. Appl. Polym. Sci., 2002, 83, 1128–1139 CrossRef CAS.
- Y. M. Lim, H. J. Gwon, J. S. Park, Y. C. Nho, J. W. Shim, I. K. Kwon, S. E. Kim and S. H. Baik, Macromol. Res., 2011, 19, 436–441 CrossRef CAS.
- R. S. Zhang, M. G. Tang, A. Bowyer, R. Eisenthal and J. Hubble, Biomaterials, 2005, 26, 4677–4683 CrossRef CAS PubMed.
- E. Akar, A. Altinisik and Y. Seki, Carbohydr. Polym., 2012, 90, 1634–1641 CrossRef CAS PubMed.
- C. M. Xiao and Y. K. Gao, J. Appl. Polym. Sci., 2008, 107, 1568–1572 CrossRef CAS.
- A. Lohani, G. Singh, S. S. Bhattacharya, R. R. Hegde and A. Verma, J. Drug Delivery Sci. Technol., 2016, 31, 53–64 CrossRef CAS.
- X. Yuan, B. Z. Ju and S. F. Zhang, Carbohydr. Polym., 2014, 114, 530–536 CrossRef CAS PubMed.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem. – Eur. J., 2015, 21, 13164–13174 CrossRef CAS PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- C. Tuncer, Y. Samav, D. Ulker, S. B. Baker and V. Butun, J. Appl. Polym. Sci., 2015, 132, 42072 CrossRef.
- Y. Zhang, T. Wu and S. Liu, Macromol. Chem. Phys., 2007, 208, 2492–2501 CrossRef CAS.
- L. Wang, M. Liu, C. Gao, L. Ma and D. Cui, React. Funct. Polym., 2010, 70, 159–167 CrossRef CAS.
- F. J. Jiang, S. Chen, Z. Q. Cao and G. J. Wang, Polymer, 2016, 83, 85–91 CrossRef CAS.
- D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073–7081 CrossRef CAS.
- W. Wu, N. Mitra, E. C. Y. Yan and S. Zhou, ACS Nano, 2010, 4, 4831–4839 CrossRef CAS PubMed.
- G. Jiang, T. Jiang, H. Chen, L. Li, Y. Liu, H. Zhou, Y. Feng and J. Zhou, Colloid Polym. Sci., 2015, 293, 209–215 CAS.
- W. F. Ma, S. A. Xu, J. M. Li, J. Guo, Y. Lin and C. C. Wang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2725–2733 CrossRef CAS.
- S. J. Kim, H. I. I. Kim, S. J. Park and S. I. Kim, Sens. Actuators, A, 2004, 115, 146–150 CrossRef CAS.
- C. A. Wei, J. Guo and C. C. Wang, Macromol. Rapid Commun., 2011, 32, 451–455 CrossRef CAS PubMed.
- D. W. Li, Y. Z. Bu, L. N. Zhang, X. Wang, Y. Y. Yang, Y. P. Zhuang, F. Yang, H. Shen and D. C. Wu, Biomacromolecules, 2016, 17, 291–300 CrossRef CAS PubMed.
- S. Kashyap, N. Singh, B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 384–398 CrossRef CAS PubMed.
- Y. Liu, X. Cao, M. Luo, Z. Le and W. Xu, J. Colloid Interface Sci., 2009, 329, 244–252 CrossRef CAS PubMed.
- C. M. Schilli, M. F. Zhang, E. Rizzardo, S. H. Thang, Y. K. Chong, K. Edwards, G. Karlsson and A. H. E. Muller, Macromolecules, 2004, 37, 7861–7866 CrossRef CAS.
- W. Zhang, L. Shi, R. Ma, Y. An, Y. Xu and K. Wu, Macromolecules, 2005, 38, 8850–8852 CrossRef CAS.
- F. F. Taktak and V. Butun, Polymer, 2010, 51, 3618–3626 CrossRef CAS.
- V. Butun, N. C. Billingham and S. P. Armes, Chem. Commun., 1997, 671–672 RSC.
- Z. S. Ge and S. Y. Liu, Macromol. Rapid Commun., 2013, 34, 922–930 CrossRef CAS PubMed.
- C. H. Li, Z. S. Ge, J. Fang and S. Y. Liu, Macromolecules, 2009, 42, 2916–2924 CrossRef CAS.
- L. Liu, P. Du, X. Zhao, J. Zeng and P. Liu, Eur. Polym. J., 2015, 69, 540–551 CrossRef CAS.
- Y. F. Zhang, W. Y. Gu, H. X. Xu and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2379–2389 CrossRef CAS.
- L. Deng, K. Shi, Y. Y. Zhang, H. M. Wang, J. G. Zeng, X. Z. Guo, Z. J. Du and B. L. Zhang, J. Colloid Interface Sci., 2008, 323, 169–175 CrossRef CAS PubMed.
- V. Butun, S. P. Armes, N. C. Billingham and Z. Tuzar, Abstr. Pap. Am. Chem. Soc., 1999, 218, U443–U443 Search PubMed.
- V. Butun, S. P. Armes and N. C. Billingham, Macromolecules, 2001, 34, 1148–1159 CrossRef.
- V. Butun, Polymer, 2003, 44, 7321–7334 CrossRef CAS.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706–5708 CrossRef CAS.
- C. S. Patrickios, A. B. Lowe, S. P. Armes and N. C. Billingham, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 617–631 CrossRef CAS.
- V. Butun, I. Bannister, N. C. Billingham, D. C. Sherrington and S. P. Armes, Macromolecules, 2005, 38, 4977–4982 CrossRef.
- J. I. Amalvy, G. F. Unali, Y. Li, S. Granger-Bevan, S. P. Armes, B. P. Binks, J. A. Rodrigues and C. P. Whitby, Langmuir, 2004, 20, 4345–4354 CrossRef CAS PubMed.
- V. Butun, R. B. Top and S. Ufuklar, Macromolecules, 2006, 39, 1216–1225 CrossRef.
- C. Tuncer and V. Butun, Eur. Polym. J., 2015, 67, 292–303 CrossRef CAS.
- N. Stavrouli, A. I. Triftaridou, C. S. Patrickios and C. Tsitsilianis, Macromol. Rapid Commun., 2007, 28, 560–566 CrossRef CAS.
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 930–937 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- C. Corten, K. Kretschmer and D. Kuckling, Beilstein J. Org. Chem., 2010, 6, 756–765 CrossRef PubMed.
- C. Zhang and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4341–4357 CrossRef CAS.
- D. Kuckling, K. Moosmann, J. E. S. Schier and A. Britze, Colloid Polym. Sci., 2013, 291, 1429–1437 CAS.
- B. Lessard and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5270–5283 CrossRef CAS.
- O. Borisova, L. Billon, M. Zaremski, B. Grassl, Z. Bakaeva, A. Lapp, P. Stepanek and O. Borisov, Soft Matter, 2012, 8, 7649–7659 RSC.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- W. M. Cai, W. M. Wan, C. Y. Hong, C. Q. Huang and C. Y. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- D. A. Z. Wever, P. Raffa, F. Picchioni and A. A. Broekhuis, Macromolecules, 2012, 45, 4040–4045 CrossRef CAS.
- D. Neugebauer and K. Matyjaszewski, Macromolecules, 2003, 36, 2598–2603 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromol. Rapid Commun., 2000, 21, 190–194 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromolecules, 1999, 32, 4826–4831 CrossRef CAS.
- A. Narumi, Y. Chen, M. Sone, K. Fuchise, R. Sakai, T. Satoh, Q. Duan, S. Kawaguchi and T. Kakuchi, Macromol. Chem. Phys., 2009, 210, 349–358 CrossRef CAS.
- P. Zhou, Y.-Y. Liu, L.-Y. Niu and J. Zhu, Polym. Chem., 2015, 6, 2934–2944 RSC.
- W. D. Zhang, W. Zhang, N. C. Zhou, J. Zhu, Z. P. Cheng and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6304–6315 CrossRef CAS.
- Y. Q. Yang, B. Zhao, Z. D. Li, W. J. Lin, C. Y. Zhang, X. D. Guo, J. F. Wang and L. J. Zhang, Acta Biomater., 2013, 9, 7679–7690 CrossRef CAS PubMed.
- S. Medel, J. M. Garcia, L. Garrido, I. Quijada-Garrido and R. Paris, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 690–700 CrossRef CAS.
- T. Gao, Q. Ye, X. Pei, Y. Xia and F. Zhou, J. Appl. Polym. Sci., 2013, 127, 3074–3083 CrossRef CAS.
- N. I. Abu-Lail, M. Kaholek, B. LaMattina, R. L. Clark and S. Zauscher, Sens. Actuators, B, 2006, 114, 371–378 CrossRef CAS.
- H. Hu, X. D. Fan, Z. L. Cao, W. X. Cheng and Y. Y. Liu, J. Appl. Polym. Sci., 2006, 101, 311–316 CrossRef CAS.
- G. F. Meijs, E. Rizzardo and S. H. Thang, Polym. Bull., 1990, 24, 501–505 CrossRef CAS.
- G. F. Meijs, E. Rizzardo and S. H. Thang, Macromolecules, 1988, 21, 3122–3124 CrossRef CAS.
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A, 1986, 23, 839–852 CrossRef.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- Y. Wang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2012, 13, 2585–2593 CrossRef CAS PubMed.
- L. Qiu, C. Y. Hong and C. Y. Pan, Int. J. Nanomed., 2015, 10, 3623–3640 CAS.
- Q. Q. Bian, Y. Xiao, C. Zhou and M. D. Lang, J. Colloid Interface Sci., 2013, 392, 141–150 CrossRef CAS PubMed.
- S. Ghosh Roy and P. De, Polym. Chem., 2014, 5, 6365–6378 RSC.
- H. Y. Tai, C. L. Duvall, A. S. Hoffman, P. S. Stayton and W. X. Wang, Macromol. Mater. Eng., 2012, 297, 1175–1183 CrossRef CAS.
- S. Demirci, S. Kinali-Demirci and T. Caykara, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2677–2685 CrossRef CAS.
- A. Zengin, G. Karakose and T. Caykara, Eur. Polym. J., 2013, 49, 3350–3358 CrossRef CAS.
- S. Demirci, S. Kinali-Demirci and T. Caykara, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1612–1619 CrossRef CAS.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446–7452 CrossRef CAS.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- D. Dupin, S. Fujii, S. P. Armes, P. Reeve and S. M. Baxter, Langmuir, 2006, 22, 3381–3387 CrossRef CAS PubMed.
- B. R. Saunders, H. M. Crowther and B. Vincent, Macromolecules, 1997, 30, 482–487 CrossRef CAS.
- S. R. Deka, A. Quarta, R. Di Corato, A. Falqui, L. Manna, R. Cingolani and T. Pellegrino, Langmuir, 2010, 26, 10315–10324 CrossRef CAS PubMed.
- W. B. Liechty, R. L. Scheuerle and N. A. Peppas, Polymer, 2013, 54, 3784–3795 CrossRef CAS.
- A. B. South and L. A. Lyon, Chem. Mater., 2010, 22, 3300–3306 CrossRef CAS.
- H. Ghandehari, P. Kopeckova and J. Kopecek, Biomaterials, 1997, 18, 861–872 CrossRef CAS PubMed.
- D. Das, P. Ghosh, S. Dhara, A. B. Panda and S. Pal, ACS Appl. Mater. Interfaces, 2015, 7, 4791–4803 CAS.
- X. Y. Gao, C. L. He, C. S. Xiao, X. L. Zhuang and X. S. Chen, Polymer, 2013, 54, 1786–1793 CrossRef CAS.
- P. Petrov, C. B. Tsvetanov and R. Jerome, Polym. Int., 2008, 57, 1258–1264 CrossRef CAS.
- T. J. Martin, K. Prochazka, P. Munk and S. E. Webber, Macromolecules, 1996, 29, 6071–6073 CrossRef CAS.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS PubMed.
- Y. S. Lee, in Self-Assembly and Nanotechnology, John Wiley & Sons, Inc., 2007, pp. 47–73 Search PubMed.
- S. Holappa, M. Karesoja, J. Shan and H. Tenhu, Macromolecules, 2002, 35, 4733–4738 CrossRef CAS.
- D. Wang, J. Yin, Z. Y. Zhu, Z. S. Ge, H. W. Liu, S. P. Armes and S. Y. Liu, Macromolecules, 2006, 39, 7378–7385 CrossRef CAS.
- X. H. Zhang, C. J. Ai, J. H. Ma, J. A. Xu and S. G. Yang, J. Colloid Interface Sci., 2011, 356, 24–30 CrossRef CAS PubMed.
- J. Zou, S. Zhang, R. Shrestha, K. Seetho, C. L. Donley and K. L. Wooley, Polym. Chem., 2012, 3, 3146–3156 RSC.
- V. Butun, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 11818–11819 CrossRef.
- S.-i. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2003, 36, 4208–4215 CrossRef CAS.
- P. D. Iddon, K. L. Robinson and S. P. Armes, Polymer, 2004, 45, 759–768 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- X. Zhao, Z. Zhang, F. Pan, Y. Ma, S. P. Armes, A. L. Lewis and J. R. Lu, Langmuir, 2005, 21, 9597–9603 CrossRef CAS PubMed.
- Y. H. Ma, Y. Q. Tang, N. C. Billingham, S. P. Armes, A. L. Lewis, A. W. Lloyd and J. P. Salvage, Macromolecules, 2003, 36, 3475–3484 CrossRef CAS.
- L. Desbaumes and A. Eisenberg, Langmuir, 1999, 15, 36–38 CrossRef CAS.
- Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642–3651 CrossRef CAS.
- C. Z. Cui, E. M. Bonder, Y. Qin and F. Jakle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2438–2445 CrossRef CAS.
- S. Y. Liu, N. C. Billingham and S. P. Armes, Angew. Chem., Int. Ed., 2001, 40, 2328–2331 CrossRef CAS.
- V. Bütün, S. Liu, J. V. M. Weaver, X. Bories-Azeau, Y. Cai and S. P. Armes, React. Funct. Polym., 2006, 66, 157–165 CrossRef.
- V. P. Torchilin, Expert Opin. Ther. Pat., 2005, 15, 63–75 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 1–9 Search PubMed.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- Y. L. Luo, W. Yu and F. Xu, Polym. Bull., 2012, 69, 597–620 CrossRef CAS.
- P. Yusan, I. Tuncel, V. Bütün, A. L. Demirel and I. Erel-Goktepe, Polym. Chem., 2014, 5, 3777–3787 RSC.
- F. Checot, J. Rodriguez-Hernandez, Y. Gnanou and S. Lecommandoux, Biomol. Eng., 2007, 24, 81–85 CrossRef CAS PubMed.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- F. T. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059–15064 CrossRef CAS PubMed.
- S. Y. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413–1416 CrossRef CAS.
- S. Y. Liu and S. P. Armes, Langmuir, 2003, 19, 4432–4438 CrossRef CAS.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- R. K. O'Reilly, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 2863–2878 CrossRef PubMed.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240 CrossRef CAS.
- M. Talelli, M. Barz, C. J. F. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed.
- Q. Jin, X. S. Liu, G. Y. Liu and J. Ji, Polymer, 2010, 51, 1311–1319 CrossRef CAS.
- Q. Jin, G. Liu and J. Ji, Eur. Polym. J., 2010, 46, 2120–2128 CrossRef CAS.
- R. X. Chang, Y. L. Tian, Y. Wang and J. L. Qin, Nanomater. Nanotechnol., 2016, 6, 1–6 CrossRef.
- A. E. Smith, X. W. Xu, T. U. Abell, S. E. Kirkland, R. M. Hensarling and C. L. McCormick, Macromolecules, 2009, 42, 2958–2964 CrossRef CAS.
- C. Chang, H. Wei, Q. A. Li, B. Yang, N. Chen, J. P. Zhou, X. Z. Zhang and R. X. Zhuo, Polym. Chem., 2011, 2, 923–930 RSC.
- J. X. Ding, W. G. Xu, Y. Zhang, D. K. Sun, C. S. Xiao, D. H. Liu, X. J. Zhu and X. S. Chen, J. Controlled Release, 2013, 172, 444–455 CrossRef CAS PubMed.
- K. Nakabayashi, D. Noda, T. Takahashi and H. Mori, Polymer, 2016, 86, 56–68 CrossRef CAS.
- V. Schmidt, R. Borsali and C. Giacomelli, Langmuir, 2009, 25, 13361–13367 CrossRef CAS PubMed.
- V. Butun, S. Liu, J. V. M. Weaver, X. Bories-Azeau, Y. Cai and S. P. Armes, React. Funct. Polym., 2006, 66, 157–165 CrossRef.
- V. Butun, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 12135–12136 CrossRef.
- S. Y. Liu, J. V. M. Weaver, M. Save and S. P. Armes, Langmuir, 2002, 18, 8350–8357 CrossRef CAS.
- X. Jiang, S. Luo, S. P. Armes, W. Shi and S. Liu, Macromolecules, 2006, 39, 5987–5994 CrossRef CAS.
- X. Xu, J. D. Flores and C. L. McCormick, Macromolecules, 2011, 44, 1327–1334 CrossRef CAS.
- J. Babin, M. Lepage and Y. Zhao, Macromolecules, 2008, 41, 1246–1253 CrossRef CAS.
- Z. W. Zhao, X. M. Yao, Z. Zhang, L. Chen, C. L. He and X. S. Chen, Macromol. Biosci., 2014, 14, 1609–1618 CrossRef CAS PubMed.
- O. Bertrand, E. Poggi, J. F. Gohy and C. A. Fustin, Macromolecules, 2014, 47, 183–190 CrossRef CAS.
- D. Kuckling and A. Wycisk, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2980–2994 CrossRef CAS.
- Z. Iatridi and C. Tsitsilianis, Polymers, 2011, 3, 1911–1933 CrossRef CAS.
- E. He, P. Ravi and K. C. Tam, Langmuir, 2007, 23, 2382–2388 CrossRef CAS PubMed.
- H. Liu, C. Li, H. Liu and S. Liu, Langmuir, 2009, 25, 4724–4734 CrossRef CAS PubMed.
- L. A. Connal, Q. Li, J. F. Quinn, E. Tjipto, F. Caruso and G. G. Qiao, Macromolecules, 2008, 41, 2620–2626 CrossRef CAS.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
- H. Hu and G. J. Liu, Macromolecules, 2014, 47, 5096–5103 CrossRef CAS.
- Y. Li, Y. Tang, R. Narain, A. L. Lewis and S. P. Armes, Langmuir, 2005, 21, 9946–9954 CrossRef CAS PubMed.
- Z. Iatridi and C. Tsitsilianis, Chem. Commun., 2011, 47, 5560–5562 RSC.
- R. Alizadeh and M. Ghaemy, Polymer, 2015, 66, 179–191 CrossRef CAS.
- B. L. Guo, J. F. Yuan and Q. Y. Gao, J. Appl. Polym. Sci., 2007, 104, 1279–1284 CrossRef CAS.
- J. Rao, Y. Zhang, J. Zhang and S. Liu, Biomacromolecules, 2008, 9, 2586–2593 CrossRef CAS PubMed.
- V. Sfika, C. Tsitsilianis, A. Kiriy, G. Gorodyska and M. Stamm, Macromolecules, 2004, 37, 9551–9560 CrossRef CAS.
- H. Yin, H. C. Kang, K. M. Huh and Y. H. Bae, Colloids Surf., B, 2014, 116, 128–137 CrossRef CAS PubMed.
- K. Wang, H. Peng, K. J. Thurecht, S. Puttick and A. K. Whittaker, Polym. Chem., 2014, 5, 1760–1771 RSC.
- R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533–1544 RSC.
- Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592–4593 CrossRef CAS.
- C. Feng, Z. Shen, L. N. Gu, S. Zhang, L. T. Li, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5638–5651 CrossRef CAS.
- Y. Y. Xu, S. Bolisetty, M. Drechsler, B. Fang, J. Y. Yuan, M. Ballauff and A. H. E. Muller, Polymer, 2008, 49, 3957–3964 CrossRef CAS.
- W. H. Chiang, V. T. Ho, W. C. Huang, Y. F. Huang, C. S. Chern and H. C. Chiu, Langmuir, 2012, 28, 15056–15064 CrossRef CAS PubMed.
- L. Wang, W. Huang, S. Wang, Y. Cui, P. Yang, X. Yang and J. V. M. Weaver, J. Appl. Polym. Sci., 2015, 132, 42183 Search PubMed.
- W. Cheng, J. N. Kumar, Y. Zhang and Y. Liu, Biomater. Sci., 2015, 3, 597–607 RSC.
- J. V. M. Weaver, R. T. Williams, B. J. L. Royles, P. H. Findlay, A. I. Cooper and S. P. Rannard, Soft Matter, 2008, 4, 985–992 RSC.
- X. B. Shi, Y. Zhao, H. Y. Gao, L. Zhang, F. M. Zhu and Q. Wu, Macromol. Rapid Commun., 2012, 33, 374–379 CrossRef CAS PubMed.
- E. Buhleier, W. Wehner and F. Vögtle, Synthesis, 1978, 155–158 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS PubMed.
- J. M. Oliveira, A. J. Salgado, N. Sousa, J. F. Mano and R. L. Reis, Prog. Polym. Sci., 2010, 35, 1163–1194 CrossRef CAS.
- D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC.
- P. S. Lai, P. J. Lou, C. L. Peng, C. L. Pai, W. N. Yen, M. Y. Huang, T. H. Young and M. J. Shieh, J. Controlled Release, 2007, 122, 39–46 CrossRef CAS PubMed.
- H. Kim, S. Lee, M. Noh, S. H. Lee, Y. Mok, G. W. Jin, J. H. Seo and Y. Lee, Polymer, 2011, 52, 1367–1374 CrossRef CAS.
- G. Pistolis, A. Malliaris, D. Tsiourvas and C. M. Paleos, Chem. – Eur. J., 1999, 5, 1440–1444 CrossRef CAS.
- M. Noh, Y. Mok, D. Nakayama, S. Jang, S. Lee, T. Kim and Y. Lee, Polymer, 2013, 54, 5338–5344 CrossRef CAS.
- C. Monteil, N. Bar, B. Moreau, R. Retoux, A. Bee, D. Talbot and D. Villemin, Part. Part. Syst. Charact., 2014, 31, 219–227 CrossRef CAS.
- I. B. Rietveld, W. G. Bouwman, M. W. P. L. Baars and R. K. Heenan, Macromolecules, 2001, 34, 8380–8383 CrossRef CAS.
- S. J. Tabatabaei Rezaei, H. S. Abandansari, M. R. Nabid and H. Niknejad, J. Colloid Interface Sci., 2014, 425, 27–35 CrossRef CAS PubMed.
- H. Huang, J. Li, L. Liao, J. Li, L. Wu, C. Dong, P. Lai and D. Liu, Eur. Polym. J., 2012, 48, 696–704 CrossRef CAS.
- Y. Yan, D. Wei, J. Li, J. Zheng, G. Shi, W. Luo, Y. Pan, J. Wang, L. Zhang, X. He and D. Liu, Acta Biomater., 2012, 8, 2113–2120 CrossRef CAS PubMed.
- H. Hui, X. D. Fan and Z. L. Cao, Polymer, 2005, 46, 9514–9522 CrossRef.
- Y. Wang, L. N. Li, J. J. Li, B. G. Yang, C. Y. Wang, W. C. Fang, F. Ji, Y. Wen and F. L. Yao, RSC Adv., 2016, 6, 17728–17739 RSC.
- J. Chen, J. Guo, B. S. Chang and W. L. Yang, Colloid Polym. Sci., 2012, 290, 517–524 CAS.
- H. i. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H. A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- L. Chen, Z. Peng, Z. Zeng, Y. She, J. Wei and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2202–2216 CrossRef CAS.
- L. Mi, M. T. Bernards, G. Cheng, Q. Yu and S. Jiang, Biomaterials, 2010, 31, 2919–2925 CrossRef CAS PubMed.
- G. W. de Groot, M. G. Santonicola, K. Sugihara, T. Zambelli, E. Reimhult, J. Voros and G. J. Vancso, ACS Appl. Mater. Interfaces, 2013, 5, 1400–1407 CAS.
- H. Y. Lei, M. M. Wang, Z. C. Tang, Y. F. Luan, W. Liu, B. Song and H. Chen, Langmuir, 2014, 30, 501–508 CrossRef CAS PubMed.
- X. Wang, R. Berger, J. I. Ramos, T. Wang, K. Koynov, G. Liu, H.-J. Butt and S. Wu, RSC Adv., 2014, 4, 45059–45064 RSC.
- Q. L. Zhang, F. Xia, T. L. Sun, W. L. Song, T. Y. Zhao, M. C. Liu and L. Jiang, Chem. Commun., 2008, 1199–1201 RSC.
- E. Stratakis, A. Mateescu, M. Barberoglou, M. Vamvakaki, C. Fotakis and S. H. Anastasiadis, Chem. Commun., 2010, 46, 4136–4138 RSC.
- H. Liu, S. Chen, H. Cui, J. Hu, H. Cai and W. Deng, RSC Adv., 2015, 5, 72444–72452 RSC.
- H. L. Liu, Y. Y. Li, K. Sun, J. B. Fan, P. C. Zhang, J. X. Meng, S. T. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- A. Brunsen, C. Diaz, L. I. Pietrasanta, B. Yameen, M. Ceolin, G. J. A. A. Soler-Illia and O. Azzaroni, Langmuir, 2012, 28, 3583–3592 CrossRef CAS PubMed.
- B. Wu, X. Wang, J. Yang, Z. Hua, K. Tian, R. Kou, J. Zhang, S. Ye, Y. Luo, V. S. J. Craig, G. Zhang and G. Liu, Sci. Adv., 2016, 2, e1600579 Search PubMed.
- N. Ayres, C. D. Cyrus and W. J. Brittain, Langmuir, 2007, 23, 3744–3749 CrossRef CAS PubMed.
- M. Motornov, T. Kin Tam, M. Pita, I. Tokarev, E. Katz and S. Minko, Nanotechnology, 2009, 20, 1–10 CrossRef PubMed.
- S. Berger, A. Synytska, L. Ionov, K. J. Eichhorn and M. Stamm, Macromolecules, 2008, 41, 9669–9676 CrossRef CAS.
- L. N. Gu, Z. Shen, C. Feng, Y. G. Li, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4056–4069 CrossRef CAS.
- L. Gu, C. Feng, D. Yang, Y. G. Li, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3142–3153 CrossRef CAS.
- M. Li, G. L. Li, Z. G. Zhang, J. Li, K. G. Neoh and E. T. Kang, Polymer, 2010, 51, 3377–3386 CrossRef CAS.
- M. R. Tomlinson, F. Cousin and M. Geoghegan, Polymer, 2009, 50, 4829–4836 CrossRef CAS.
- K. T. Oh, T. K. Bronich, L. Bromberg, T. A. Hatton and A. V. Kabanov, J. Controlled Release, 2006, 115, 9–17 CrossRef CAS PubMed.
- C. Chen, M. Z. Liu, C. M. Gao, S. Y. Lu, J. C. Chen, X. Y. Yu, E. Y. Ding, C. M. Yu, J. Guo and G. J. Cui, Carbohydr. Polym., 2013, 92, 621–628 CrossRef CAS PubMed.
- J. X. Ding, C. L. He, C. S. Xiao, J. Chen, X. L. Zhuang and X. S. Chen, Macromol. Res., 2012, 20, 292–301 CrossRef CAS.
- X. J. Wu and C. M. Dong, J. Controlled Release, 2015, 213, E121–E121 CrossRef PubMed.
- S. B. Lee, D. I. Ha, S. K. Cho, S. J. Kim and Y. M. Lee, J. Appl. Polym. Sci., 2004, 92, 2612–2620 CrossRef CAS.
- H. Q. Bao, L. Li, W. C. Leong and L. H. Gan, J. Phys. Chem. B, 2010, 114, 10666–10673 CrossRef CAS PubMed.
- H. Q. Bao, J. H. Hu, L. H. Gan and L. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6682–6692 CrossRef CAS.
- J. H. Kim, S. B. Lee, S. J. Kim and Y. M. Lee, Polymer, 2002, 43, 7549–7558 CrossRef CAS.
- J. Aleman, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvil, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1827 CrossRef CAS.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- O. E. Philippova, D. Hourdet, R. Audebert and A. R. Khokhlov, Macromolecules, 1997, 30, 8278–8285 CrossRef CAS.
- M. F. Abou Taleb, Int. J. Biol. Macromol., 2013, 62, 341–347 CrossRef CAS PubMed.
- K. E. Christodoulakis and M. Vamvakaki, Langmuir, 2010, 26, 639–647 CrossRef CAS PubMed.
- M. M. Ei-Masry, M. M. M. EInashar and H. M. El-Sherif, J. Appl. Polym. Sci., 2007, 106, 3571–3580 CrossRef.
- T. Sawai, S. Yamazaki, Y. Ikariyama and M. Aizawa, Macromolecules, 1991, 24, 2117–2118 CrossRef CAS.
- J. C. Leroux, R. M. Cozens, J. L. Roesel, B. Galli, E. Doelker and R. Gurny, Pharm. Res., 1996, 13, 485–487 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- J. Moselhy, T. Vira, F. F. Liu and X. Y. Wu, Int. J. Nanomed., 2009, 4, 153–164 CrossRef CAS.
- H. Hayashi, M. Iijima, K. Kataoka and Y. Nagasaki, Macromolecules, 2004, 37, 5389–5396 CrossRef CAS.
- S. A. Dergunov and G. A. Mun, Radiat. Phys. Chem., 2009, 78, 65–68 CrossRef CAS.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed.
- H. Ghandehari, P. Kopeckova, P. Y. Yeh and J. Kopecek, Macromol. Chem. Phys., 1996, 197, 965–980 CrossRef CAS.
- H. Brondsted and J. Kopecek, Biomaterials, 1991, 12, 584–592 CrossRef CAS PubMed.
- P. Y. Wang, Y. Y. Zhang, L. Cheng and W. G. Liu, Macromol. Chem. Phys., 2015, 216, 164–171 CrossRef CAS.
- L. H. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- C. J. Chang and G. Swift, J. Macromol. Sci., Part A: Pure Appl. Chem., 1999, 36, 963–970 CrossRef.
- Y. Zhao, H. J. Su, L. Fang and T. W. Tan, Polymer, 2005, 46, 5368–5376 CrossRef CAS.
- M. Sen and A. Yakar, Int. J. Pharm., 2001, 228, 33–41 CrossRef CAS PubMed.
- B. E. Rodriguez, M. S. Wolfe and M. Fryd, Macromolecules, 1994, 27, 6642–6647 CrossRef CAS.
- K. D. Yao, M. X. Xu, Y. J. Yin, J. Y. Zhao and X. L. Chen, Polym. Int., 1996, 39, 333–337 CrossRef CAS.
- A. C. Foss, T. Goto, M. Morishita and N. A. Peppas, Eur. J. Pharm. Biopharm., 2004, 57, 163–169 CrossRef CAS PubMed.
- A. J. Morse, J. Madsen, D. J. Growney, S. P. Armes, P. Mills and R. Swart, Langmuir, 2014, 30, 12509–12519 CrossRef CAS PubMed.
- M. Oishi and Y. Nagasaki, React. Funct. Polym., 2007, 67, 1311–1329 CrossRef CAS.
- J. Ding, X. Zhuang, C. Xiao, Y. Cheng, L. Zhao, C. He, Z. Tang and X. Chen, J. Mater. Chem., 2011, 21, 11383–11391 RSC.
- C. A. Schoener, H. N. Hutson and N. A. Peppas, Polym. Int., 2012, 61, 874–879 CrossRef CAS PubMed.
- Y. X. Zhang, Y. F. Chen, X. Y. Shen, J. J. Hu and J. S. Jan, Polymer, 2016, 86, 32–41 CrossRef CAS.
- H. L. Cao, Y. X. Dong, L. Bre, C. Tapeinos, W. X. Wang and A. Pandit, RSC Adv., 2016, 6, 9604–9611 RSC.
- X. Yang, L. Chen, B. Huang, F. Bai and X. Yang, Polymer, 2009, 50, 3556–3563 CrossRef CAS.
- H. Chen and Y. L. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6331–6339 CrossRef CAS.
- T. Li, X. C. Yin, W. Z. Zhai, Y. F. He and R. M. Wang, Aust. J. Chem., 2016, 69, 191–197 CrossRef CAS.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine, 2010, 6, 714–729 CAS.
- M. P. Liudvikas Segewicz, Polymer Aging, Stabilizers and Amphiphilic Block Copolymers, 2009 Search PubMed.
- V. Butun, A. Atay, C. Tuncer and Y. Bas, Langmuir, 2011, 27, 12657–12665 CrossRef CAS PubMed.
- M. Koenig, F. Simon, P. Formanek, M. Muller, S. Gupta, M. Stamm and P. Uhlmann, Macromol. Chem. Phys., 2013, 214, 2301–2311 CrossRef CAS.
- J. G. Zhang, S. Q. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS PubMed.
- A. Mayer and M. Antonietti, Colloid Polym. Sci., 1998, 276, 769–779 CAS.
- S. H. Anastasiadis and M. Vamvakaki, Int. J. Nanotechnol., 2009, 6, 46–70 CrossRef CAS.
- K. E. Christodoulakis, D. Palioura, S. H. Anastasiadis and M. Vamvakaki, Top. Catal., 2009, 52, 394–411 CrossRef CAS.
- D. Palioura, S. P. Armes, S. H. Anastasiadis and M. Vamvakaki, Langmuir, 2007, 23, 5761–5768 CrossRef CAS PubMed.
- L. M. Bronstein, M. Vamvakaki, M. Kostylev, V. Katsamanis, B. Stein and S. H. Anastasiadis, Langmuir, 2005, 21, 9747–9755 CrossRef CAS PubMed.
- M. Vamvakaki, L. Papoutsakis, V. Katsamanis, T. Afchoudia, P. G. Fragouli, H. Iatrou, N. Hadjichristidis, S. P. Armes, S. Sidorov, D. Zhirov, V. Zhirov, M. Kostylev, L. M. Bronstein and S. H. Anastasiadis, Faraday Discuss., 2005, 128, 129–147 RSC.
- S. H. Yu, H. Colfen and M. Antonietti, Chem. – Eur. J., 2002, 8, 2937–2945 CrossRef CAS.
- M. Al-Hussein, M. Koenig, M. Stamm and P. Uhlmann, Macromol. Chem. Phys., 2014, 215, 1679–1685 CrossRef CAS.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS PubMed.
- K. Esumi, A. Suzuki, N. Aihara, K. Usui and K. Torigoe, Langmuir, 1998, 14, 3157–3159 CrossRef CAS.
- Z. H. Li, H. Sai, S. C. Warren, M. Kamperman, H. Arora, S. M. Gruner and U. Wiesner, Chem. Mater., 2009, 21, 5578–5584 CrossRef CAS PubMed.
- A. B. R. Mayer, Polym. Adv. Technol., 2001, 12, 96–106 CrossRef CAS.
- M. Hamidi, K. Rostamizadeh and M. A. Shahbazi, in Intelligent Nanomaterials: Processes, Properties, and Applications, 2012, pp. 583–624 Search PubMed.
- Y. Liu, W. Wang, J. Yang, C. Zhou and J. Sun, Asian J. Pharm. Sci., 2013, 8, 159–167 CrossRef.
- K. H. Min, J.-H. Kim, S. M. Bae, H. Shin, M. S. Kim, S. Park, H. Lee, R.-W. Park, I.-S. Kim, K. Kim, I. C. Kwon, S. Y. Jeong and D. S. Lee, J. Controlled Release, 2010, 144, 259–266 CrossRef CAS PubMed.
- J. W. Cui, Y. Yan, Y. J. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- O. Sedláček, M. Hrubý, M. Studenovský, D. Větvička, J. Svoboda, D. Kaňková, J. Kovář and K. Ulbrich, Bioorg. Med. Chem., 2012, 20, 4056–4063 CrossRef PubMed.
- G. F. Walker, C. Fella, J. Pelisek, J. Fahrmeir, S. Boeckle, M. Ogris and E. Wagner, Mol. Ther., 2005, 11, 418–425 CrossRef CAS PubMed.
- H. Y. Li, M. Li, C. Chen, A. P. Fan, D. L. Kong, Z. Wang and Y. J. Zhao, Int. J. Pharm., 2015, 495, 572–578 CrossRef CAS PubMed.
- K. Ulbrich, T. Etrych, P. Chytil, M. Pechar, M. Jelinkova and B. Rihova, Int. J. Pharm., 2004, 277, 63–72 CrossRef CAS PubMed.
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, J. Controlled Release, 2003, 87, 33–47 CrossRef CAS PubMed.
- Y. Gu, Y. Zhong, F. Meng, R. Cheng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 2772–2780 CrossRef CAS PubMed.
- E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254–1263 CrossRef CAS PubMed.
- Y. Chan, T. Wong, F. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826–1836 CrossRef CAS PubMed.
- S. Jiang, Y. Yao, Y. Z. Nie, J. J. Yang and J. Yang, J. Colloid Interface Sci., 2011, 364, 264–271 CrossRef CAS PubMed.
- J. H. Park, Y. W. Cho, Y. J. Son, K. Kim, H. Chung, S. Y. Jeong, K. Choi, C. R. Park, R.-W. Park, I.-S. Kim and I. C. Kwon, Colloid Polym. Sci., 2006, 284, 763–770 CAS.
- F.-Q. Hu, L.-N. Liu, Y.-Z. Du and H. Yuan, Biomaterials, 2009, 30, 6955–6963 CrossRef CAS PubMed.
- H. S. Yoo, E. A. Lee and T. G. Park, J. Controlled Release, 2002, 82, 17–27 CrossRef CAS PubMed.
- S. Zhu, M. Hong, G. Tang, L. Qian, J. Lin, Y. Jiang and Y. Pei, Biomaterials, 2010, 31, 1360–1371 CrossRef CAS PubMed.
- M. C. D. Clochard, S. Rankin and S. Brocchini, Macromol. Rapid Commun., 2000, 21, 853–859 CrossRef CAS.
- C. Ding, J. Gu, X. Qu and Z. Yang, Bioconjugate Chem., 2009, 20, 1163–1170 CrossRef CAS PubMed.
- Y. H. Kim, J. H. Park, M. Lee, Y. H. Kim, T. G. Park and S. W. Kim, J. Controlled Release, 2005, 103, 209–219 CrossRef CAS PubMed.
- V. F. Patel, J. N. Hardin, J. M. Mastro, K. L. Law, J. L. Zimmermann, W. J. Ehlhardt, J. M. Woodland and J. J. Starling, Bioconjugate Chem., 1996, 7, 497–510 CrossRef CAS PubMed.
- V. F. Patel, J. N. Hardin, G. B. Grindey and R. M. Schultz, Bioorg. Med. Chem. Lett., 1995, 5, 513–518 CrossRef CAS.
- J. Zou, F. Zhang, S. Zhang, S. F. Pollack, M. Elsabahy, J. Fan and K. L. Wooley, Adv. Healthcare Mater., 2014, 3, 441–448 CrossRef CAS PubMed.
- X. N. Huang, F. S. Du, J. Cheng, Y. Q. Dong, D. H. Liang, S. P. Ji, S. S. Lin and Z. C. Li, Macromolecules, 2009, 42, 783–790 CrossRef CAS.
- C. C. Song, R. Ji, F. S. Du, D. H. Liang and Z. C. Li, ACS Macro Lett., 2013, 2, 273–277 CrossRef CAS.
- W. Chen, F. H. Meng, F. Li, S. J. Ji and Z. Y. Zhong, Biomacromolecules, 2009, 10, 1727–1735 CrossRef CAS PubMed.
- X. Ke, D. J. Coady, C. Yang, A. C. Engler, J. L. Hedrick and Y. Y. Yang, Polym. Chem., 2014, 5, 2621–2628 RSC.
- C. Alvarez-Lorenzo and A. Concheiro, Mini-Rev. Med. Chem., 2008, 8, 1065–1074 CrossRef CAS PubMed.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- Y. Zhao, Y. X. Zhou, D. S. Wang, Y. J. Gao, J. W. Li, S. J. Ma, L. Zhao, C. Zhang, Y. Liu and X. R. Li, Acta Biomater., 2015, 17, 182–192 CrossRef CAS PubMed.
- M. Licciardi, E. F. Craparo, G. Giammona, S. P. Armes, Y. Tang and A. L. Lewis, Macromol. Biosci., 2008, 8, 615–626 CrossRef CAS PubMed.
- C. Giacomelli, L. Le Men, R. Borsali, J. Lai-Kee-Him, A. Brisson, S. P. Armes and A. L. Lewis, Biomacromolecules, 2006, 7, 817–828 CrossRef CAS PubMed.
- Y. Tang, S. Y. Liu, S. P. Armes and N. C. Billingham, Biomacromolecules, 2003, 4, 1636–1645 CrossRef CAS PubMed.
- Y. Shen, Y. Zhan, J. Tang, P. Xu, P. A. Johnson, M. Radosz, E. A. Van Kirk and W. J. Murdoch, AIChE J., 2008, 54, 2979–2989 CrossRef CAS.
- N. M. Oh, K. T. Oh, H. J. Baik, B. R. Lee, A. H. Lee, Y. S. Youn and E. S. Lee, Colloids Surf., B, 2010, 78, 120–126 CrossRef CAS PubMed.
- C. Duan, D. Zhang, F. Wang, D. Zheng, L. Jia, F. Feng, Y. Liu, Y. Wang, K. Tian, F. Wang and Q. Zhang, Int. J. Pharm., 2011, 409, 252–259 CrossRef CAS PubMed.
- J. Li, W. Q. Hu, Y. J. Zhang, H. Tan, X. J. Yan, L. L. Zhao and H. Z. Liang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1235–1244 CrossRef CAS.
- R. P. Johnson, Y. I. Jeong, E. Choi, C. W. Chung, D. H. Kang, S. O. Oh, H. Suh and I. Kim, Adv. Funct. Mater., 2012, 22, 1058–1068 CrossRef CAS.
- J. M. Zhou, W. J. Zhang, C. Y. Hong and C. Y. Pan, ACS Appl. Mater. Interfaces, 2015, 7, 3618–3625 CAS.
- A. Pourjavadi, Z. M. Tehrani and S. Jokar, J. Ind. Eng. Chem., 2015, 28, 45–53 CrossRef CAS.
- S. Pavlukhina and S. Sukhishvili, Adv. Drug Delivery Rev., 2011, 63, 822–836 CrossRef CAS PubMed.
- K. Sakai, E. G. Smith, G. B. Webber, E. J. Wanless, V. Butun, S. P. Armes and S. Biggs, J. Colloid Interface Sci., 2006, 303, 372–379 CrossRef CAS PubMed.
- K. Sakai, E. G. Smith, G. B. Webber, C. Schatz, E. J. Wanless, V. Butun, S. P. Armes and S. Biggs, Langmuir, 2006, 22, 5328–5333 CrossRef CAS PubMed.
- K. Sakai, E. G. Smith, G. B. Webber, C. Schatz, E. J. Wanless, V. Butun, S. P. Armes and S. Biggs, J. Phys. Chem. B, 2006, 110, 14744–14753 CrossRef CAS PubMed.
- K. Sakai, G. B. Webber, C. D. Vo, E. J. Wanless, M. Vamvakaki, V. Butun, S. P. Armes and S. Biggs, Langmuir, 2008, 24, 116–123 CrossRef CAS PubMed.
- S. Biggs, K. Sakai, T. Addison, A. Schmid, S. P. Armes, M. Vamvakaki, V. Butun and G. Webber, Adv. Mater., 2007, 19, 247–250 CrossRef CAS.
- T. Addison, O. J. Cayre, S. Biggs, S. P. Armes and D. York, Langmuir, 2008, 24, 13328–13333 CrossRef CAS PubMed.
- T. Addison, O. J. Cayre, S. Biggs, S. P. Armes and D. York, Langmuir, 2010, 26, 6281–6286 CrossRef CAS PubMed.
- I. Erel, H. E. Karahan, C. Tuncer, V. Butun and A. L. Demirel, Soft Matter, 2012, 8, 827–836 RSC.
- I. Erel, V. Butun and A. L. Demirel, Abstr. Pap. Am. Chem. Soc., 2011, 242 Search PubMed , Pub. no. 336-Coll.
- C. Hippius, V. Butun and I. Erel-Goktepe, Mater. Sci. Eng., C, 2014, 41, 354–362 CrossRef CAS PubMed.
- B. Onat, V. Butun, S. Banerjee and I. Erel-Goktepe, Acta Biomater., 2016, 40, 293–309 CrossRef CAS PubMed.
- V. S. Joseph, S. Kim, Q. L. Zhang, R. Hoogenboom and J. D. Hong, Polymer, 2013, 54, 4894–4901 CrossRef CAS.
- M. G. Kellum, C. A. Harris, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1104–1111 CrossRef CAS.
- I. Erel, Z. C. Zhu, A. Zhuk and S. A. Sukhishvili, J. Colloid Interface Sci., 2011, 355, 61–69 CrossRef CAS PubMed.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS PubMed.
- W. T. Wu, J. Shen, Z. Gai, K. L. Hong, P. Banerjee and S. Q. Zhou, Biomaterials, 2011, 32, 9876–9887 CrossRef CAS PubMed.
- A. S. Timin, E. V. Balantseva, S. Y. Khashirova, E. V. Rumyantsev and T. Y. Osadchaya, Colloids Surf., A, 2015, 477, 26–34 CrossRef CAS.
- X. R. Li, P. C. Du and P. Liu, Colloids Surf., B, 2014, 122, 99–106 CrossRef CAS PubMed.
- Y. Shang, F. Y. Ding, L. Xiao, H. B. Deng, Y. M. Du and X. W. Shi, Carbohydr. Polym., 2014, 102, 413–418 CrossRef CAS PubMed.
- G. Unsoy, R. Khodadust, S. Yalcin, P. Mutlu and U. Gunduz, Eur. J. Pharm. Sci., 2014, 62, 243–250 CrossRef CAS PubMed.
- L. H. Zhao, Y. Y. Chen, W. Li, M. L. Lu, S. S. Wang, X. D. Chen, M. X. Shi, J. D. Wu, Q. P. Yuan and Y. Li, Carbohydr. Polym., 2015, 121, 276–283 CrossRef CAS PubMed.
- D. Suhag, R. Bhatia, S. Das, A. Shakeel, A. Ghosh, A. Singh, O. P. Sinha, S. Chakrabarti and M. Mukherjee, RSC Adv., 2015, 5, 53963–53972 RSC.
- F. Reyes-Ortega, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. S. Román, Woodhead Publishing, 2014, pp. 45–92 Search PubMed.
- R. Das, D. Das, P. Ghosh, A. Ghosh, S. Dhara, A. B. Panda and S. Pal, Cellulose, 2015, 22, 313–327 CrossRef CAS.
- L. Z. Zhao, C. L. Wu, F. Wang, A. G. Ying, C. D. Xu and S. F. Liu, Colloid Polym. Sci., 2014, 292, 1675–1683 CAS.
- M. Yigitoglu, G. Aydin and N. Isiklan, Polym. Bull., 2014, 71, 385–414 CrossRef CAS.
- T. K. Georgiou, M. Vamvakaki, C. S. Patrickios, E. N. Yamasaki and L. A. Phylactou, Biomacromolecules, 2004, 5, 2221–2229 CrossRef CAS PubMed.
- T. K. Georgiou, M. Vamvakaki, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2005, 6, 2990–2997 CrossRef CAS PubMed.
- T. K. Georgiou, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3505–3512 CrossRef CAS PubMed.
- M. Kurisawa, M. Yokoyama and T. Okano, J Control Release, 2000, 69, 127–137 CrossRef CAS PubMed.
- M. Kurisawa, M. Yokoyama and T. Okano, J Control Release, 2000, 68, 1–18 CrossRef CAS PubMed.
- S. Asayama, A. Maruyama, C. S. Cho and T. Akaike, Bioconjugate Chem., 1997, 8, 833–838 CrossRef CAS PubMed.
- C. Zhao, S. Nie, M. Tang and S. Sun, Prog. Polym. Sci., 2011, 36, 1499–1520 CrossRef CAS.
- D. Wandera, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2010, 357, 6–35 CrossRef CAS.
- J. F. Hester, S. C. Olugebefola and A. M. Mayes, J. Membr. Sci., 2002, 208, 375–388 CrossRef CAS.
- T. Peng and Y. L. Cheng, J. Appl. Polym. Sci., 2000, 76, 778–786 CrossRef CAS.
- N. S. Terefe, O. Glagovskaia, K. De Silva and R. Stockmann, Food Bioprod. Process., 2014, 92, 208–225 CrossRef CAS.
- A. Venault, C. W. Huang, J. Zheng, A. Chinnathambi, S. A. Alharbi, Y. Chang and Y. Chang, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 65–74 CrossRef CAS.
- F. Zhou and W. T. S. Huck, Chem. Commun., 2005, 5999–6001 RSC.
- M. T. Fenske, W. Meyer-Zaika, H.-G. Korth, H. Vieker, A. Turchanin and C. Schmuck, J. Am. Chem. Soc., 2013, 135, 8342–8349 CrossRef CAS PubMed.
- R. Liu, P. Liao, J. Liu and P. Feng, Langmuir, 2011, 27, 3095–3099 CrossRef CAS PubMed.
- X. J. Gao, W. Y. Yang, P. F. Pang, S. T. Liao, Q. Y. Cai, K. F. Zeng and C. A. Grimes, Sens. Actuators, B, 2007, 128, 161–167 CrossRef CAS.
- L. V. Sigolaeva, U. Gunther, D. V. Pergushov, S. Y. Gladyr, I. N. Kurochkin and F. H. Schacher, Macromol. Biosci., 2014, 14, 1039–1051 CrossRef CAS PubMed.
- C. C. Yang, Y. Q. Tian, A. K. Y. Jen and W. C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5495–5504 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |