DOI:
10.1039/C6QO00119J
(Research Article)
Org. Chem. Front., 2016,
3, 971-974
Reaction of alkenecarboxylic acids with isocyanates via rhodium(III)-catalyzed C–H activation: a versatile route to cyclic imides†
Received
24th March 2016
, Accepted 11th June 2016
First published on 13th June 2016
Abstract
Under rhodium(III) catalysis, substituted propenoic acids successfully reacted with aryl isocyanates to provide cyclic imides via direct functionalization of the β-alkenyl C–H bond followed by intramolecular cyclization. The cascade process has been corroborated by the isolated intermediate. The reaction provides direct access to cyclic imides from readily available starting materials.
Introduction
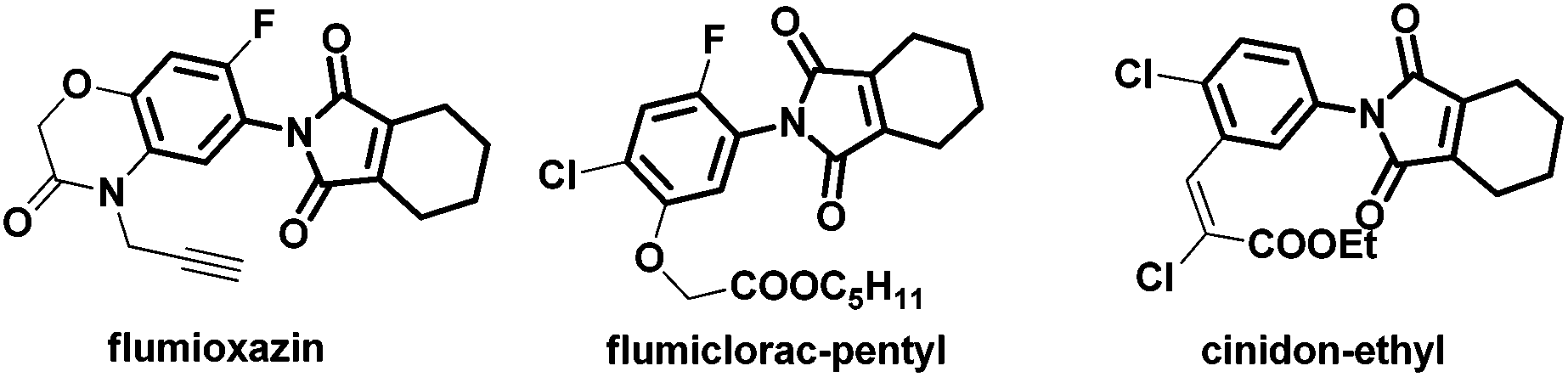
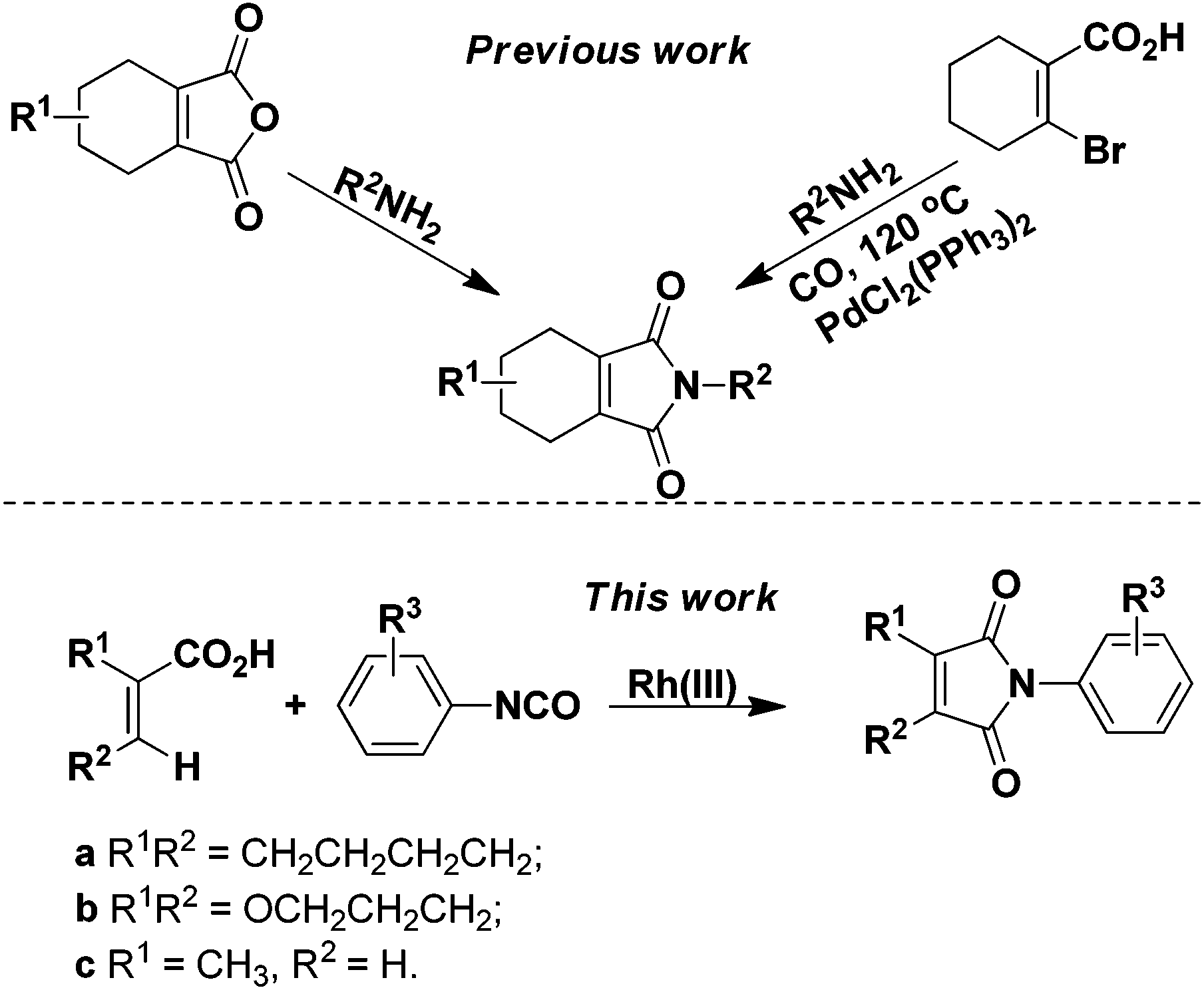
Since the discovery that protoporphyrinogen oxidase (PPO, E.C. 1.3.3.4) is the last enzyme in the common tetrapyrrole biosynthesis pathway before it branches toward chlorophyll (in plant) and heme (in animal) synthesis,1 many classes of herbicides against PPO have been developed. Among them, 2-aryl-4,5,6,7-tetrahydro-1H-isoindole-1,3(2H)-dione derivatives were paid much more attention due to their low toxicity, low dosage (10–50 g ai per ha), broad herbicidal spectrum, and long lasting effect,2 which resulted in the discovery of many structurally diverse commercial compounds, such as cinidon-ethyl,3 flumiclorac-pentyl,4,5 and flumioxazin (Fig. 1).6,7 The most commonly used strategy for their synthesis involves reactions between the corresponding cyclohex-1-ene-1,2-dicarboxylic anhydrides8–11 or prefunctionalized β-halo cyclohex-1-ene-1-carboxylic acids12 with amines (Scheme 1). However, the very limited availability of differently substituted cyclohex-1-ene-1,2-dicarboxylic acids, mostly due to the harsh reaction conditions in their preparations, calls for alternative protocols. Recently, rhodium complexes have emerged as a powerful transition-metal catalyst in C–H activation chemistry.13,14 For instance, phthalimides can be synthesized by Rh(III)-catalyzed ortho-carbonylation of benzoic acids via a C–H activation mechanism.14a However, the analogous β-vinyl C–H bond of alkenoic acids is more difficult to activate than the ortho-aryl C–H bond of benzoic acid and, hence, most recent research studies were mainly directed to the synthesis of aryl phthalimides by metal-catalyzed ortho-carbonylation of benzamides/benzoic acids respectively.14 In addition, although a similar cascade mechanism has been proposed for forming the phthalimides, relatively little mechanistic information is known. Herein, we report the synthesis of 2-aryl-4,5,6,7-tetrahydro-1H-isoindole-1,3(2H)-dione derivatives from readily available 1-cyclohexene-1-carboxylic acid and isocyanates through β-vinyl C–H functionalization for the first time. Furthermore, experimental results substantiated the cascade mechanism for forming the cyclic imides.
 |
| | Fig. 1 Selected bioactive tetrahydrophthalimide derivatives. | |
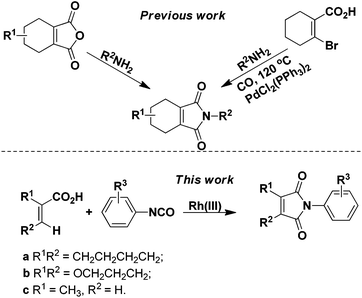 |
| | Scheme 1 Strategies for the synthesis of 2-aryl-4,5,6,7-tetrahydro-1H-isoindole-1,3(2H)-dione derivatives. | |
Results and discussion
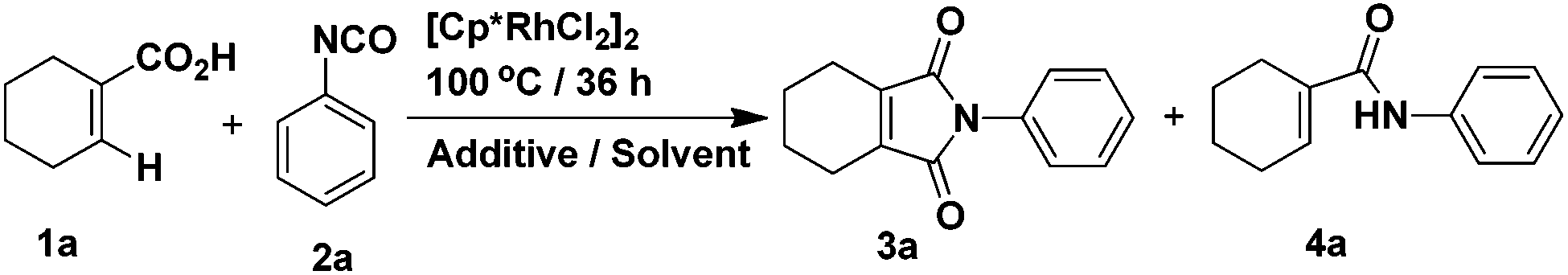
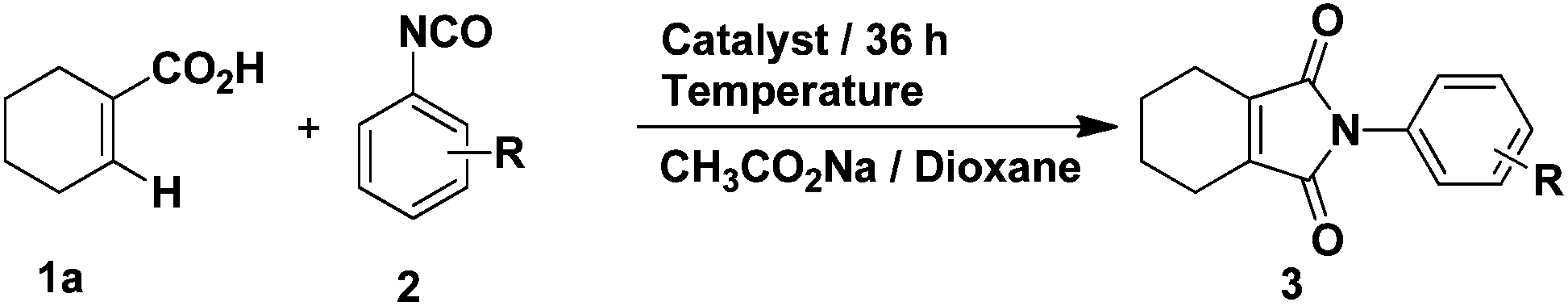
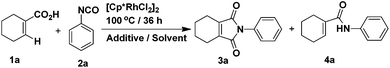
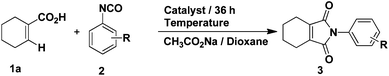
Our studies began with conditions which were used successfully in the formation of phthalimides from benzoic acids and phenyl isocyanate. Firstly, the reaction between 1-cyclohexene-1-carboxylic acid and phenyl isocyanate proceeded under the same conditions as in the previous report.14 The yield of target product 3a was 50% (Table 1, entry 1), while 4a was formed in ca. 24% yield. Then, we attempted to improve the yield of 3a by adding various inorganic salts, without observing any positive effect on the reaction (Table 1, entries 2–9). In order to eliminate or minimize the effect of the by-product water on the reaction, anhydrous sodium sulfate, anhydrous magnesium sulfate and 3 Å molecular sieves (dried at 400 °C) were used respectively, which only led to decreased product yield (Table 1, entries 10–12). Finally, various solvents were examined (Table 1, entries 13–20) and only THF led to a similar yield as dioxane. Further optimization of the catalyst and reaction temperature (Table 2, entries 1–9) showed that it was preferable to run the reaction using Cp*Rh(MeCN)3(SbF6)2 as the catalyst at 100 °C when the substituent R was hydrogen. However, when phenyl isocyanate was replaced by 3-methoxyphenyl isocyanate, only a small amount of product 3 was formed whether the catalyst was Cp*Rh(MeCN)3(SbF6)2 or [Cp*RhCl2]2 (Table 2, entries 10 and 11). In the end, it was found that by carrying out the reaction (R = 3-methoxy) at 120 °C, the yield could be improved significantly (Table 2, entries 12–14) and the catalyst [Cp*RhCl2]2 was preferable.
Table 1 Effect of additive and solventa
|
|
| Entry |
Additives (equiv.) |
Solvent |
Yieldb [%] |
|
Reaction conditions: 1a (0.1 mmol), 2a (0.3 mmol), [Cp*RhCl2]2 (5 mol%), additive (0.1 mmol), solvent (0.5 mL), 100 °C, 36 h, argon.
The yield was determined by 1H NMR analysis of the crude reaction mixture using mesitylene as the internal standard.
|
| 1 |
CH3CO2Na (1.0) |
Dioxane |
50 |
| 2 |
NaHCO3 (1.0) |
Dioxane |
26 |
| 3 |
Na2CO3 (1.0) |
Dioxane |
31 |
| 4 |
Sodium pivalate (1.0) |
Dioxane |
0 |
| 5 |
CF3CO2Ag (1.0) |
Dioxane |
0 |
| 6 |
CH3CO2Ag (1.0) |
Dioxane |
0 |
| 7 |
CH3CO2Na (1.0) + 20% AgOTf |
Dioxane |
40 |
| 8 |
Cs2CO3 (1.0) |
Dioxane |
0 |
| 9 |
NaH (1.0) |
Dioxane |
0 |
| 10 |
CH3CO2Na (1.0) + 1 equiv. Na2SO4 |
Dioxane |
42 |
| 11 |
CH3CO2Na (1.0) + 1 equiv. MgSO4 |
Dioxane |
35 |
| 12 |
200% (w/w) 3 Å molecular sieves |
Dioxane |
43 |
| 13 |
CH3CO2Na (1.0) |
Toluene |
23 |
| 14 |
CH3CO2Na (1.0) |
Chlorobenzene |
35 |
| 15 |
CH3CO2Na (1.0) |
THF |
48 |
| 16 |
CH3CO2Na (1.0) |
o-Xylene |
25 |
| 17 |
CH3CO2Na (1.0) |
Acetonitrile |
18 |
| 18 |
CH3CO2Na (1.0) |
DMF |
0 |
| 19 |
CH3CO2Na (1.0) |
DMA |
0 |
| 20 |
CH3CO2Na (1.0) |
DME |
0 |
Table 2 Effect of catalyst and temperaturea
|
|
| Entry |
R |
Catalyst |
Temperature (°C) |
Yieldb [%] |
|
Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), [Cp*RhCl2]2 (5 mol%), CH3CO2Na (0.1 mmol), dioxane (0.5 mL), 36 h, argon.
The yield was determined by 1H NMR analysis of the crude reaction mixture using mesitylene as the internal standard.
|
| 1 |
H |
Chloro(1,5-cyclooctadiene) rhodium(I) dimer |
100 |
Trace |
| 2 |
H |
Chloronorbornadiene rhodium(I) dimer |
100 |
6 |
| 3 |
H |
Rhodium dicarbonyl chloride dimer |
100 |
8 |
| 4 |
H |
(Ph3P)3RhCl |
100 |
Trace |
| 5 |
H |
[Cl(cod)Rh]2 |
100 |
Trace |
| 6 |
H |
Acetylacetonatobis(cyclooctene)rhodium(I) |
100 |
10 |
| 7 |
H |
Cp*Rh(MeCN)3(SbF6)2 |
100 |
65 |
| 8 |
H |
Cp*Rh(MeCN)3(SbF6)2 |
150 |
50 |
| 9 |
H |
Cp*Rh(MeCN)3(SbF6)2 |
80 |
0 |
| 10 |
3-OCH3 |
Cp*Rh(MeCN)3(SbF6)2 |
100 |
13 |
| 11 |
3-OCH3 |
[Cp*RhCl2]2 |
100 |
21 |
| 12 |
3-OCH3 |
Cp*Rh(MeCN)3(SbF6)2 |
120 |
60 |
| 13 |
3-OCH3 |
[Cp*RhCl2]2 |
120 |
65 |
| 14 |
3-OCH3 |
[Cp*RhCl2]2 |
150 |
45 |
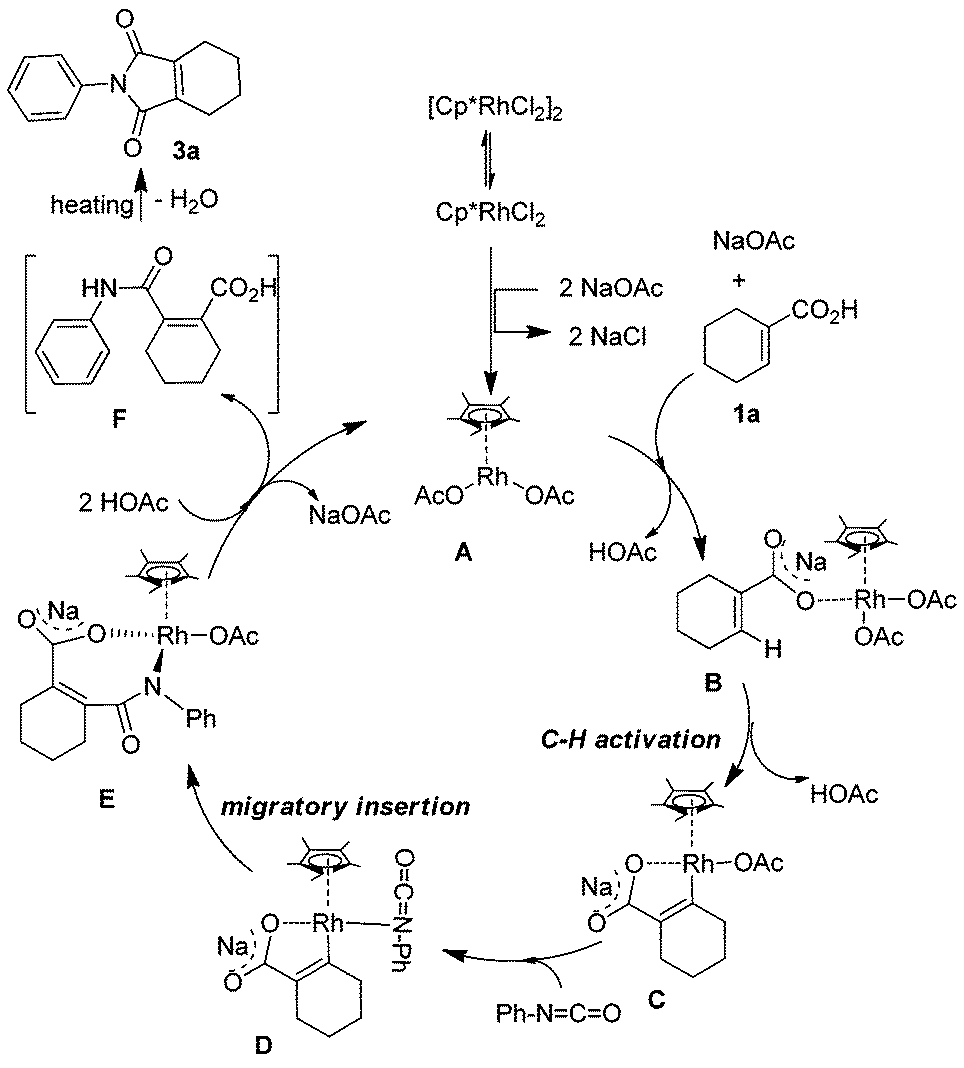
In order to further understand the reaction mechanism, a series of experiments was performed between phenyl isocyanate and 1-cyclohexene-1-carboxylic acid with different reaction times. When the corresponding reaction mixture was characterized by 1H NMR at different reaction times, an interesting phenomenon was observed. The signal strength of the peak at about 2.57 ppm decreased with the reaction time. After the reaction was run for 48 h, the signal nearly disappeared. Based on this result, we postulated that the peak belongs to the intermediate of the reaction. Since this intermediate is very unstable and sensitive to temperature, many experiments were performed in order to characterize it. Finally, the intermediate was isolated at a low temperature with higher purity by TLC and determined by 1H NMR. These peaks at 2.61, 2.43 and 1.86 showed that the two carbon atoms directly connected with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of cyclohexene were substituted by different groups. It can be evidenced from the chemical shift that one of them is a stronger electron-withdrawing group in comparison with C(O)N. This analysis was also supported by 13C NMR (21.39, 21.37, 21.24 and 20.43 ppm) and high resolution mass spec. According to the above, the structure of the intermediate was concluded to be 2-(phenylcarbamoyl)cyclohex-1-enecarboxylic acid (F) (Scheme 2). When it was analyzed by GCMS, the molecular weight was 227.2, which was resulted from the reaction of intramolecular dehydration. Based on the above results and the knowledge of analogous chelation-assisted C–H bond activation reactions, a plausible mechanism for the rhodium-catalyzed reaction of 1-cyclohexene-1-carboxylic acid and phenyl isocyanate is outlined in Scheme 2. At first, the dimeric precatalyst [Cp*RhCl2]2 dissociated to give the coordinatively unsaturated monomer. Next, the monomer reacted with sodium acetate, 1-cyclohexene-1-carboxylic acid and isocyanate with the help of NaOAc respectively.15,16 A rhodium alkoxide was generated through several steps. Finally, protonation of intermediate E by acetic acid gave the intermediate F and regenerated the rhodium catalyst.
C bond of cyclohexene were substituted by different groups. It can be evidenced from the chemical shift that one of them is a stronger electron-withdrawing group in comparison with C(O)N. This analysis was also supported by 13C NMR (21.39, 21.37, 21.24 and 20.43 ppm) and high resolution mass spec. According to the above, the structure of the intermediate was concluded to be 2-(phenylcarbamoyl)cyclohex-1-enecarboxylic acid (F) (Scheme 2). When it was analyzed by GCMS, the molecular weight was 227.2, which was resulted from the reaction of intramolecular dehydration. Based on the above results and the knowledge of analogous chelation-assisted C–H bond activation reactions, a plausible mechanism for the rhodium-catalyzed reaction of 1-cyclohexene-1-carboxylic acid and phenyl isocyanate is outlined in Scheme 2. At first, the dimeric precatalyst [Cp*RhCl2]2 dissociated to give the coordinatively unsaturated monomer. Next, the monomer reacted with sodium acetate, 1-cyclohexene-1-carboxylic acid and isocyanate with the help of NaOAc respectively.15,16 A rhodium alkoxide was generated through several steps. Finally, protonation of intermediate E by acetic acid gave the intermediate F and regenerated the rhodium catalyst.
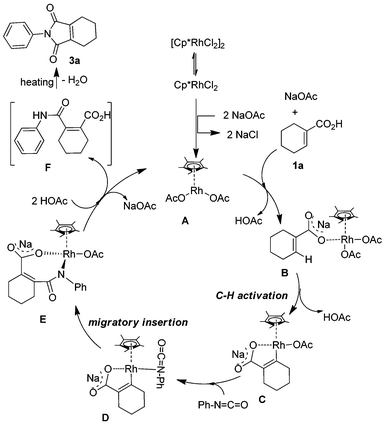 |
| | Scheme 2 Plausible mechanism. | |
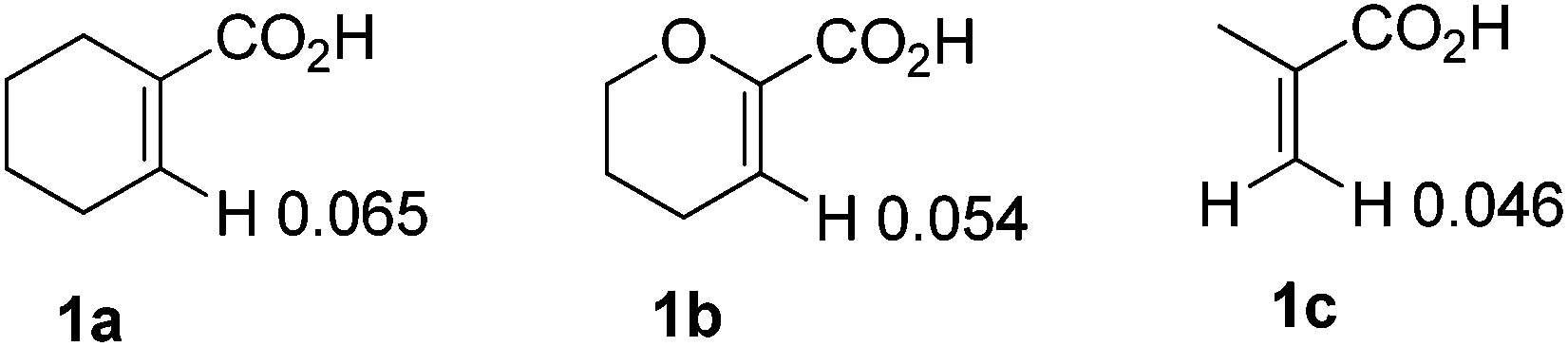
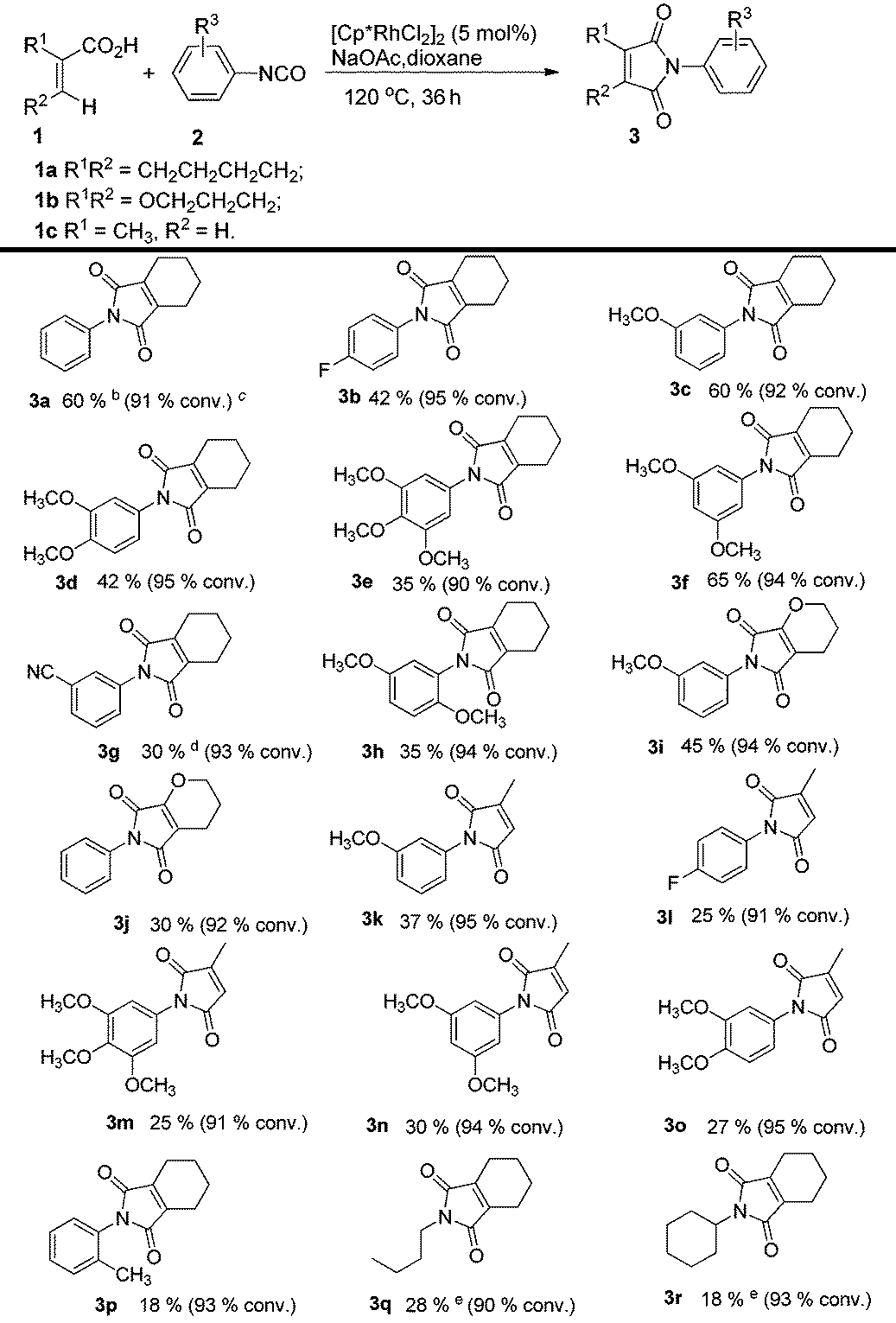
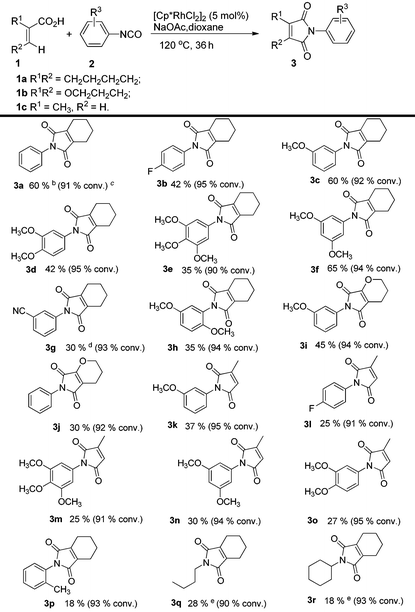
To further explore the substrate scope and limitations, we tested the reactions of 1-cyclohexene-1-carboxylic acid, 3,4-dihydro-2H-pyran-6-carboxylic acid, and 2-methylpropenoic acid with various isocyanates respectively (Table 3). As listed in Table 3, the conversions range from 90% to 95%. The main by-products were N-aryl alkenecarboxamides. For this reaction, 1-cyclohexene-1-carboxylic acid showed higher reactivity than the latter ones (3fvs.3n; 3cvs.3k). In order to explain the reactivity difference, the charges of β-H of the three alkenoic acids were calculated using SYBYL6.9 (Tripos Inc.) (Fig. 2). Based on this calculation, the rank of β-H's positive charge was: 1a > 1b > 1c. This indicated that the β-H of compound 1a possessed stronger acidity and was easier to be activated by rhodium in comparison with 1b and 1c.
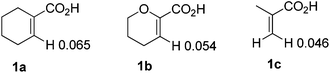 |
| | Fig. 2 The charge of compounds 1a, 1b and 1c. | |
Table 3 Substrate scope of 2-aryl-4,5,6,7-tetrahydro-1H-isoindole-1,3(2H)-dionesa
|
Reaction conditions: acid (0.2 mmol), isocyanate (0.6 mmol), [Cp*RhCl2]2 (5 mol%), NaOAc (0.2 mmol), dioxane (0.5 mL), 120 °C, 36 h, argon. Yield of the purified product is reported; the lower yield of product resulted from the formation of the by-product alkenecarboxamide.
The catalyst is Cp*Rh(MeCN)3(SbF6)2 (5 mol%).
The values in bracket were the acid conversions and measured by GCMS.
Acetic acid (10 mol%) was used.
The yield was determined by 1H NMR analysis of the crude reaction mixture using mesitylene as the internal standard.
|
|
Conclusions
We have reported the synthesis of cyclic imides by directly reacting alkenoic acids and various isocyanates under rhodium(III) catalysis. This is the first time that the intermediate was isolated and determined. This experimental result further demonstrated that the reaction involved a cascade amidation of acid and isocyanate via Rh(III)-catalyzed C–H bond functionalization, followed by intramolecular dehydration. It provides direct access to cyclic imides in one pot, and uses readily available starting materials. In view of its advantages, this procedure will be useful in finding novel protoporphyrinogen oxidase-inhibiting herbicides and in other organic syntheses.
Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J. Li), the CFI, FQRNT Center for Green Chemistry and Catalysis, NSERC, and McGill University for support of our research. Dr You-Quan Zhu is grateful for the support from the China Scholarship Council (CSC) and the National Natural Science Foundation of China (no. 21372134).
Notes and references
- H. B. Li, Y. Q. Zhu, X. W. Song, F. Z. Hu, B. Liu, Y. H. Li, Z. X. Niu, P. Liu, Z. H. Wang, H. B. Song, X. M. Zou and H. Z. Yang, J. Agric. Food Chem., 2008, 56, 9535 CrossRef CAS PubMed.
-
G. Theodoridis, Protoporphyrinogen-IX-oxidase Inhibitors, in Modern Crop Protection Compounds, ed. W. Kraomer, U. Schirmer, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007, p. 153 Search PubMed.
- K. Grossmann and H. Schiffer, Pestic. Sci., 1999, 55, 687 CrossRef CAS.
- H. Matsunaga, N. Isobe, H. Kaneko, I. Nakatsuka and H. Yamada, J. Agric. Food Chem., 1996, 44, 1910 CrossRef CAS.
- H. Matsunaga, N. Isobe, H. Kaneko, I. Nakatsuka and S. Yamane, J. Agric. Food Chem., 1997, 45, 501 CrossRef CAS.
- T. Katagi, Pestic. Sci., 2003, 28, 44 CrossRef CAS.
- J. W. Kwon, K. L. Armbrust and T. L. Grey, Pest Manage. Sci., 2004, 60, 939 CrossRef CAS PubMed.
- J. W. Lyga, R. M. Patera, G. Theodoridis, B. P. Halling, F. W. Hotzman and M. J. Plummer, J. Agric. Food Chem., 1991, 39, 1667 CrossRef CAS.
- Y.-P. Luo, L.-L. Jiang, G.-D. Wang, Q. Chen and G.-F. Yang, J. Agric. Food Chem., 2008, 56, 2118 CrossRef CAS PubMed.
- M.-Z. Huang, F.-X. Luo, H.-B. Mo, Y.-G. Ren, X.-G. Wang, X.-M. Ou, M.-X. Lei, A.-P. Liu, L. Huang and M.-C. Xu, J. Agric. Food Chem., 2009, 57, 9585 CrossRef CAS PubMed.
- L.-L. Jiang, Y. Zuo, Z.-F. Wang, Y. Tan, Q.-Y. Wu, Z. Xi and G.-F. Yang, J. Agric. Food Chem., 2011, 59, 6172 CrossRef CAS PubMed.
- H. K. Lee and C. S. Cho, Appl. Organomet. Chem., 2012, 26, 185 CrossRef CAS.
- A. C. Denise, G. B. Robert and A. E. Jonathan, Chem. Rev., 2010, 110, 624 CrossRef PubMed.
- For rhodium(III)-catalyzed addition of C–H bonds to isocyanates, see:
(a) X.-Y. Shi, A. Renzetti, S. Kundu and C.-J. Li, Adv. Synth. Catal., 2014, 356, 723 CrossRef CAS;
(b) B. Zhou, W. Hou, Y. Yang and Y. Li, Chem. – Eur. J., 2013, 19, 4701 CrossRef CAS PubMed;
(c) W. Hou, B. Zhou, Y. Yang, H. Feng and Y. Li, Org. Lett., 2013, 15, 1814 CrossRef CAS PubMed;
(d) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed;
(e) B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., 2012, 124, 4014 (
Angew. Chem., Int. Ed.
, 2012
, 51
, 3948
) CrossRef. For ruthenium(I)-catalyzed addition, see:
(f) X.-F. Dong, J. Fan, X.-Y. Shi, K.-Y. Liu, P.-M. Wang and J.-F. Wei, J. Organomet. Chem., 2015, 779, 55 CrossRef CAS;
(g) S. D. Sarkar and L. Ackermann, Chem. – Eur. J., 2014, 20, 13932 CrossRef PubMed;
(h) S. Sueki, Y. Guo, M. Kanai and Y. Kuninobu, Angew. Chem., 2013, 125, 12095 (
Angew. Chem., Int. Ed.
, 2013
, 52
, 11879
) CrossRef;
(i) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; X.-Y. Shi, X.-F Dong, J. Fan, K.-Y. Liu, J.-F. Wei and C.-J. Li, Sci. Chin. Chem., 2015, 58, 1286 Search PubMed.
- K. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00119j |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.