
Aromatic C–H bond cleavage by using a Cu(I) ate-complex†
Guanghui
Zhang‡
 a,
Hong
Yi‡
a,
Jie
Xin
a,
Yi
Deng
a,
Ruopeng
Bai
a,
Zhiliang
Huang
a,
Jeffrey T.
Miller
bc,
A. Jeremy
Kropf
b,
Emilio E.
Bunel
b,
Xiaotian
Qi
d,
Yu
Lan
d and
Aiwen
Lei
*abe
a,
Hong
Yi‡
a,
Jie
Xin
a,
Yi
Deng
a,
Ruopeng
Bai
a,
Zhiliang
Huang
a,
Jeffrey T.
Miller
bc,
A. Jeremy
Kropf
b,
Emilio E.
Bunel
b,
Xiaotian
Qi
d,
Yu
Lan
d and
Aiwen
Lei
*abe
aCollege of Chemistry and Molecular Sciences, Wuhan University, the Institute of Advanced Studies, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn
bChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, Illinois 60439, USA
cSchool of Chemical Engineering, Purdue University, West Lafayette, Indiana 47907, USA
dSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400030, P. R. China
eState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 13th June 2016
Abstract
In situ X-ray absorption spectroscopy (XAS), infrared (IR) and nuclear magnetic resonance (NMR) techniques were used to identify the structures and reactivity of copper-containing active intermediates in the sp2 C–H bond cleavage reaction of electron-deficient aromatics. An ate-complex [Cu(OtBu)2]Na was found to be able to cleave the C–H bond of benzothiazole (ArH) producing [ArCuI(OtBu)]Na with a rate constant of 3.2 × 10−2 mol−1 L s−1 at −50 °C and with an activation enthalpy of 0.73 kcal mol−1 at room temperature.
Introduction
Selective functionalization of C–H bonds is considered as a great challenge in organic chemistry.1–4 Great success has been achieved using transition metal salts and complexes as catalysts in C–C, C–N and C–O bond formation reactions involving C–H functionalization.5–7 Recent advances highlighted the potential to achieve highly efficient functionalization of C–H bonds using Cu catalysts.8–14 Investigations into the reaction mechanism, especially the identification of key intermediates, will be helpful for understanding the chemistry and designing new Cu catalysts for practical applications. With the CuX/tBuOM (M = Li, K, and Na) catalytic system, several highly efficient functionalization reactions of electron-poor aromatic C–H bonds have been achieved including hydroxylation, alkynylation, arylation, trifluoromethylation, halogenation, etc.15–20 The versatility of the CuX/tBuOM catalytic system prompted us to further investigate the intermediates involved in the C–H bond cleavage step. Recently, an efficient copper-catalyzed oxidative functionalization of aromatic C–H bonds has been achieved with O2 at room temperature (Scheme 1).15 The high efficiency, ligand-free and mild reaction conditions prompted us to use this reaction as the model to further investigate the intermediates involved in the C–H bond cleavage.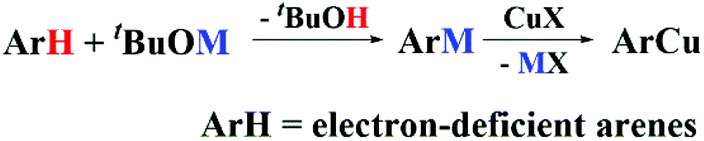 | ||
| Scheme 1 Generally accepted C–H cleavage mechanism of the CuX/tBuOM system. | ||
It is generally believed that in the CuX/tBuOM catalyst system tBuOM functions as a strong base which deprotonates the C–H bond of the electron-poor aromatic compound forming Ar–M, followed by transmetallation with CuX generating Ar–Cu (Scheme 1). In this study, we demonstrate a completely new understanding toward the CuX/tBuOM system in the benzothiazole C–H cleavage reaction, clarifying that the Cu ate complex is responsible for the C–H bond cleavage via identification of intermediates under realistic reaction conditions by utilization of in situ X-ray absorption spectroscopy (XAS),21,22 and nuclear magnetic resonance (NMR) spectroscopy. The kinetics of the C–H bond cleavage of benzothiazole is revealed by in situ IR spectroscopy.
Results and discussion
In the copper-catalyzed functionalization of electron-poor aromatic C–H bonds, CuCl could be used as a pre-catalyst in the presence of excess tBuONa.15 Our initial effort was focused on the reaction of 10 eq. of tBuONa with CuCl under a N2 atmosphere trying to mimic the catalytic reaction conditions in the absence of an oxidant. The structure of the obtained Cu species was studied by in situ XAS spectroscopy. As shown in the X-ray absorption near-edge structure (XANES) spectra (Fig. 1A), the peak at 8982.0 eV, which could be assigned to 1s to 4p transition, has long been recognized as a characteristic feature of Cu(I) species.23–26 The edge energy of 8980.9 eV, determined by the inflection point of the first peak, was also in the typical range of Cu(I) species (Table S1†). The intense peak of Cu(I) typically suggests a two-coordination environment. The fitting results of the extended X-ray absorption fine structure (EXAFS) spectrum suggested that the obtained Cu(I) species was two-oxygen bonded at an average distance of 1.83 Å, and no chloride anion was found bonded to Cu. It has been shown that CuCl2 could also be used in the copper-catalyzed oxidative functionalization of aromatic C–H bonds. We have shown very recently that in the presence of excess tBuONa, CuCl2 could be reduced to [CuI(OtBu)2]Na.27 Herein, by comparing the XANES and EXAFS data, as well as the EXAFS fitting results, we concluded that the reaction between CuCl and excess tBuONa generated the same Cu(I) ate-complex, which is also consistent with NMR analysis (Fig. S1 and S2†).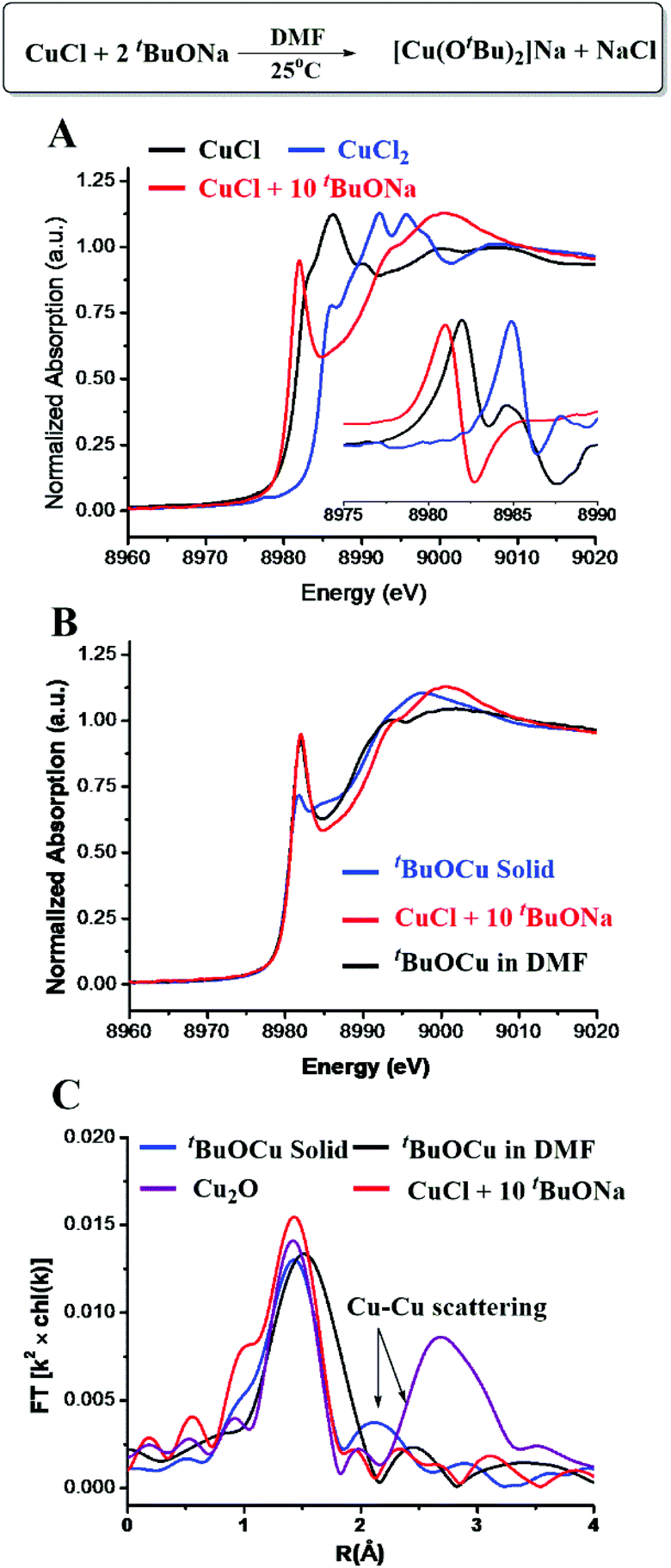 | ||
| Fig. 1 Formation of the Cu(I) ate-complex. (A) XANES spectra. The inset shows the normalized first derivatives. (B) XANES spectra. (C) Comparison of the FT magnitude of [CuI(OtBu)2]Na with tBuOCu and Cu2O (FT: 2.51–11.55 Å−1). | ||
XAS spectra of solid tBuOCu28 and Cu2O samples were collected for further comparison. As shown in Fig. 1B, the XANES spectra showed the same oxidation state but a different coordination environment. In the single crystal, tBuOCu adopts a tetramer structure, and each Cu atom is bonded to two tert-butyl oxide groups with an average bond distance of 1.85 Å.29 The EXAFS first-shell fitting results of the solid tBuOCu sample is consistent with the crystal structure showing that each Cu(I) has two oxygen bonded at a distance of 1.83 Å (Table S2 and Fig. S4†). In bulk Cu2O, each Cu has 12 Cu second-shell neighbors at a distance of 3.02 Å.30 While in the tBuOCu crystal structure, each Cu atom has 2 Cu neighbors at a distance of 2.71 Å.29 The X-ray scattering of the Cu–Cu pair in the EXAFS spectra was observed for both samples (Fig. 1C). However, the Cu–Cu scattering peak is absent in the EXAFS spectrum of [CuI(OtBu)2]Na confirming its monomeric structure in DMF (dimethylformamide) solution.
After an equal molar amount of [CuI(OtBu)2]Na was added to benzothiazole (ArH) at room temperature, more than 95% deprotonation of 2-H was observed by 1H NMR in 5 minutes (Fig. 2A and S3†). Additionally, the formation of tBuOH was observed by IR. The XANES analysis of the new Cu species showed an edge energy of 8980.2 eV, consistent with CuI (Fig. 2B). Compared with [CuI(OtBu)2]Na, the 0.9 eV shift toward lower energy suggests the coordination of a more electron-rich ligand. The first-shell fitting of the R-space EXAFS spectrum gave a coordination number of 2 with an average bond distance of 1.83 Å. The structure was thus proposed to be [ArCuI(OtBu)]Na.
 | ||
| Fig. 2 CuI-promoted C–H bond cleavage. (A) 1H NMR spectra of the reaction of [Cu(OtBu)2]Na with ArH. (B) XANES spectra with normalized first derivatives. | ||
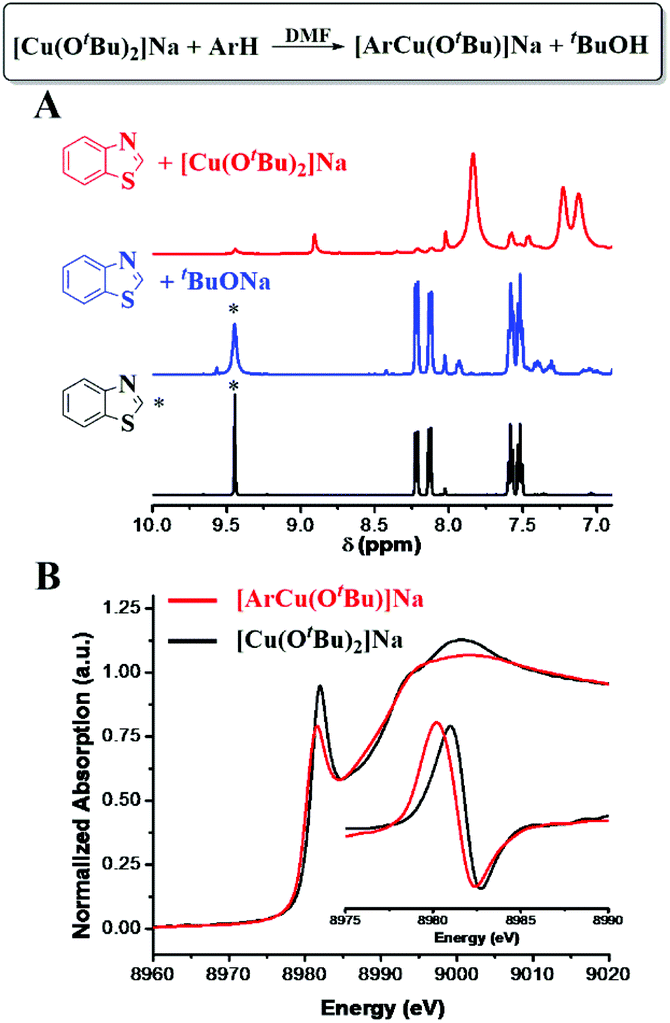
[ArCuI(OtBu)]Na did not form when tBuOCu was mixed with ArH under a N2 atmosphere nor did the latter give the hydroxylation product in air. However, when an additional eq. of tBuONa was added to the latter mixture, the hydroxylation product was obtained in 84% yield in 5 minutes (Table 1). The control experiment of ArH with 1 eq. of tBuONa displayed little deprotonation (∼5% based on the 1H NMR spectra), while under the same conditions, [CuI(OtBu)2]Na deprotonated 95% of the ArH in 5 minutes (Fig. 2A). These results suggested that [CuI(OtBu)2]Na could deprotonate ArH much faster than tBuONa and tBuOCu. It is noteworthy that the deprotonation of ArH by [CuI(OtBu)2]Na was complete within 10 minutes even at −50 °C, corresponding to the rate constant of 3.2 × 10−2 mol−1 L s−1. As shown in Fig. 3A, reaction rates were also measured at various temperatures, and the enthalpy of activation Δ≠rH⊖m was calculated to be 0.73 kcal mol−1. The low activation entropy illustrates that the ate-complex [Cu(OtBu)2]Na promoted C–H bond cleavage is facile. Under the catalytic reaction conditions, tBuONa is in excess, and [CuI(OtBu)2]Na would be the major Cu species. We believe that [CuI(OtBu)2]Na instead of tBuOCu was the active intermediate for the C–H bond cleavage of ArH.
 | ||
| Fig. 3 Kinetics of [CuI(OtBu)2]Na promoted C–H cleavage of benzothiazole. (A) tBuOH formation rate. (B) Arrhenius plot. | ||
|
|
||||
|---|---|---|---|---|
| Entry | [Cu] | t BuONa | t (min) | Isolated yield (%) |
| 1 | CuCl | 2 | 10 | 74 |
| 2 | t BuOCu | 0 | 60 | 0 |
| 3 | t BuOCu | 1 | 5 | 84 |
Different C–H bond cleavage rates were observed in different solvents. A high reaction rate was observed in polar solvents, such as DMF and NMP (N-methyl-2-pyrrolidone), while less polar solvents, such as dioxane and toluene led to lower reaction rates (Fig. 4A), which could possibly be attributed to the stabilization of the ate-complex in polar solvents. As shown in Fig. 4B, lower pKa of the C–H bond (weaker C–H bonds) led to a higher C–H cleavage rate; while higher pKa (stronger C–H bonds) gave a lower rate. Possible coordination of the ArH (such as benzothiazole and thiazole) to Cu through either the formation of Cu–N or Cu–S coordination bonds does not seem to dominantly increase the C–H cleavage rates, since pentafluorobenzene showed highest C–H cleavage rates.
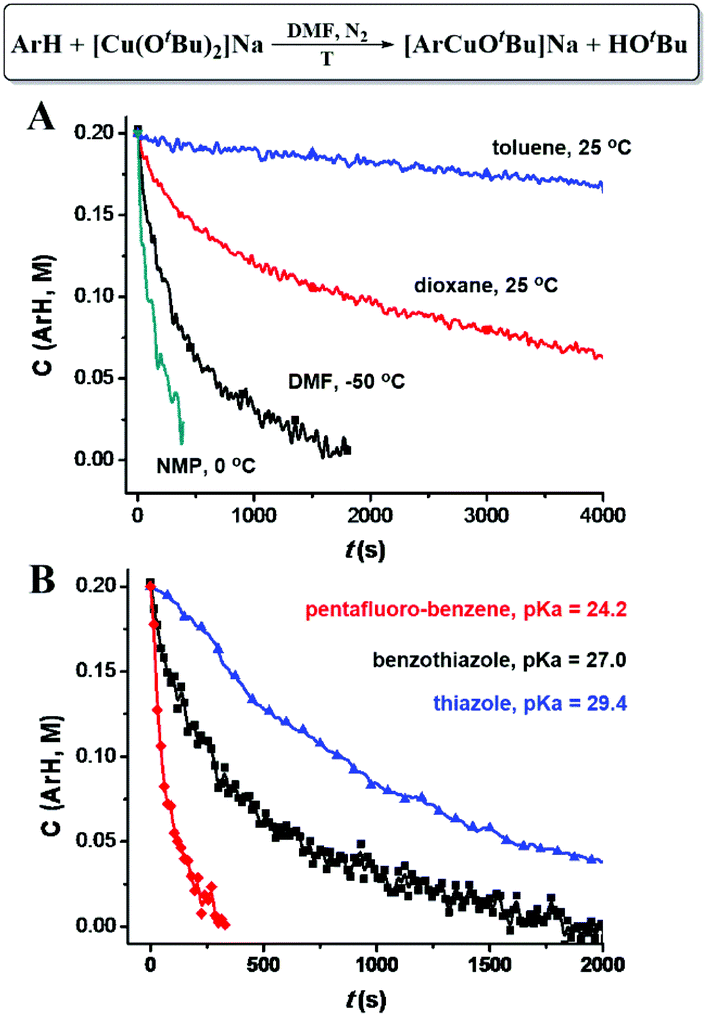 | ||
| Fig. 4 (A) [CuI(OtBu)2]Na promoted C–H bond cleavage of benzothiazole in various solvents. (B) [CuI(OtBu)2]Na promoted C–H cleavage of various aromatic C–H bonds. | ||
Experimental
X-ray absorption measurements were acquired on the insertion device beam line of the Materials Research Collaborative Access Team (MRCAT) at the Advanced Photon Source, Argonne National Laboratory. The monochromator was scanned continuously during the measurements with data points integrated over 0.5 eV for 0.03 s per data point. The ionization chambers were optimized for the maximum current with linear response with 10% absorption (N2) in the incident ion chamber and 70% absorption (60% N2 and 40% Ar) in the transmission detector. A Cu foil spectrum (edge energy 8979 eV) was acquired simultaneously with each measurement for energy calibration. Solution samples were prepared in a glove box and placed in a sample holder made of PEEK (polyether ether ketone) equipped with a screw top and O-ring fitting to prevent exposure to air and water. NMR spectra were recorded on a Bruker Avance spectrometer at 298 K (500 MHz). All samples were prepared in a glovebox using DMF (D7) as the solvent. For the ReactIR kinetic experiments, all spectra were recorded on a Mettler Toledo React IR™ 15 spectrometer using a diamond comb.Conclusions
An ate-complex [CuI(OtBu)2]Na can be generated by the reaction of CuCl with 2 eq. of tBuONa and it readily cleaves the C–H bond of electron-deficient ArH producing [ArCu (OtBu)]Na. The kinetic study suggested that the C–H cleavage rate of benzothiazole was 3.2 × 10−2 mol−1 L s−1 with the enthalpy of activation Δ≠rH⊖m of 0.73 kcal mol−1. More polar solvents and lower pKa of the C–H bonds tend to give a higher C–H cleavage rate.Acknowledgements
This work was supported by the 973 Program (2012CB725302), the National Natural Science Foundation of China (21390400, 21520102003, 21272180 and 21302148), the Hubei Province Natural Science Foundation of China (2013CFA081), the Research Fund for the Doctoral Program of Higher Education of China (20120141130002), and the Ministry of Science and Technology of China (2012YQ120060). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-06CH11357. MRCAT operations are supported by the Department of Energy and the MRCAT member institutions. This work was also funded by the Chemical Sciences and Engineering Division, Argonne National Laboratory.Notes and references
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854–3867, 10.1039/B902868d.
- N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358–2361 CrossRef CAS PubMed.
- A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS.
- K. M. Gligorich and M. S. Sigman, Angew. Chem., Int. Ed., 2006, 45, 6612–6615 CrossRef CAS PubMed.
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed.
- S. S. Stahl, Science, 2005, 309, 1824–1826 CrossRef CAS PubMed.
- P. Gamez, P. G. Aubel, W. L. Driessen and J. Reedijk, Chem. Soc. Rev., 2001, 30, 376–385 RSC.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- E. Nakamura and S. Mori, Angew. Chem., Int. Ed., 2000, 39, 3750–3771 CrossRef CAS PubMed.
- L. A. Zhang, J. H. Cheng, T. Ohishi and Z. M. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670–8673 CrossRef CAS PubMed.
- Y. Li, J. Jin, W. Qian and W. Bao, Org. Biomol. Chem., 2010, 8, 326–330 Search PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed.
- Q. Liu, P. Wu, Y. H. Yang, Z. Q. Zeng, J. Liu, H. Yi and A. W. Lei, Angew. Chem., Int. Ed., 2012, 51, 4666–4670 CrossRef CAS PubMed.
- Y. Wei, H. Q. Zhao, J. Kan, W. P. Su and M. C. Hong, J. Am. Chem. Soc., 2010, 132, 2522–2523 CrossRef CAS PubMed.
- H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 12404–12405 CrossRef CAS PubMed.
- H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2009, 131, 17052–17053 CrossRef CAS PubMed.
- L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2012, 134, 1298–1304 CrossRef CAS PubMed.
- H.-Q. Do and O. Daugulis, Org. Lett., 2009, 11, 421–423 CrossRef CAS PubMed.
- G. Zhang, J. Li, Y. Deng, J. T. Miller, A. J. Kropf, E. E. Bunel and A. W. Lei, Chem. Commun., 2014, 50, 8709–8711 RSC.
- Q. Lu, J. Zhang, P. Peng, G. Zhang, Z. Huang, H. Yi, J. T. Miller and A. Lei, Chem. Sci., 2015, 6, 4851–4854 RSC.
- L. S. Kau, D. J. Spirasolomon, J. E. Pennerhahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- G. Zhang, H. Yi, G. T. Zhang, Y. Deng, R. P. Bai, H. Zhang, J. T. Miller, A. J. Kropf, E. E. Bunel and A. W. Lei, J. Am. Chem. Soc., 2014, 136, 924–926 CrossRef CAS PubMed.
- R. Bai, G. Zhang, H. Yi, Z. Huang, X. Qi, C. Liu, J. T. Miller, A. J. Kropf, E. E. Bunel, Y. Lan and A. Lei, J. Am. Chem. Soc., 2014, 136, 16760–16763 CrossRef CAS PubMed.
- Y. Deng, G. Zhang, X. T. Qi, C. Liu, J. T. Miller, A. J. Kropf, E. E. Bunel, Y. Lan and A. W. Lei, Chem. Commun., 2015, 51, 318–321 RSC.
- H. Yi, G. Zhang, J. Xin, Y. Deng, J. T. Miller, A. J. Kropf, E. E. Bunel, X. Qi, Y. Lan, J.-F. Lee and A. Lei, Chem. Commun., 2016, 52, 6914–6917 RSC.
- A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901–20913 CrossRef CAS PubMed.
- T. Greiser and E. Weiss, Chem. Ber., 1976, 109, 3142–3146 CrossRef CAS.
- R. Restori and D. Schwarzenbach, Acta Crystallogr., Sect. B: Struct. Sci., 1986, 42, 201–208 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, further XAS, NMR and IR spectra. See DOI: 10.1039/c6qo00201c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |