
Dioxygen binding at a four-coordinate cobaltous porphyrin site in a metal–organic framework: structural, EPR, and O2 adsorption analysis†
Audrey T.
Gallagher‡
,
Margaret L.
Kelty‡
,
Jesse G.
Park
,
John S.
Anderson
,
Jarad A.
Mason
,
James P. S.
Walsh
,
Shenell L.
Collins
and
T. David
Harris
*
Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA
First published on 22nd January 2016
Abstract
The study of cobalt porphyrin interactions with O2 is important owing largely to its relevance to biological heme-mediated O2 transport and storage. The immobilization of a metalloporphyrin site in a metal–organic framework (MOF) enables the study of these interactions without interference from solution effects such as bimolecular reactions, axial ligation, and solvent/solute interactions involving the porphyrin ligand. Here, we investigate the reaction of O2 with the four-coordinate cobaltous porphyrin complex in the MOF PCN-224Co. Single-crystal X-ray diffraction and electron paramagnetic resonance (EPR) spectroscopy of the oxygenated form, PCN-224CoO2 reveals a five-coordinate low-spin CoIII center coordinated to an S = 1/2 superoxo (O2−˙) ligand. Moreover, O2 adsorption measurements on PCN-224Co reveal two distinct binding sites with adsorption enthalpies of hads = −15.2(6) and −10.2(3) kJ mol−1. The former binding event is ascribed to ligation of O2 at the open Co site, and the obtained value is considerably lower than those observed for cobalt porphyrin units in substituted proteins and model complexes that feature axial ligands. These results provide the first structurally-characterized five-coordinate Co–O2 species, further highlight the importance of axial ligation in biological O2 transport and storage, and demonstrate the ability of a MOF to enable isolation and study of a species that is highly unstable in molecular form.
 T. David Harris | T. David Harris was born in Poca, West Virginia, in 1981. He received a B.S. in Chemistry from Marshall University in 2004, where he worked in the laboratory of Prof. Michael P. Castellani. He then carried out graduate research with Prof. Jeffrey R. Long at the University of California, Berkeley, where he received his Ph.D. in Chemistry in 2010. Following postdoctoral work with Prof. Theodore A. Betley at Harvard University, he joined the Department of Chemistry faculty at Northwestern University in 2012. His research group is focused on the synthesis of new metal–organic coordination complexes and solids that feature programmable physical and chemical properties, with an emphasis on magnetic molecules and materials. |
Introduction
Dioxygen adducts of molecular metalloporphyrin complexes have generated much interest for decades, owing to their relevance as models of proteins that carry out biological processes such as O2 transport and catalytic oxidation chemistry.1–3 In particular, porphyrin iron, or heme, complexes serve as molecular models of the O2-binding proteins hemoglobin and myoglobin.4,5 In addition to hemes, cobalt porphyrin complexes have also garnered significant interest, owing largely to the fact that the doublet electronic ground state of both their deoxy- and oxy-forms lends itself to electron paramagnetic resonance (EPR) analysis.6–8Studies of molecular metalloporphyrin dioxygen adducts, in particular those involving Fe, have been limited by the propensity of these complexes to undergo deleterious bimolecular condensation reactions to form thermodynamically favored and kinetically inert oxo-bridged dinuclear species.9,10 These challenges have been partially overcome through introduction of bulky substituents onto the porphyrin scaffold in order to block access to one or both axial coordination sites of the metal center. Nevertheless, even in the presence of sterically encumbered porphyrin ligands, an axial ligand such as imidazole is necessary in order to prevent bimolecular condensation or dioxygen dissociation. Consequently, the characterization of five-coordinate, base-free oxyheme11–14 and oxycobalt porphyrin15–21 complexes in molecular form has been largely limited to spectroscopic studies in frozen solvent matrices at low temperature.
We recently reported the post-synthetic metalation of the porphyrinic zirconium MOF PCN-224 with FeII to give a four-coordinate ferrous heme complex within the compound PCN-224FeII.22 Subsequent addition of dry O2 to this species at −78 °C gave a five-coordinate heme dioxygen adduct that was characterized by single-crystal X-ray diffraction and several spectroscopic methods. Moreover, O2 adsorption measurements on activated PCN-224FeII revealed an Fe–O2 binding enthalpy of −34(4) kJ mol−1. This value is nearly half of that commonly observed in ferrous heme model complexes and in myoglobin,2,23–25 and demonstrates the importance of an axial ligand in biological O2 binding. Herein, we extend this work to cobalt by examining the O2 binding of a four-coordinate cobaltous porphyrin within PCN-224 through single-crystal X-ray diffraction, EPR spectroscopy, and O2 adsorption measurements. Specifically, we show that O2 binds the coordinatively unsaturated Co center to give a five-coordinate CoIII superoxo species and that O2 binding at a four-coordinate CoII center is considerably weaker than has been observed in analogues with axial ligands, in line with our previous findings regarding four-coordinate FeII.
Results and discussion
The compound PCN-224Co was reported previously, as synthesized by carrying out the MOF assembly reaction from 5,10,15-20-tetrakis(carboxyphenyl)porphyrin cobalt(II).26 We synthesized PCN-224Co through a different route, by post-synthetic metalation of the free-base porphyrin-containing PCN-224.26 Here, soaking cubic single-crystals of PCN-224 with excess anhydrous CoCl2 in DMF in the presence of excess 2,6-lutidine at 150 °C for 12 h resulted in the insertion of a CoII ion into the porphyrin cavity to give, after activation, the compound PCN-224Co (1). Quantitative Co metalation of this material was confirmed by diffuse-reflectance UV/Visible spectroscopy and trace metals analysis (see Experimental section and Fig. S1†). In addition, an N2 adsorption isotherm collected for 1 at 77 K provided a BET surface area of 3070(70) m2 g−1 (see Fig. S2–S4†). This surface area is similar to other values reported for PCN-224 derivatives,22,26 and therefore confirms that microporosity is retained upon post-synthetic metalation. This result provides another example of post-synthetic metalation of a MOF with cobalt.27–32Upon metalation of PCN-224 with Co, the compound retains its single crystallinity, enabling characterization of 1 by single-crystal X-ray diffraction. The structure of 1 features a four-coordinate CoII center that lies squarely within the N4 plane formed by the four pyrrole nitrogen atoms of the porphyrin ligand, along a crystallographic four-fold rotation axis (see Fig. 1 and S5 and Table S1†). The Co–N distance of 1.936(5) Å is in the range of 1.931–1.944 Å observed for molecular four-coordinate cobalt porphyrin complexes,33 although considerably shorter than the distance of 2.156(1) Å reported in the related MOF Hf-PCN-221(Co).34 No significant residual electron density was present in the difference Fourier map, confirming the absence of ligation in the Co axial coordination sites.
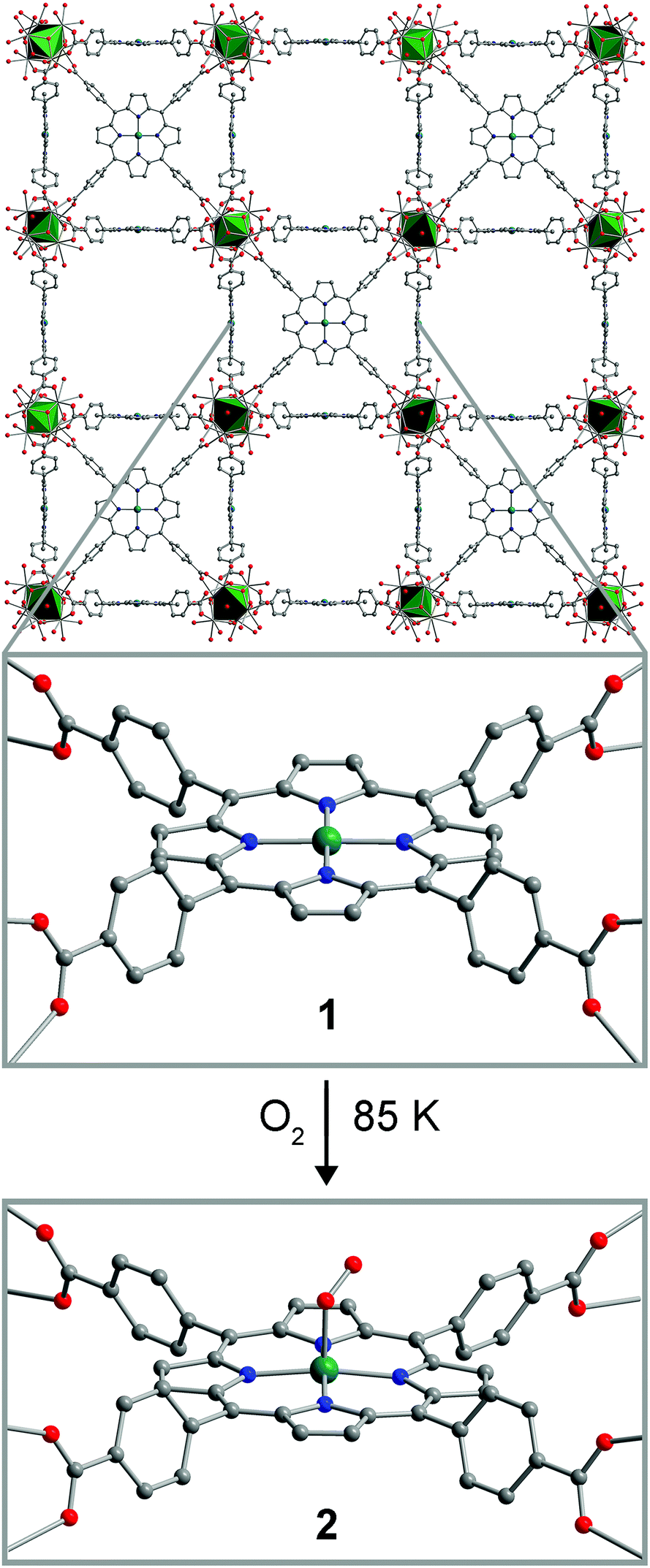 | ||
| Fig. 1 Reaction of PCN-224Co (1) with O2 at 85 K to form PCN-224CoO2 (2). Green octahedra represent Zr atoms; teal, blue, red, and gray spheres represent Co, N, O, and C atoms, respectively; hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (°) for 2: Co–O 1.93(4), Co–N 1.974(5), O–O 1.30(4), Co⋯N4 plane 0.15(4), Co–O–O 121(2). | ||
A thin-walled boron-rich capillary containing a single crystal of PCN-224Co was exposed to 1 atm of dry O2, cooled to 77 K and sealed under reduced pressure. Subsequent X-ray analysis of data collected at 85 K revealed the formation of a new species, PCN-224CoO2 (2). The structure of 2 displays a five-coordinate Co center in a square-pyramidal coordination environment, with the O2 ligand coordinated to the Co center in an η1, end-on binding mode (see Fig. 1, and S5 and Table S2†). The Co–O distance of 1.93(4) Å falls in the range of 1.92–1.93 Å previously reported for six-coordinate molecular Co–O2 adducts, which have been described as CoIII superoxo (O2−˙) species.35,36 The O–O distance of 1.30(4) Å and the Co–O–O angle of 121(2)° are also consistent with previously reported molecular species. Note, however, that these values should be regarded with caution owing to disorder associated with the crystallographic four-fold symmetry at the Co center. The CoIII center is displaced from the mean plane of the four pyrrole nitrogen atoms by 0.15(4) Å, with a corresponding elongated Co–N distance of 1.974(5) Å, and these metrics are consistent with five-coordinate, low-spin CoIII porphyrin species in molecular form.37,38 Notably, the displacement of the Co center is significantly smaller than that previously observed in the analogous MOF-based Fe species, which featured a displacement of 0.526(2) Å. This difference may be attributed to the smaller ionic radius of low-spin CoIII relative to low-spin FeIII.33 To our knowledge, 2 provides the first example of a structurally-characterized Co–O2 adduct with a Co coordination number less than six.
In order to further probe the electronic structure of 1 and 2, continuous-wave X-band EPR spectra were collected on activated crystalline samples at 15 K. In a quartz tube under static vacuum, 1 exhibits an axial spectrum where each feature is split into an eight-line pattern, with this splitting arising due to hyperfine coupling of the unpaired electron to the I = 7/2 59Co nucleus (see Fig. 2, upper). To model these data, spectral simulations were carried out using the program Easyspin39 and the Hamiltonian Ĥ = μBH·g·S + ICo·ACo·S, where μB is the Bohr magneton, H is the applied dc magnetic field, g the g-tensor, S and ICo are the electronic and 59Co nuclear spins, respectively, and ACo is the tensor for hyperfine coupling to the 59Co nucleus. The spectrum was best modeled with values of S = 1/2, g⊥ = 3.271, g‖ = 1.783, A⊥Co = 1122 MHz, and A‖Co = 480 MHz, with the parallel direction taken to be the axis normal to the porphyrin N4 plane. Here, the S = 1/2 ground state and large g⊥/g‖ ratio are consistent with previous reports of four-coordinate CoII porphyrin complexes that were doped into solid-state diamagnetic matrices to prevent axial ligation.18–21 Moreover, the deviation of g⊥ from the free electron value of 2.0023 in cobalt porphyrin complexes is directly correlated to the axial perturbation of the Co center along the parallel direction,3,40–43 and the large deviation observed here for 1 corroborates the four-coordinate environment of the Co center as indicated by X-ray crystallography.
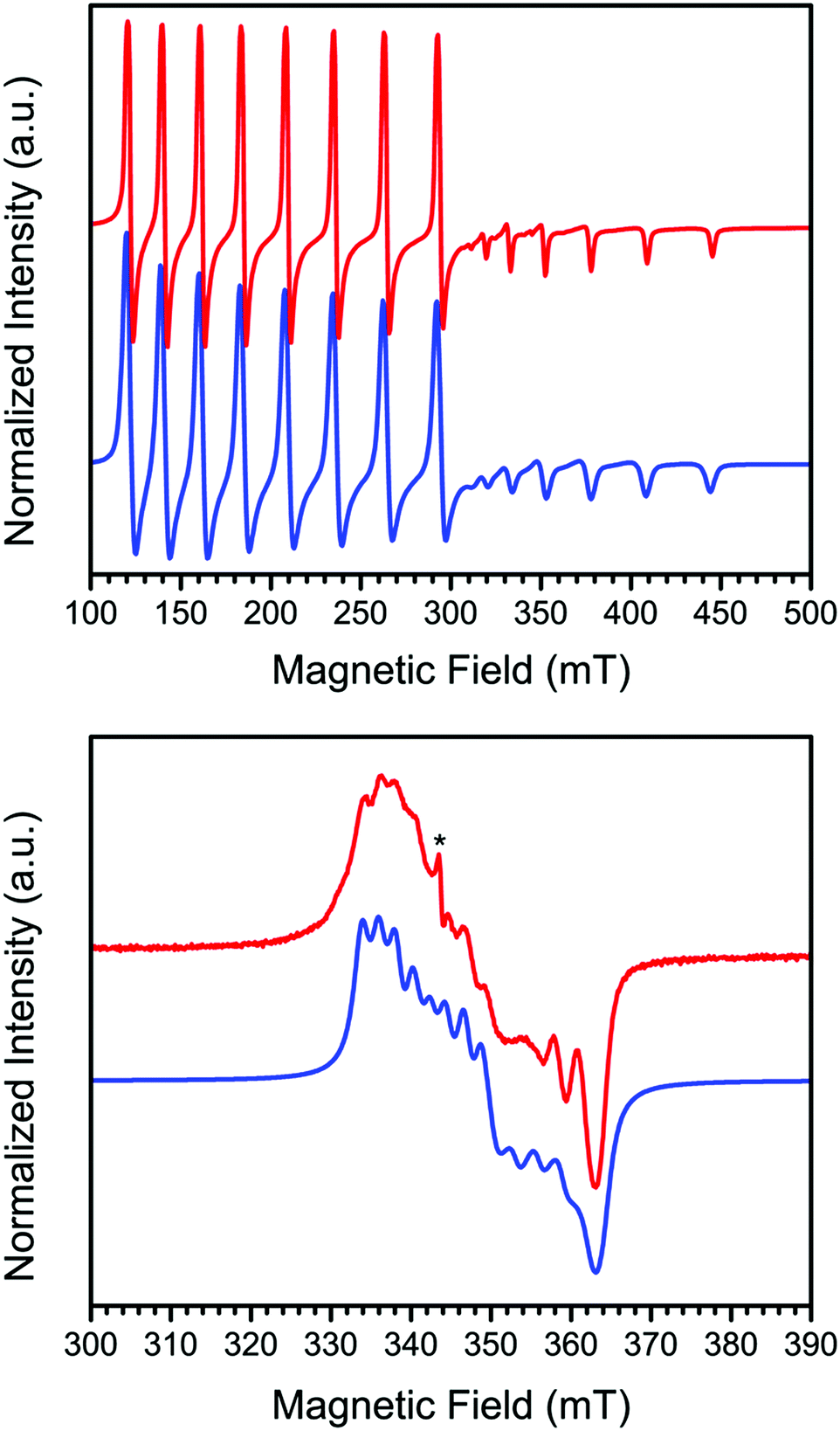 | ||
| Fig. 2 X-Band EPR spectra for 1, collected at 15 K, under static vacuum (upper) and dosed with 1 atm O2 at ambient temperature (lower). Red and blue lines correspond to experimental data and simulations, respectively, and the asterisk denotes a small amount of S = 1/2 impurity with g = 2.00. Microwave frequency = 9.632 GHz; microwave power = 6.31 mW. | ||
The lower panel of Fig. 2 shows the spectrum of the oxygenated derivative 2, formed by dosing a sample of 1 with 1 atm of O2 at ambient temperature followed by cooling to 15 K, which exhibits a rhombic spectrum with a significantly decreased degree of hyperfine coupling. The spectral features exist over a much smaller field range than in 1, indicative of reduced g-anisotropy. Simulating the spectrum according to the spin Hamiltonian given above provides the following parameters: S = 1/2, gx = 2.016, gy = 1.973, gz = 1.900, AxCo = 60 MHz, AyCo = 80 MHz, and AzCo = 0 MHz. The rhombicity of this spectrum is consistent with a bent Co–O–O angle, which precludes a symmetry rotation axis.
The significant decrease of hyperfine coupling to the Co nuclear spin suggests that the unpaired electron of the dioxygen adduct resides primarily on the oxy ligand. Accordingly, 2 is best described as containing the species CoIII–O2−˙, in other words a low-spin CoIII center bound by an S = 1/2 superoxo ligand. Indeed, this electronic structure is consistent with previous studies of dioxygen adducts of molecular porphyrin Co complexes that feature axial ligands at the Co center or solute and/or solvent molecules that engage in π interactions with the porphyrin ligand.44 More specifically, one of the two singly occupied π* orbitals of O2 forms a σ bond with the Co dz2 orbital to give a doubly occupied molecular orbital, while the other remains a singly occupied and non-bonding orbital that is predominantly an oxygen p orbital in character.7,45
In order to examine the thermodynamics of O2 binding in PCN-224Co, O2 adsorption data were collected at selected temperatures. As depicted in Fig. 3 and S6,† the O2 isotherm for 1 collected at 113 K exhibits an initial steep uptake at low pressure. As temperature is increased, the slope of this steep region decreases until the isotherm becomes nearly linear at 195 K. In order to quantitate the O2 binding, isotherm data at temperatures of 113, 141, 156, and 195 K were each fit to a dual-site Langmuir–Freundlich model (see Table S3†).22,46 Subsequent treatment of the variable-temperature isothermal data with the Clausius–Clapeyron equation revealed a differential enthalpy of adsorption of hads = −15.2(6) kJ mol−1 at low O2 loading, followed by a gradual drop near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 O2/Co to a plateau at hads = −10.2(3) kJ mol−1. We respectively assign these distinct values to O2 binding at the four-coordinate CoII center and physisorption to the remainder of the MOF surface. The adsorption enthalpy of −15.2(6) kJ mol−1 is slightly lower than that of −17.8 kJ mol−1 reported for O2 binding at a five-coordinate (μ4-O)CoII tetracarboxylate unit in PCN-9.47
:1 O2/Co to a plateau at hads = −10.2(3) kJ mol−1. We respectively assign these distinct values to O2 binding at the four-coordinate CoII center and physisorption to the remainder of the MOF surface. The adsorption enthalpy of −15.2(6) kJ mol−1 is slightly lower than that of −17.8 kJ mol−1 reported for O2 binding at a five-coordinate (μ4-O)CoII tetracarboxylate unit in PCN-9.47
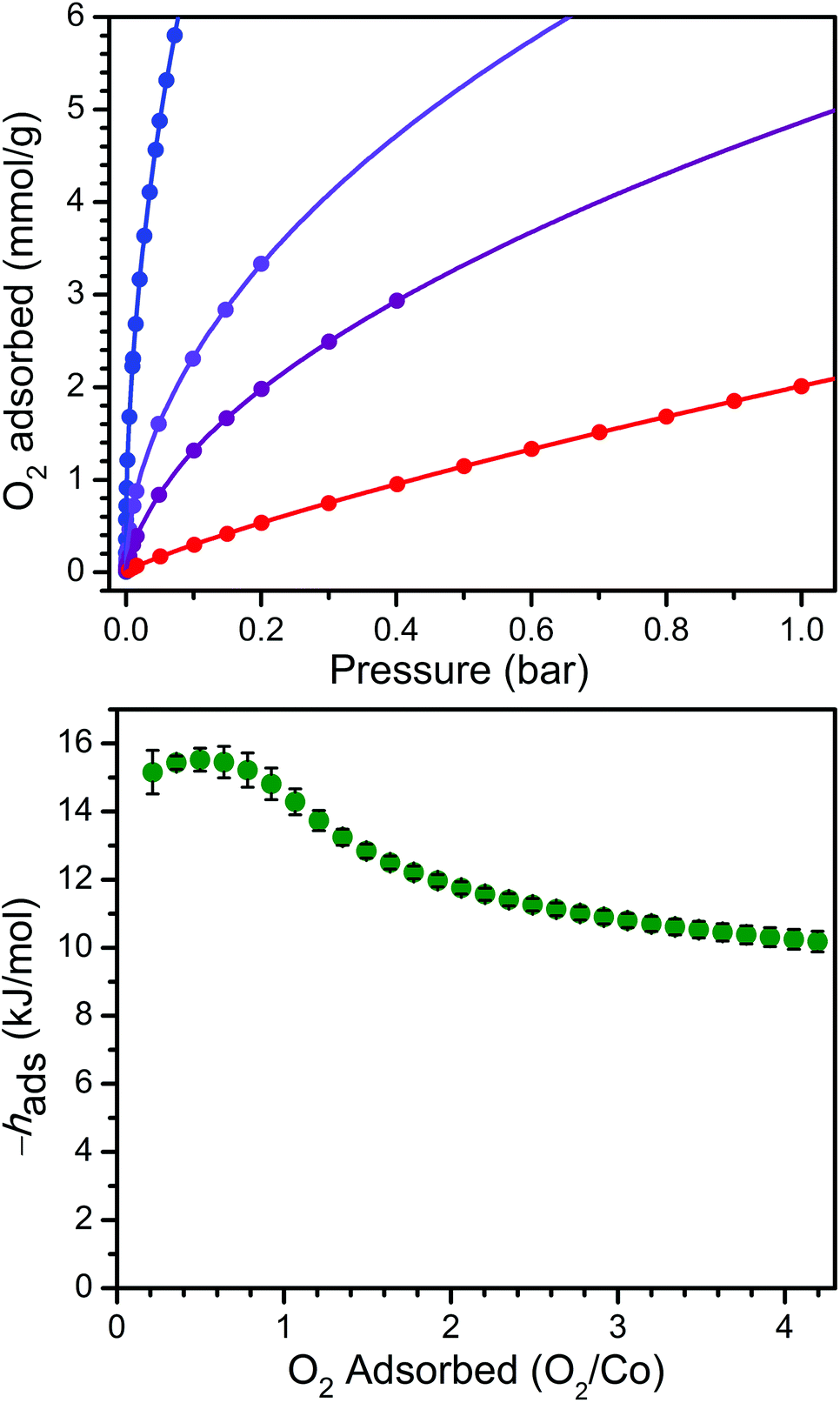 | ||
| Fig. 3 Upper: O2 adsorption data for 1 at 113, 141, 156, and 195 K (blue to red gradient). Circles represent data, and solid lines correspond to fits using a dual-site Langmuir–Freundlich model. Lower: O2 differential enthalpy of adsorption curve for 1 as a function of amount adsorbed. Green circles represent data, and error bars are shown in black. | ||
The Co–O2 binding enthalpy of hads = −15.2(6) kJ mol−1 at low coverage is considerably weaker than values previously reported for cobalt porphyrins that feature axial ligands, both in Co-substituted globin proteins and in molecular model complexes.48–59 These values range from −33 kJ mol−1 for a 1-methylimidazole-bound capped Co complex56 to −68 kJ mol−1 for cobalt octaethylporphyrin supported on a highly oriented pyrolytic graphite (HOPG) surface, where the HOPG acts as an axial ligand.59 Indeed, the binding enthalpy of hads = −15.2(6) kJ mol−1 observed for 1 + O2 falls in the range of 22–46% of these values. This difference is similar but even more pronounced than that observed for the O2 binding of the four-coordinate heme in PCN-224FeII, which was approximately half of the values commonly observed for hemes with axial ligands,22 and further underscores the importance of axial ligand electron donation to the metal center to enable O2 transport and storage.
Acknowledgements
This research was funded by the U. S. Army Research Office through agreement numbers W911NF-14-1-0168, W911NF-15-1-0119, and W911NF-15-1-0331, the U. S. Air Force Office of Scientific Research through agreement number FA9550-14-1-0274, the Institute for Sustainability and Energy at Northwestern, and Northwestern University. A.T.G. is supported primarily by the National Science Foundation through the Graduate Research Fellowship Program. M.L.K. is supported primarily by the Chemistry of Life Processes through a Lambert Fellowship. J.A.M. is supported by the Northwestern University International Institute for Nanotechnology. S.L.C. was supported primarily by the National Science Foundation under NSF Award Number EEC-1359004. We thank Prof. B.M. Hoffman for a helpful discussion.References
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841 CrossRef PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919 CrossRef CAS PubMed.
- J. P. Collman, Acc. Chem. Res., 1977, 10, 265 CrossRef CAS.
- B. Giardina, I. Messana, R. Scatena and M. Castagnola, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 165 CrossRef PubMed.
- B. M. Hoffman and D. H. Petering, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 637 CrossRef.
- T. D. Smith and J. R. Pilbrow, Coord. Chem. Rev., 1981, 39, 295 CrossRef.
- R. D. Jones, D. A. Summerville and F. Basolo, Chem. Rev., 1979, 79, 139 CrossRef CAS.
- A. B. Hoffman, D. M. Collins, V. W. Day, E. B. Fleischer, T. S. Srivastava and J. L. Hoard, J. Am. Chem. Soc., 1972, 94, 3620 CrossRef CAS PubMed.
- D.-H. Chin, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1980, 102, 4344 CrossRef CAS.
- L. Latos-Grazynski, R. J. Cheng, G. N. La Mar and A. L. Balch, J. Am. Chem. Soc., 1982, 104, 5992 CrossRef CAS.
- K. Nakamoto, T. Watanabe, T. Ama and M. W. Urban, J. Am. Chem. Soc., 1982, 104, 3744 CrossRef CAS.
- K. Nakamoto, Coord. Chem. Rev., 1990, 100, 363 CrossRef CAS.
- L. M. Proniewicz, I. R. Paeng and K. Nakamoto, J. Am. Chem. Soc., 1991, 113, 3294 CrossRef CAS.
- M. Kozuka and K. Nakamoto, J. Am. Chem. Soc., 1981, 103, 2162 CrossRef CAS.
- A. Wesełucha-Birczyńska, K. Nakamoto and L. M. Proniewicz, J. Mol. Struct., 1992, 275, 95 CrossRef.
- L. M. Proniewicz, A. Kulczycki, A. Wesełucha-Birczyńska, H. Majcherczyk and K. Nakamoto, New J. Chem., 1999, 23, 71 RSC.
- J. M. Assour, J. Chem. Phys., 1965, 43, 2477 CrossRef CAS.
- F. A. Walker, J. Am. Chem. Soc., 1970, 92, 4235 CrossRef CAS.
- S. Van Doorslaer and A. Schweiger, Phys. Chem. Chem. Phys., 2001, 3, 159 RSC.
- A. Ozarowski, H. M. Lee and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 12606 CrossRef CAS PubMed.
- J. S. Anderson, A. T. Gallagher, J. A. Mason and T. D. Harris, J. Am. Chem. Soc., 2014, 136, 16489 CrossRef CAS PubMed.
- K. S. Suslick and T. J. Reinert, J. Chem. Educ., 1985, 62, 974 CrossRef CAS.
- M. Momenteau and C. A. Reed, Chem. Rev., 1994, 94, 659 CrossRef CAS.
- J. P. Collman and L. Fu, Acc. Chem. Res., 1999, 32, 455 CrossRef CAS.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105 CrossRef CAS PubMed.
- A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 13252 CrossRef CAS PubMed.
- X.-S. Wang, M. Chrzanowski, L. Wojitas, Y.-S. Chen and S. Ma, Chem. – Eur. J., 2013, 19, 3297 CrossRef CAS PubMed.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906 CrossRef CAS PubMed.
- K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182 CrossRef CAS PubMed.
- C.-W. Kung, T.-H. Chang, L.-Y. Chou, J. T. Hupp, O. K. Farha and K.-C. Ho, Chem. Commun., 2015, 51, 2414 RSC.
- M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Inorg. Chem., 2015, 54, 2995 CrossRef CAS PubMed.
- According to a search of the Cambridge Structural Database Version 5.35.0 CrossRef; F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661 CrossRef CAS PubMed.
- J. Li, B. C. Noll, A. G. Oliver and W. R. Scheidt, J. Am. Chem. Soc., 2012, 134, 10595 CrossRef CAS PubMed.
- P. Doppelt, J. Fischer, L. Ricard and R. Weiss, New J. Chem., 1987, 11, 357 CAS.
- T. Sakurai, K. Yamamoto, J. Naito and N. Nakamoto, Bull. Chem. Soc. Jpn., 1976, 49, 3042 CrossRef CAS.
- J. Li, B. C. Noll, A. G. Oliver, G. Ferraudi, A. G. Lappin and W. R. Scheidt, Inorg. Chem., 2010, 49, 2398 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- J. S. Griffith, Discuss. Faraday Soc., 1958, 26, 81 RSC.
- A. H. Maki, N. Edelstein, A. Davidson and R. H. Holm, J. Am. Chem. Soc., 1964, 86, 4580 CrossRef CAS.
- L. M. Engelhardt and M. Green, J. Chem. Soc., Dalton Trans., 1972, 4724 Search PubMed.
- B. R. McGarvey, Can. J. Chem., 1975, 53, 2498 CrossRef CAS.
- F. A. Walker, J. Magn. Reson., 1974, 15, 201 CAS.
- R. S. Drago and B. B. Corden, Acc. Chem. Res., 1980, 13, 353 CrossRef CAS.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 11734 CrossRef CAS PubMed.
- H. C. Stynes and J. A. Ibers, J. Am. Chem. Soc., 1972, 94, 1559 CrossRef CAS PubMed.
- H. C. Stynes and J. A. Ibers, J. Am. Chem. Soc., 1972, 94, 5125 CrossRef CAS PubMed.
- C. A. Spilburg, B. M. Hoffman and D. H. Petering, J. Biol. Chem., 1972, 247, 4219 CAS.
- F. A. Walker, J. Am. Chem. Soc., 1973, 95, 1154 CrossRef CAS PubMed.
- D. V. Stynes, H. C. Stynes, B. R. James and J. A. Ibers, J. Am. Chem. Soc., 1973, 95, 1796 CrossRef CAS PubMed.
- T. J. Beugelsdijk and R. S. Drago, J. Am. Chem. Soc., 1975, 97, 6466 CrossRef CAS PubMed.
- J. P. Collman, J. I. Brauman, K. M. Doxsee, T. R. Halbert, S. E. Hayes and K. S. Suslick, J. Am. Chem. Soc., 1978, 100, 2761 CrossRef CAS.
- M.-Y. R. Wang, B. M. Hoffman, S. J. Shire and F. R. N. Gurd, J. Am. Chem. Soc., 1979, 101, 7394 CrossRef CAS.
- J. E. Linard, P. E. Ellis Jr., J. R. Budge, R. D. Jones and F. Basolo, J. Am. Chem. Soc., 1980, 102, 1896 CrossRef CAS.
- B. Steiger, J. S. Baskin, F. C. Anson and A. H. Zewail, Angew. Chem., Int. Ed., 2000, 39, 257 CrossRef CAS.
- S. Zou, J. S. Baskin and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9625 CrossRef CAS PubMed.
- B. A. Friesen, A. Bhattarai, U. Mazur and K. W. Hipps, J. Am. Chem. Soc., 2012, 134, 14897 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, additional spectroscopic and crystallographic data. Crystallographic information of 1 and 2. CCDC 1439664 and 1447556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00275c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |