
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of bis(arylimino)acenaphthene (BIAN) copper complexes as visible light harvesters for potential photovoltaic applications†
J. W.
Kee
a,
Y. Y.
Ng
a,
S. A.
Kulkarni
b,
S. K.
Muduli
ab,
K.
Xu
a,
R.
Ganguly
a,
Y.
Lu
*a,
H.
Hirao
*a and
H. S.
Soo
*acd
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371. E-mail: yplu@ntu.edu.sg; hirao@ntu.edu.sg; hansen@ntu.edu.sg
bEnergy Research Institute@NTU (ERI@N), Nanyang Technological University, Research Techno Plaza, Singapore 637553
cSingapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE), 1 Create Way, Singapore 138602
dSolar Fuels Laboratory, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798
First published on 8th February 2016
Abstract
Photovoltaics with dye-sensitized solar cells have been recognized as being promising for the utilization of sunlight to produce electricity and ‘solar chemicals’. One of the remaining unsolved challenges is the development of an affordable, robust dye that has a panchromatic light harvesting range and efficiently provides separated charges for the desired photochemistry. The most commonly employed molecular photosensitizers include the noble metal-based ruthenium and iridium complexes, synthetically challenging porphyrin derivatives, and expensive, functionalized polypyridine compounds. Here, we describe the development of Cu(I) dyes supported by bis(arylimino)acenaphthene (Ar-BIAN) ligands, which can be synthesized in fewer than three steps from affordable, commercially available reagents. The diamagnetic, homoleptic complexes have been characterized by a suite of spectroscopic and analytical methods and exhibit panchromatic light absorption extending to the near infrared (NIR) region. Remarkably, the crystal structure of a complex bearing an ortho-iodoarylimino substituent displays a unique, rhombically distorted square planar geometry around the Cu(I) center, for crystals isolated from two disparate solvent combinations. Density functional theory (DFT) calculations were performed to provide insights into the spectroscopic features and the unusual coordination sphere around the metal center, and allude to non-covalent interactions between the aromatic groups and among the iodide atoms. Preliminary studies were conducted to explore the application of these copper photosensitizers in dye-sensitized solar cells.
 H. S. Soo | Han Sen Soo graduated from the Massachusetts Institute of Technology (MIT) with S.B. and S.M. degrees in 2003 under the tutelage of Prof. Christopher C. Cummins. He subsequently joined the research group of Prof. Christopher J. Chang at the University of California, Berkeley, working on small molecule activation using earth-abundant elements. After completing his Ph.D. studies in 2009, Han Sen journeyed up the hill to become a post-doctoral fellow with Dr Heinz Frei in the Helios Solar Energy Research Center (SERC). He employed X-ray absorption and time-resolved optical and FT-IR spectroscopic methods to study organic–nanomaterial hybrid systems applied in artificial photosynthesis. Han Sen embarked on his independent career as a Nanyang Assistant Professor in the Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, at Nanyang Technological University in 2012. The overarching theme of his research program is the creation of artificial photosynthetic systems. His current research interests include the development of molecular photosensitizers and catalysts to harvest sunlight for CO2 and proton reduction to produce solar fuels and chemicals. His team is also preparing photocatalysts and mesoporous materials derived from earth-abundant elements to valorize biomass lignin and degrade environmental pollutants. |
Introduction
There has been growing interest in the use of photoredox catalysis as a sustainable means to mediate organic chemical transformations by harvesting visible light as a source of energy.1–13 Numerous seminal studies have been documented recently for novel, chemoselective, photodriven, and mild routes to C–C bond formation.1–15 However, the adopted photocatalysts have been predominantly expensive Ru- and Ir-based photosensitizers,1–8 although there have been sporadic reports on the employment of organic or Cu(I) dyes.9–13 Likewise, for dye-sensitized solar cells (DSSCs) and the nascent field of dye-sensitized photoelectrosynthesis cells (DSPECs), functionalized Ru(II) polypyridyl photosensitizers have remained the workhorse for several decades, despite the high cost and low abundance of Ru.16–19 Cognizant of this issue, our team has been exploring the use of (photo)catalysts comprising earth-abundant elements for C–C activation,20,21 bioinspired oxidation,22,23 pollutant degradation,24 and proton reduction reactions, in an effort to create artificial photosynthetic units.25–30 Inspired by the paucity of research on photosensitizers containing earth-abundant first-row transition metals,9–13,31–39 we sought to develop new Cu(I) visible light harvesters.Similar to the ubiquitous Ru(II) dyes, Cu(I) photosensitizers with polypyridine ligands in the form of 2,2′-bipyridine and phenanthroline derivatives have received the most attention.9,34,36,40–46 Cu(I) polypyridyl complexes have d10 electronic configurations and possess comparable photophysical characteristics as Ru(II) chromophores since they are not plagued by as many non-radiative relaxation losses through low-energy d–d transitions that are common among first row transition metals. By using suitable π-acceptor ligands such as pyridines, Cu(I) compounds can absorb visible light through metal-to-ligand charge transfer (MLCT) processes.9,34,36,40–46 However, a transient Cu(I) to Cu(II) conversion typically results in significant Jahn–Teller (J–T) distortions,34,36,38,40 unlike the minimal reorganization after a Ru(II) to Ru(III) transition, which can lead to undesired non-radiative losses. Although some of these Cu(I) polypyridyl complexes have notably long lifetimes, the more successful, bulky polypyridyl ligands are notoriously expensive, require demanding multi-step syntheses, and typically yield Cu(I) complexes that do not absorb much red and longer wavelength irradiation.9,34,36,40–46 In contrast, bis(arylimino)-acenaphthene (Ar-BIAN) ligands are highly modular and readily accessible by facile condensation reactions between commercially available and affordable substituted anilines and acenaphthenequinone.39,47–54 Ar-BIAN ligands have been explored and reviewed in the context of transition metal and main-group molecular compounds.39,47–54 They have been established as redox non-innocent ligands with low-lying π* orbitals that can behave as ‘capacitors’ for multi-electron reductions.55–57 Moreover, Ar-BIAN ligands are acknowledged as robust scaffolds for catalysis and even photovoltaic devices.51–53,58 Critically, pioneering work on Cu(I) Ar-BIAN complexes verified that judicious selection of the arylimino component or an appropriate co-ligand can generate panchromatic Cu(I) dyes, which can absorb in the NIR region.39,50–54
In this paper, we present the synthesis, experimental characterization, and DFT studies on new panchromatic Ar-BIAN Cu(I) compounds. Heavier halogens have been introduced on the ligand periphery to harness the heavy-atom effect in our efforts to prolong the lifetime of the MLCT photoexcited state.39 In anticipation of potential applications in DSSCs and DSPECs, we have installed sulfonate59 and ester groups60 on the ligands to serve as anchoring groups and charge conduits into semiconductors. We have tested these photosensitizers in DSSCs and intend to use the insights from the DFT calculations to develop the next generation of Cu(I) compounds that will perform as light harvesters in artificial photosynthetic and photoredox units as well. In particular, the observation of an unusually flat coordination sphere of Cu(I) may have interesting implications on the photochemistry as discussed previously by Iwamura et al.36
Results and discussion
Design and synthesis of ligands
The synthetic steps for the two substituted Ar-BIAN ligands are illustrated in Scheme 1 and detailed characterization data can be found in the ESI.† Briefly, template-free methods, namely the acid-catalyzed synthesis61 of Na(ArSO3-Br-BIAN) (1) and the TiCl4-promoted condensation62 leading to ArI,COOMe-BIAN (2), were adapted to give moderate yields. We decided against the more popular ZnCl2-templated route to avoid complications with hydrolysis and purification issues during the extraction of zinc with sodium oxalate.47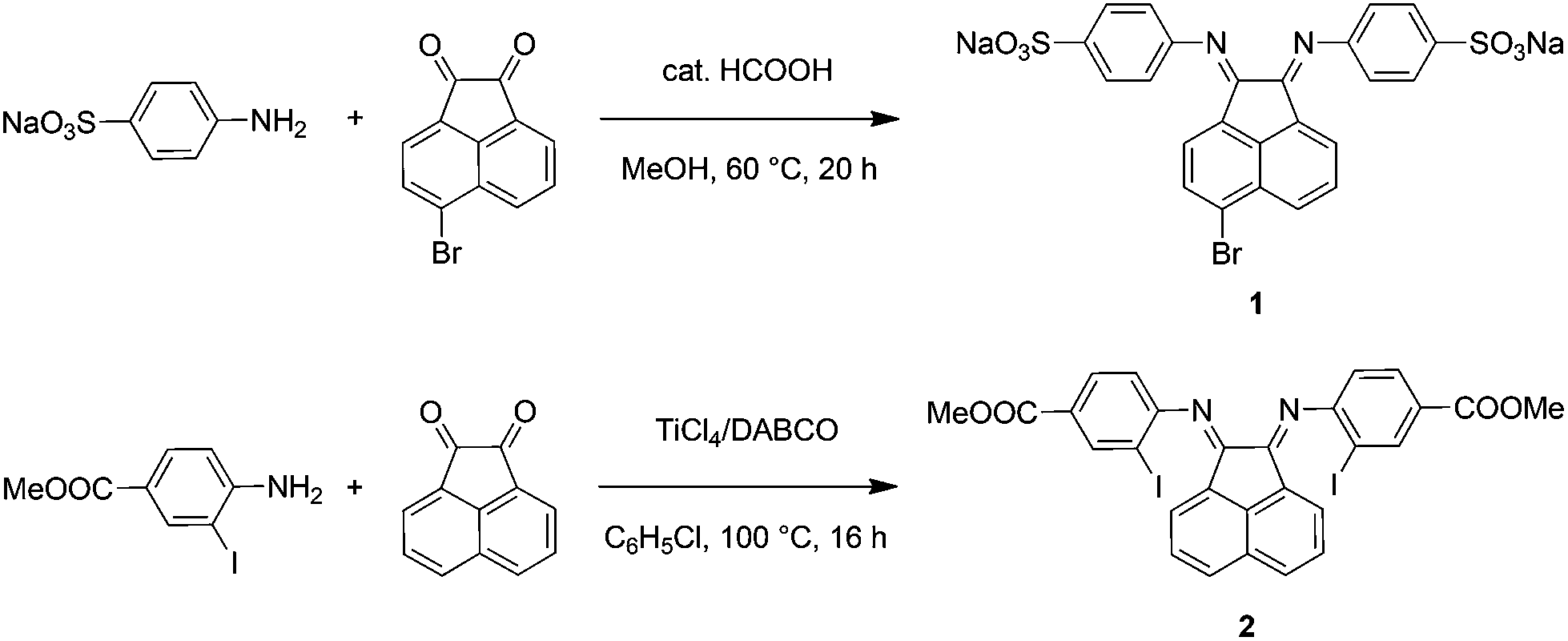 | ||
| Scheme 1 Synthetic routes for ligands 1 and 2. | ||
Both the (E,E)- and (E,Z)-isomers for 2 (Fig. 1) were present in the CDCl3 solution in a ratio of around 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 respectively, as suggested by the broad peaks in the 1H NMR spectrum of 2 at room temperature. The broadening is likely due to the thermal exchange between the two isomers under ambient conditions. Variable temperature (VT) NMR experiments at low (Fig. S1, ESI†) and high (Fig. S2, ESI†) temperatures were conducted to obtain thermodynamic parameters of this isomeric equilibrium of 2. This isomerism has been previously reported by Gasperini et al.47 The solid-state structure was obtained to confirm the identity of 2 with single crystals grown from methanol/dichloromethane (DCM) and the (E,Z)-isomer of 2 was isolated (Fig. 1). Notably, the (E,Z)-isomerism does not impede the coordination of Cu(I), since we have been able to isolate and characterize the Cu(I) complexes bound to the thermally more stable (E,E) form (Fig. S1 and S2, ESI†) in high yields, presumably due to facile isomerization between the two isomers at room temperature.
:1 respectively, as suggested by the broad peaks in the 1H NMR spectrum of 2 at room temperature. The broadening is likely due to the thermal exchange between the two isomers under ambient conditions. Variable temperature (VT) NMR experiments at low (Fig. S1, ESI†) and high (Fig. S2, ESI†) temperatures were conducted to obtain thermodynamic parameters of this isomeric equilibrium of 2. This isomerism has been previously reported by Gasperini et al.47 The solid-state structure was obtained to confirm the identity of 2 with single crystals grown from methanol/dichloromethane (DCM) and the (E,Z)-isomer of 2 was isolated (Fig. 1). Notably, the (E,Z)-isomerism does not impede the coordination of Cu(I), since we have been able to isolate and characterize the Cu(I) complexes bound to the thermally more stable (E,E) form (Fig. S1 and S2, ESI†) in high yields, presumably due to facile isomerization between the two isomers at room temperature.
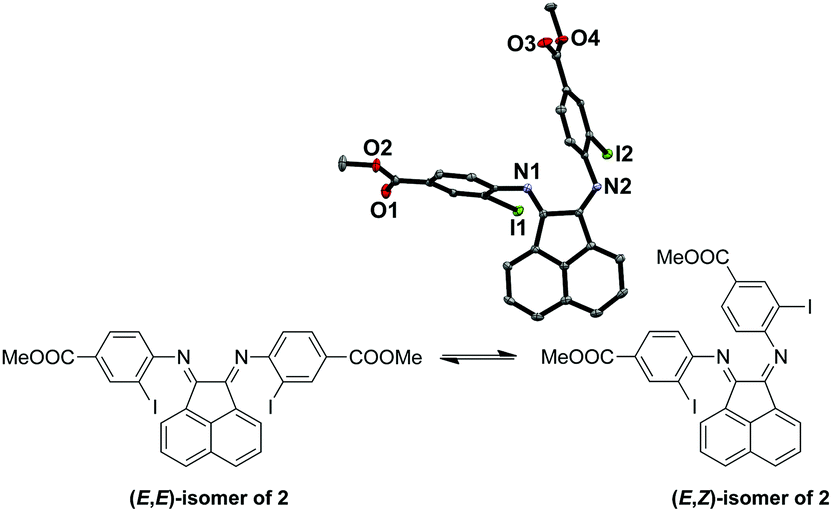 | ||
| Fig. 1 (E,E)- and (E,Z)-isomeric forms of 2. | ||
Preparation of Cu(I) complexes
Upon mixing two equivalents of 1 with CuCl in dimethyl sulfoxide (DMSO), a dark blue-colored solution formed within 1 h. The solution was filtered and the product was isolated by precipitation from the filtrate by adding acetonitrile (CH3CN). The residue was purified by recrystallization from DMSO/CH3CN to obtain Na3[(ArSO3-Br-BIAN)2Cu] (3). We did not succeed in growing single crystals of 3 to confirm the structure. However, the 1H and 13C NMR spectra, as well as the electrospray ionization-mass spectrum (ESI-MS) were consistent with the assignment of 3 as a homoleptic cuprate complex supported by two units of the ligand 1.With the more soluble 2, the complex [(ArI,COOMe-BIAN)2Cu]PF6 (4) could be prepared by using another Cu(I) precursor, namely [Cu(CH3CN)4]PF6, in tetrahydrofuran (THF) and the product was precipitated with pentane as a dark green material. Through recrystallization from THF/toluene, dark green crystals suitable for X-ray structural analyses were obtained (Fig. 2). Complex 4 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Tables S11 and S16, ESI†).
(Tables S11 and S16, ESI†).
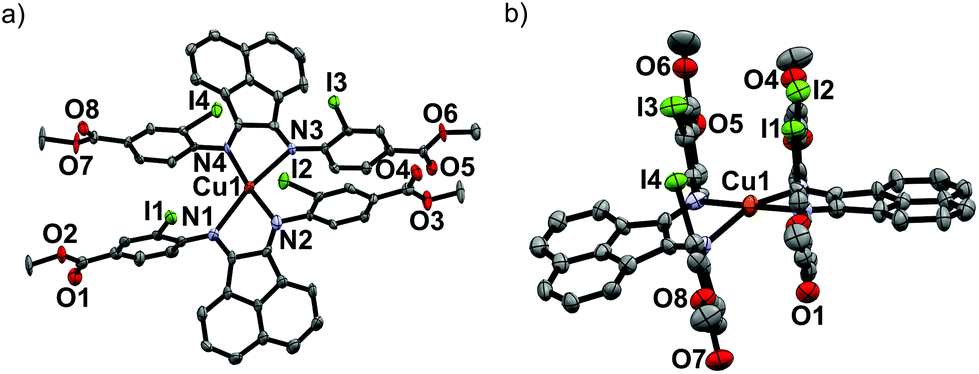 | ||
| Fig. 2 Oak Ridge Thermal Ellipsoid Plots (ORTEPs) from single crystal X-ray diffraction experiments of 4. Top (a) and side views (b) of crystals grown from THF/toluene. The PF6-counteranion and the calculated hydrogen atoms are omitted for clarity. | ||
The crystal structure reveals the expected Cu(I) cation chelated by two molecules of the ligand 2, with a PF6− counteranion. Intriguingly, 4 displays a rhombically distorted square planar coordination sphere (Fig. 2a) around the Cu(I) center. This is in stark contrast to the typical distorted tetrahedral geometry of Cu(I) Ar-BIAN complexes reported previously.51,63 One measure of the deviation from a tetrahedral geometry about Cu(I) is the dihedral angle51 between the two five-membered chelate rings and this was found to be 21.2(3)° in 4, distinct from those reported previously (55.3–89.4°).51,63 When crystals of 4 were grown out of DCM/diethyl ether (Et2O), the coordination sphere still approximates a distorted square planar geometry (Fig. S5a and S5b, ESI†), albeit with a larger dihedral angle of 32.0(3)°. Other salient parameters are summarized in Table 1.
| Parameter | THF/toluene | DCM/Et2O |
|---|---|---|
| Cu1–N1 | 1.938(11) | 1.907(9) |
| Cu1–N2 | 2.463(11) | 2.460(9) |
| Cu1–N3 | 1.931(11) | 1.892(7) |
| Cu1–N4 | 2.517(13) | 2.645(9) |
| I1–I2 | 4.354(2) | 4.984(2) |
| I2–I3 | 4.276(2) | 4.857(2) |
| I3–I4 | 4.621(2) | 5.144(2) |
| I1–I4 | 4.309(2) | 4.524(2) |
| I1–I3 | 4.829(2) | 4.110(1) |
| N1–Cu1–N2 | 76.5(4) | 76.1(4) |
| N3–Cu1–N4 | 75.8(4) | 74.6(3) |
| Dihedral angle between chelate rings | 21.2(3) | 32.0(3) |
| Dihedral angles between acenaphthene and o-iodoaryl rings | 89.5(4), 84.9(3), 82.6(3), 89.7(4) | 71.2(3), 79.9(3), 82.7(3), 87.9(3) |
| o-Iodoaryl centroid–centroid distances | 3.615, 3.622 | 3.719, 4.381 |
In addition, dramatic asymmetry in the way each unit of 2 binds to Cu(I) is evident regardless of the crystal growth solvent combination employed. For the sake of clarity, the discussions below correspond to the crystals grown out of THF/toluene, unless stated otherwise. The structures we present are drastically different from the more symmetric homoleptic Cu(I) Ar-BIAN complexes in prior publications.
We observe that the Cu–N bond lengths are not equivalent for each ligand. For example, the Cu–N bond distances are 1.931(11) and 2.517(13) Å for one of the ligands, while the other distances are 1.938(11) Å and 2.463(11) Å. In the previously documented homoleptic Cu(I) Ar-BIAN complexes, the typical Cu–N bond lengths are in a narrower range of 1.98–2.06 Å.51,63 Evidently, for each coordinated 2, one nitrogen atom has an almost expected value, whereas the second nitrogen atom is only weakly coordinated. The coordination geometry surrounding the Cu(I) can arguably be considered as being linear. A survey of complexes with Cu–N bond distances longer than 2.40 Å suggests that the majority involved Cu(II) centers with axially lengthened octahedral geometries due to a first-order Jahn–Teller effect. Six examples of Cu(I) complexes with at least one Cu–N bond length above 2.40 Å were found in the Cambridge Structural Database (CSD), all of which feature neutral amine or heterocycle donors.64–69 Out of these instances, one consists of a distorted octahedral Cu(I),68 two possess trigonal monopyramidal Cu centers with long axial bonds,65,67 while the remaining two have been described as distorted square planar or sawhorse geometries.64,66 In particular, the bis(amino-oxazoline) Cu(I) complex reported by Doherty et al. has exceptionally long Cu–N bonds of 2.640(5) and 2.838(5) Å, which have been aptly considered as weak interactions between the Cu and aniline ligands.66 Since the sum of the van der Waals radii of Cu(I) and N is 2.90 Å, 4 joins a class of rare Cu(I) complexes containing weak Cu–N interactions.66
The pendant o-iodoaryl rings appear to be almost perpendicular to the acenaphthene ring, and the dihedral angles among the two contiguous aryl rings varied between 71.2(3) and 89.7(4)°. This is unlike most of the known homoleptic Cu(I) Ar-BIAN complexes. Furthermore, the four ortho-iodides on the two ligands are in close proximity with one another on the same face of 4, with distances between 4.276 and 4.621 Å. The intriguing steric pocket surrounding the Cu(I) is most patently observed in the space-filling model (Fig. S5, ESI†). This alludes to some non-covalent electronic interactions that may favor this unusual arrangement among these iodide atoms or between the iodides and Cu, since the geometry is mostly independent of the crystal growth solvent. Steric effects are unlikely to be the sole reason because crystal structures of Cu(I) compounds with (o-isopropyl)aryl-BIAN ligands have shown tetrahedral coordination spheres, and the isopropyl groups orientate themselves to minimize steric hindrance by being far apart.51
Another factor that might affect the orientation of the o-iodoaryl rings, and hence the overall coordination sphere of Cu(I), is the π–π interactions between the aryl rings (Fig. 2; Fig. S5, ESI†). The o-iodoaryl moieties within 4 are aligned parallel to each other with offset centroid–centroid distances of 3.615 and 3.622 Å between the aryl groups, for the crystal grown from THF/toluene. However, longer centroid–centroid distances (3.719 and 4.381 Å) were observed in the crystal grown out of DCM/Et2O, which is inconsistent with a dominant π–π interaction between the aryl rings to account for the remarkably flat coordination geometry around the Cu(I) center. The structural data suggests that novel electronic or steric reasons originating from the ortho-iodides are contributing to the unusual coordination geometry of 4, and the effect does not solely arise from crystal-packing forces in disparate solvents. In combination with the exceptionally long Cu–N bond distances in the complex, 4 possesses a truly unique rhombically distorted square planar Cu(I) center. To explain this unusual phenomenon, and also several remarkable UV-visible features, we sought insights from DFT calculations (vide infra). Additional metrical parameters and information about these crystal structures are described in the ESI.†
In addition, we attempted to isolate heteroleptic compounds containing 1 and 2 as ligands to explore the photophysical and electrochemical effects of mixed ligand systems. However, owing to a combination of the lability of Cu(I) complexes and the poor solubility of the homoleptic complexes, the homoleptic products typically precipitated out instead. Current efforts are directed towards developing ligands with improved solubilities in organic solvents to enable us to investigate Cu(I) compounds with mixed Ar-BIAN ligand systems.
Electrochemical measurements
Cyclic voltammetric techniques were employed to probe the redox behavior of the Ar-BIAN ligands and the complexes in DMSO/N,N-dimethylformamide (DMF) 1:1 (v:v), containing 0.10 M tetrabutylammonium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (n-Bu4NBArF4) as the supporting electrolyte. Compounds 1 and 3 were unable to dissolve in non-coordinating solvents such as 1,2-difluorobenzene or CH2Cl2. All potentials (Table 2) reported and discussed herein are referenced against the ferrocenium ion/ferrocene redox couple (Fc+/Fc).
| Compound | E oxp/Eredp (V vs. Fc+/Fc) | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| a E oxp and Eredp represent the oxidation and reduction peak potentials respectively and are reported in V versus Fc+/Fc. b An additional oxidation wave at −0.86 V is associated with this ligand reduction. c An additional oxidation wave at −0.72 V is associated with this ligand reduction. d An additional oxidation wave at −0.02 V is associated with this ligand reduction. | ||||
| Ligand oxidations | +0.38/— | — | — | — |
| +0.71/— | ||||
| Ligand reductions | −1.20/−1.68 | −1.36/−1.41b | −1.08/−1.16c | −0.62/−0.96 |
| —/−2.24 | −2.18/−2.27 | −1.37/−1.46 | —/−1.21 | |
| −2.35/−2.40 | −1.59/−1.84 | −1.30/−1.37d | ||
| —/−2.05 | −2.10/−2.28 | |||
| −2.34/−2.39 | ||||
| CuI oxidation | — | — | −0.08/−0.38 | −0.33/−0.42 |
A survey of the electrochemistry reported for similar Ar-BIAN ligands revealed that at least two reductions (−1.78 to −1.93 V and −2.37 to −2.55 V) and two oxidations (+0.22 to +0.97 V and +1.07 to +1.82 V) were commonly observed.47,48,53 The electrochemical behavior of 1 in DMF/DMSO consists of a series of irreversible reductions and oxidations. The cathodic scan of 1 reveals two irreversible reduction waves at −1.68 V and at −2.24 V (Fig. S6a, ESI†). When an anodic scan was performed, two irreversible oxidations appeared at +0.38 and +0.71 V and upon the reverse cathodic scan up to −2.6 V, an additional reduction wave at −0.76 V appeared (Fig. S6b, ESI†), indicating that significant structural change or decomposition occurred during the oxidation of 1. However, this additional reduction wave may also be due to unavoidable solvent oxidation by-products, due to the limited solvent choices for the ionic 1.
The electrochemistry of 2 in DMF/DMSO can be compared to that reported for the structurally similar p-(methylester)aryl-BIAN.48 When the cathodic scan of 2 was performed, three irreversible reduction waves appeared at −1.41, −2.27 and −2.40 V before the onset of solvent reduction (Fig. S7a, ESI†). The first wave at −1.41 V appears to be a two-electron reduction process and has two oxidation waves at −0.86 and −1.36 V associated with it. In addition, these three waves are similar to that reported for p-(methylester)aryl-BIAN (−1.68 and −2.23 V). Within the electrochemical window accessible for the DMF/DMSO combination (+0.8 to −2.6 V) before solvent redox events occur, we were unable to access the electrochemical oxidation potentials of 2.
The electrochemical oxidation is accessible when the cyclic voltammetry is conducted in THF instead. An irreversible oxidation wave was observed at +0.94 V before the oxidation of the THF solvent at higher potentials (Fig. S7d, ESI†). This potential is slightly higher than that reported for p-(methylester)aryl-BIAN (+0.82 V) and could be due to the electron-withdrawing effect of the ortho-iodides, thus raising the oxidation potential slightly. An additional reduction wave at −2.36 V was observed in the return cathodic scan (Fig. S7d, ESI†) suggesting that oxidation of 2 in THF results in significant chemical transformations and changes to the electrochemical properties of 2. In addition, the cathodic scan in THF (Fig. S7c, ESI†) also reveals a more negative potential of −2.86 V for the second reduction instead. It is likely that this electrochemical reduction is complicated by the reductive de-iodination of the iodoaryl moiety, since it is close to the reported reduction potential of −2.64 V for iodobenzene.70
The Cu(I) complexes exhibit even more complicated voltammograms than the ligands, with additional reduction and oxidation processes owing to the presence of the Cu(I) center.53 For 3, the anodic scan revealed two overlapping oxidation peaks before the onset of solvent oxidation (Fig. 3a). The redox wave was quasi-reversible at −0.08 V with a cathodic return wave at −0.38 V (Fig. 3a). These potentials are similar to those previously reported for Cu(I) Ar-BIAN homoleptic complexes and have been ascribed to Cu(I)/Cu(II) oxidations that involve donation from the iminyl moiety of the Ar-BIAN ligands.53
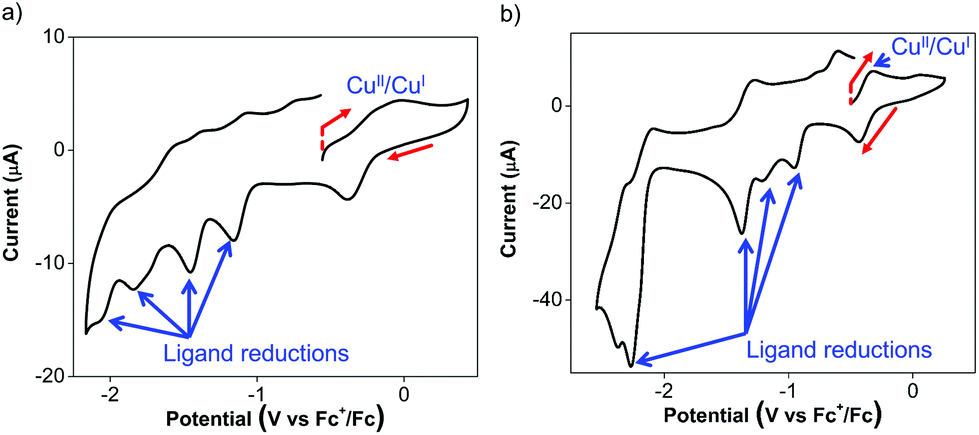 | ||
| Fig. 3 Cyclic voltammograms of (a) 1.0 mM of 3 and (b) 1.0 mM of 4 in DMF/DMSO 1:1 (v:v), with 0.10 M n-Bu4NBArF4 as the electrolyte, each at a scan rate of 100 mV s−1 with its scan direction and initial potential indicated by the red arrows and red vertical dashed line respectively. The data are reported relative to Fc+/Fc. | ||
During the cathodic scans of 3, two quasi-reversible reduction waves at −1.16 and −1.46 V, and two irreversible reduction waves at −1.84, and −2.05 V were observed (Fig. S8a, ESI†). The electrochemical behavior in the cathodic scan in a smaller potential window (Fig. S8b, ESI†) is identical to that observed during the anodic scan, suggesting that the Cu(II)/Cu(I) redox process probably does not result in ligand dissociation or decomposition of the complex. Instead, the quasi-reversible behavior of the Cu(II)/Cu(I) process may be due to structural reorganization or reversible, transient solvent coordination by DMSO or DMF. The reduction potentials for 3 are slightly less negative than those observed in the ligand 1. This could be due to the stabilization by distribution of electron density between both Ar-BIAN ligands within the complex. On the other hand, the heteroleptic Cu(I) Ar-BIAN phosphane complexes reported previously display only irreversible reduction processes, since the anionic charge is concentrated on only one ligand.47
Similarly, the cyclic voltammetry of 4 in DMF/DMSO is more complicated than that of its ligands while still displaying electrochemical behavior associated with ligand reduction processes. The anodic scan of 4 in DMF/DMSO (Fig. 3b) exhibits a quasi-reversible oxidation at −0.33 V, which is significantly lower than that for 2 and other previously reported Cu(I) Ar-BIAN homoleptic complexes.53 The Cu(I)/Cu(II) oxidation process could have become more accessible as a result of stabilization from solvent coordination on the Cu(I) metal center, as facilitated by the rhombically distorted square planar geometry. It is especially noteworthy that the Cu(I) complex did not suffer from ligand dissociation issues, since its purple color remained upon dissolution in DMF/DMSO. This is in contrast to the lability of 4 in CH3CN, which led to the dissociation of 2 in solution, as suggested by UV-visible spectroscopic measurements (vide infra).
The cathodic scan of 4 in DMF/DMSO yields three ligand-centered reduction waves (Fig. S9a, ESI†) at −1.37, −2.28 and −2.39 V as evident by their similarities in potentials to those observed during the reduction of 2. Additional irreversible reduction waves at −0.96 and −1.21 V are likely due to ligand reductive processes that involve stabilization by electron density distribution between Cu and the two ligands, similar to that found in 3. As a result of the reduction process at −1.37 V, the oxidation peak current at −0.33 V appears to be diminished and a new anodic wave was observed at −0.02 V (Fig. S9b, ESI†). This could be due to dissociation of Cu upon excessive ligand reduction, which results in solvated Cu ions being subsequently oxidized.
On the other hand the anodic scan of 4 in the relatively less coordinating THF exhibits two irreversible oxidations at +0.26 and +0.78 V (Fig. S9c, ESI†). The first oxidation likely corresponds to the Cu(I)/Cu(II) oxidation process, while the second oxidation can be assigned to the ligand oxidation, due to its comparable oxidation potential as 2 in THF. The cathodic scan of 4 in THF produces two quasi-reversible reduction waves at −0.98 and −1.63 V (Fig. S9d, ESI†), which is distinct from the electrochemical behavior in DMF/DMSO. In addition, the reduction wave at −1.63 V appears to be a two-electron reduction process, with double the current drawn compared to the reduction at −0.98 V. This solvent-dependence in the electrochemical behavior is likely due to the stronger coordination by DMF and DMSO than THF.
Spectroscopic features of Cu(I) photosensitizers
UV-visible spectroscopy was employed to study the absorption characteristics of 1–4 and the results are summarized in Table 3. Details about the fitted parameters can be found in the ESI.† The UV-visible spectra of ligands 1 and 2 (Fig. 4a) reveal strong absorption in the UV region and a weaker, broad band that extends to about 550 nm. These UV-visible absorptions have been previously attributed to π–π* transitions while the visible bands could be assigned to iodide n–π* transitions or intraligand π–π* charge transfer (ILCT) from the aryl rings to the naphthyl backbone.63 The visible absorption peaks are similar to those observed for Ar-BIAN with meta substituents63 and suggest that their effect on the π system of the acenaphthene ring may not be too significant.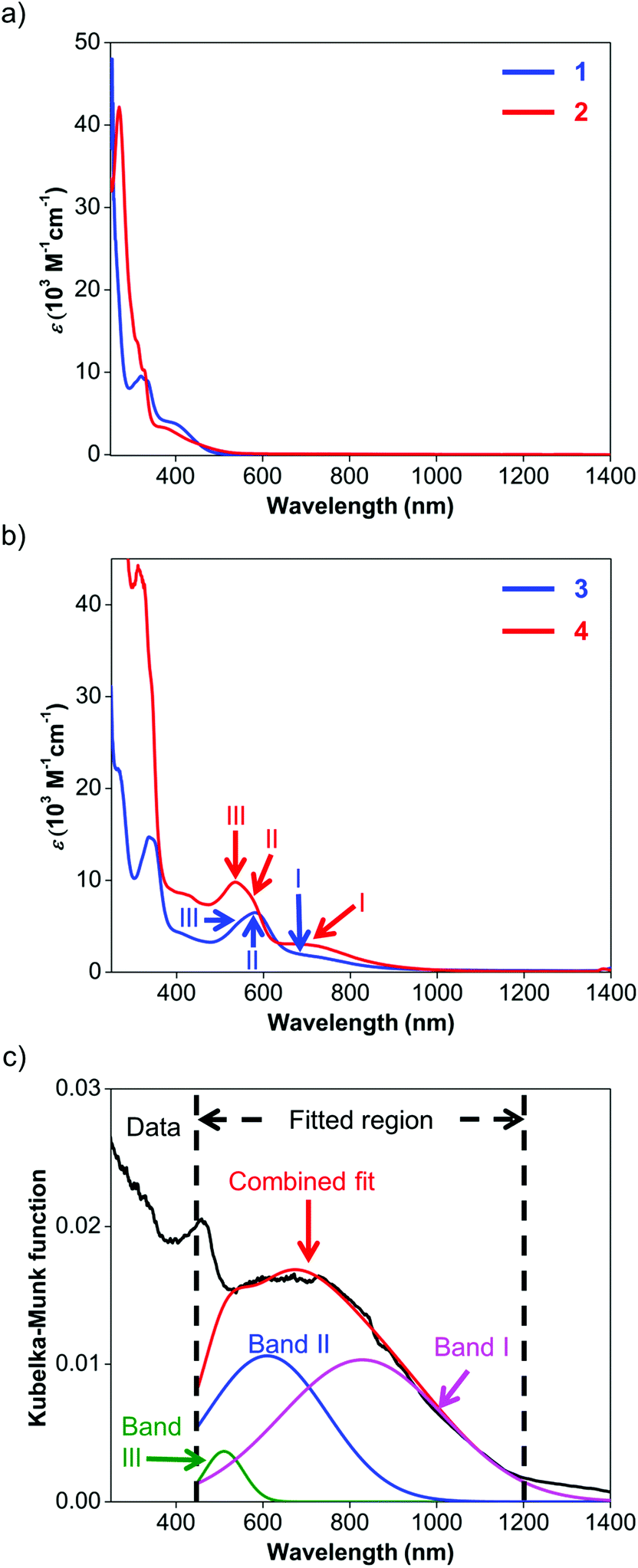 | ||
| Fig. 4 (a) UV-visible spectra of 1 (blue) and 2 (red) in methanol and DCM respectively. (b) UV-visible spectra of 3 (blue) and 4 (red) in methanol and DCM respectively. Roman numerals and arrows indicate the wavelength for the various fitted absorption bands of 3 (blue) and 4 (red). (c) Diffuse reflectance spectrum of 4 with fits for bands I (pink), II (blue), and III (green). The black line corresponds to the experimental data, while the red line is the superposition of the fitted bands. | ||
| Compound | Band | Wavelength (nm) | ε (M−1 cm−1) |
ε ratio of bands I:II |
|---|---|---|---|---|
| 1 a | — | 367 | 4190 | |
| 2 b | — | 364 | 3070 | |
| 3 a | III | 529 | 2970 | 0.59 |
| II | 581 | 2930 | ||
| I | 686 | 1750 | ||
| 4 b | III | 510 | 7730 | 0.57 |
| II | 569 | 5340 | ||
| I | 700 | 3040 |
Similarly, the spectroscopic features for 3 and 4 are characterized by similar ILCT transitions in the UV region, but exhibit additional MLCT bands in the visible region (Fig. 4b). The molar extinction coefficients (ε) of the bands in the UV regions of 3 and 4 are similar to their corresponding transitions in 1 and 2 respectively, as observed for Cu(I) Ar-BIAN previously reported.63 However, as established for Cu(I) phenanthroline complexes,35,71 the MLCT bands can provide information about the degree of distortion in the ground-state geometry from a tetrahedral coordination environment towards a flatter rhombically distorted square planar geometry. Three bands are typically observed. Band I is a D2d-symmetry forbidden low-energy band above 700 nm, corresponding to the lowest energy MLCT transition to the S1 state. The main part of the spectrum consists of band II, which has been attributed to MLCT to the S3 excited state. As a result, band I typically has low extinction coefficients and the ratios of band I to band II are lower for structures with ideal tetrahedral Cu(I) centers. Band III is an absorption between 500 and 600 nm, often enveloped within band II, which corresponds to excitation to higher MLCT singlet states.71 The intensity ratios of band I to band II for both complexes 3 and 4 in solution are similar to those reported previously (Fig. 4b and Table 3).53 This observation suggests that in solution, 3 and 4 adopt slightly distorted tetrahedral geometries. However, the solution colors of 3 (dark green) and 4 (dark purple) differ significantly from the colors in the solid state (dark blue for 3; dark green for 4, vide supra). This prompted us to investigate the solid-state absorption characteristics through diffuse reflectance UV-visible spectroscopy (DRS). These measurements would also confirm the X-ray crystal structural features and provide information about the dyes when they are immobilized in DSSCs.
As anticipated, the DRS spectra (Fig. 4c; Fig. S11 and S12, ESI†) differ significantly from the solution UV-visible spectra. In general, the MLCT bands from 500 nm and longer wavelengths bathochromically shift into the NIR region. For 3, the two bands of the solution spectra split into the three distinct bands III, II and I (Fig. S11, ESI†) at 493, 602, and 869 nm respectively, and yielded a ratio of 1.2 for the fitted Kubelka–Munk functions. Similarly, the absorption bands of 4 converge into a broad band centered around 660 nm in the DRS spectrum (Fig. 4c; Fig. S12, ESI†). This broad absorption could be deconvoluted into the constituent bands III, II and I at 510 (green), 610 (blue), and 829 (pink) nm respectively, giving a ratio of 0.97 for the fitted Kubelka–Munk functions (Table S3, ESI†). These results concur with the severely distorted geometry in the X-ray structures of 4 (Fig. 2; Fig. S5, ESI†), which leads to absorptions in the NIR region.
In addition, ligand dissociation has been discussed as one of the possible degradation pathways for Cu(I) complexes in coordinating solvents,43 and 4 behaved similarly. When 4 was dissolved in CH3CN, the UV-visible spectrum obtained was devoid of the MLCT bands, whereas only the absorption bands due to the constituent ligand 2 were observed instead. We propose that 4 is labile in the presence of small, coordinating solvents, which can be visually detected by a change in solution color from an intensely dark purple (in DCM) to yellow (in CH3CN). In combination with the solvent dependence during the electrochemical measurements, the UV-visible data suggest that judicious selection of the solvents will be necessary for applications of 4.
DFT calculations
To further our understanding of the unusual structural features of 4 and its observed UV-visible spectral behavior, a series of DFT calculations have been conducted. First, screening of various DFT methods and basis sets were performed to find the best agreement, for both the Cu–N bond lengths and the dihedral angles between the two chelated rings, among the optimized and solid-state structures (Table S21, ESI†). Some of the frontier orbitals for the structure of 4 optimized in toluene as the solvent are illustrated in Fig. 5.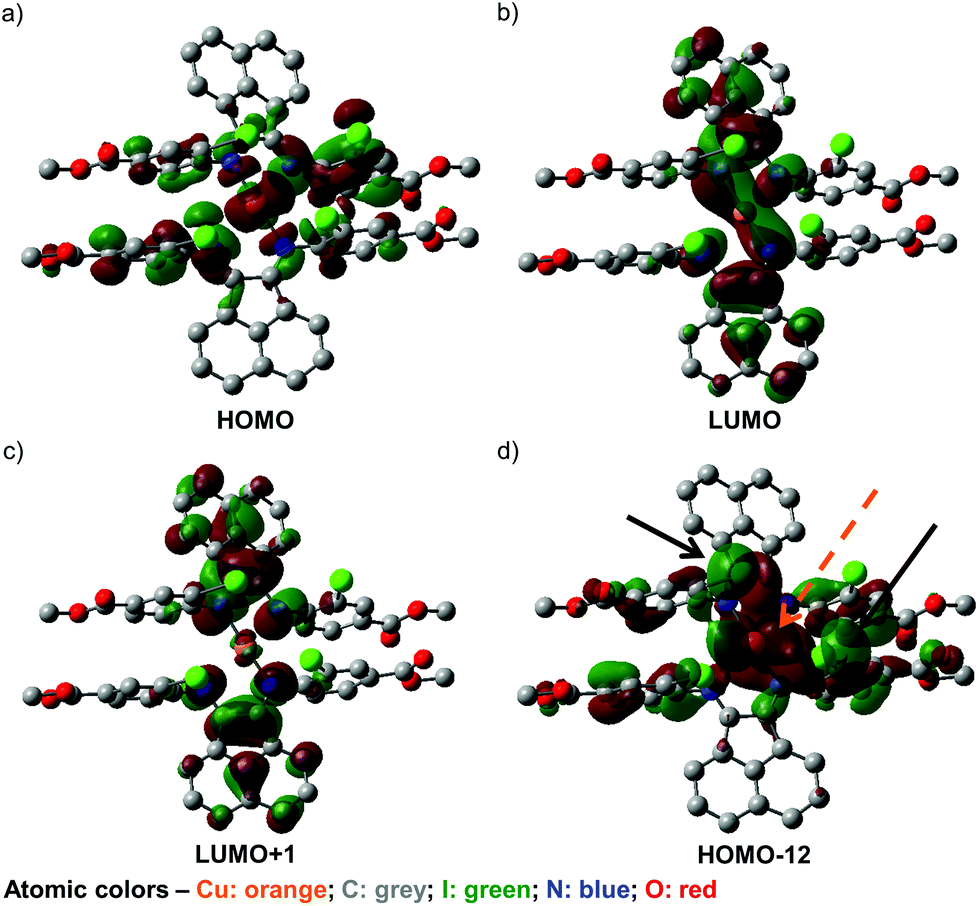 | ||
| Fig. 5 (a) HOMO, (b) LUMO, and (c) LUMO+1 of geometry-optimized 4 in toluene, set at isovalues of 0.02, and (d) HOMO−12 set at an isovalue of 0.01. The orange arrows in (d) indicate the d orbital lobe contributions from Cu(I), while the black arrows point to the p-orbital contributions from two of the iodides in 2. | ||
As anticipated, the HOMO (Fig. 5a) appears to consist predominantly of a d orbital from the Cu(I) center, with spin density distributed over the iminoaryl parts of ligand 2, and almost no contribution from the acenaphthene fragment. In contrast, the LUMO (Fig. 5b) and LUMO+1 (Fig. 5c) are mainly comprised of orbital components from the acenaphthene fragments. In fact, inter-ligand lobes connecting both the imino moieties of the ligands can be observed in the LUMO, alluding to ligand-to-ligand charge transfer characteristics in 4. Most intriguingly, we identified a low energy orbital (HOMO−12) that consists of overlapping lobes between two opposite iodide p orbitals and a copper d orbital (Fig. 5d), despite the long copper–iodide distances (3.904(2) and 4.051(2) Å for crystals grown from THF/toluene; and 3.481(2) and 4.273(2) Å for crystals grown from DCM/Et2O), as determined from the crystal structures. We propose that these weak copper–iodide interactions, some iodide–iodide interactions, and the π–π stacking between the iminoaryl motifs in Fig. 5d are responsible for the unusual geometrical arrangement of the ligands surrounding the Cu(I) center.
Non-covalent interactions within the molecule were further analysed by the Atoms-in-Molecules (AIM) approach.72,73 As shown in Fig. 6, the AIM analysis yielded several bond critical points (BCPs) between the aromatic rings of the ligands, indicative of the presence of π–π stacking effects. Furthermore, BCPs are also seen between the iodine atoms, which imply the presence of halogen–halogen interactions, even though the arrangement of iodine atoms does not appear to strictly fulfill the geometric criteria set by Desiraju.74 We therefore propose that these combined non-covalent effects facilitate the establishment of the unique coordination sphere around the metal center observed in this compound. We also performed an analysis using the non-covalent interactions (NCI) index.75,76 The NCI analysis also implicates the involvement of these non-covalent interactions (Fig. S26†).
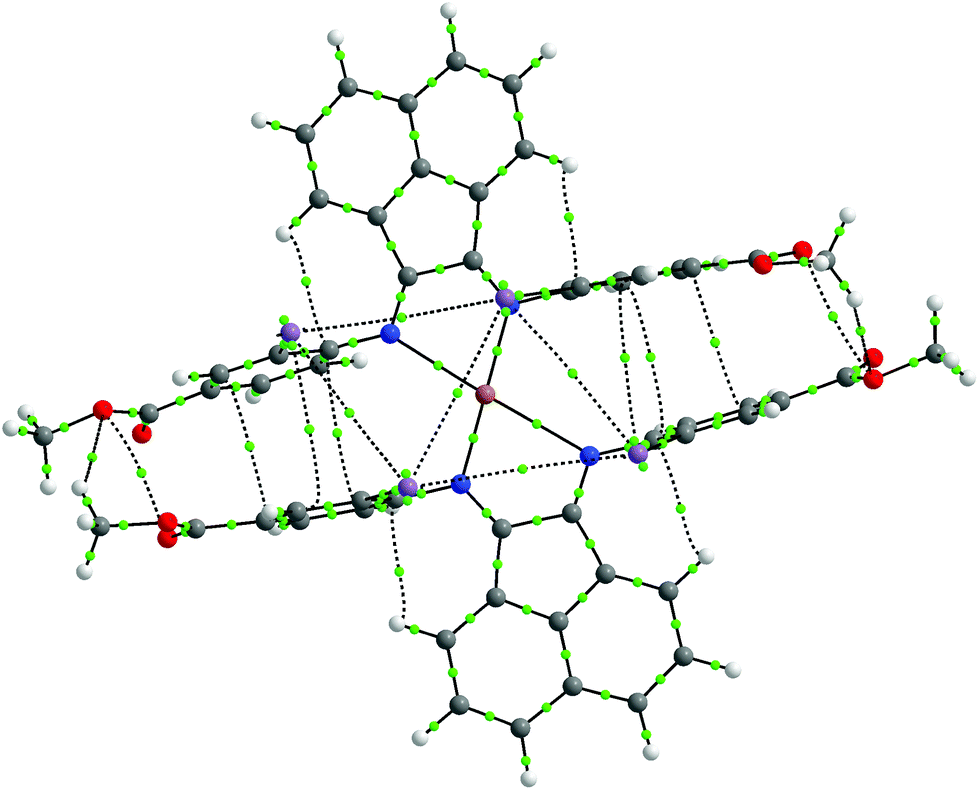 | ||
| Fig. 6 AIM plot for 4 in toluene, with the light-green points being BCPs. | ||
The TD-DFT calculations of 4 geometry-optimized in DCM suggested that the most intense vertical transitions in the visible region should correspond to the absorption bands around 440 nm. These transitions arise from combinations of MLCT with iodide n to π* and π to π* excitations (Tables S22–S24, ESI†). We overlaid the predicted transitions from the TD-DFT calculations on our UV-visible data for 4, by normalizing the oscillator strengths to the extinction coefficient at 440 nm (Fig. S25†). An MLCT transition between the HOMO and LUMO+1, with an excitation energy of 2.18 eV (567 nm), is also present. However, the calculated oscillator strength is not as intense as that for the bands at 440 nm. This could be due to an underestimation of the weak copper–iodide and iodide–iodide interactions, effects arising from the lower symmetry of the Cu(I) complex, and the well-known failure of TD-DFT in calculating electronic transitions with high charge-transfer characters.77 Use of more computationally intensive multi-configurational self-consistent field methods, which is beyond the scope of this manuscript, may be necessary to provide more accurate predictions of our electronic spectroscopic data.
Dye-sensitized solar cells with Cu(I) light harvesters
To test the viability of Cu(I) Ar-BIANs in DSSCs, we adopted a method reported previously to incorporate 3 and 4 into such devices.33,78 Sulfonates are known to serve as attachment groups on TiO2 surfaces through electrostatic or covalent linkages. Thus, 3 can be conveniently grafted by dipping the TiO2 into a DMSO solution of 3.79,80 However, the anchoring of 4 is less straightforward, since the methyl esters do not effectively bind to metal oxides. To circumvent this issue, the TiO2 was pre-treated with a THF solution of 1.0 M potassium tert-butoxide for 2 days to deprotonate the surface titanol groups and give “Ti–O−” anions prior to dipping the substrate into a DCM solution of 4.81 The activated TiO2 surface can then react with the methyl esters to facilitate attachment of 4. After 24 h, the TiO2 films acquired the solid-state colors of the Cu(I) complexes. The dyes appeared to be retained successfully since they were not rinsed off the TiO2 films (DMSO for 3; DCM for 4). Each TiO2 film was then incorporated into a DSSC according to previously published protocols.33,78 Despite reports that I−/I3− electrolytes have been found to be incompatible with Cu(I) photosensitizers,82 we sought to collect preliminary data as a proof-of-concept by using the standard I−/I3− electrolyte. Table 4 summarizes measurements made on freshly sealed cells. The low, but unoptimized efficiencies obtained from the measured J–V curves (Fig. 7a; Fig. S24a, ESI†) demonstrate that the Cu(I) Ar-BIAN complexes are capable of electron injection into TiO2 for rudimentary DSSCs (Fig. 7c).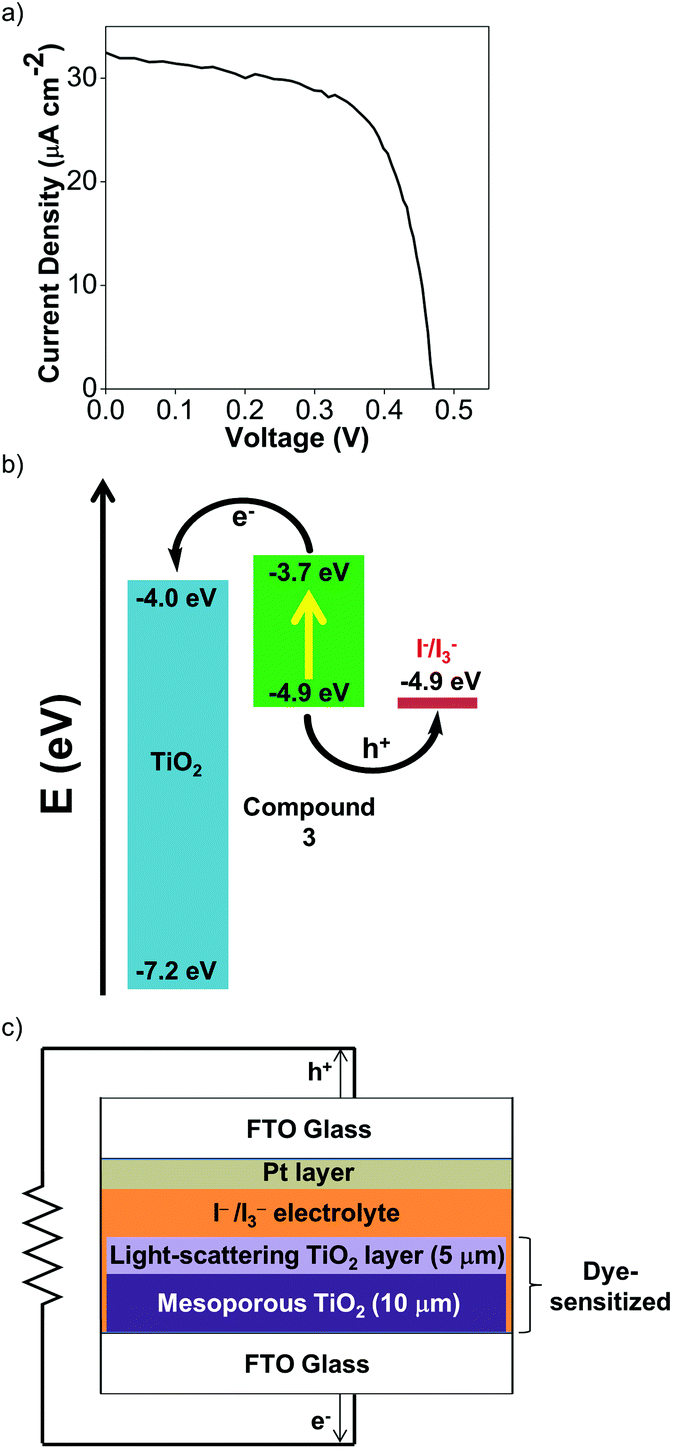 | ||
| Fig. 7 (a) J–V plot for DSSCs after 3 has been adsorbed for dye-sensitization. (b) Band alignment for DSSC incorporated with 3, showing the relative energy levels (in eV against vacuum) for 3, TiO2, and the I−/I3− electrolyte. (c) General device structure for the DSSCs prepared in this study. | ||
| Compound | J SC (mA cm−2) | V OC (mV) | FF | η (%) |
|---|---|---|---|---|
| 3 | 0.0325 | 470 | 0.66 | 0.0101 |
| 4 | 0.0338 | 339 | 0.40 | 0.0046 |
In addition, the low efficiencies of these DSSCs sensitized by 3 and 4 can be explained by considering the charge transfer kinetics and band alignment (Fig. 7b; Fig. S24b, ESI†) for the devices. In 3, the LUMO is localized around the iminyl and acenaphthyl moieties of the BIAN ligand (Fig. 5b), whereas the sulfonate anchoring group is on the aniline ring. This may result in less efficient electron injection into TiO2, since the LUMO containing the photoexcited electron will not have an effective overlap with the conduction band of TiO2. For 4, the low efficiency can be further attributed to poor dye regeneration as a result of its HOMO being higher in energy than that of the I−/I3− electrolyte.
The conditions for these Cu(I) Ar-BIAN DSSCs can be fine-tuned to improve the stability and response of the Cu(I) photosensitizers. For instance, future measurements with tris(bipyridyl) cobalt electrolytes will be attempted to circumvent CuI formation and also improve the band alignment with the dyes. Moreover, it was observed that after the cell made with 4 was sealed, the Cu(I) dye appeared to leach into the CH3CN electrolyte solution. We intend to replace CH3CN with bulkier, less-coordinating solvents such as DMSO or γ-butyrolactone to overcome this problem. Finally, the Ar-BIAN ligands are being further derivatized to explore their utility in DSSCs. Long hydrocarbon chains can be introduced to the periphery of the aryl-imino motif to obstruct the approach of the iodide electrolyte through hydrophobic effects, similar to that reported previously.32,83 This approach will limit the formation of CuI, as well as minimize recombination processes.
Experimental section
Synthesis of ligands
Synthesis of Cu(I) complexes
UV-visible spectroscopic experiments
UV-visible spectra were recorded with a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer with a resolution of 1 nm. Each sample was dissolved in methanol or DCM in a cuvette with a path length of either 1 cm or 0.5 cm. The samples were dissolved in their respective solvents before serial dilution was performed to achieve the desired concentrations for measurements. Concentrations between 25 and 400 μM for 1 and 2 were used, while concentrations between 10 and 100 μM were employed for 3 and 4. To locate the absorption maxima between 450 nm and 800 nm, digitized spectra of 3 and 4 were treated by a Peakfit Autofit algorithm using the second-derivative technique. The parameters were set up to fit three bands with the initial wavelengths set to 560, 620, and 720 nm, before optimizing the amplitudes and widths of the Gaussian curves until a satisfactory fit was obtained (Fig. S10–S12, ESI†). The molar extinction coefficients (ε) were then obtained from the individual band amplitudes of the Gaussian curves fitted into each spectrum. Each simulation was optimized to try to achieve an R2 value of 0.995.Fabrication of DSSCs and device measurements
The device fabrication was carried out as reported elsewhere.86 Freshly cleaned FTO glass (2.2 mm thickness, 14 Ω sq−1 sheet resistance, Pilkington) was used as the current collector. The FTO glass was immersed in a 40 mM aqueous TiCl4 solution at 70 °C for 30 min and rinsed with water and ethanol. The 10 μm-thick transparent TiO2 layers (Dyesol 18NR-T, average nanoparticle size: 20 nm) and 5 μm-thick scattering TiO2 layers (Dyesol WER2-O paste, average nanoparticle size: 150–250 nm) were then printed on the FTO glass plates and the plates were annealed under air at 125 °C for 10 min, at 325 °C for 5 min, at 375 °C for 5 min, at 450 °C for 15 min, and finally, at 500 °C for 15 min. The FTO plates were further treated with a 40 mM aqueous TiCl4 solution at 70 °C for 30 min and subsequently annealed at 450 °C for 20 min. The TiO2 photoanodes were then made by immersing the plates into either a dye solution of 3 (500 μM in DMSO) for 24 h, or a dye solution of 4 (500 μM in DCM) for 24 hours after pretreatment with 1.0 M potassium tert-butoxide in THF for 48 hours.The dye-sensitized TiO2 photoanode and the Pt-coated FTO glass counter electrode were sandwiched together using a 25 μm-thick transparent Surlyn® film (Meltonix 1170-25, Solaronix). The electrolyte was injected through a hole at the back of the counter electrode via vacuum backfilling. The electrolyte employed was a solution containing 1.0 mM 1,3-dimethylimidazolium iodide (DMII), 50 mM LiI, 30 mM I2, 0.50 mM tert-butylpyridine, and 0.10 mM guanidinium thiocyanate (GNCS) in a mixed solvent of CH3CN and valeronitrile (v/v, 85/15). Finally, the hole was sealed using a 25 μm-thick Surlyn® film and a cover glass (0.1 mm thickness) to avoid leakage of the electrolyte.
Photocurrent density–photovoltage (J–V) curves were measured under AM 1.5 (100 mW cm−2) illumination using a solar simulator (San-EI Electric, XEC-301S) equipped with a 450 W xenon lamp, which was coupled with an Agilent semiconductor parameter analyzer (4155C). The power of the simulated light was calibrated to 100 mW cm−2 by using a silicon reference cell (Fraunhofer) and monitored using a power meter throughout the testing. A black mask (6 mm × 6 mm) was used in the subsequent photovoltaic studies to avoid the effects of diffusive light on the cell performance. The reported values are calculated based on the average of three batches of devices with identical compositions and fabrication procedures.
Conclusions
We have developed two Cu(I) dyes bearing new Ar-BIAN ligands that can be expediently synthesized from affordable commercial reagents. For complex 4, the solid-state crystal structures derived from single crystal X-ray diffraction experiments using crystals grown from two different solvent combinations both displayed remarkable rhombically distorted square planar geometries around the Cu(I) center. This unique coordination sphere around the Cu(I) nucleus is manifest in the broad, panchromatic light absorption extending to the NIR region, as determined by DRS measurements. DFT and AIM calculations suggest that weak, non-covalent interactions between the aromatic rings and among the iodides may be responsible for the rare coordination geometry and photophysical features. The new Cu(I) complexes have been preliminarily applied as photosensitizers in DSSCs and give solar cells with unoptimized efficiencies of up to 0.010%. Further studies include the judicious design of new derivatives of these Cu(I) Ar-BIAN photosensitizers, time-resolved spectroscopic experiments, and exploration of their applications in artificial photosynthesis.Acknowledgements
H. S. S. is supported by a NTU start-up grant (M4081012), the Nanyang Assistant Professorship (M4081154), and an MOE Tier 1 grant (M4011144). The authors acknowledge the support from the Solar Fuels Laboratories at NTU and the Singapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE) CREATE Programme. This research programme is funded by the National Research Foundation (NRF), Prime Minister's Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE). H. H. is grateful for the Nanyang Assistant Professorship.Notes and references
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed.
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999–7002 CrossRef CAS PubMed.
- G. Fumagalli, P. T. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481–11484 CrossRef CAS PubMed.
- A. C. Hernandez-Perez and S. K. Collins, Angew. Chem., Int. Ed., 2013, 52, 12696–12700 CrossRef CAS PubMed.
- X. J. Tang and W. R. Dolbier Jr., Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- M. Gratzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef CAS PubMed.
- D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 134, 12354–12357 CrossRef CAS PubMed.
- K. C. D. Robson, K. Hu, G. J. Meyer and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1961–1971 CrossRef CAS PubMed.
- K. Hu, K. C. D. Robson, P. G. Johansson, C. P. Berlinguette and G. J. Meyer, J. Am. Chem. Soc., 2012, 134, 8352–8355 CrossRef CAS PubMed.
- S. Gazi, W. K. H. Ng, R. Ganguly, A. M. P. Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- H. S. Soo, S. Gazi and M. Dokic, Selective Carbon–Carbon Bond Cleavage By Earth Abundant Vanadium Compound Under Visible Light Photocatalysis, PCT Application NumberPCT/SG2016/050056, 2016 Search PubMed.
- H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024–10035 CrossRef CAS PubMed.
- H. S. Soo, M. T. Sougrati, F. Grandjean, G. J. Long and C. J. Chang, Inorg. Chim. Acta, 2011, 369, 82–91 CrossRef CAS.
- S. K. Muduli, S. Wang, S. Chen, C. F. Ng, C. H. A. Huan, T. C. Sum and H. S. Soo, Beilstein J. Nanotechnol., 2014, 5, 517–523 CrossRef CAS PubMed.
- H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Chem. Commun., 2016, 52, 2948–2951 RSC.
- M. L. Macnaughtan, H. S. Soo and H. Frei, J. Phys. Chem. C, 2014, 118, 7874–7885 CAS.
- H. S. Soo, A. Agiral, A. Bachmeier and H. Frei, J. Am. Chem. Soc., 2012, 134, 17104–17116 CrossRef CAS PubMed.
- H. S. Soo, M. L. Macnaughtan, W. W. Weare, J. Yano and H. M. Frei, J. Phys. Chem. C, 2011, 115, 24893 CAS.
- H. S. Soo, P. L. Diaconescu and C. C. Cummins, Organometallics, 2003, 23, 498–503 CrossRef.
- H. S. Soo, J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2004, 126, 11370–11376 CrossRef CAS PubMed.
- T. C. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca Jr., P. Huang, P. Chabera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundstrom and K. Warnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed.
- B. Bozic-Weber, V. Chaurin, E. C. Constable, C. E. Housecroft, M. Meuwly, M. Neuburger, J. A. Rudd, E. Schonhofer and L. Siegfried, Dalton Trans., 2012, 41, 14157–14169 RSC.
- B. Bozic-Weber, E. C. Constable, S. O. Furer, C. E. Housecroft, L. J. Troxler and J. A. Zampese, Chem. Commun., 2013, 49, 7222–7224 RSC.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS PubMed.
- A. K. Ichinaga, J. R. Kirchhoff, D. R. McMillin, C. O. Dietrich-Buchecker, P. A. Marnot and J. P. Sauvage, Inorg. Chem., 1987, 26, 4290–4292 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- E. A. Juban, A. L. Smeigh, J. E. Monat and J. K. McCusker, Coord. Chem. Rev., 2006, 250, 1783–1791 CrossRef CAS.
- X. Q. Lu, S. X. Wei, C. M. L. Wu, S. R. Li and W. Y. Guo, J. Phys. Chem. C, 2011, 115, 3753–3761 CAS.
- P. A. Papanikolaou and N. V. Tkachenko, Phys. Chem. Chem. Phys., 2013, 15, 13128–13136 RSC.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- L. N. Ashbrook and C. M. Elliott, J. Phys. Chem. C, 2013, 117, 3853–3864 CAS.
- C. Bizzarri, C. Strabler, J. Prock, B. Trettenbrein, M. Ruggenthaler, C. H. Yang, F. Polo, A. Iordache, P. Bruggeler and L. De Cola, Inorg. Chem., 2014, 53, 10944–10951 CrossRef CAS PubMed.
- B. A. Gandhi, O. Green and J. N. Burstyn, Inorg. Chem., 2007, 46, 3816–3825 CrossRef CAS PubMed.
- R. S. Khnayzer, C. E. McCusker, B. S. Olaiya and F. N. Castellano, J. Am. Chem. Soc., 2013, 135, 14068–14070 CrossRef CAS PubMed.
- J. V. Lockard, S. Kabehie, J. I. Zink, G. Smolentsev, A. Soldatov and L. X. Chen, J. Phys. Chem. B, 2010, 114, 14521–14527 CrossRef CAS PubMed.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2013, 52, 8114–8120 CrossRef CAS PubMed.
- M. Gasperini, F. Ragaini, E. Gazzola, A. Caselli and P. Macchi, Dalton Trans., 2004, 3376–3382 RSC.
- K. Hasan and E. Zysman-Colman, J. Phys. Org. Chem., 2013, 26, 274–279 CrossRef CAS.
- N. J. Hill, I. Vargas-Baca and A. H. Cowley, Dalton Trans., 2009, 240–253 RSC.
- T. Kern, U. Monkowius, M. Zabel and G. Knor, Eur. J. Inorg. Chem., 2010, 4148–4156 CrossRef CAS.
- L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Aviles and P. T. Gomes, Dalton Trans., 2012, 41, 5144–5154 RSC.
- L. D. Li, P. S. Lopes, C. A. Figueira, C. S. B. Gomes, M. T. Duarte, V. Rosa, C. Fliedel, T. Aviles and P. T. Gomes, Eur. J. Inorg. Chem., 2013, 1404–1417 CrossRef CAS.
- P. Papanikolaou, P. D. Akrivos, A. Czapik, B. Wicher, M. Gdaniec and N. Tkachenko, Eur. J. Inorg. Chem., 2013, 2418–2431 CrossRef CAS.
- V. Rosa, C. I. M. Santos, R. Welter, G. Aullon, C. Lodeiro and T. Aviles, Inorg. Chem., 2010, 49, 8699–8708 CrossRef CAS PubMed.
- I. L. Fedushkin, V. A. Chudakova, A. A. Skatova, N. M. Khvoinova, Y. A. Kurskii, T. A. Glukhova, G. K. Fukin, S. Dechert, M. Hummert and H. Schumann, Z. Anorg. Allg. Chem., 2004, 630, 501–507 CrossRef CAS.
- I. L. Fedushkin, A. A. Skatova, V. K. Cherkasov, V. A. Chudakova, S. Dechert, M. Hummert and H. Schumann, Chem. – Eur. J., 2003, 9, 5778–5783 CrossRef CAS PubMed.
- I. L. Fedushkin, A. A. Skatova, V. A. Chudakova and G. K. Fukin, Angew. Chem., Int. Ed., 2003, 42, 3294–3298 CrossRef CAS PubMed.
- W. K. Chan, C. S. Hui, K. Y. K. Man, K. W. Cheng, H. L. Wong, N. Y. Zhu and A. B. Djurisic, Coord. Chem. Rev., 2005, 249, 1351–1359 CrossRef CAS.
- J. Huang, O. Buyukcakir, M. W. Mara, A. Coskun, N. M. Dimitrijevic, G. Barin, O. Kokhan, A. B. Stickrath, R. Ruppert, D. M. Tiede, J. F. Stoddart, J.-P. Sauvage and L. X. Chen, Angew. Chem., Int. Ed., 2012, 51, 12711–12715 CrossRef CAS PubMed.
- J. He, A. Hagfeldt, S.-E. Lindquist, H. Grennberg, F. Korodi, L. Sun and B. Åkermark, Langmuir, 2001, 17, 2743–2747 CrossRef CAS.
- J. Zhou, X. Li and H. Sun, Can. J. Chem., 2008, 86, 782–790 CrossRef CAS.
- L. Li, M. Jeon and S. Y. Kim, J. Mol. Catal. A: Chem., 2009, 303, 110–116 CrossRef CAS.
- P. Papanikolaou, P. D. Akrivos, A. Czapik, B. Wicher, M. Gdaniec and N. Tkachenko, Eur. J. Inorg. Chem., 2013, 2013, 2418–2431 CrossRef CAS.
- V. Madhu, Y. Diskin-Posner and R. Neumann, Eur. J. Inorg. Chem., 2011, 1792–1796 CrossRef CAS.
- M. G. B. Drew, C. J. Harding, O. W. Howarth, Q. Lu, D. J. Marrs, F. F. Morgan, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1996, 3021–3030 RSC.
- S. Doherty, J. G. Knight, C. H. Smyth, N. T. Sore, R. K. Rath, W. McFarlane, R. W. Harrington and W. Clegg, Organometallics, 2006, 25, 4341–4350 CrossRef CAS.
- M. Stollenz, M. John, H. Gehring, S. Dechert, C. Grosse and F. Meyer, Inorg. Chem., 2009, 48, 10049–10059 CrossRef CAS PubMed.
- A. L. Spek, A. J. M. Duisenberg, G. C. van Stein and G. van Koten, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 374–377 CrossRef.
- U. Monkowius, Y. N. Svartsov, T. Fischer, M. Zabel and H. Yersin, Inorg. Chem. Commun., 2007, 10, 1473–1477 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, 2007, ch. 128, vol. 280, pp. 69–115 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, USA, 2013 (aim.tkgristmill.com) Search PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- F. Neese, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS PubMed.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Gratzel, M. K. Nazeeruddin and M. Gratzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- C. Bauer, P. Jacques and A. Kalt, Chem. Phys. Lett., 1999, 307, 397–406 CrossRef CAS.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 40, 271–286 CrossRef CAS.
- J. J. He, A. Hagfeldt, S. E. Lindquist, H. Grennberg, F. Korodi, L. C. Sun and B. Akermark, Langmuir, 2001, 17, 2743–2747 CrossRef CAS.
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089–3106 CrossRef CAS.
- M. K. Nazeeruddin, E. Baranoff and M. Grätzel, Solar Energy, 2011, 85, 1172–1178 CrossRef CAS.
- C. D. Grant, S. O. Kang and B. P. Hay, J. Org. Chem., 2013, 78, 7735–7740 CrossRef CAS PubMed.
- B. Patel, G. Saviolaki, C. Ayats, M. A. E. Garcia, T. Kapadia and S. T. Hilton, RSC Adv., 2014, 4, 18930–18932 RSC.
- L. H. Nguyen, H. K. Mulmudi, D. Sabba, S. A. Kulkarni, S. K. Batabyal, K. Nonomura, M. Gratzel and S. G. Mhaisalkar, Phys. Chem. Chem. Phys., 2012, 14, 16182–16186 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization, electrochemistry, and DFT calculations. CCDC 1431314–1431316. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00221d |
| This journal is © the Partner Organisations 2016 |