
Stereoselective synthesis of 3-amino-2-oxindoles from isatin imines: new scaffolds for bioactivity evaluation
Jasneet Kaur
a,
Swapandeep Singh Chimni
*a,
Suhel Mahajan
a and
Akshay Kumar
b
aDepartment of Chemistry, U.G.C. Centre of Advance Studies in Chemistry, Guru Nanak Dev University, Amritsar, India. E-mail: sschimni@yahoo.com; sschimni.chem@gndu.ac.in; Fax: +91-183-2258820
bDepartment of Chemistry, DAV University, Jalandhar, India
First published on 26th May 2015
Abstract
3-Substituted-3-aminooxindoles have attracted the attention of organic and medicinal chemists because these motifs constitute the core structure of a number of natural products and drug candidates. The catalytic potential of chiral organocatalysts and metal catalysts has been successfully exploited for the synthesis of enantioenriched 3-amino-2-oxindoles via the addition of various nucleophiles to isatin imines. This review focuses on the catalytic asymmetric synthesis of chiral 3-amino-3-substituted-2-oxindoles.
 Jasneet Kaur | Jasneet Kaur was born in 1989 in Vain Poin village of Tarn Taran, Punjab, India. She obtained her BSc (Hons. Sch.) and MSc (Hons. Sch.) in Chemistry from Guru Nanak Dev University, Amritsar. To pursue a research career in Organic Synthesis she joined the research group of Prof. Swapandeep Singh Chimni in the same University. Her research interests include synthesis of new chiral bifunctional organocatalysts and their applications for enantioselective carbon–carbon, carbon–heteroatom bond formations and domino reactions. |
 Swapandeep Singh Chimni | Swapandeep Singh Chimni was born in 1962 at Amritsar, India. He received his MSc (Hons. Sch.) in Chemistry in 1985 and PhD in 1991 from Guru Nanak Dev University, Amritsar. After two years as a lecturer at Regional Engineering College (now NIT) Jalandhar, he joined the Department of Chemistry, Guru Nanak Dev University as Lecturer in 1992. He is presently working as a Professor in the same department. He has a research experience of 25 years and published over 107 publications. He works in the area of synthetic organic chemistry with emphasis on asymmetric organocatalysis, biocatalysis and phase transfer catalysis as well as green chemistry. |
 Suhel Mahajan | Suhel Mahajan was born in 1985 in Gurdaspur. He studied in Hindu College, Amritsar for Bachelor of Science. After completing his MSc (I.A.) from Guru Nanak Dev University, Amritsar in 2008, he started his PhD under the supervision of Prof. Swapandeep Singh Chimni. He is interested in developing new synthetic methodologies using dual catalysis. |
 Akshay Kumar | Akshay Kumar was born in 1983 at Maslana Kalan, a small village in Hamirpur District of Himachal Pradesh, India. He obtained his BSc from Himachal Pradesh University, Shimla in 2004 and MSc Chemistry from Guru Nanak Dev University, Amritsar in 2007. He earned his PhD under the supervision of Prof. Swapandeep Singh Chimni from Guru Nanak Dev University, Amritsar, India, in 2013 and after that he worked as a research associate in the same research group. Presently, he is working as Assistant Professor in Chemistry at DAV University, Jalandhar where he is pursuing research interest on the development of organocatalytic processes for the synthesis of chiral bio-relevant molecules. |
(1) Introduction
3-Substituted-3-amino-2-oxindole is a privileged core structure found in a variety of natural products and biologically active compounds1 such as the gastrin/CCK-B receptor antagonist AG-041R,2 the vasopressin VIb receptor antagonist SSR-1494153 (ref. 3) and antimalarial drug candidate NITD609 (Fig. 1).4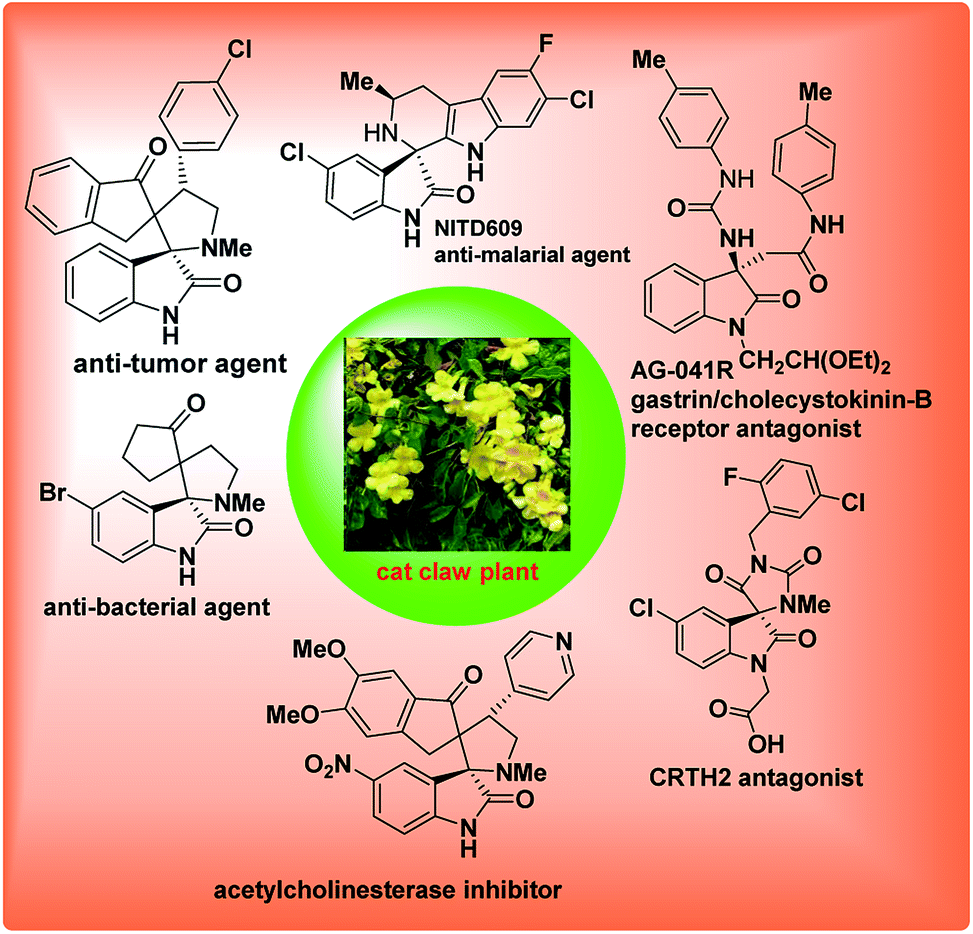 | ||
| Fig. 1 Examples of bioactive quaternary aminooxindoles and related natural compounds. | ||
The bioactivity of these compounds is greatly affected by the nature of the substituent at the C-3 position as well as the absolute configuration of the stereogenic centre.5 Therefore, the development of efficient and practical methods to synthesize such molecules is of great importance and is the current area of research in asymmetric catalysis. In the past few years, a variety of methods for preparing these compounds have been explored.6
Among these the enantioselective addition of nucleophile to isatin imines is one of the most efficient and straightforward methods. Organocatalytic enantioselective addition reactions such as aza-Friedel–Crafts reaction, Mannich reaction, Henry reaction, Strecker reaction, Morita–Baylis–Hillman reaction have been developed to construct 3-substituted-3-amino-2-oxindoles with a chiral quaternary carbon center. The representative examples of these catalytic strategies reported since 2009 involves isatin derived ketimine as a substrate are shown in Fig. 2.
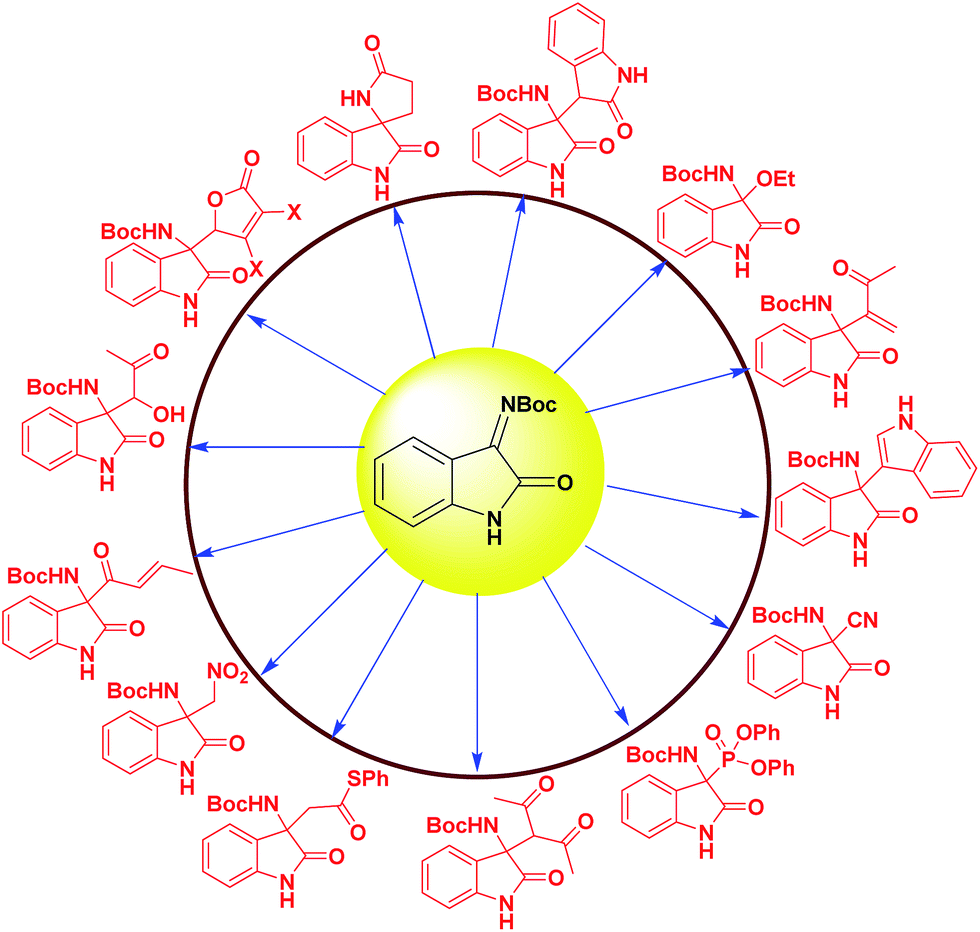 | ||
| Fig. 2 Representative examples from isatin imine framework. | ||
For convenience in presentation and easy understanding, this review has been classified according to the type of reaction catalyzed by organocatalysts and metal catalysts.
(2) Enantioselective aza-Morita–Baylis–Hillman reaction
The Morita–Baylis–Hillman/aza-Morita–Baylis–Hillman reaction has received increasing interest since it combines two important requirements, atom economy and generation of functional groups.7 The last decade has seen exponential growth of Morita–Baylis–Hillman (MBH) reaction and its applications. This is a reaction between electron deficient olefins and imines to provide densely functionalized chiral amines. Min Shi et al. have developed a highly enantioselective aza-Morita–Baylis–Hillman reaction of isatin imines (1) with methyl vinyl ketones (MVK) 2 catalyzed by β-isocupreidine I and chiral phosphines II (Scheme 1).8 Similar results were obtained affording 3-amino-2-oxindoles 3 bearing a C-3 tetra-substituted stereogenic centre with excellent enantioselectivity (up to 99% ee) and excellent yield up to 98%. In addition to this, the absolute configuration was also found to be same (R) in both cases.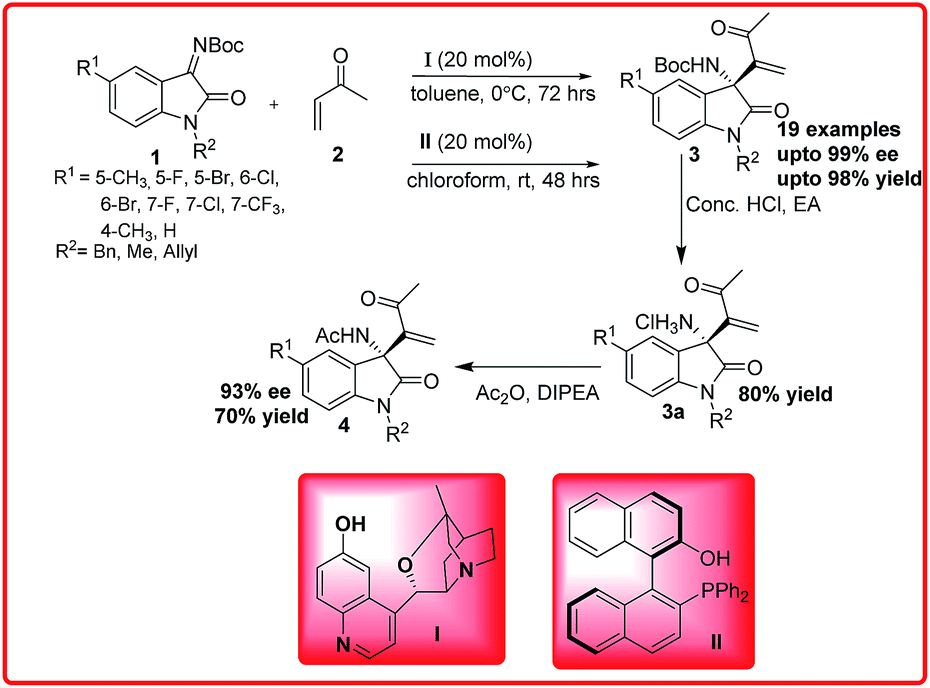 | ||
| Scheme 1 Enantioselective aza-Morita–Baylis–Hillman reaction of isatin imines with MVK. | ||
The synthetic use of this methodology was illustrated by the synthesis of N-acyl-3-aminooxindole 4 after treating with acetic anhydride, in 70% yield.
Subsequent to this report, Feng Sha et al. reported the phosphine squaramide catalyzed enantioselective aza-MBH reaction of isatin imine 1 with acrylates 2b to provide 3-substituted-3-amino-2-oxindole 5 in moderate to excellent yields (43–99%) and good enantioselectivity (70–91% ee).
In the proposed transition state TS 1, the catalyst activates the ketimine through H-bonding whereas the cyclohexyl scaffold of the catalyst III provides a favourable orientation to the phosphinoyl associated enolate to attack the activated ketimine from Si-face to form the adduct with S-configuration (Scheme 2).9
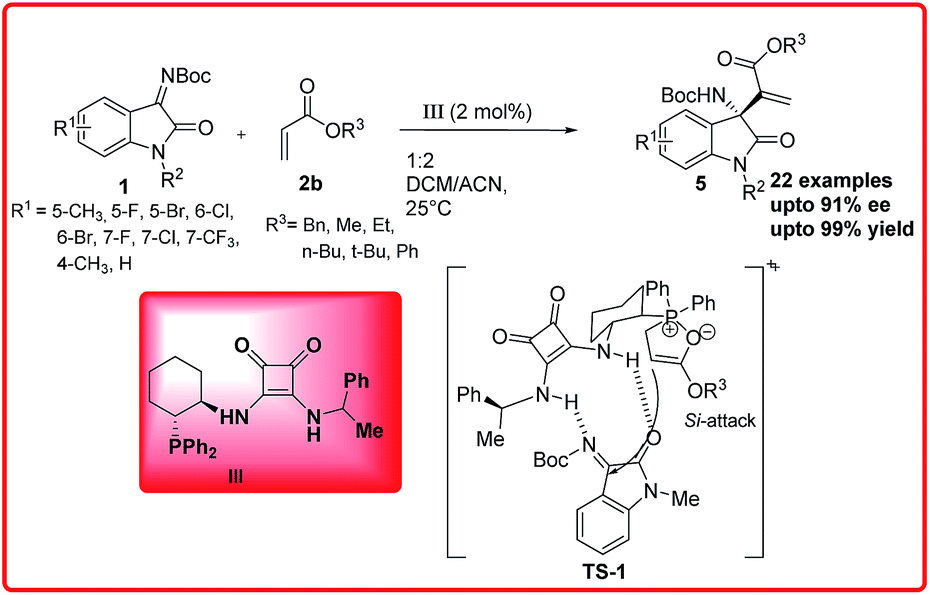 | ||
| Scheme 2 Asymmetric aza-MBH reaction of isatin imines with acrylates catalyzed by phosphine-squaramide organocatalyst. | ||
Recently, our group reported an organocatalyzed aza-Morita–Baylis–Hillman reaction of maleimides 6 with isatin imines 1 using β-isocupreidine I as an organocatalyst (Scheme 3).10 Maleimide as a MBH donor is more challenging task because maleimides are traditionally Michael acceptors. A wide variety of 3-substituted-3-aminoindolin-2-ones 7 were synthesized in good yield (up to 77%) and excellent enantioselectivities (up to 99%).
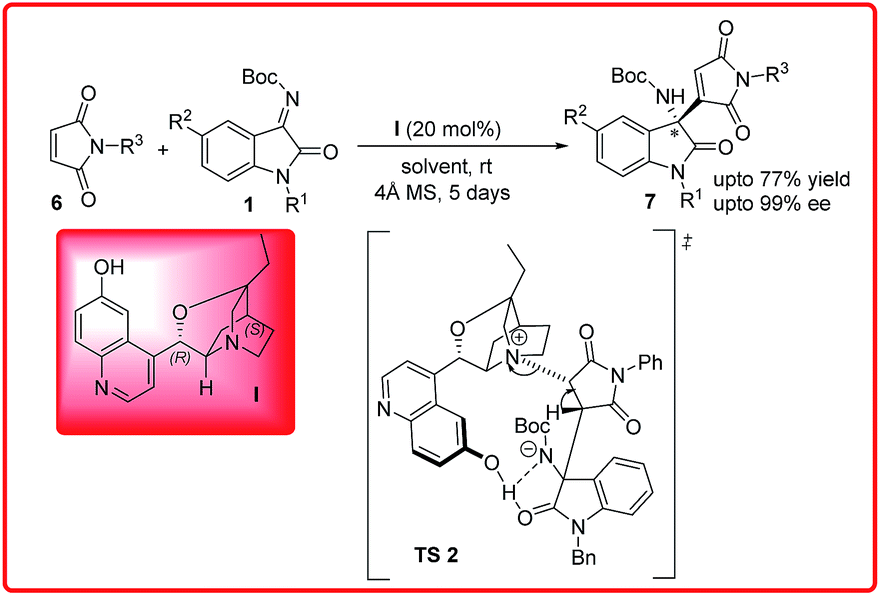 | ||
| Scheme 3 Enantioselective aza-Morita–Baylis–Hillman reaction of isatin imines with maleimide. | ||
In the proposed transition state TS 2, the tertiary amine of the catalyst form enolate of the maleimide which simultaneously attacks on the isatin imine to form the favourable R enantiomer.
(3) Enantioselective Friedel–Crafts reaction
The Friedel–Crafts reaction is an important reaction for the construction of carbon–carbon and carbon–nitrogen bond which can provide important building blocks for pharmaceutically applicable compounds.11 To promote these transformations, significant progress has been made by employing both chiral Brønsted acid and Lewis acid catalysts. The first asymmetric aza-Friedel–Crafts reaction of indoles 8 and pyrroles 10 with isatin imines 1 was reported by Wang et al. in 2012 (Scheme 4).12a The phosphoric acid catalyst IV efficiently catalyzed the aza-Friedel–Crafts reaction of N-methylindoles with isatin imines to afford the desired product. In addition to this, the introduction of an indole moiety into an oxindole scaffold produces 3-indolyl-3-amino-2-oxindoles which are promising substrates for the study of their biological activity as well as useful synthetic intermediates for drug candidates and alkaloids. On the basis of the observed stereochemistry, a plausible transition state TS 3 was proposed in which the phosphoric acid proton activates the ketimines and the N-methylindole attacks ketimine from Re-face leading to the S-configured product. The phosphoric acid catalyzed reaction of indole with isatin imine provides the desired product in only 64% ee which suggests that the hydrogen bond between indole and the phosphoryl oxygen could have a negative effect on the enantioselectivity. However with 2 mol% of catalyst, the pyrrole reacts rapidly with various ketimines to afford the adducts in up to 98% yield and 64–98% enantioselectivity. The reaction of N-methylpyrrole 10 gives only moderate enantioselectivity (70% ee) and lower yield (49%). This shows that the bifunctional nature of the chiral phosphoric acid is responsible for concurrent activation of both the isatin imine and the pyrrole through hydrogen bond interactions. Recently, Cinchona derived organocatalyst XXI has been used for catalyzing the reaction of isatin imine with phenol derivatives providing 3-amino-2-oxindoles in 51–99% yield and up to 99% enantiomeric excess.12c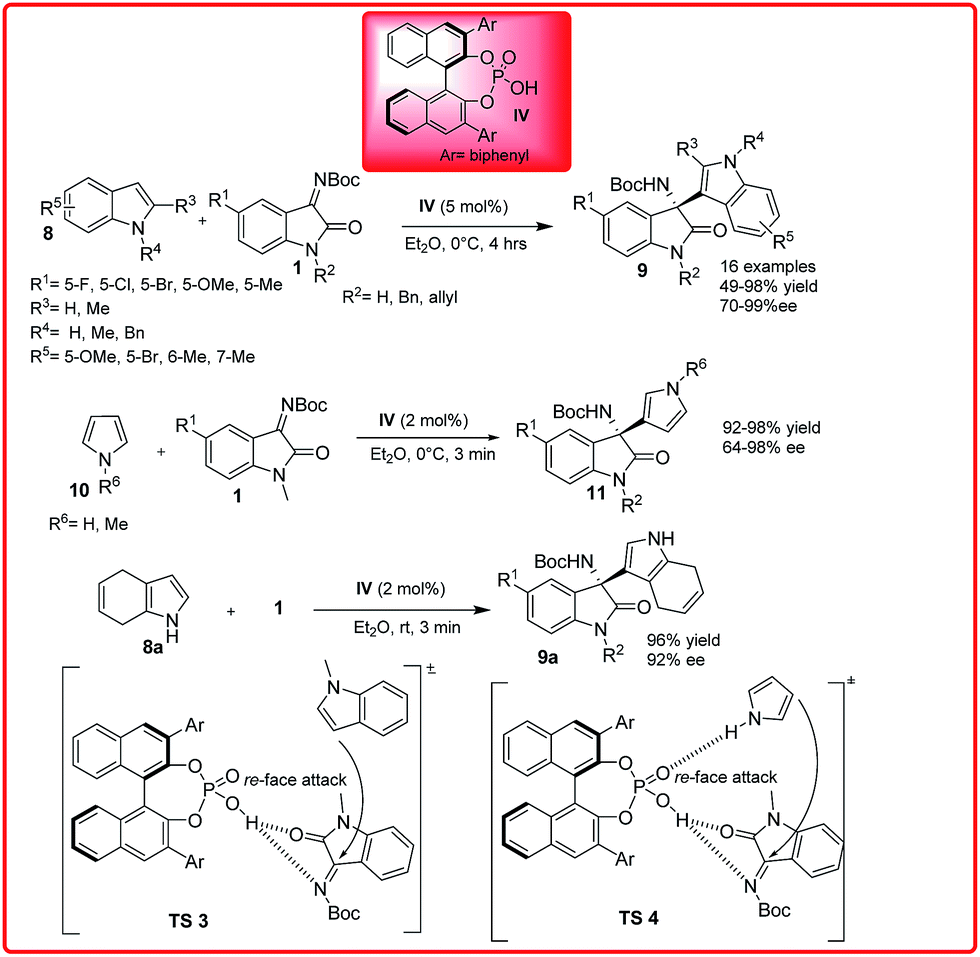 | ||
| Scheme 4 Chiral phosphoric acids catalyzed enantioselective addition of indole and pyrrole derivatives to isatin imine. | ||
Recently, Xu et al. reported the same reaction using only 5 mol% of Bi(OTf)3 (Scheme 5).12b The reaction completes in 24 hours providing the desired adduct in excellent yields (up to 99%) with good to excellent enantioselectivities ranging from 90–98% ee. In the proposed transition state TS 5, the Lewis acidic metal coordinates both with the imine nitrogen and carbonyl oxygen in such a way that the bulky tertiary group blocks the Si-face of ketimine molecule, thus the indole attacks from Re face of ketimine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond to give S-configured product.
N bond to give S-configured product.
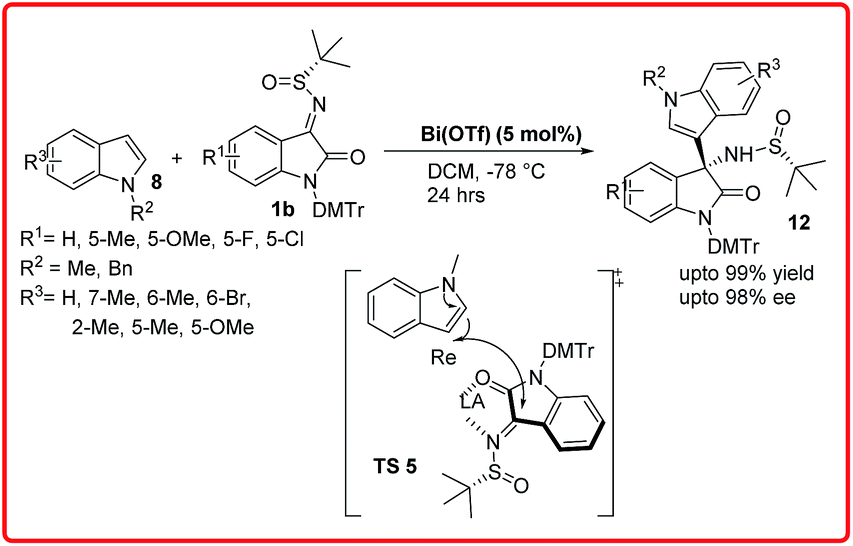 | ||
| Scheme 5 Diastereoselective Friedel–Crafts reaction of N-sulfinyl isatin imines with indoles. | ||
(4) Enantioselective hydrophosphonylation reaction
Phosphonic esters have received significant attention because of their important role as metabolic intermediates, regulatory switches for proteins and backbones for genetic information.13In particular, α-aminophosphonic acid derivatives14 are known to exhibit a broad spectrum of biological activities such as peptide mimetics, antibacterial,15 antiviral agents16 and enzyme inhibitor.17 Inspite of this, there are only a few reports on asymmetric addition of phosphite to simple ketimines.18 The asymmetric addition to functionalized ketimines is more challenging task because of their low reactivity and difficulty in enantiofacial discrimination.
Only two successful enantioselective examples of this reaction has been reported. B. V. Reddy et al. reported the first asymmetric organocatalytic hydrophosphonylation reaction of isatin imines (Scheme 6).19 Quinine squaramide V was identified as the best catalyst for the reaction of isatin imines 1 with diphenylphosphite 13 to provide 3-amino-oxindoles 14 in 80–96% yield and 52–97% ee. The reaction was unsuccessful with diethyl/dimethyl-phosphites due to their low reactivity. The proposed transition state TS 6 for this enantioselective hydrophosphonylation reaction involves a hydrogen bonding activation of ketimine 1 through the NH of the squaramide and simultaneous generation of phosphonate anion from diphenylphosphite 13 by the tertiary amine of the catalyst. In addition to this synergic activation, the catalyst V also provides a favourable orientation to both substrates to provide the desired adduct in high enantioselectivity.
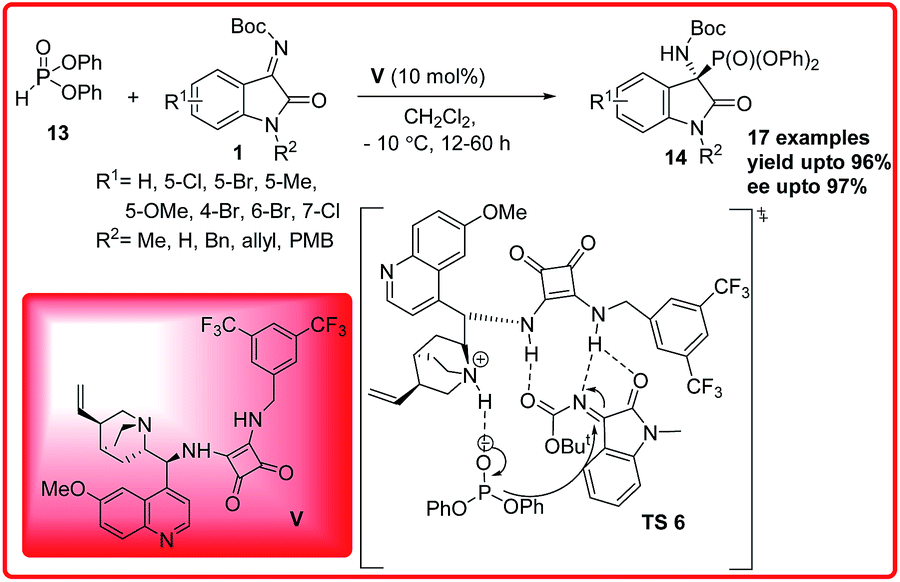 | ||
| Scheme 6 Quinine-squaramide catalyzed enantioselective addition of diphenylphosphite to isatin imines. | ||
Soon after this, our group published the same reaction catalyzed by Cinchonidine derived thiourea VI for the synthesis of 3-amino-2-oxindoles 14 in good to excellent yields (72–88%) and excellent enantioselectivity up to 97% (Scheme 7).20 To improve the synthetic efficiency, the combination of aza-Wittig and phospha-Mannich one pot sequential protocol was developed resulting in α-amino phosphonates in good yield and good enantioselectivity. The proposed transition state TS 7 involves the activation and orientation of isatin imine for face selective attack through H-bonding. Out of the two possible orientations of ketimines the Re face orientation is favoured which avoids the steric interaction between the isatin imine benzene ring and aryl group of phosphate.
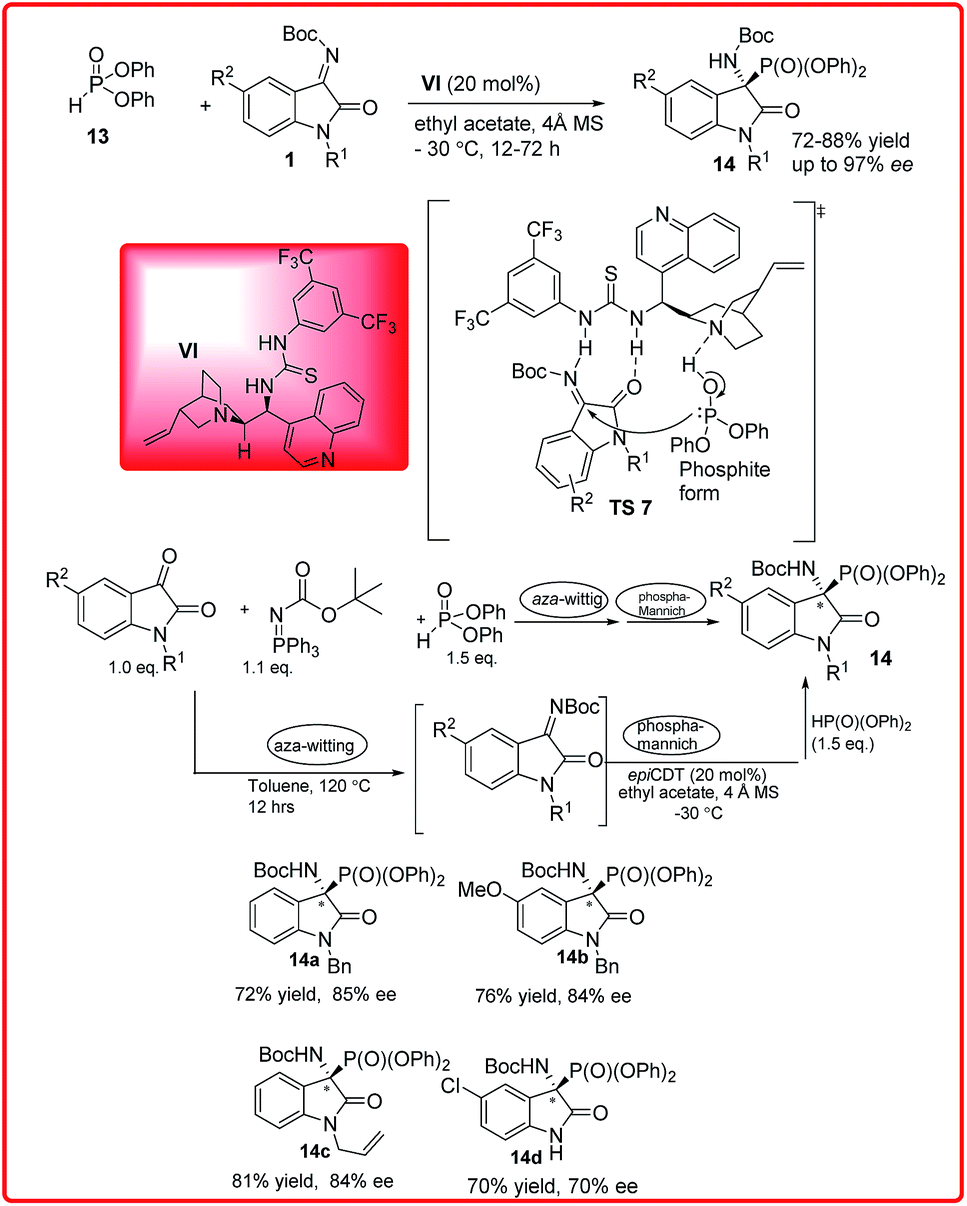 | ||
| Scheme 7 Cinchonidine derived thiourea catalyzed hydrophosphonylation reaction of diphenylphosphite and isatin imines. | ||
(5) Enantioselective Mannich reaction
Mannich reaction is a classic method for the preparation of optically active β-amino carbonyl units, which are useful chiral building blocks for number of biologically active and pharmaceutically important compounds.21The versatility and potential to create both functional and structural diversity using this reaction have long stimulated the creativity of chemists. Inspired by this, Wang et al. synthesized N-alkoxycarbonyl isatin imines via aza-Wittig reaction and used them for the synthesis of chiral 3-aminooxindoles by the enantioselective addition of 1,3-dicarbonyl compounds (Scheme 8).22
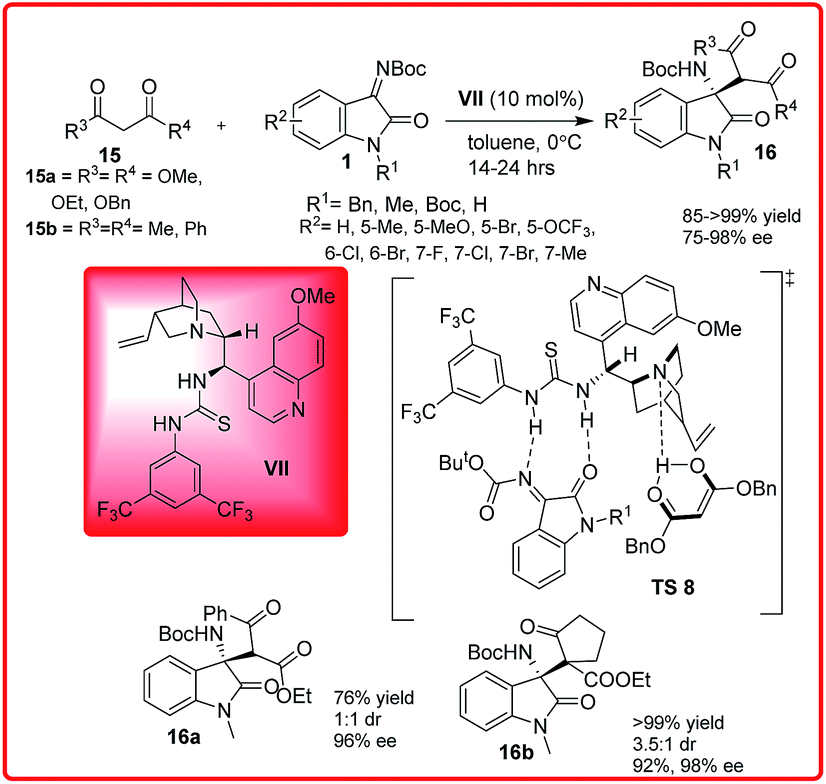 | ||
| Scheme 8 Cinchona-derived thiourea catalyzed enantioselective addition of 1,3-dicarbonyl compounds to N-Boc isatin imines. | ||
Quinidine derived thiourea VII catalyzes the Mannich reaction of malonates 15a and diketones 15b with isatin imines 1 providing 3-amino-2-oxindoles 16 in high yield (>99%) and excellent enantioselectivities (up to 98%). In the proposed transition state TS 8, the bifunctional organocatalyst VII deprotonates the dicarbonyl compound with quinuclidine nitrogen, thus stabilizing the enol, while the thiourea moiety binds and activates the ketimines I through double hydrogen bonding with nitrogen and oxygen atoms. The activated enol approaches the ketimine from one preferred enantioface to afford the desired product with S stereochemistry.
Shibata et al. reported an organocatalytic enantioselective decarboxylative Mannich reaction of malonic acid half thioesters 17 with isatin imines 1 (Scheme 9).23 Out of various catalysts examined, the best results were obtained with N-heteroarenesulfonyl Cinchona alkaloid VIII as the catalyst which affords the final products 18 in good to high yield (58–92%) and good enantioselectivity (75–83%). This protocol was used efficiently for the synthesis of optically active AG-041R 19, a gastrin/cholecystokinin-B-receptor.
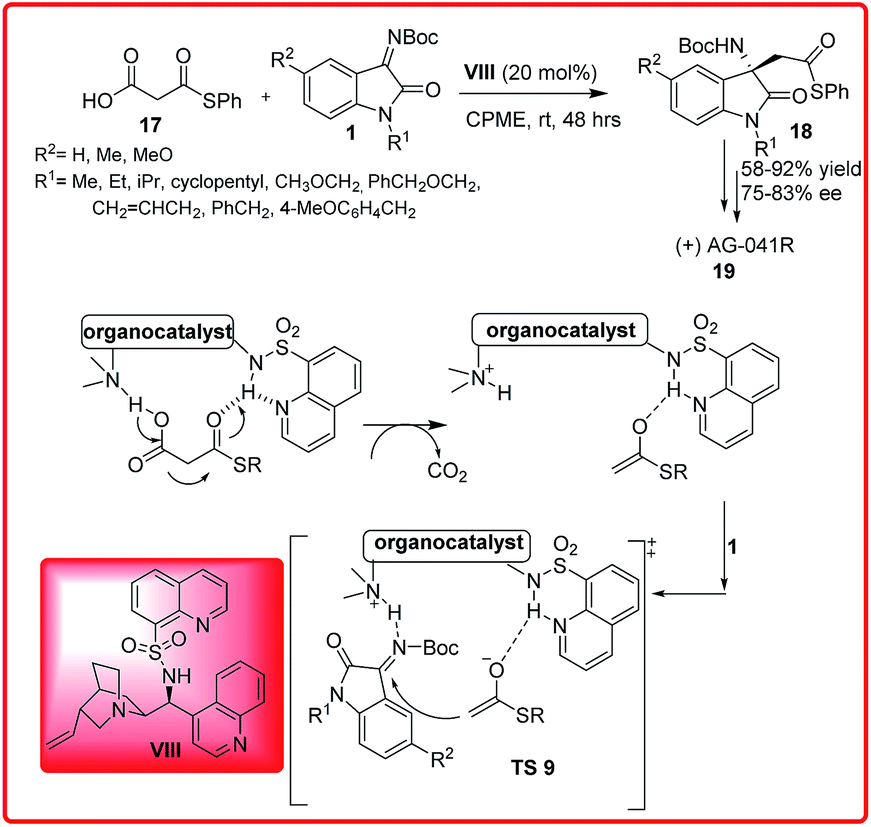 | ||
| Scheme 9 Organocatalytic enantioselective decarboxylation Mannich-type reaction of malonic half thioesters with isatin imines. | ||
The proposed transition state TS 9 reveals that the hydrogen on the sulfonamide forms H-bonds with the nitrogen atoms in quinoline and quinuclidine. The sulfonamide functionality activates through H-bonding with the thiocarbonyl oxygen and the quinuclidine assists the deprotonation and decarboxylation of 17 to give the thioester enolate. The ketimine 1 is activated by the protonated quinuclidine through H-bonding. The reaction of thioester enolate with ketimine 1 in the chiral environment of VIII gives 18 with high enantioselectivity. The proposed transition state TS 9 involves a ternary complex of substrates and catalyst in which the thioester enolate approaches the Re face of ketimine to provide the observed S enantiomer of the product 18.
Recently, Enders group reported the enantioselective organocatalytic addition reaction of ethyl nitroacetate 20 to isatin imine 1 (Scheme 10).24 A wide variety of desired adduct 21 was formed in good to excellent yields (51–91%) and excellent enantioselectivities (92–99%). The desired product 21 could be efficiently transformed into AG-041R 19 and hexahydrofurano[2,3-b]indole 22 which is the skeleton of the natural product physoveninel.
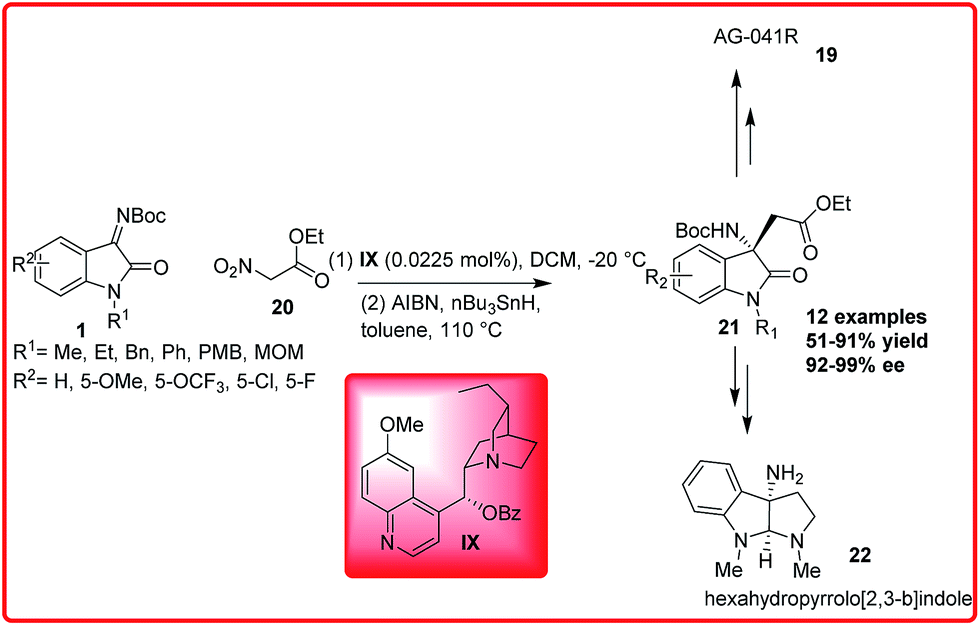 | ||
| Scheme 10 Organocatalytic Mannich/denitration reaction for the asymmetric synthesis of ethyl 2-((S)-3-amino-2-oxoindolin-3-yl)acetate derivatives. | ||
Primary amino acid catalyzed Mannich reaction of N-substituted isatin imines 1 with hydroxyacetone 23 was reported by Peng and co-workers (Scheme 11).25 The chiral amino acid in diethyl ether catalyzed this reaction to afford the anti-Mannich adduct 24b in good to high yield (72–98%) and low to good diastereoselectivity (69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31–88:12 dr) and good to excellent enantioselectivity (79–99%). However, the imine derived from 7-chloroisatin affords the desired adduct in a ratio of 47:53 in favour of syn-diastereomer. When toluene was used as solvent, it provides syn-Mannich adduct 24a in high yield (82–99%) and moderate diastereoselectivity (75:25–83:17 dr) and good enantioselectivity (86–91%). On using isatin imines in toluene, only anti-Mannich product 24b was formed. The reason for this behaviour was explained on the basis of differential E/Z ratio of isatin imine in different solvents. It was assumed that, when this reaction was carried out in ether, the amount of E-isomer was greater than the Z-isomer and the reaction proceeded through transition state TS 10 to provide anti product. But in toluene, the major component was Z-isomer, which afforded syn adduct via TS 11.
:31–88:12 dr) and good to excellent enantioselectivity (79–99%). However, the imine derived from 7-chloroisatin affords the desired adduct in a ratio of 47:53 in favour of syn-diastereomer. When toluene was used as solvent, it provides syn-Mannich adduct 24a in high yield (82–99%) and moderate diastereoselectivity (75:25–83:17 dr) and good enantioselectivity (86–91%). On using isatin imines in toluene, only anti-Mannich product 24b was formed. The reason for this behaviour was explained on the basis of differential E/Z ratio of isatin imine in different solvents. It was assumed that, when this reaction was carried out in ether, the amount of E-isomer was greater than the Z-isomer and the reaction proceeded through transition state TS 10 to provide anti product. But in toluene, the major component was Z-isomer, which afforded syn adduct via TS 11.
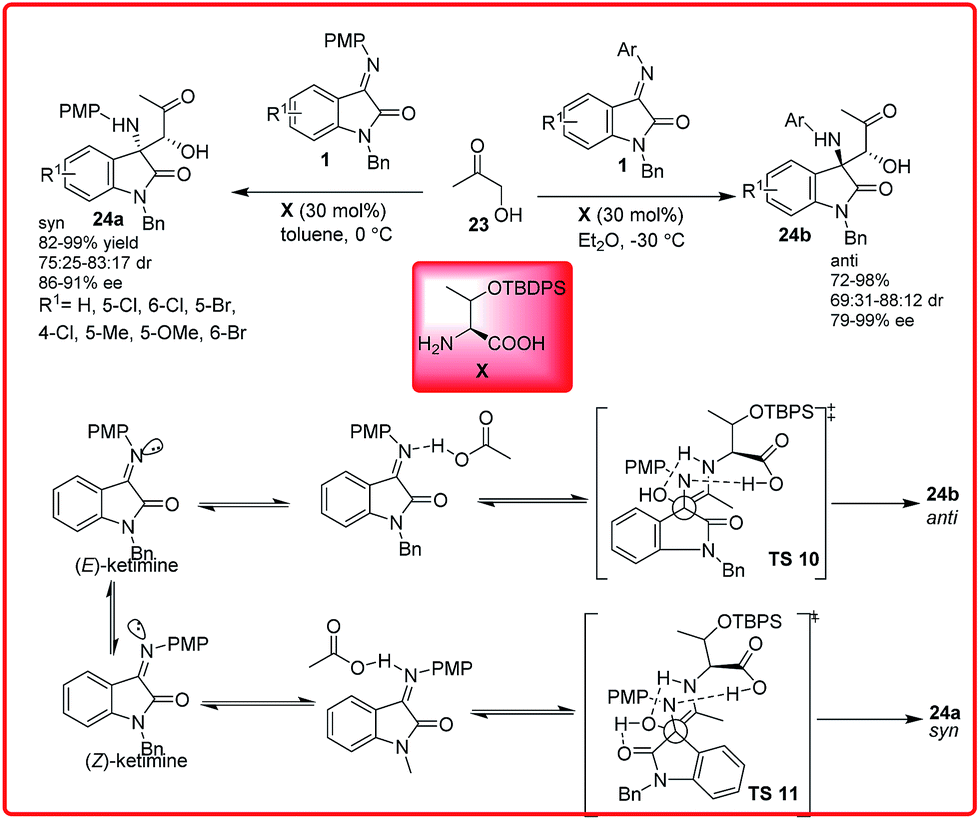 | ||
| Scheme 11 Diastereo-divergent enantioselective Mannich reaction of hydroxyacetone with isatin imines. | ||
Recently Wu et al. reported the Cinchona alkaloid derived thiourea XI catalyzed enantioselective Mannich reaction of pyrazoleamides 25 with isatin imines 1 (Scheme 12).26
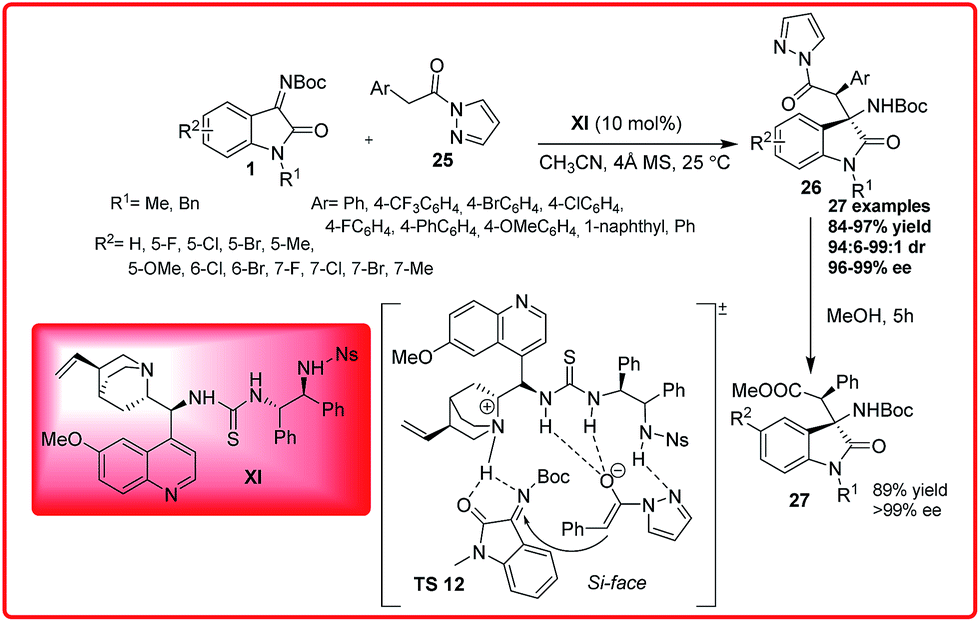 | ||
| Scheme 12 Organocatalytic enantioselective Mannich reaction of pyrazoleamide with isatin imines. | ||
Using 10 mol% of the catalyst XI and molecular sieves as additive, the product with tetra-substituted stereogenic carbon centre at the C-3 position of oxindoles 26 was obtained in good to excellent yield (84–97%) with excellent dr 99:1 and perfect enantioselectivity (96–99%). The pyrazoleamide could easily be transformed to β-amino ester 27 by one pot alcoholysis with high yield (89%) and with excellent enantioselectivity (>99% ee). Methyl 2-phenylacetate and (2-phenyl-1-pyrrolidin-1-yl)ethanone were unreactive nucleophiles with isatin imine suggesting the importance of pyrazole group in facilitating the Mannich reaction. Based on the observed stereochemistry, a plausible transition state TS 12 was proposed in which bifunctional organocatalyst XI deprotonates the pyrazoleamide 25 with quinuclidine nitrogen, while the thiourea moiety binds and activates the ketimine 1 through double hydrogen bond formation with the nitrogen and oxygen atoms. The activated enolized pyrazoleamide approaches the ketimine from one preferred enantioface i.e. Si face to afford the desired product with (3R,1′S) configuration.
Soon after this Wang et al. reported an organocatalyzed asymmetric vinylogous Mannich reaction of γ-butenolides 28 with isatin imines 1 (Scheme 13).27
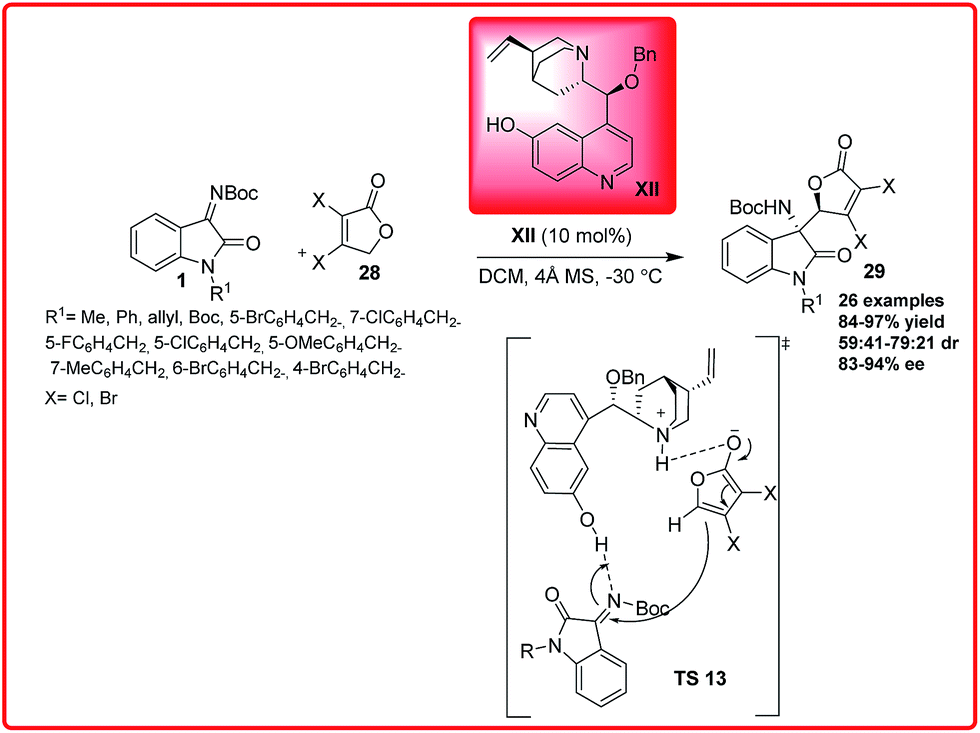 | ||
| Scheme 13 Organocatalytic direct asymmetric vinylogous Mannich reaction of γ-butenolides with isatin imines. | ||
Using Quinidine derived organocatalyst XII, the 3-amino-2-oxindoles 29 bearing adjacent quaternary and tertiary stereocentres were synthesized in good to excellent yield (56–97%) and with moderate to good enantioselectivity (83–96%) with moderate dr of 79:21. On the basis of the experimental results, the transition state TS 13 was proposed in which γ-butenolide is activated by tertiary amine of the catalyst XII and ketimine I is activated by hydroxyl group of catalyst. So, the activated butenolide approaches from one preferred enantioface to afford the desired product 29 with observed stereochemistry.
Wu et al. successfully disclosed an enantioselective Mannich-cyclization reaction of isatin imine 1 with 4-bromo-3-oxobutanoates 30 (Scheme 14).28 Cinchona alkaloid derived squaramide XIII catalyzed the reaction of various isatin imines I with 4-bromo-3-oxobutanoates 30 to provide the 3-substituted-3-amino-2-oxindoles 31 in 90–97% yield and 94–98% enantioselectivity.
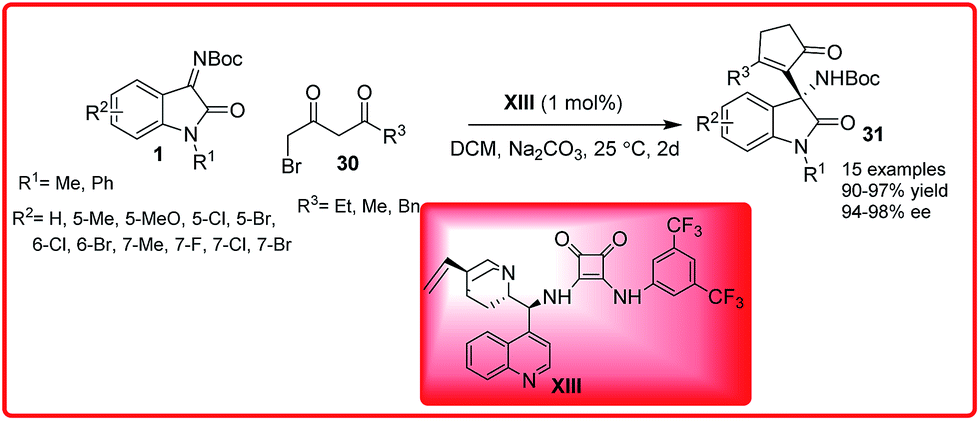 | ||
| Scheme 14 Organocatalytic enantioselective squaramide catalyzed domino-Mannich-cyclization reaction of isatin imines. | ||
Another study on vinylogous Mannich reaction was reported by Deng and co-workers in which AgOAc was used to promote the reaction of isatin imine 1 and trimethylsilyloxyfuran (TMSOF) 32 to provide 3-aminooxindoles 33, which leads to desired products in excellent yields (94–99%) and excellent anti-diastereoselectivity (>99%) under mild conditions, is a frequent core unit in natural products (Scheme 15).29 The ability of AgOAc to promote this reaction was higher than that of other metal salts, such as Cu, Zn, Ni.
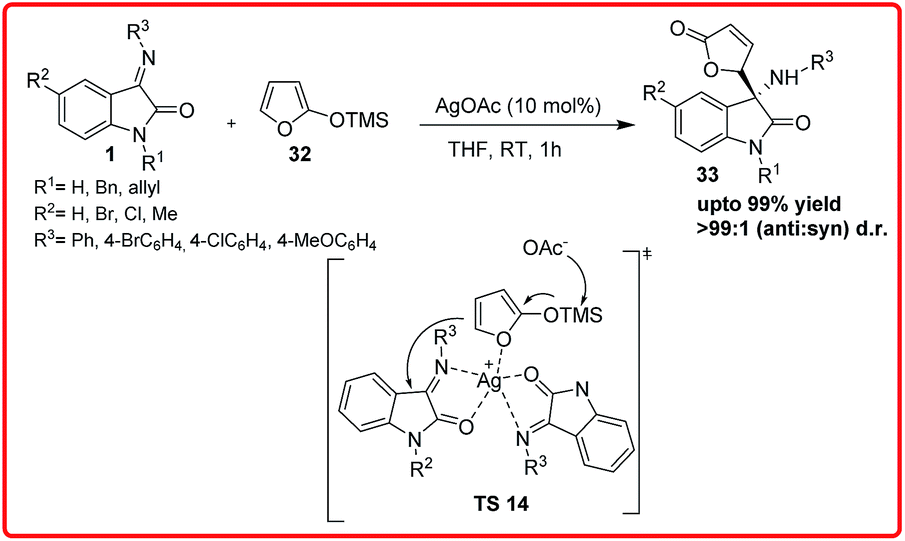 | ||
| Scheme 15 AgOAc catalyzed vinylogous Mannich reaction of isatin imine and trimethylsiloxyfuran (TMSOF). | ||
On the basis of proposed transition state TS 14, the Ag(I) is coordinated as a Lewis acid to two equivalents of the substrate and also acts as a base after coordinating to TMSOF and releasing −OAc, which is the real base that captures the TMS group and consequently promotes the attack of TMSOF over the isatin imine.
Recently, Nakamura et al. have reported the Cinchona alkaloid sulphonamide XIV catalyzed enantioselective addition of thiols 34 to isatin imine 1 to provide N,S-acetals 35 with excellent yield (91–99%) and excellent enantioselectivities up to 97% (Scheme 16).30 Interestingly, the authors demonstrated that a three component version of this reaction could be achieved. The one pot Mannich sequence allowed products to be obtained with comparable enantiomeric excess and yields. In the proposed transition state TS 15, the tertiary amine of the catalyst activates the thiol via H-bonding and simultaneously the ketimine gets activated by pyridine-sulfonamide through H-bonding providing the desired adduct with high enantioselectivity.
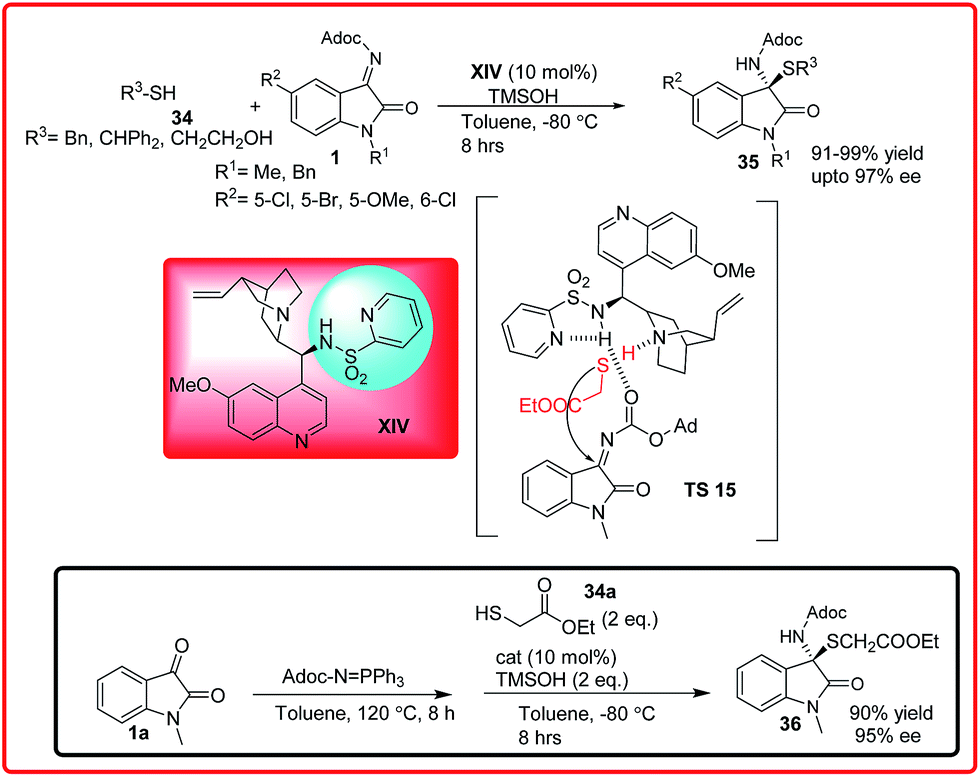 | ||
| Scheme 16 Enantioselective synthesis of N,S-acetals through the addition of thiols to imines and its one-pot asymmetric version. | ||
Ender's group published simultaneously the same reaction. The reaction leads to the formation of chiral isatin derived N,S-acetals 37 in excellent yield up to 98% with excellent enantioselectivities up to 93% (Scheme 17).31 The reaction with non-protected isatin imine resulted in low yields. Both ketimines bearing electron withdrawing and electron donating substituents were well tolerated.
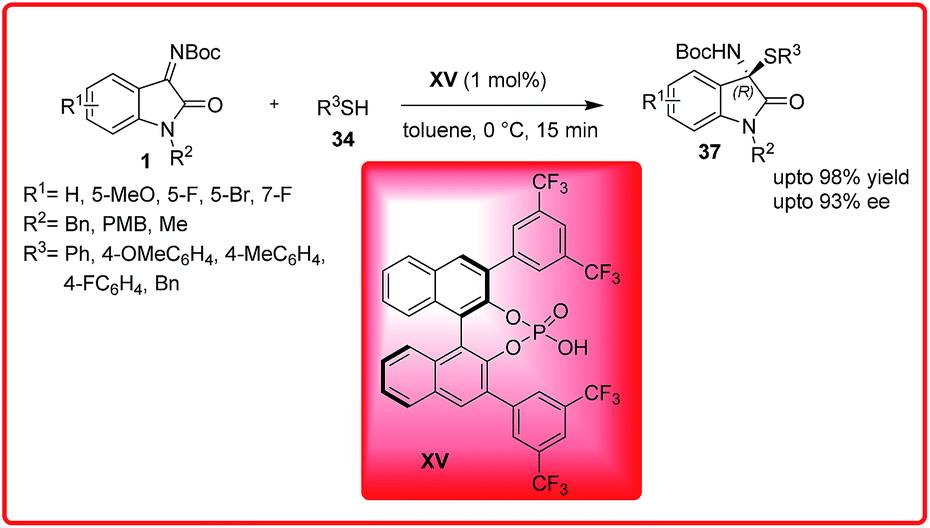 | ||
| Scheme 17 Brønsted acid-catalyzed enantioselective synthesis of isatin-derived N,S-acetals. | ||
An organocatalytic reaction of silyl ketene imines 38 with isatin imines 1 has been recently reported by Feng and co-workers (Scheme 18).32 A variety of β-amino nitriles containing vicinal tetrasubstituted stereocentres 39 were synthesized via N,N′-dioxide/ZnII XVI catalyzed Mannich reaction in excellent yield upto 98% with dr up to >19:1 and with excellent enantioselectivity ranging from 91–99% ee.
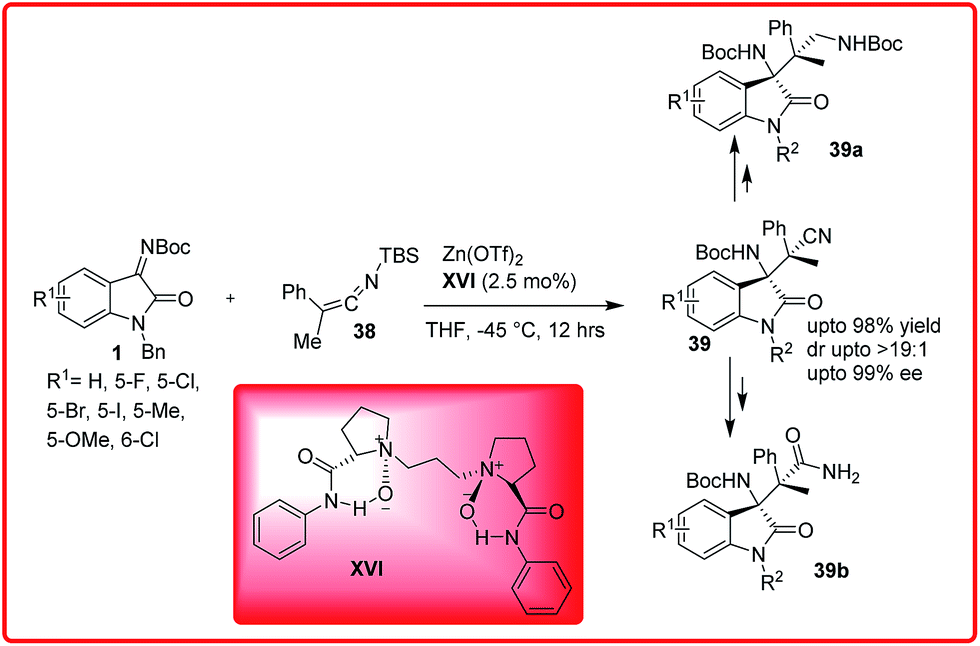 | ||
| Scheme 18 Enantioselective Mannich reaction of silyl ketene imines with isatin imine. | ||
The desired product 39 could be efficiently transformed into 1,3-diamine 39a and β-amino amide 39b which is analogous structure of AG-041R.
(6) Enantioselective Henry reaction
The asymmetric Henry reaction has emerged as a powerful transformation to procure chiral β-nitro alcohols.33 Both chiral organocatalysts and chiral metallic complexes have been developed for this reaction. Isatin and various aldehydes have been used as substrates for asymmetric Henry reaction. The use of isatin imines has been less explored as Henry acceptors for the enantioselective construction of chiral centres.Our group reported the BnCPN XVII catalyzed asymmetric Henry reaction of N-Boc ketimines 1 and nitromethane 40 to provide the aminooxindoles 41 (Scheme 19).34 This method works efficiently with several ketimines to produce the corresponding 3-substituted 3-amino-oxindoles 41 in good yield (68–82%) and moderate to good enantiomeric excess (65–86%).
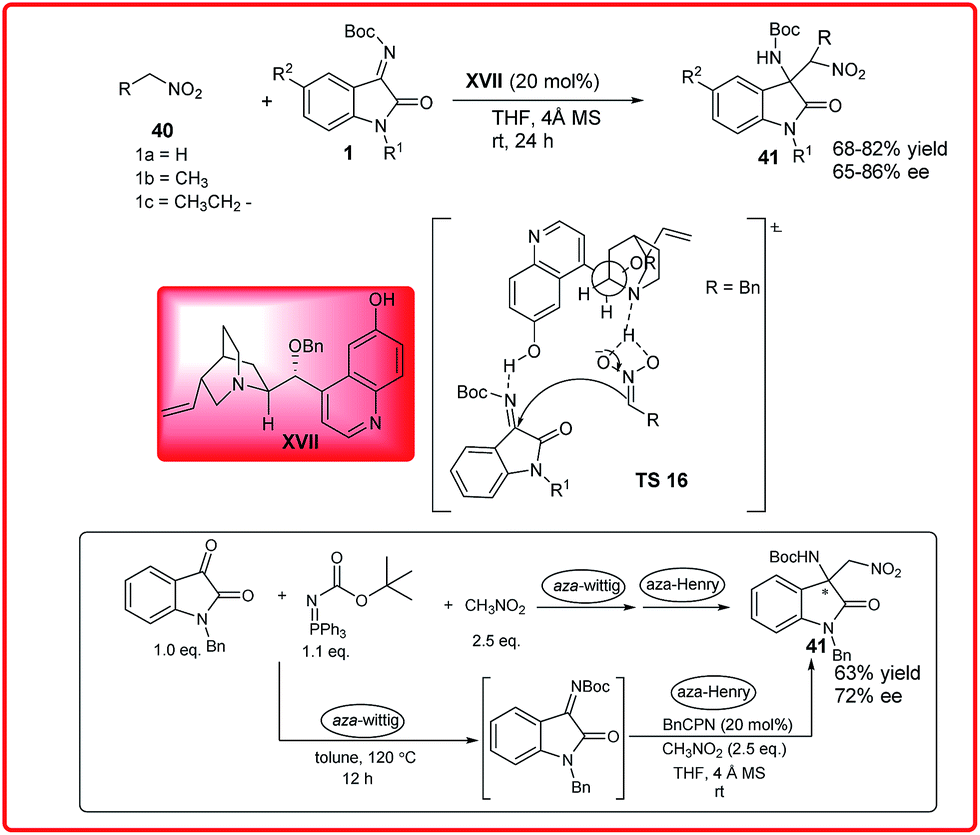 | ||
| Scheme 19 BnCPN catalyzed asymmetric Henry reaction of N-Boc isatin imines with different nitroalkanes. | ||
To improve the synthetic utility, the multicomponent version of this reaction was also studied by combination of the aza-Wittig and aza-Henry reaction in a one-pot sequential protocol. The product 41 was isolated in 63% yield with lower enantioselectivity (72% ee). The mechanism of this reaction was illustrated with the help of designed experiments which indicated the importance of 6′-OH-group of BnCPN for introducing high enantioselectivity.
The catalyst having no free amine moiety failed to catalyze the model reaction, suggesting the role of free amine moiety in deprotonation of nitroalkanes. These results show that the tertiary amine present in the catalyst is a prerequisite for this reaction to occur along with the C6′-OH group, which provides favourable orientation for high enantioinduction. As shown in the transition state TS 16 the catalyst behaves as a bifunctional catalyst by providing favourable orientation and synergistic activation of both the substrate via deprotonation of nitroalkane with quinuclidine nitrogen and activation of ketimine by phenolic –OH of the catalyst.
Subsequently, in the same year, Zhou and coworkers had reported the asymmetric Henry reaction of nitromethane 40 to N-Boc isatin imine 1 and α-ketoester derived N-tosyl isatin imine catalyzed by DBU (Scheme 20).35 Only 5 mol% of catalyst in the presence of molecular sieves efficiently catalyzes the reaction of nitromethane 40 with various isatin imine derivatives 1 to provide an easy access to chiral 3-substituted-3-aminooxindole derivatives 41 in excellent yields (94–99%). They had also reported an enantioselective reaction catalyzed by BnCPN XVII to afford the desired adduct 41 in moderate enantioselectivity (71% ee) but with excellent yield of 93%.
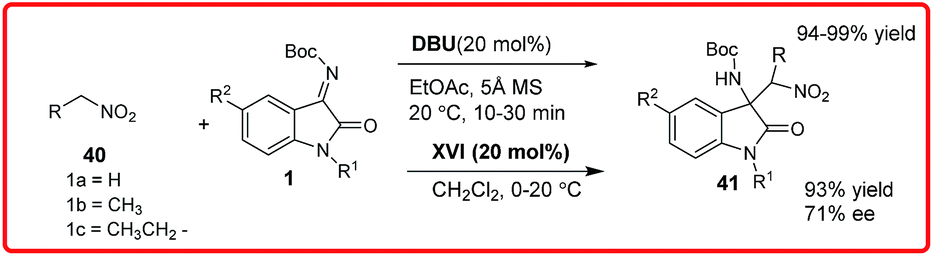 | ||
| Scheme 20 Catalytic enantioselective addition of nitroalkanes to isatin imines. | ||
Then, Arai and co-workers have developed the reaction of nitromethane 40 with isatin imine 1 catalyzed by bis(imidazoline)pyridine–NiCl2 XVIII (Scheme 21).36
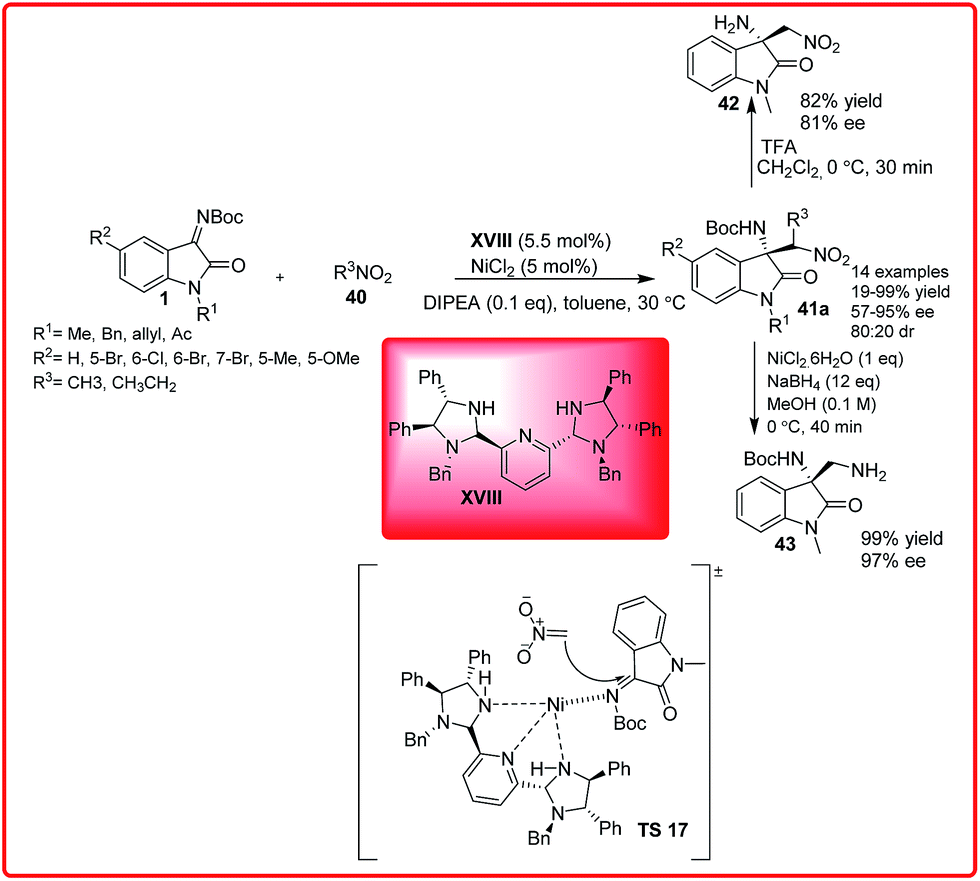 | ||
| Scheme 21 PyBidine–NiCl2 catalyzed asymmetric reaction of nitroalkanes with N-Boc isatin imine. | ||
The 3-amino-2-oxindole 41a was formed with excellent yield (up to 99%) with excellent enantioselectivity (up to 95%) under mild conditions. To illustrate the synthetic utility of the product, the reduction of nitro group leading to 43 and deprotection of Boc group leading to 42 was achieved without loss of enantioselectivity. In the proposed transition state TS 17, the pyridine–NiCl2 complex acts as Lewis acid which activates the ketimines through coordination to the nickel centre through the lone pair of isatin imine which is then attacked by nitronate carbanion to provide the desired adduct.
A similar work was reported by Pedro et al. with Cu(II) BOX XIX complex as a catalyst (Scheme 22).37a The catalyst efficiently catalyzes the reaction to gave the desired Henry adduct 41 in excellent enantioselectivity up to 99.9% with good to excellent yield (84–99%). The nitroamines 41 can be easily transformed to amino nitriles that are useful synthetic intermediates for spirocyclic oxindoles. On treatment with dry HCl in methanol, the amino ester 44 was obtained in 61% yield with 92% enantioselectivity.
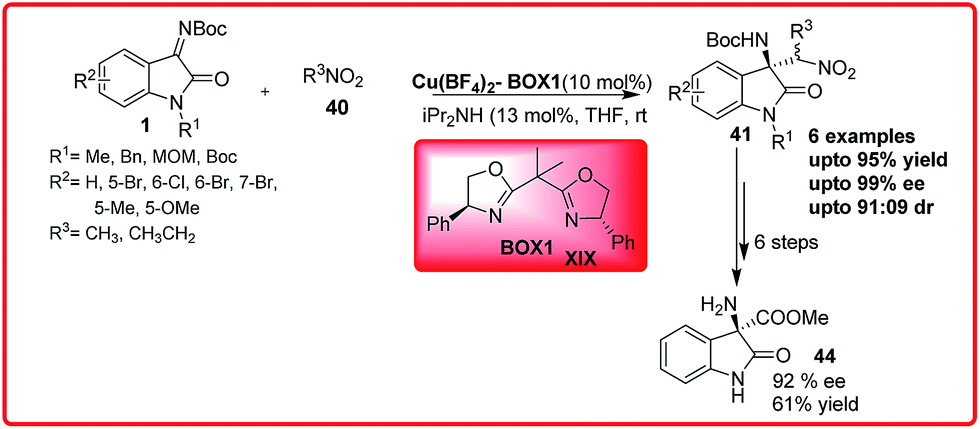 | ||
| Scheme 22 Highly enantioselective aza-Henry reaction of N-Boc isatin imines and nitroalkanes using a Cu(II)–BOX complex as a catalyst. | ||
Recently, the same reaction was reported by Feng and co-workers using chiral guanidine-amide XIX A as a catalyst. The product 41 was formed in excellent yield up to 99% and excellent enantioselectivity up to 94%. The nitro group can be further transformed either to an amine 43a or nitrile 43b in a good yield and enantioselectivity (Scheme 23).37b
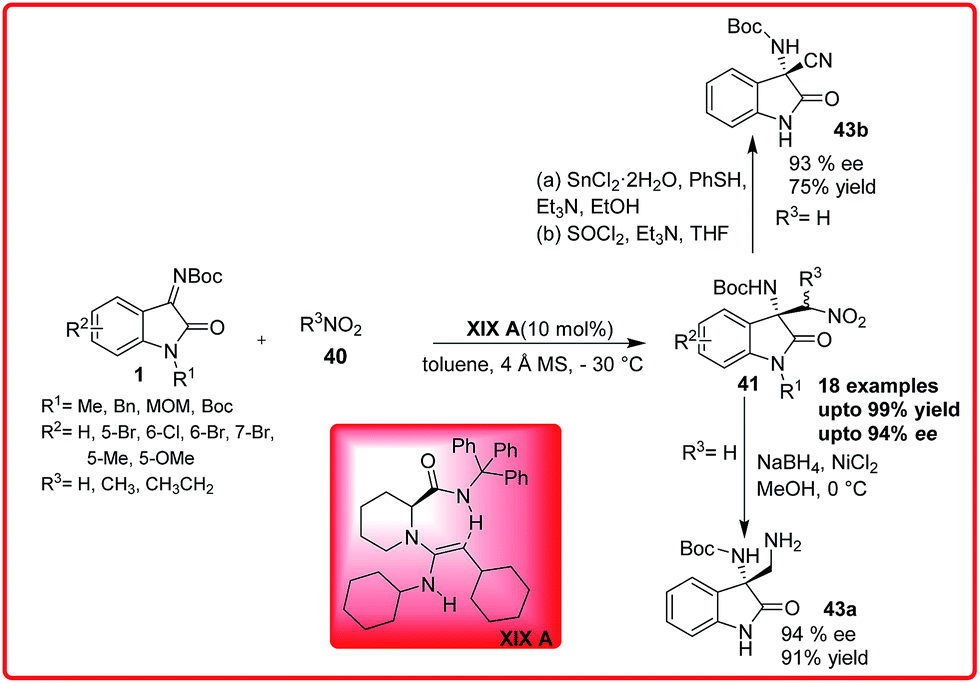 | ||
| Scheme 23 Enantioselective aza-Henry reaction of isatin imine with nitroalkanes catalyzed by chiral bifunctional guanidine catalyst. | ||
(7) Enantioselective Zn-mediated allylation of isatin derived ketimines
Xu et al. reported the zinc-mediated diastereoselective allylation and propargylation of isatin N-tert-butanesulfinyl imines 1 for the synthesis of tetrasubstituted 3-aminooxindoles 46 (Scheme 24).38 The product 46 was formed in good yield (up to 85%) and de of up to 99% 46 could be easily transformed into chiral spirocyclic oxindoles 47 under mild conditions.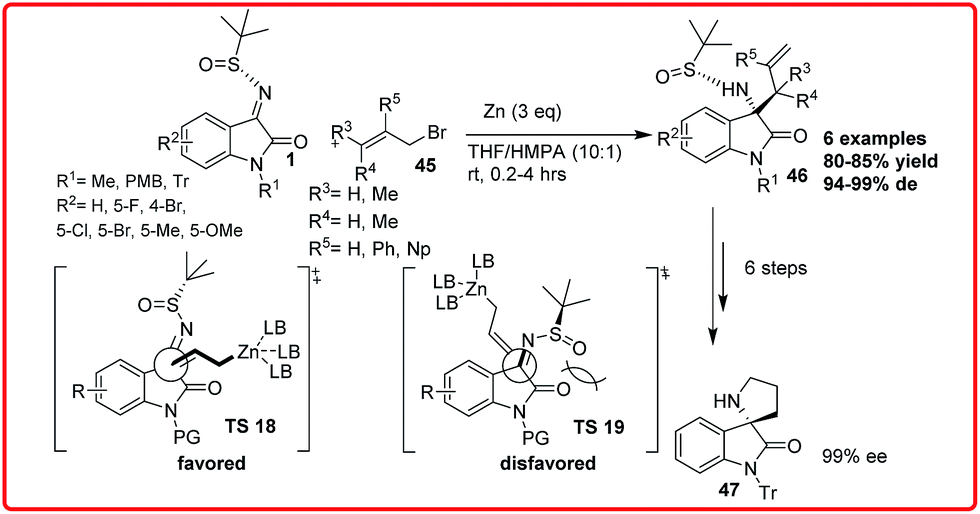 | ||
| Scheme 24 Zn-mediated diastereoselective allylation of N-sulfinyl isatin imine. | ||
In order to rationalize the observed diastereofacial selectivity, the transition state model TS 18 was proposed where the chelation of allyl zinc to imine nitrogen or carbonyl oxygen was less favoured in solution due to the strong metal cation coordination ability of HMPA. The bulky tert-butyl group is positioned at the Si-face of ketimine molecule due to the adoption of synperiplanar configuration by uncoordinated N-sulfinyl group. Thus, the allyl zinc reagent preferentially attacks from the Re-face of ketimine facilitating (S)-amine formation.
(8) The Strecker reaction
The asymmetric addition of cyanide ions to imines to give α-amino nitriles is a useful methodology in organic chemistry.39 The optically active α-amino nitriles are versatile intermediates for the preparation of both natural and unnatural amino acids that have importance in the synthesis of drugs and pharmaceuticals. In the past years, there has been a tremendous growth in the field of asymmetric Strecker reaction, of which, the stereoselective synthesis of 3-aminooxindoles bearing quaternary carbon centre has received great attention due to bioactivity associated with 3-aminooxindoles. The first report on the asymmetric addition of TMSCN 48 to isatin imines 1 was given by Zhou et al. using 10 mol% of newly developed phosphoramide catalyst XX derived from Cinchona alkaloids, 3-cyano-3-amino-2-oxindole derivatives 49 were obtained in low to good yield (27–72%) and moderate enantioselectivity of 39–74% ee (Scheme 25).40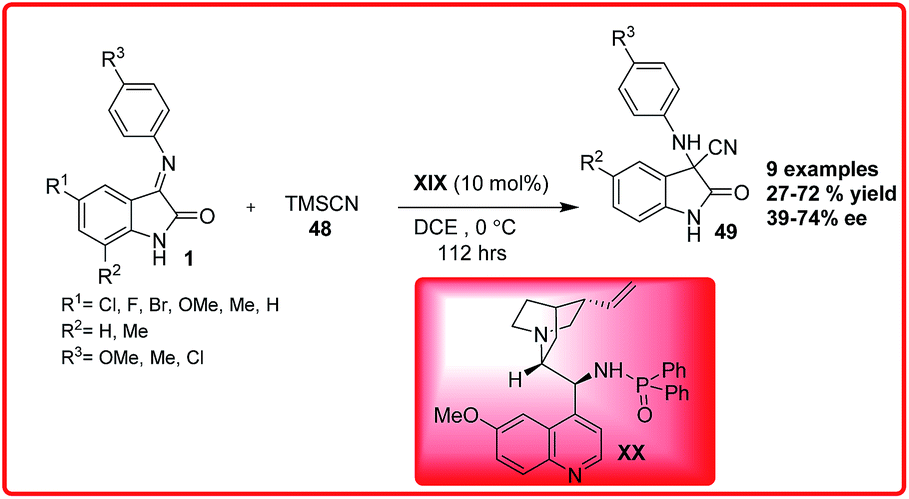 | ||
| Scheme 25 Cinchona derived phosphoramide catalyzed asymmetric Strecker reaction of isatin imines. | ||
An improved version of the asymmetric Strecker reaction of isatin imine was developed by Wang et al. using quinine thiourea XXI as a catalyst. 3-Amino-3-cyanooxindoles 49a was obtained in moderate to excellent yields (65–98%) and with moderate to high enantioselectivity (60–94%) (Scheme 26).41
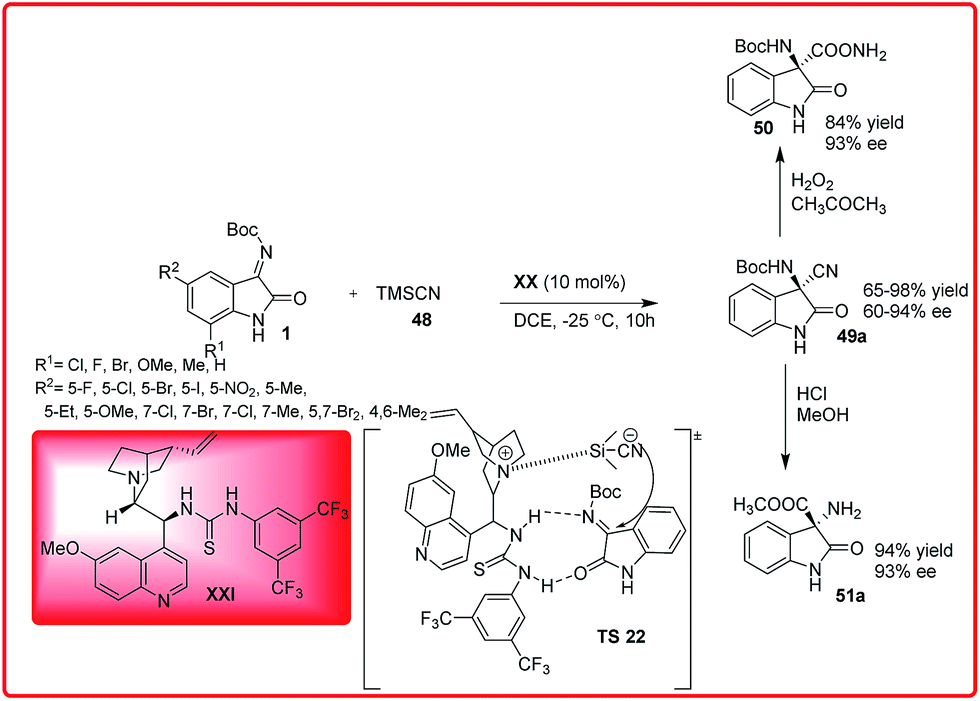 | ||
| Scheme 26 Quinine thiourea catalyzed asymmetric Strecker reaction. | ||
The synthetic utility of this protocol was demonstrated by the conversion of cyano group into amide (50) and ester (51a) without loss of enantioselectivity.
The proposed transition state TS 20 for the enantioselective cyanation reaction involves H-bonding activation of the ketimine through thiourea moiety and simultaneous generation of nucleophilic cyanide from TMSCN by tertiary amine of the Cinchona alkaloid (Scheme 26).
The diastereoselective synthesis of 3-amino-3-cyano-oxindoles 49b was reported by Sacchetti and coworkers using auxillary-based approach for the addition of TMSCN 48 to the chiral ketimines 1, the reaction was performed in the presence of Lewis acid (Scheme 27).42 The desired products 49b were obtained in moderate to good yields (26–88%) and with moderate diastereomeric ratios (55:45–74–26 dr). The cyanide functionality was transformed to obtain amino esters 51 and spirohydantoin 52.
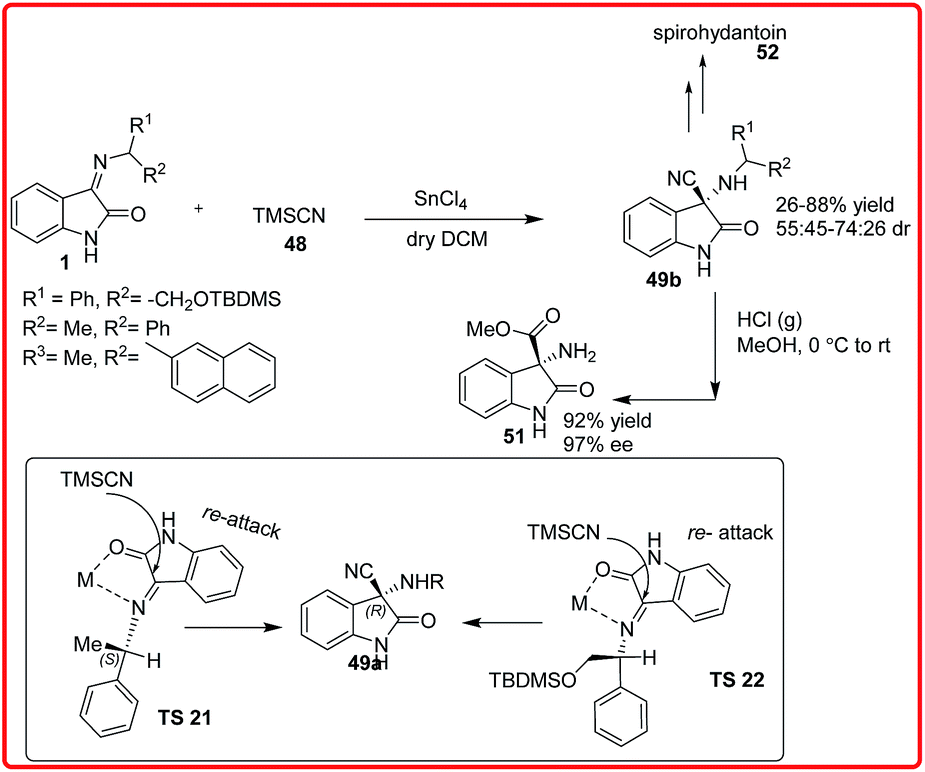 | ||
| Scheme 27 Addition of TMSCN to chiral isatin imines. | ||
Shortly after this, Zhou et al. reported the asymmetric Strecker reaction of isatin imines catalyzed by Cinchonidine derived thiourea VI for the asymmetric synthesis of 3-amino-3-cyanooxindoles 49 in high yield (81–95%) and with excellent enantioselectivity (90–99% ee) (Scheme 28).43
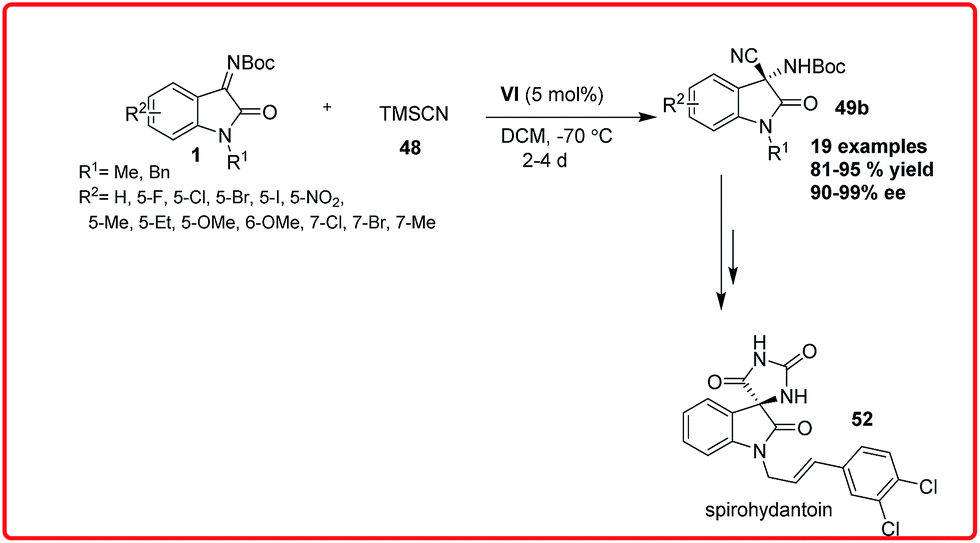 | ||
| Scheme 28 Cinchona derived thiourea catalyzed asymmetric Strecker reaction of N-Boc isatin imines. | ||
A tandem aza-Wittig/Strecker reaction was carried out involving the in situ formation of isatin imine from isatin followed by enantioselective cyanation mediated by catalyst VI to provide the product 49 in 41–86% yield and 76–96% enantioselectivity. The synthetic utility of this methodology was demonstrated by transformation of adducts into spirohydantoin 52. Very recently, Xu and coworkers used chiral auxillary based approach for highly diastereoselective Strecker reaction of isatin sulfinyl ketimines (Scheme 29).44 The addition of TMSCN 48 to isatin sulfinyl imines 1 catalyzed by magnesium bromide diethyl ether and potassium fluoride as cocatalyst gave the desired product in high yield (up to 87%) and with high diastereoselectivity (up to 99%). The synthetic utility of the product was shown by its conversion to an ester 51a without loss of enantioselectivity.
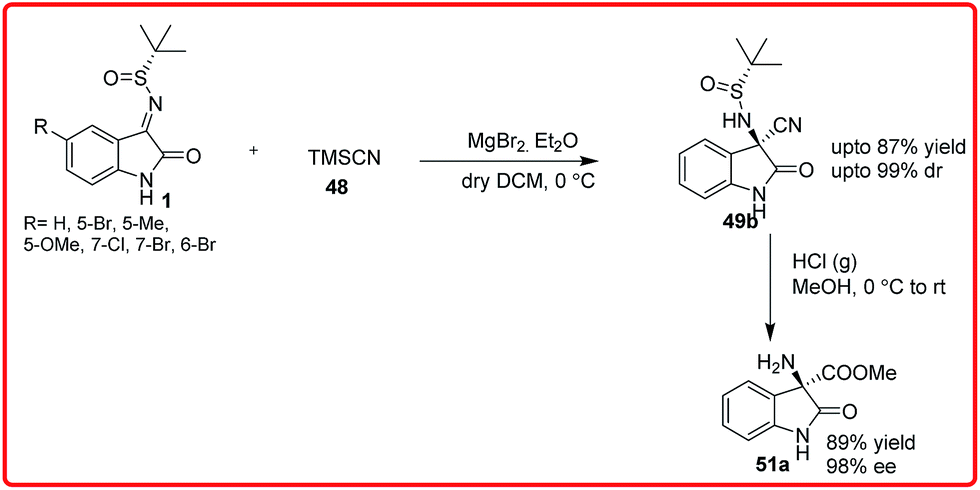 | ||
| Scheme 29 Lewis acid catalyzed diastereoselective addition of TMSCN to N-sulfinyl isatin imines. | ||
(9) Michael reaction
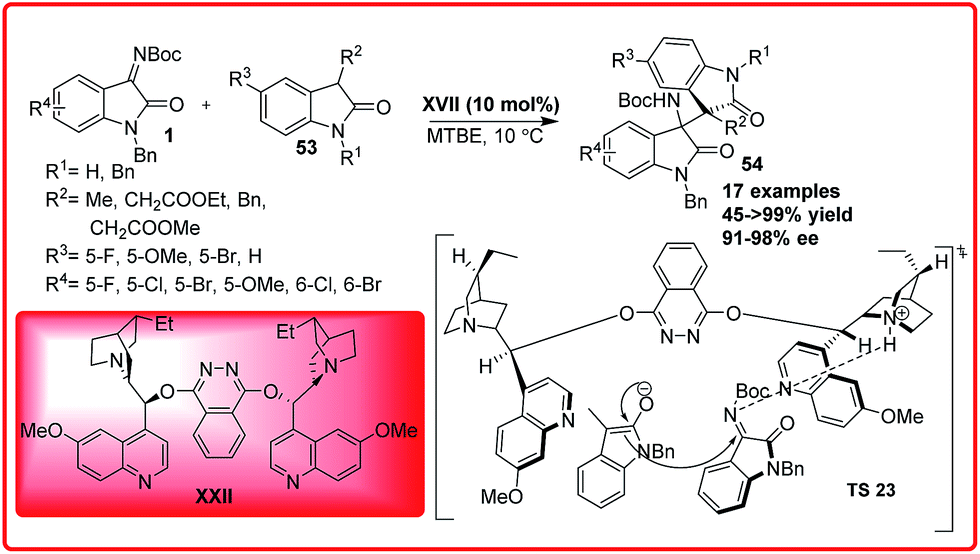
The Michael addition reaction plays an important role among the numerous carbon–carbon bond forming reactions45 which involves the addition of various active methylene compounds to the electron deficient alkenes. In the past few years, numerous classes of nucleophiles have been extensively used in conjugate addition.In 2014, Zhu et al. reported an organocatalytic addition of 3-substituted oxindoles to isatin imines catalyzed by chiral Lewis base to provide bisoxindole 54 with two chiral vicinal quaternary carbon centres in good to excellent yield (45 to >99%), excellent enantioselectivity (91–98%) and >99:1 dr under mild conditions (Scheme 30).46 In addition to this N-benzyl isatin was also found to be reactive providing the desired adduct in good yield (81–88%) but with low enantioselectivity.
 | ||
| Scheme 30 Organocatalytic asymmetric addition of 3-substituted oxindoles to isatin imines. | ||
Based on the experimental results, the transition state TS 23 was proposed where the isatin imine is activated by quinuclidine nitrogen via H-bonding followed by the deprotonation of oxindole enolate which then preferably attacks from Re face of the ketimine to give the desired product in high enantiomeric excess. Sha et al. reported the first asymmetric organocatalytic addition of alcohols 55 to isatin imines 1 using quinine based bifunctional catalyst XXI (Scheme 31).47 3-Substituted-3-amino-2-oxindoles 56 were obtained in good to excellent yield (92–98%) and 63–78% enantioselectivity. The substituent at 6-position had positive effect on the reaction but substituent at 7-position resulted in decrease in enantioselectivity of the product 56. In the proposed transition state TS 24, the ketimine 1 is activated by the thiourea moiety through H-bond interaction while the alcohol gets activated by the tertiary-amine which attacks the ketimine from Re-face to generate the product with (R)-configuration.
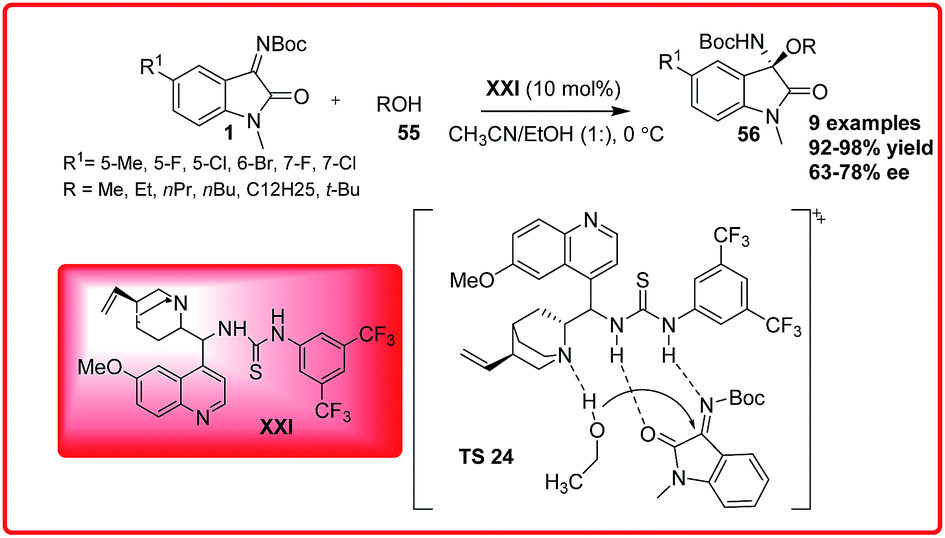 | ||
| Scheme 31 Quinine catalyzed asymmetric addition of alcohols to isatin imines. | ||
(10) N-Heterocyclic carbenes catalyzed reaction
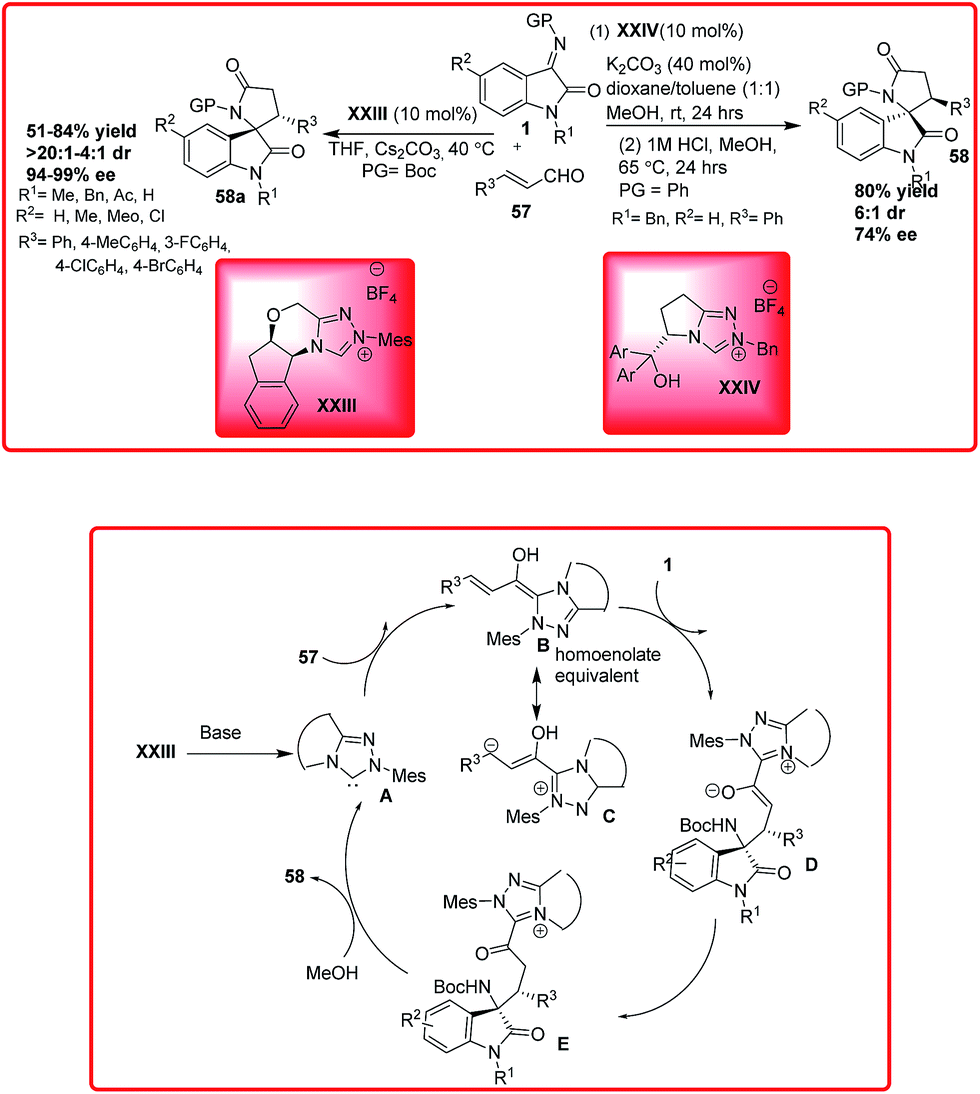
In the past few years, there has been a significant growth in the field of N-heterocyclic carbenes due to their ability to introduce umplong of reaction in aldehydes.48 The classic N-heterocyclic carbene catalyzed reactions such as benzoin condensation and Stetter reaction proceed via nucleophilic Breslow intermediates which are generated by the addition of N-heterocyclic carbenes to aldehydes.49 However, the reaction of enals by N-heterocyclic carbenes has received great attention. In 2012, Jiao and coworkers reported the asymmetric synthesis of spirocyclic-γ-lactam oxindoles 58 using N-heterocyclic carbenes XXIV for the addition of enals 57 to N-aryl isatin imines 1 (Scheme 32).50 Using 10 mol% of NHC XXIV homoenolate equivalents of enals were added to isatin imines 1 and subsequent acid hydrolysis of the addition product generated various spirocyclic oxindoles in 35–88% yield and 1:1–1:6 dr. The catalytic enantioselective version of this reaction was also developed using chiral NHC catalyzed addition of cinnamaldehyde to N-phenyl isatin imine to provide spirocyclic oxindole 58 in 80% yield, 6:1 dr and 74% enantioselectivity.
 | ||
| Scheme 32 NHCs catalyzed enantioselective addition of enals to isatin imines. | ||
Recently, Chi and coworkers have demonstrated the application of N-heterocyclic carbenes XXIII for the reaction of enals 57 to isatin imines 1 for the synthesis of spirocyclic-γ-lactams 58a in moderate to good yield (51–84%), good to high diastereoselectivity (4:1 to >20:1 dr) and excellent enantioselectivity (94–99% ee) (Scheme 32).51 The mechanism of the reaction involves the generation of NHC XXIII by the deprotonation of imidazolium salt in the presence of base which undergoes addition to enal 57 resulting in the formation of Breslow intermediates B and C. The attack of intermediate B on isatin ketimine 1 produces intermediate D which after tautomerization generates intermediate E. Subsequent cyclization of E generates spirocyclic oxindoles γ-lactams 58a with regeneration of NHC to complete the catalytic cycle.
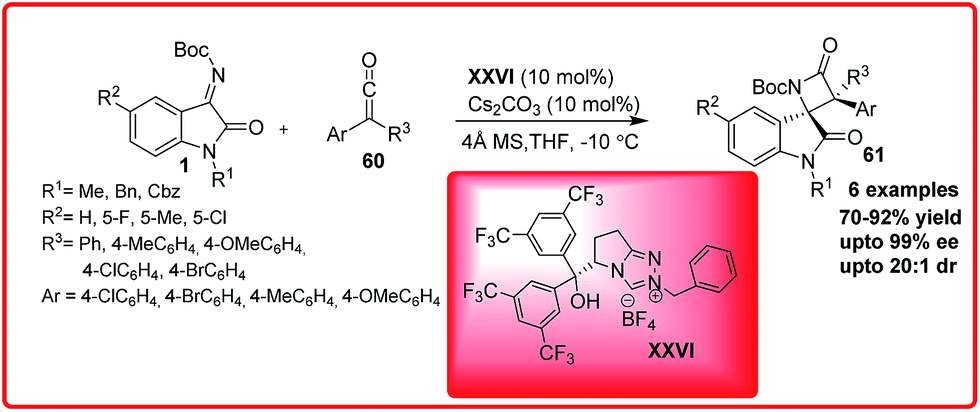
After this pioneering example, the same group reported the chemoselective cross-aza benzoin reaction of isatin imines 1 with enals 57 proceeded via the formation of acyl anion catalyzed by the N-heterocyclic carbene XXV (Scheme 33).52 But the reaction to provide the chiral quaternary aminooxindoles 59 in poor to good yield (10–76%) and er up to 98:2. Ye and co-workers developed a bifunctional N-heterocyclic carbene XXVI catalyzed enantioselective synthesis of spirocyclic oxindolo-β-lactams 61 by performing the reaction of isatin imines 1 with ketenes 60 (Scheme 34).53
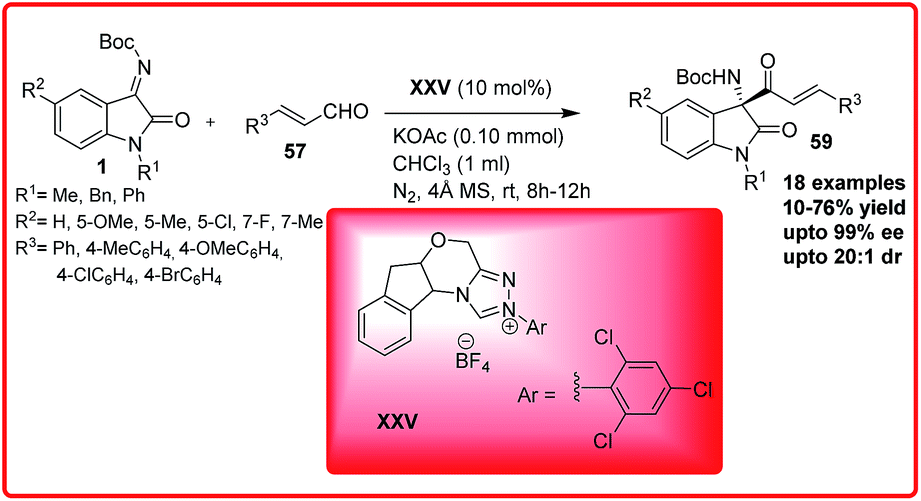 | ||
| Scheme 33 NHC catalyzed chemoselective cross-aza-Benzoin reaction of enals with isatin imines. | ||
 | ||
| Scheme 34 Enantioselective Staudinger reaction of ketenes with imines. | ||
The product 61 having chiral tetrasubstituted stereocentres was obtained in moderate to good yield (70–92%) with excellent enantioselectivity up to 99% and dr up to 20:1. The resulting N-Boc protected β-lactam 61 could be easily deprotected to afford the free β-lactam in high yield without any loss of enantioselectivity.
(11) Ugi reaction
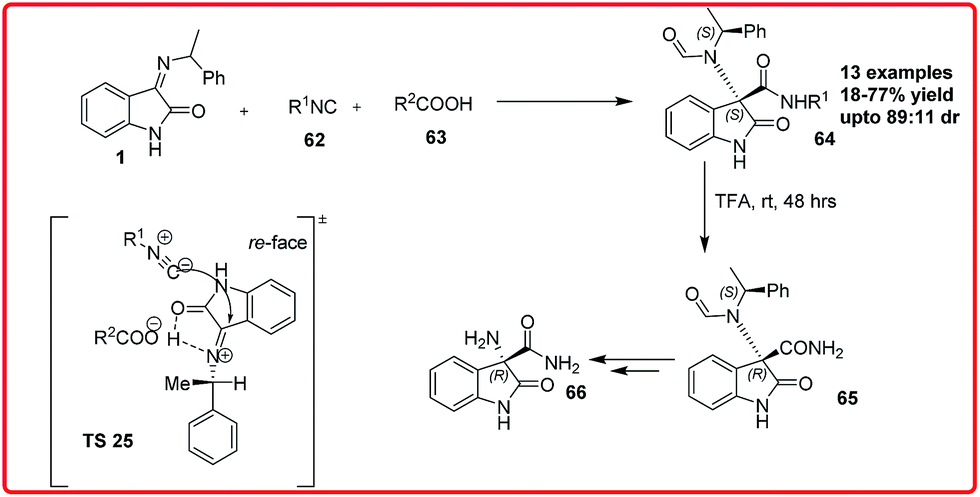
The Ugi reaction was first reported by Ivar Ugi in 1959. This multicomponent reaction with one of its component as an isocyanide results in the formation of α-N-acylamino amide.54 This reaction has received much attention due to its ability to construct complex heterocyclic scaffolds via tandem reaction.Recently Silvani et al. reported the synthesis of optically active 3,3-disubstituted-3-aminooxindoles 64 by means of a three component Ugi reaction of isatin imines 1 with isonitrile 62 and trifluoroacetic acid 63 (Scheme 35).55
 | ||
| Scheme 35 Asymmetric Ugi-three-component reaction with isatin imines. | ||
(12) Miscellaneous reaction
Rhodium-catalyzed reactions are the most versatile for carbon–carbon bond formation among the transition-metal mediated reactions owing to their ability to tolerate a variety of functional groups. The rhodium catalyzed diastereoselective synthesis of 3-amino-3-substituted-2-oxindoles has been reported by three independent groups. B. V. Reddy et al. reported a highly diastereoselective reaction of α-diazoesters 68 with aryl alcohols 67 and isatin imine 1 catalyzed by Rh2(OAc)4 to obtain β-amino-α-hydroxyesters 69 in 80–92% yield, which on deprotection gave 70 in 92% yield (Scheme 36).56 The proposed transition state involves the formation of oxonium ylide from a rhodium carbenoid and alcohol which then attacked the isatin imine 1 through the formation of five-membered transition state. The transition state is stabilized by the intramolecular H-bonding between the H-attached to oxonium ylide and imine N-atom and Π–Π interaction between the aryl group of diazoester and oxindole resulting in the formation of erythro isomer.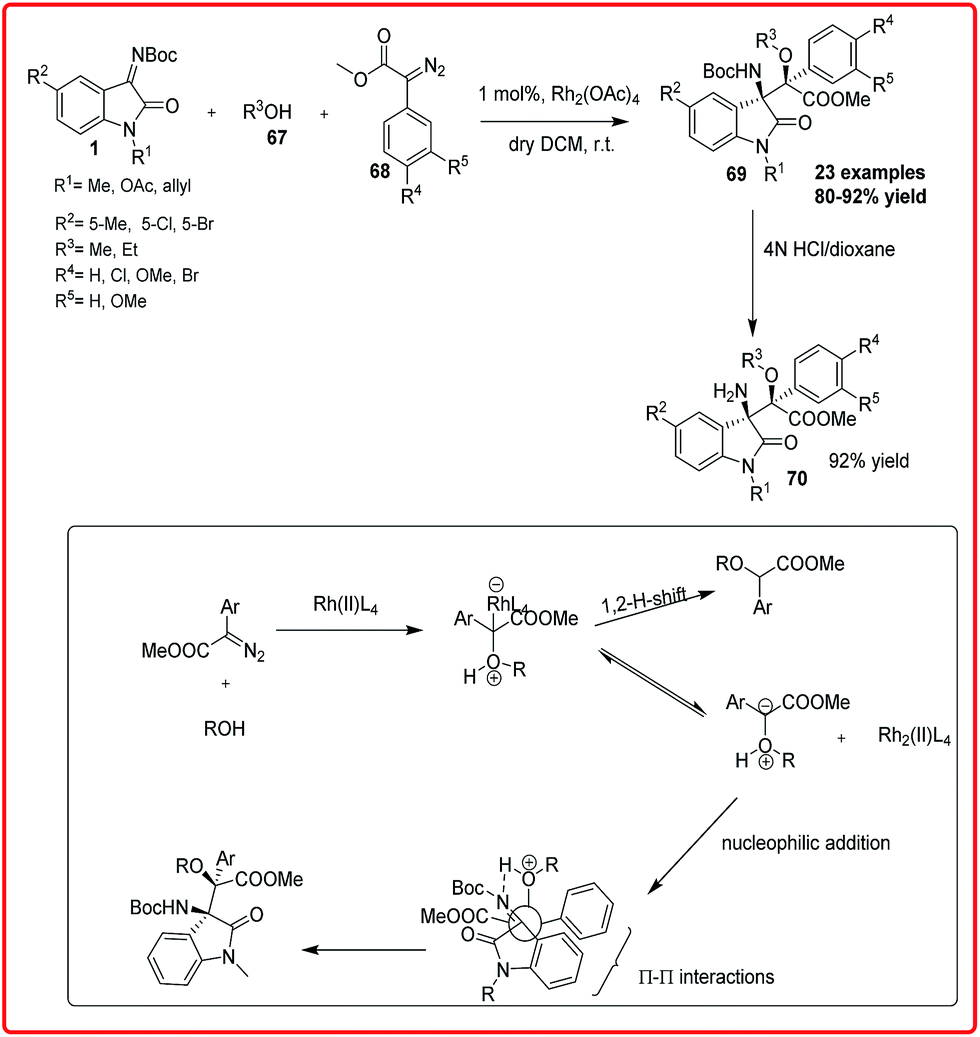 | ||
| Scheme 36 Highly diastereoselective reaction of α-diazoesters with aryl alcohols and isatin imines. | ||
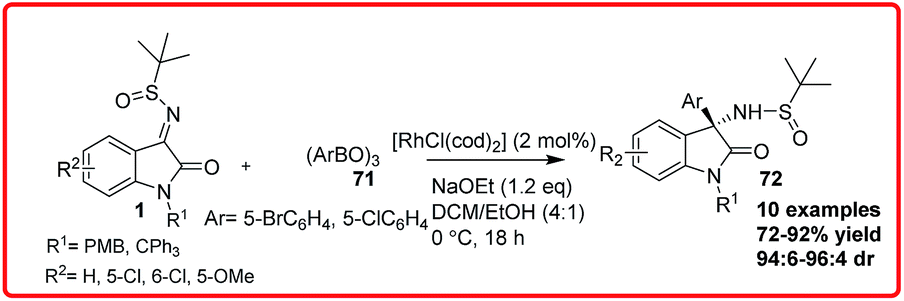
Ellman et al. realized the diastereoselective synthesis of 3-amino-2-oxindoles 72 bearing a tetrasubstituted stereogenic centre in moderate to good yield (72–92%) with good dr (94:6–96:4) via RhCl(cod)2 catalyzed reaction of arylboroxines 69 to isatin imine 1 (Scheme 37).57
 | ||
| Scheme 37 RhCl(cod)2 catalyzed reaction of arylboroxines to isatin imines. | ||
In 2014, Min Shi and coworkers reported the three component reaction of 3-diazooxindoles 73 with indoles 6 and isatin imines 1. Using Rh2(OAc)4 as catalyst, the functionalized 3,3′,3′′-trisindoles 74 were synthesized in moderate to good yield (70–97%) with good dr (5:1 to >20:1) via in situ generation of an active zwitterionic intermediate 75 (Scheme 38).58
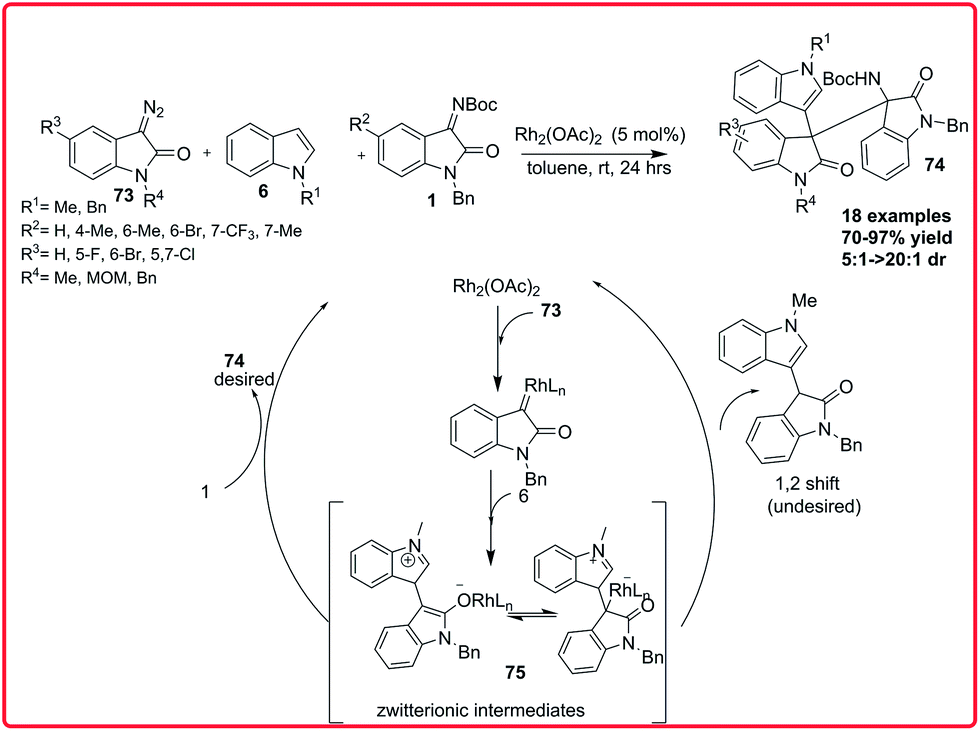 | ||
| Scheme 38 Rhodium catalyzed 3-component reaction of 3-diazooxindoles with indoles and isatin imines. | ||
Liu et al. reported an organo-metal catalyzed one-pot sequential Mannich/hydroamination reaction of isatin imine and propargylated malononitrile (Scheme 39).59 The product was isolated in good yield (up to 91%) and with excellent enantioselectivity (up to 97% ee). The deprotection of the final cyclic adduct resulted into spiro[pyrrolidin-3,2′-oxindole] derivative in 75% yield and 95% enantioselectivity.
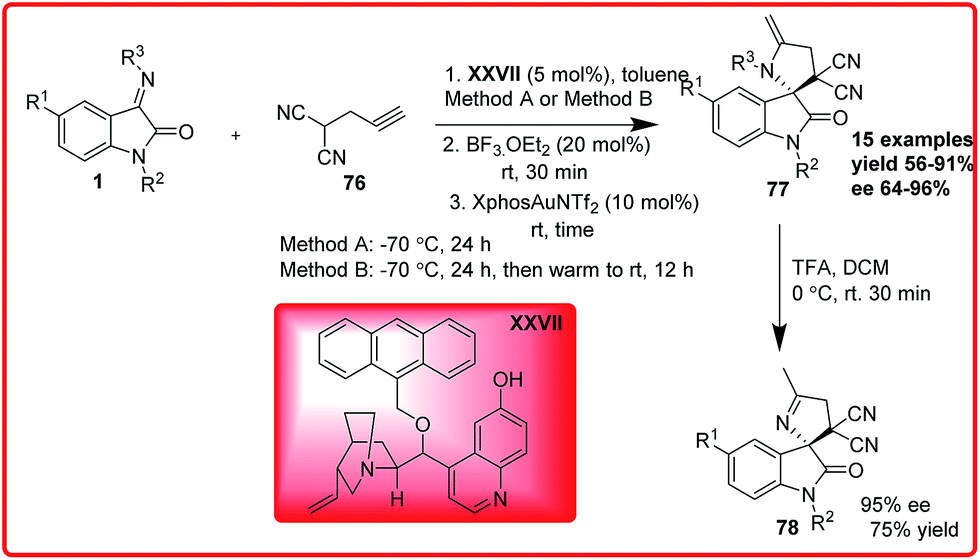 | ||
| Scheme 39 Organocatalyzed and metal catalyzed one-pot sequential Mannich/hydroamination reaction of isatin imine. | ||
(DHQ)2AQN catalyzed asymmetric substitution of isatin derived hydrazones was reported by Shi group. Azo compounds incorporating an oxindole scaffold were obtained in up to 91% yield and 93% ee (Scheme 40).60
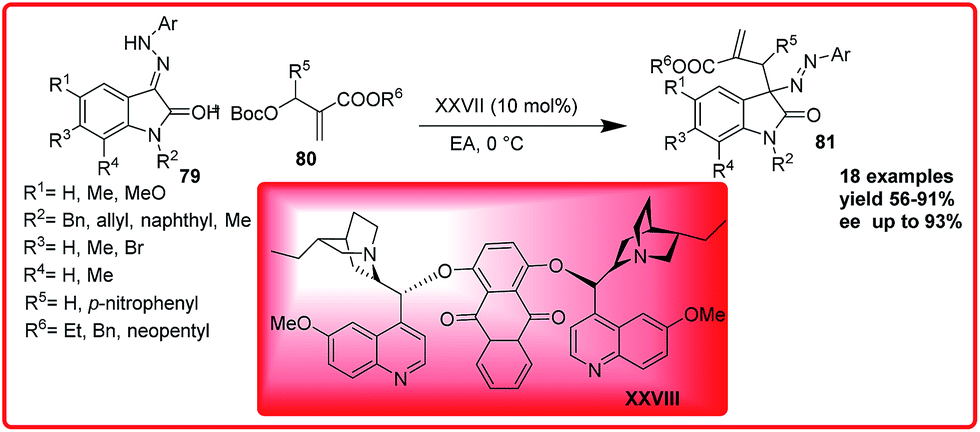 | ||
| Scheme 40 (DHQ)2AQN catalyzed asymmetric reaction of isatin derived hydrazones with O-Boc-protected aza-Morita–Baylis–Hillman adducts. | ||
Sacchetti and co-workers reported the addition of Grignard reagent to isatin imine 82 to obtain 3-substituted-3-aminooxindoles 83 in yield up to 77% and dr up to 95:5. In the proposed transition state TS 26, the MgBr2 coordinates both with the imine nitrogen and the carbonyl oxygen in such a way that the attack of nucleophile is oriented from the less hindered Re face of the imine (Scheme 41).61
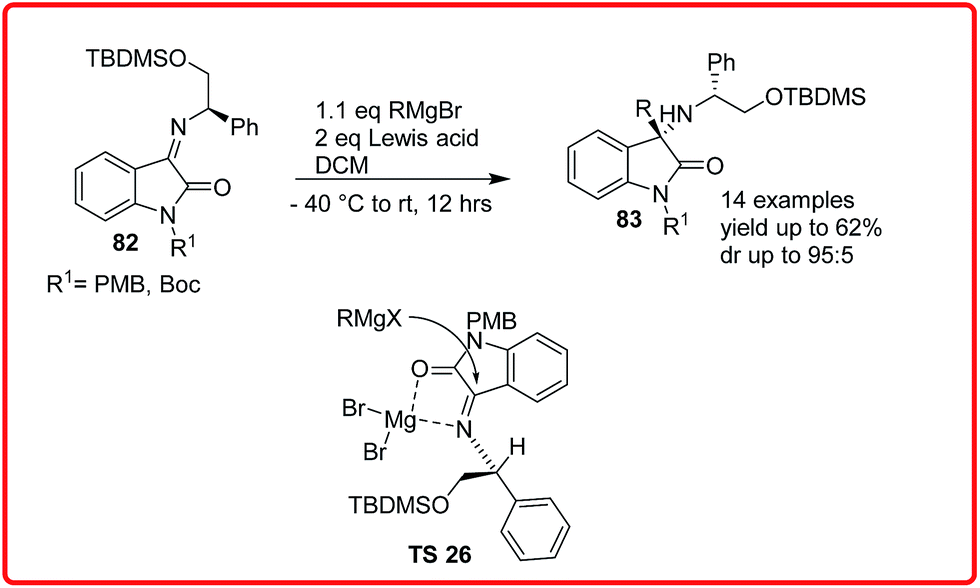 | ||
| Scheme 41 Addition of Grignard reagent to isatin imines. | ||
Copper(I) catalyzed one pot, three component diastereoselective synthesis of 3-spiroazetidinimine-2-oxindoles was reported by Shanmugam group. The product 87 was obtained in up to 77% yield, which undergoes a facile ring opening reaction of the spiroazetidinimine unit on treatment with p-thiocresol or KOH/MeOH to yield products 88 and 89, respectively (Scheme 42).62
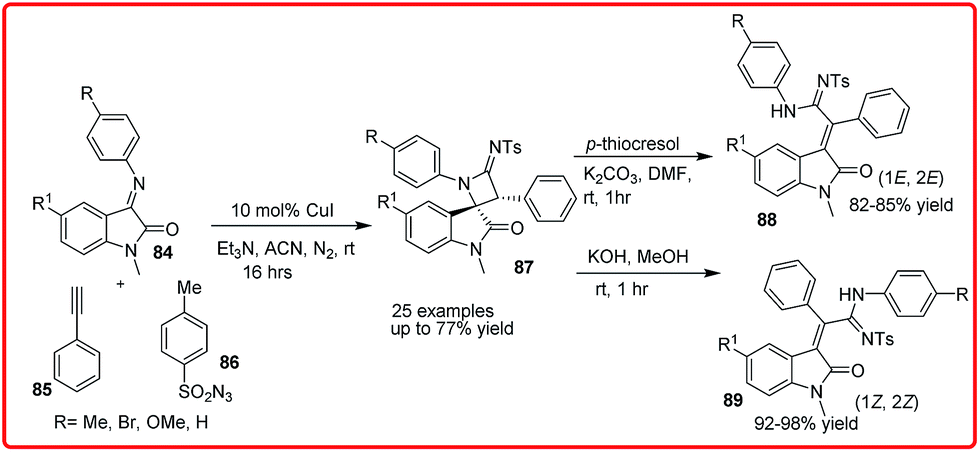 | ||
| Scheme 42 Copper(I) catalyzed one-pot, three-component diastereoselective synthesis of 3-spiroazetidinimine-2-oxindoles. | ||
(13) Summary and outlook
Quaternary 3-amino-2-oxindoles are important and ubiquitous motifs in many natural products and pharmaceuticals. In the past few years, a variety of methods like enantioselective addition of nucleophiles to CN bonds, Mannich reaction, Morita–Baylis–Hillman reaction, Strecker reaction are used to construct 3-substituted-3-amino-2-oxindoles with a chiral quaternary carbon centre. The synthesis of these derivatives has been achieved by using different catalyst providing the product in moderate to good yields and enantioselectivity. This collection provides a broad vision of the current status of research in the synthesis of 3-amino-3-substituted-2-oxindoles using both metal and organocatalysts. Still, there is an extensive room for the addition of various other nucleophiles to isatin imines. We believe that much more can still be done in this area.
Acknowledgements
We are thankful to UGC for SRF(NET) fellowship to JK. This work was supported by the research project (SR/S1/OC-35/2011) sanctioned to SSC by the DST. Financial support from the Department of Science and Technology (DST), India under FIST programme and UGC, India, under CAS-I and UPE programme.Notes and references
- For reviews, see: (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (c) G. S. Singh, M. D'hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed; (d) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS PubMed; (e) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327 RSC; (f) J. E. M. N. Klein and R. J. K. Taylor, Eur. J. Org. Chem., 2011, 6821 CrossRef CAS PubMed; (g) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (h) F. Shi, Z.-L. Tao, S.-W. Luo, S.-J. Tu and L.-Z. Gong, Chem.–Eur. J., 2012, 18, 6885 CrossRef CAS PubMed; (i) F. Shi, G.-J. Xing, R.-Y. Zhu, W. Tan and S. Tu, Org. Lett., 2013, 15, 128 CrossRef CAS PubMed; (j) Y. Wang, F. Shi, X.-X. Yao, M. Sun, L. Dong and S.-J. Tu, Chem.–Eur. J., 2014, 20, 15047 CrossRef CAS PubMed; (k) W. Dai, H. Lu, X. Li, F. Shi and S.-J. Tu, Chem.–Eur. J., 2014, 20, 11382 CrossRef CAS PubMed; (l) F. Shi, R.-Y. Zhu, X. Liang and S.-J. Tu, Adv. Synth. Catal., 2013, 355, 2447 CrossRef CAS PubMed.
- M. Ochi, K. Kawasaki, H. Kataoka and Y. Uchio, Biochem. Biophys. Res. Commun., 2001, 283, 1118 CrossRef CAS PubMed.
- (a) G. Decaux, A. Soupart and G. Vassart, Lancet, 2008, 371, 1624 CrossRef CAS; (b) T. Shimazaki, M. Iijima and S. Chaki, Eur. J. Pharmacol., 2006, 543, 63 CrossRef CAS PubMed.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H. P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed.
- (a) P. Hewawasam, M. Erway, S. L. Moon, J. Knipe, H. Weiner, C. G. Boissard, D. J. Post-Munson, Q. Gao, S. Huang, V. K. Gribkoff and N. A. Meanwell, J. Med. Chem., 2002, 45, 1487 CrossRef CAS PubMed; (b) T. Tokunaga, W. E. Hume, J. Nagamine, T. Kawamura, M. Taiji and R. Nagata, Bioorg. Med. Chem. Lett., 2005, 15, 1789 CrossRef CAS PubMed.
- For a comprehensive review on catalytic asymmetric synthesis of 3-amino-2-oxindoles see: P. Chauhan and S. S. Chimni, Tetrahedron: Asymmetry, 2013, 24, 343 CrossRef CAS PubMed.
- For recent reviews on the Morita–Baylis–Hillman (MBH) reaction, see: (a) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed; (b) C. G. Lima-Junior and M. L. A. A. Vasconcellos, Bioorg. Med. Chem., 2012, 20, 3954 CrossRef CAS PubMed; (c) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (d) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614 CrossRef CAS PubMed; (e) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1 CrossRef CAS PubMed; (f) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (g) Y. Wei and M. Shi, Chin. Sci. Bull., 2010, 55, 1699 CrossRef CAS.
- F. L. Hu, Y. Wei, M. Shi, S. Pindi and G. Li, Org. Biomol. Chem., 2013, 11, 1921 CAS.
- X. Zhao, T. Z. Li, J. Y. Qian, F. Sha and X. Y. Wu, Org. Biomol. Chem., 2014, 12, 8072 CAS.
- A. Kumar, V. Sharma, J. Kaur, N. Kumar and S. S. Chimni, Org. Biomol. Chem., 2015, 13, 5629 CAS.
- For selected reviews on organocatalytic asymmetric Friedel–Crafts reaction, see: (a) S. L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (b) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CrossRef CAS PubMed; (c) M. Zen and S. L. You, Synlett, 2010, 1289 Search PubMed; (d) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 6117 RSC; (e) M. Bandini and A. U. Ronchi, Catalytic Asymmetric Friedel-Crafts Alkylations, Wiley-VCH, Weinheim, 2009 Search PubMed.
- (a) J. Feng, W. Yan, D. Wang, P. Li, Q. Sun and R. Wang, Chem. Commun., 2012, 48, 8003 RSC; (b) J. P. Chen, W. W. Chen, Y. Li and M. H. Xu, Org. Biomol. Chem., 2015, 13, 3363 RSC; (c) M. M. Magraner, C. Vila, R. Cantón, G. Blay, I. Fernández, M. C. Muñoz and J. R. Pedro, Angew. Chem., Int. Ed., 2015, 54, 6320 CrossRef PubMed.
- (a) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS PubMed , For reviews on the enantioselective addition of P-H bond to C-C/C-X double bond, see:; (b) H. Groger and B. Hammer, Chem.–Eur. J., 2000, 943 CrossRef CAS; (c) L. Albrecht, A. Albrecht, H. Krawczyk and K. A. Jorgensen, Chem.–Eur. J., 2010, 16, 28 CrossRef CAS PubMed; (d) D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095 RSC.
- A. B. Smith III, K. M. Yager and C. M. Taylor, J. Am. Chem. Soc., 1995, 117, 10879 CrossRef.
- (a) F. R. Atherton, C. H. Hassall and R. W. Lambert, J. Med. Chem., 1986, 29, 29 CrossRef CAS; (b) J. G. Allen, F. R. Atherton, M. J. Hall, C. H. Hassal, S. W. Holmes, R. W. Lambert, L. J. Nisbet and P. S. Ringrose, Nature, 1978, 272, 56 CrossRef CAS PubMed.
- (a) M. C. Allen, W. Fuhrer, B. Tuck, R. Wade and J. M. Wood, J. Med. Chem., 1989, 32, 1652 CrossRef CAS; (b) J. Bird, R. C. De Mello, H. P. Harper, D. J. Hunter, E. H. Karran, E. R. Markwell, A. J. Miles-Williams, S. S. Rahman and R. W. Ward, J. Med. Chem., 1994, 37, 158 CrossRef CAS; (c) W. M. Smith and P. A. Bartlett, J. Am. Chem. Soc., 1998, 120, 4622 CrossRef CAS.
- D. V. Patel, K. R. Gauvin, D. E. Ryono, A. F. Charles, W. L. Rogers, S. A. Smith, J. M. Deforrest, R. S. Oehl and E. W. Petrillo Jr, J. Med. Chem., 1995, 38, 4557 CrossRef CAS.
- For hydrophosphonylation of ketimines, see: (a) S. Nakamura, M. Hayashi, Y. Hiramatsu, N. Shibata, Y. Funahashi and T. Toru, J. Am. Chem. Soc., 2009, 131, 18240 CrossRef CAS PubMed; (b) L. Yin, Y. Bao, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2013, 135, 10338 CrossRef CAS PubMed , For cyclic imines, see:; (c) H. Xie, A. Song, X. Zhang, X. Chen, H. Li, C. Sheng and W. Wang, Chem. Commun., 2013, 49, 928 RSC.
- J. George, B. Sridhar and B. V. S. Reddy, Org. Biomol. Chem., 2014, 12, 1595 CAS.
- A. Kumar, V. Sharma, J. Kaur, V. Kumar, S. Mahajan, N. Kumar and S. S. Chimni, Tetrahedron, 2014, 70, 7044 CrossRef CAS PubMed.
- For reviews, see: (a) N. R. Candeias, F. P. Montalbano, M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed; (b) A. Córdova, Acc. Chem. Res., 2004, 37, 102 CrossRef PubMed.
- W. Yan, D. Wang, J. Feng, P. Li, D. Zhao and R. Wang, Org. Lett., 2012, 14, 2512 CrossRef CAS PubMed.
- N. Hara, S. Nakamura, M. Sano, R. Tamura, Y. Funahashi and N. Shibata, Chem.–Eur. J., 2012, 18, 9276 CrossRef CAS PubMed.
- K. Zhao, T. Shu, J. Jia, G. Raabe and D. Enders, Chem.–Eur. J., 2015, 21, 3933 CrossRef CAS PubMed.
- Q. X. Guo, Y. W. Liu, X. C. Li, L. Z. Zhong and Y. G. Peng, J. Org. Chem., 2012, 77, 3589 CrossRef CAS PubMed.
- T. Z. Li, X. B. Wang, F. Sha and X. Y. Wu, J. Org. Chem., 2014, 79, 4332 CrossRef CAS PubMed.
- Y. Guo, Y. Zhang, L. Qi, F. Tiana and L. Wang, RSC Adv., 2014, 4, 27286 RSC.
- X. B. Wang, T. Z. Li, F. Sha and X. Y. Wu, Eur. J. Org. Chem., 2014, 739 CrossRef PubMed.
- Y. H. Shi, Z. Wang, Y. Shi and W. P. Deng, Tetrahedron, 2012, 68, 3649 CrossRef CAS PubMed.
- S. Nakamura, S. Takahashi, D. Nakane and H. Masuda, Org. Lett., 2015, 17, 106 CrossRef CAS PubMed.
- C. Beceño, P. Chauhan, A. Rembiak, A. Wang and D. Enders, Adv. Synth. Catal., 2015, 357, 672 CrossRef PubMed.
- J. Zhao, B. Fang, W. Luo, X. Hao, X. Liu, L. Lin and X. Feng, Angew. Chem., 2015, 127, 243 CrossRef PubMed.
- For reviews on the catalytic asymmetric Henry reactions, see: (a) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS PubMed; (b) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315 CrossRef CAS PubMed; (c) C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS PubMed.
- A. Kumar, J. Kaur and S. S. Chimni, RSC Adv., 2014, 4, 24816 RSC.
- Y. H. Wang, Y. L. Liu, Z. Y. Cao and J. Zhou, Asian J. Org. Chem., 2014, 3, 429 CrossRef CAS PubMed.
- T. Arai, E. Matsumura and H. Masu, Org. Lett., 2014, 16, 2768 CrossRef CAS PubMed.
- (a) M. Holmquist, G. Blay and J. R. Pedro, Chem. Commun., 2014, 50, 9309 RSC; (b) B. Fang, X. Liu, J. Zhao, Y. Tang, L. Lin and X. Feng, J. Org. Chem., 2015, 80, 3332 CrossRef CAS PubMed.
- D. Chen and M. H. Xu, Chem. Commun., 2013, 49, 1327 RSC.
- For a review, see: D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC.
- Y. L. Liu, F. Zhou, J. J. Cao, C. B. Ji, M. Ding and J. Zhou, Org. Biomol. Chem., 2010, 8, 3847 CAS.
- D. Wang, J. Liang, J. Feng, K. Wang, Q. Sun, L. Zhao, D. Li, W. Yan and R. Wang, Adv. Synth. Catal., 2013, 355, 548 CAS.
- A. Sacchetti, A. Silvani, F. G. Gatti, G. Lesma, T. Pilati and B. Trucchi, Org. Biomol. Chem., 2011, 9, 5515 CAS.
- Y. L. Liu and J. Zhou, Chem. Commun., 2013, 49, 4421 RSC.
- D. Chen and M. H. Xu, J. Org. Chem., 2014, 79, 7746 CrossRef CAS PubMed.
- For recent reviews of asymmetric Michael addition reactions, see: (a) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688 CrossRef CAS PubMed; (b) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (c) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS; (d) D. Almasi, D. A. Alonso and C. Najera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS PubMed; (e) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS PubMed; (f) S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123 RSC; (g) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS PubMed; (h) S. Jautze and R. Peters, Synthesis, 2010, 365 CAS.
- Z. Tang, Y. Shi, H. Mao, X. Zhu, W. Li, Y. Cheng, W. H. Zheng and C. Zhu, Org. Biomol. Chem., 2014, 12, 6085 CAS.
- T. Z. Li, X. B. Wang, F. Sha and X. Y. Wu, Tetrahedron, 2013, 69, 7314 CrossRef CAS PubMed.
- For reviews: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS PubMed; (c) V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130 CrossRef CAS PubMed.
- For reviews, see: (a) M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 CrossRef CAS PubMed; (b) J. R. de Alaniz and T. Rovis, Synlett, 2009, 1189 Search PubMed.
- B. Zhang, P. Feng, L. H. Sun, Y. Cui, S. Ye and N. Jiao, Chem.–Eur. J., 2012, 18, 9198 CrossRef CAS PubMed.
- H. Lv, B. Tiwari, J. Mo, C. Xing and Y. R. Chi, Org. Lett., 2012, 14, 5412 CrossRef CAS PubMed.
- J. Xu, C. Mou, T. Zhu, B. A. Song and Y. R. Chi, Org. Lett., 2014, 16, 3272 CrossRef CAS PubMed.
- H. M. Zhang, Z. H. Gao and S. Ye, Org. Lett., 2014, 16, 3079 CrossRef CAS PubMed.
- For reviews: (a) I. Ugi, S. Lohberger and R. Karl, Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon Press, New York, 1991, vol. 2, part 2, sect. 4.6, p. 1083 Search PubMed; (b) I. Ugi, A. Demharter, W. Horl and T. Schmid, Tetrahedron, 1996, 52, 11657 CrossRef CAS; (c) A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef CAS.
- G. Lesma, F. Meneghetti, A. Sacchetti, M. Stucchi and A. Silvani, Beilstein J. Org. Chem., 2014, 10, 1383 CrossRef PubMed.
- T. Rajasekaran, G. Karthik, B. Sridhar, S. K. Kumar and B. V. S. Reddy, Eur. J. Org. Chem., 2014, 2221 CrossRef CAS PubMed.
- H. H. Jung, A. W. Buesking and J. A. Ellman, Org. Lett., 2011, 13, 3912 CrossRef CAS PubMed.
- L. Y. Mei, X. Y. Tang and M. Shi, Org. Biomol. Chem., 2014, 12, 1149 CAS.
- X. J. Chen, H. Chen, X. Ji, H. L. Jiang, Z. J. Yao and H. Liu, Org. Lett., 2013, 15, 1846 CrossRef CAS PubMed.
- H.-B. Yang, Y.-Z. Zhao, R. Sang and M. Shi, J. Org. Chem., 2014, 79, 3519 CrossRef CAS PubMed.
- G. Lesma, N. Landoni, T. Pilati, A. Sacchetti and A. Silvani, J. Org. Chem., 2009, 74, 4537 CrossRef CAS PubMed.
- S. Periyaraja, P. Shanmugam and A. B. Mandal, Eur. J. Org. Chem., 2014, 954 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |