
Reverse-hydroformylation: a missing reaction explored
Avanashiappan
Nandakumar
a,
Manoj K.
Sahoo
ab and
Ekambaram
Balaraman
*ab
aCatalysis Division, CSIR-National Chemical Laboratory (CSIR-NCL), Dr. Homi Bhabha Road, Pune - 411008, India
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi - 110025, India. E-mail: eb.raman@ncl.res.in; balaraman2002@yahoo.com
First published on 19th August 2015
Abstract
Recent progress in transition-metal catalysed acceptor- and acceptorless-reverse hydroformylation of aldehydes for the conversion of olefins has been discussed. The aldehyde feedstock serves as a source for production of syngas and valuable alkenes.
 Avanashiappan Nandakumar | Avanashiappan Nandakumar received his Bachelor's degree in Chemistry (2006) from Gobi Arts & Science College, Gobichettipalayam. He received his Master's degree in Applied Chemistry (2009) from Anna University, Chennai, and subsequently joined the CSIR-Central Leather Research Institute as a Ph.D. student with Dr P. T. Perumal. He was awarded a Ph.D. in 2014 by the University of Madras and is a recipient of a SERB-Young Scientist Award, India. He will be joining Dr E. Balaraman's research group, focusing on transition-metal-catalyzed annulation reactions. |
 Manoj K. Sahoo | Manoj Kumar Sahoo received his Bachelor's degree in Chemistry (2008) from Narasingha Choudhury (Autonomous) College, Jajpur affiliated to Utkal University. He received his Master's degree in Chemistry (2011) from Utkal University, Orissa. Currently he is working as a Junior Research Fellow under the guidance of Dr E. Balaraman at CSIR-National Chemical Laboratory. His research focuses on photoredox catalysis and green chemistry. |
 Ekambaram Balaraman | Ekambaram Balaraman received his M.Sc degree in Chemistry from the University of Madras, India (2002). He was awarded a Ph.D in Chemistry from the Central University of Hyderabad, India (2008). He was an FGS-Post Doctoral Fellow at the Weizmann Institute of Science (2008–2012). Currently he is working as a Senior Scientist at CSIR-National Chemical Laboratory in the Catalysis Division. His research interests are focused on metal-mediated organic transformations and photoredox catalysis. |
Development of new and sustainable catalytic systems for fundamentally important reactions is an important and intellectually stimulating challenge. Hydroformylation, the so-called “oxo-process”, is a very important and widely used reaction in the chemical industry for synthesizing value-added bulk and fine chemicals and represents one of the leading applied homogeneously catalysed reactions (Fig. 1a).1 However, the reverse reaction, “dehydroformylation”, is a thermodynamically uphill process and has been gaining much attention of chemists in recent years. It was previously observed as a side reaction in the decarbonylation and related reactions.2b,c Inspired by the biosynthesis of sterols via dehydroformylation (Fig. 1b), several transition-metal catalysed dehydroformylation of aldehydes have been attempted in the past few decades.2 However, the dehydroformylation either with the concomitant generation of synthesis gas (gaseous CO + H2) or transfer of the formyl group (–CHO) to an olefin acceptor is a missing reaction in chemical science. Thus, in the development of a more appropriate catalytic system for dehydroformylation reaction under mild conditions, one can efficiently utilize the renewable oxygenated biomass derivatives to produce value-added chemicals, such as monomers, solvents, and intermediates for bulk and fine chemicals.
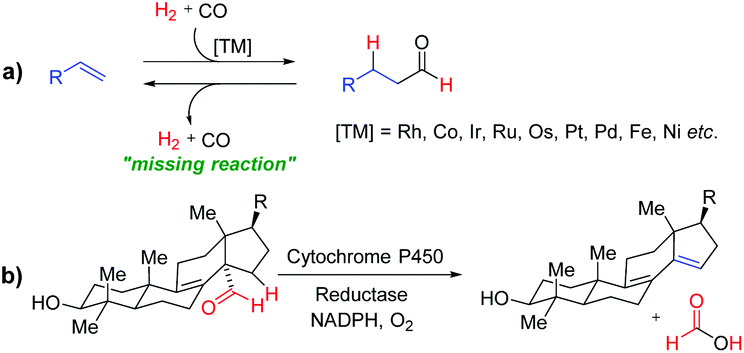 | ||
| Fig. 1 (a) Transition-metal catalyzed hydroformylation of olefins. (b) Dehydroformylation during biosynthesis of sterols. | ||
In 1990, Watanabe et al. reported the Ru-complex catalysed intermolecular hydroacylation reaction (addition of aldehydic C–H to the olefinic bond).2a However, the reaction of the aliphatic aldehyde n-heptanal with cyclohexene by using the same catalytic conditions resulted in 29% yield of cyclohexanecarboxaldehyde along with a mixture of esters (∼9%) at 200 °C. This reaction is termed ‘transfer-hydroformylation’, i.e. transfer of CO and H2 takes place from the donor aldehyde to the acceptor olefin at the metal centre in the absence of synthesis gas (Fig. 2). It was observed that the aldehyde is the source of hydrogen and carbon monoxide3 and the lower yield of the formylated product clearly indicated that the transformation is kinetically controlled by CO liberation from the metal centre before the hydroformylation of the acceptor olefin takes place. Notably, in the seminal work of Dong and co-workers, the kinetically as well as thermodynamically balanced ‘transfer-hydroformylation’ of an aliphatic aldehyde to an olefin has been successfully achieved by using the Rh(Xantphos) complex as a catalyst (Fig. 2). The reaction is facilitated by the introduction of a strained olefin as a sacrificial carbonyl as well as a hydrogen acceptor enabling the formation of a thermodynamically more stable olefin from the aliphatic aldehyde.4a
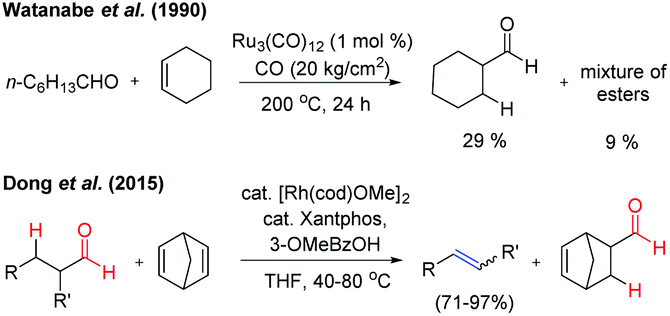 | ||
| Fig. 2 Transfer-hydroformylation reaction. | ||
The mechanistic insights into transfer-hydroformylation reveal that the benzoate counter ion acts as a proton shuttle via a reversible redox process and plays a crucial role in the conversion of aliphatic aldehydes into thermodynamically favourable olefins (Fig. 3). The initial coordination of the benzoate ion triggers the oxidative addition of the aldehydic C–H bond at the Rh-centre. This is followed by the reductive elimination as benzoic acid allowing the conversion of the aldehyde into an olefin via decarbonylation and β-hydride elimination. The kinetically more favourable hydroformylation of the strained olefin can precede the liberation of gases CO and hydrogen from the metal centre. Notably, the hydroacylation of the sacrificial olefin and the decarbonylation of the aldehyde at the Rh-metal center are found as equally critical steps in the catalytic transformation. Thus, the choice of donor aldehyde–acceptor olefin determines the success of transfer-hydroformylation reaction.
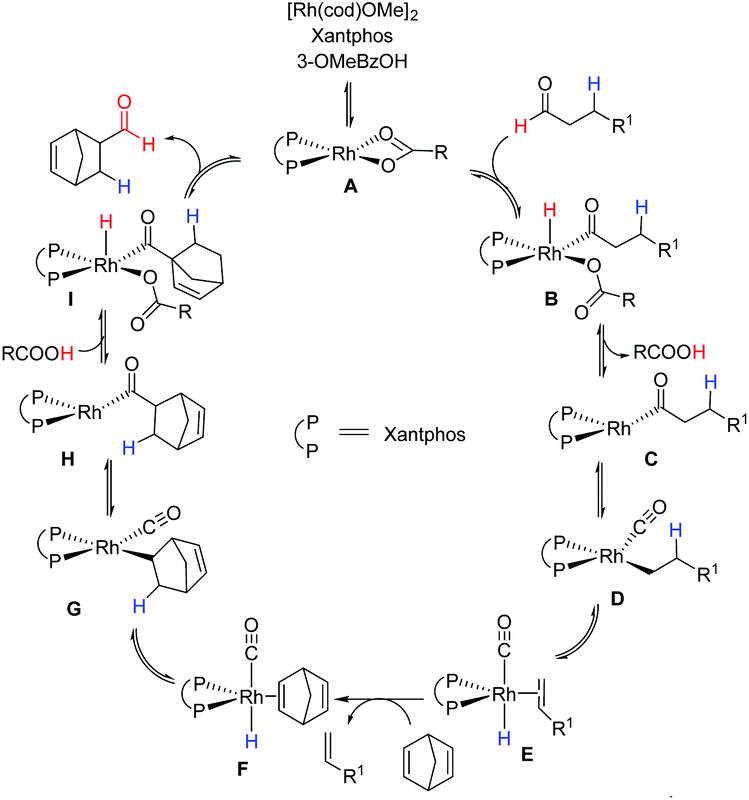 | ||
| Fig. 3 A proposed mechanism for the transfer-hydroformylation.4a | ||
The transfer-hydroformylation reaction has a broad substrate scope as well as functional group tolerance. Importantly, many synthetically valuable groups such as dienes, hydroxyl, amines, ethers, esters and acetals could be tolerated under standard reaction conditions. Noteworthy progress in the conversion of a commercially available inexpensive (+)-yohimbine to a natural product (+)-yohimbenone with high chemoselectivity was achieved in three steps utilizing transfer-hydroformylation as one of the key steps (Fig. 4). This novel strategy might open a new avenue for the development of completely gasless (avoiding the use of syngas) hydroformylation reaction to access complex and valuable alkenes from the aldehyde feedstock by utilizing an inexpensive norbornadiene (nbd) as a sacrificial acceptor.4
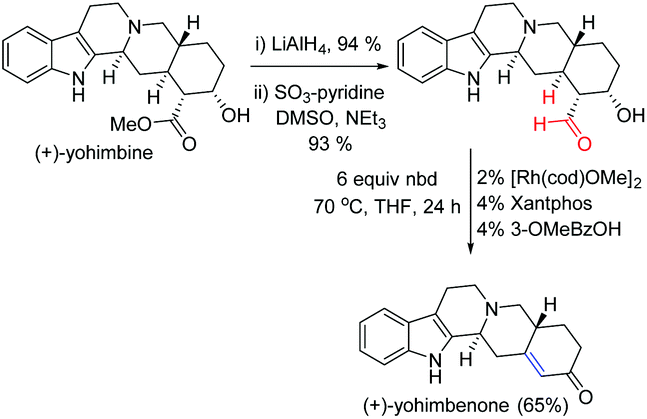 | ||
| Fig. 4 Synthesis of (+)-yohimbenone using the Dong's transfer-hydrogenation as one of the key steps.4a | ||
In 2008, Tsuji et al. reported the Ir-catalyzed decarbonylation of aldehydes.2c The aldehydes containing β-hydrogen on sp3-carbon resulted in the decarbonylation product besides a trace amount of alkenes owing to β-hydride elimination (Fig. 5). The formation of alkenes is due to reversible hydroformylation reaction of aldehydes. Very recently, Nozaki and co-workers have reported an unprecedented retro-hydroformylation reaction5 catalysed by a well-defined iridium-based catalyst for the direct, selective conversion of aliphatic aldehydes into the corresponding alkenes with the concomitant generation of syngas (Fig. 5). This is the first report on the utilization of the hydroxycyclopentadienyl iridium complex in association with N-heterocyclic carbene ligands, such as N,N-bis(2,6-diisopropylphenyl)imidazolylidene (IPr), as an efficient catalytic system for the acceptorless dehydroformylation reaction, though the cyclopentadienyl based metal complexes are well explored in the metal–ligand cooperation mechanism.6 Based on the mechanistic insights a possible sequential decarbonylation–dehydrogenation or dehydrogenation–decarbonylation pathway was completely ruled-out. The initial oxidative addition of the aldehydic C–H bond followed by simultaneous liberation of CO and H2 resulted in the relatively more stable olefins.
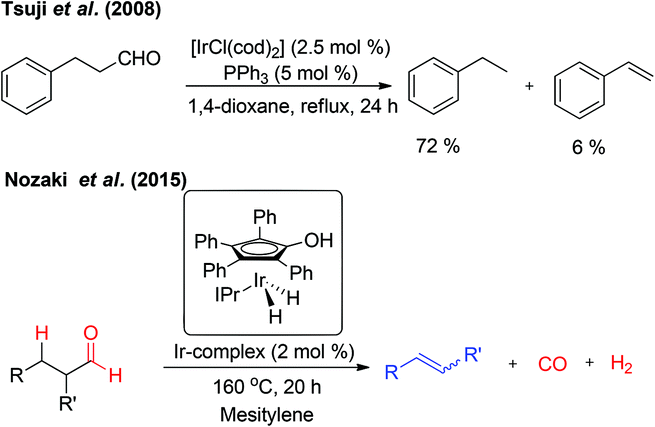 | ||
| Fig. 5 Retro-hydroformylation reaction. | ||
Cyclic and acyclic aliphatic aldehydes were successfully converted into the corresponding olefins with the concomitant elimination of syngas enabling a sustainable catalytic approach to syngas production from the aldehyde feedstock (Fig. 6). Cyclic aldehydes yielded the corresponding olefins along with a minor amount of reduced alcohols whereas acyclic olefins were more susceptible to hydrogenation of as-yielded olefins. In order to overcome the undesired hydrogenation reaction, acyclic aldehyde transformation was carried out under continuous argon bubbling. This fundamentally important reaction might provide a new strategy for the production of syngas and gasification of biomass derivatives under very mild conditions.
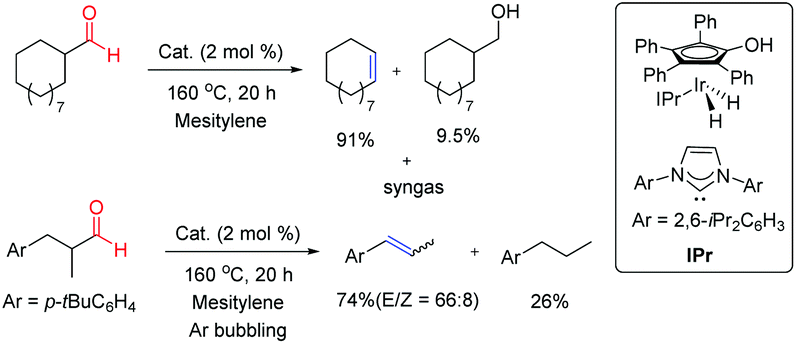 | ||
| Fig. 6 Retro-hydroformylation of aliphatic aldehydes.5 | ||
In summary, this report reflects the remarkable progress in the acceptor- and acceptorless-dehydroformylation process for the conversion of a large feedstock of aliphatic aldehydes into more valuable olefins marking an important milestone in the area of chemical synthesis. Future work will be directed towards the utilization of more economical first row transition-metals for the dehydroformylation process and renewable biomass derivatives as an alternative for the effective production of synthesis gas enabling a sustainable approach for synthesis gas production.
This research was supported by the SERB (SB/FT/CS-065/2013) and CSIR-NCL (MLP028726) and M. K. Sahoo acknowledges the CSIR fellowship.
Notes and references
- (a) J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2012, 52, 2852 CrossRef PubMed; (b) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed.
- (a) T. Kondo, M. Akazome, Y. Tsuji and Y. Watanabe, J. Org. Chem., 1990, 55, 1286 CrossRef CAS; (b) M. Kreis, A. Palmelund, L. Bunch and R. Madsen, Adv. Synth. Catal., 2006, 348, 2148 CrossRef CAS PubMed; (c) T. Iwai, T. Fujihara and Y. Tsuji, Chem. Commun., 2008, 6215 RSC.
- Paraformaldehyde as a C1 source for carbonylation reaction, see: (a) L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310 CrossRef CAS PubMed; (b) B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 3267 CrossRef CAS.
- (a) S. K. Murphy, J. Park, F. A. Cruz and Vy M. Dong, Science, 2015, 347, 56 CrossRef CAS PubMed; (b) C. R. Landis, Science, 2015, 347, 29 CrossRef CAS PubMed.
- S. Kusumoto, T. Tatsuki and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 8458 CrossRef CAS PubMed.
- (a) S. Kusumoto, M. Akiyama and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 18726 CrossRef CAS PubMed; (b) S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |