
Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations
Jun
Gu
a,
Xuan
Wang
a,
Weichao
Xue
b and
Hegui
Gong
*ab
aSchool of Materials Science and Engineering, Shanghai University, 99 Shang-Da Road, Shanghai 200444, China
bDepartment of Chemistry, Shanghai University, 99 Shang-Da Road, Shanghai 200444, China. E-mail: hegui_gong@shu.edu.cn
First published on 20th August 2015
Abstract
The formation of C–C bonds directly by catalytic reductive cross-coupling of two different electrophiles represents one of the practical synthetic protocols that differs from the conventional nucleophile/electrophile coupling methods. Particularly the reductive coupling of alkyl electrophiles with other electrophiles is still a challenge. This report summarizes the advances in the formation of C(sp3)–C(sp3) bonds between two alkyl electrophiles, with emphasis on the control of chemoselectivity that is exceedingly challenging to achieve due to similar structures and reactivities of two unactivated alkyl halides. The coupling of alkyl halides with aryl or acyl electrophiles was also discussed based on the chemical approach developed by our group, followed by a brief overview of the reactions of tertiary alkyl halides. In the end, a brief overview of the challenges in this exciting field was illustrated. Whereas the reaction mechanisms generating alkyl–alkyl products are proposed to involve reactions of Ni(I) species with alkyl halides to generate Ralkyl–Ni(III)–Ralkyl intermediates through a radical/Ni cage-rebound process, the obtained evidence seemingly supports that the radical chain mechanism governs the acylation and arylation of alkyl halides. The latter features a cage-escaped alkyl radical.
 Jun Gu | Jun Gu was born in Shanghai, China. He obtained his B.S. degree in chemistry from Shanghai University in 2006, and then worked in the Shanghai Record Pharmaceuticals Co., Ltd for three years. He joined Prof. Gong's Lab to pursue his Ph.D in 2010. His current research interests focus on coupling chemistry and H-bond mediated supramolecular chemistry in water. |
 Xuan Wang | Wang Xuan was born in Hubei province, China. After receiving his B.S. degree from Shanxi University in 2012, he moved to Shanghai University to pursue a PhD degree under the supervision of Prof. Hegui Gong. His research interests focus on the development of reductive cross coupling of tertiary halides with other electrophiles. |
 Weichao Xue | Weichao Xue was born in 1989 in Henan Province, China. He received his B.S. degree from Henan University in 2012 under the supervision of Professor Feng Shi. Then he moved to Shanghai University and obtained his M.S. degree in 2015 under the supervision of Professor Hegui Gong. Now he is going to pursue his PhD study. His research focused on Ni-catalyzed reductive coupling reactions, including cyclization, reductive methylation as well as esterification. |
 Hegui Gong | Hegui Gong was born in Jiangxi, China. He received a Bachelor's degree in 1995 from Zhengzhou University of Light Industry and a Master's degree from Tsinghua University in 1998 (both in P. R. China). In 2005 he obtained his Ph.D. degree from the University of Texas at Austin (USA) under the supervision of Professor Michael Krische. After postdoctoral research at the University of North Carolina at Chapel Hill in Professor Michel Gagné's laboratories, he was appointed as a Full Professor at Shanghai University (P. R. China) in 2008. His research focuses on supramolecular chemistry and organic synthesis. |
1. Introduction
The construction of C–C bonds through transition metal-catalyzed reactions of a nucleophile with an electrophile represents the main research directions in the field of cross-coupling chemistry, wherein the catalytic coupling of alkyl electrophiles or alkyl nucleophiles is generally more challenging than that of the aryl counterparts.1,2 It was not until early this century that the Pd- and Ni-catalyzed cross coupling of a variety of organometallic nucleophiles with alkyl electrophiles achieved rapid development after enormously innovative studies by Fu and many others (Scheme 1).3 Alternatively, coupling of alkyl nucleophiles particularly tertiary ones with different electrophiles has also evolved as an important protocol for making alkyl compounds.4 Both strategies generally necessitate the prepreparation of the nucleophiles, which may become problematic for those bearing β-functional groups susceptible to elimination.5 | ||
| Scheme 1 Conventional coupling vs. reductive coupling of alkyl electrophiles. | ||
Unlike the conventional coupling method that requires organometallic nucleophiles, direct coupling of two readily accessible electrophiles under reductive conditions provides an alternative yet more straightforward means to the construction of C–C bonds. Not only preparation of organometallic nucleophilic reagents is avoided, but the reaction mechanisms may be distinctive from a classic scenario pertaining to transmetallation of a nucleophile to a metal center.
The concept of homocoupling of two electrophiles can be dated back to the Wurtz and Ullmann reactions that were discovered 160 and 100 years ago for the generation of alkyl–alkyl and aryl–aryl compounds, respectively.6 The Wurtz reactions are characterized by poor dimerization efficiency and harsh reducing conditions, e.g. the use of Na, which tolerates a limited number of substrates with very narrow functionalities. Consequently, the development of more important cross-coupling between two different alkyl electrophiles, especially the unactivated ones using the Wurtz protocol, is not appealing when further considering the poor chemoselectivity between the alkyl coupling partners; competitive homocoupling may become a severe issue due to the statistically controlled process.
The challenges in the development of efficient cross-coupling between two unactivated alkyl electrophiles prompted us to question whether a transition-metal-catalyzed reductive cross-coupling protocol is feasible to afford C(sp3)–C(sp3) bonds. Further, could the same concept be extended to the coupling of alkyl halides with a wide range of other electrophiles? Third, how would the reaction mechanisms operate for these novel types of reductive coupling reactions? In this regard, we emphasize our discussion on these three perspectives based upon our group's recent discoveries on the Ni-catalyzed reductive coupling chemistry of alkyl halides (Scheme 1). The concurrent or earlier discoveries from other groups through reductive coupling, including electrochemical coupling or in situ formation of organometallics/coupling between two electrophiles, have been nicely reviewed,7 and will not be detailed. Overview of the challenges in the reductive coupling chemistry will be addressed in the end of this minireview.
2. C(sp3)–C(sp3) bond formation
In order to achieve effective cross-coupling between two different yet structurally similar alkyl electrophiles, the catalytic conditions should first be able to overcome the intrinsically poor chemoselectivities of the coupling partners, which may induce severe competition with homocoupling. Second, the catalyst should competently inhibit possible β-H elimination by paths that are often found for alkyl substrates and may become problematic.Encouraged by the success in Fu's Ni-catalyzed Negishi coupling of alkyl zincs with alkyl halides,8 we were curious whether two alkyl halides were suitable for Ni-catalyzed conditions using Zn as the terminal reductant. In the mechanistic studies for the Ni-catalyzed Negishi coupling, Vicic has indicated that a Terpy–NiI–R1alkyl (Terpy = 1a, Fig. 1) complex is able to transfer one electron to an alkyl halide (R2alkyl–X) which results in an alkyl radical.9 A rapid combination of the resultant Terpy–NiII–R1alkyl with an R2alkyl radical yields R1alkyl–NiIII(Terpy)–R2alkyl (a cage-rebound process). Reductive coupling gives an R1alkyl–R2alkyl product and a Terpy–NiI–X intermediate which is readily converted into Terpy–NiI–R1alkyl by transmetallation with R1alkyl–Zn, allowing the catalytic process to continue (Scheme 2, red box).
 | ||
| Fig. 1 The structures of ligands. | ||
 | ||
| Scheme 2 Proposed reaction mechanisms for reductive coupling and Negishi reactions involving a cage-rebound process for the formation of NiIII. | ||
To turn the Negishi reaction process to a reductive coupling process whereby an organozinc reagent is replaced with an alkyl halide and a reductant such as Zn, a rational design would require a suitable reductant that effectively reduces the R1alkyl–NiII–Ln intermediate to R1alkyl–NiI–Ln. The former species is generated by oxidative addition of R1alkyl–X (X = halide) to Ni0–Ln, which can be generated by reduction of Ln–NiI–X with Zn (Scheme 2, blue box). The formation of the Ln–NiI–X complex follows the scenario in Negishi coupling shown in the yellow box (Scheme 2), which would otherwise undergo a transmetallation process with an organozinc reagent. This reductive coupling proposal features double oxidative addition of alkyl halides to low valent Ni complexes. The lifetime of the Ni intermediates, the rates of oxidative addition of alkyl halides to the Ni intermediates and reduction of the Ni complexes should be highly matched. In particular, possible reduction of R1alkyl–NiI and R1alkyl–NiIII–R2alkyl by Zn should be negligible, allowing their involvement in the oxidative addition of alkyl halides or reductive elimination steps to be viable.
The proof-of-concept studies were established in 2009 based upon the coupling of 4-bromo-1-tosylpiperidine with n-butyl bromide. The optimized reaction conditions provided the C(sp3)–C(sp3) product in 67% yield, which required the secondary alkyl bromide to be the limiting reagents (conditions 1, Fig. 2).10a To ensure good selectivity, n-BuBr needed to be 3 equivalents in excess with 4-Cl-PyBox (2) being the ligand. Likewise, coupling of 4-bromo-1-tosylpiperidine with 3 equiv. of n-PrI also afforded the product in 67% yield encompassing (s)-sBu-Pybox (3a) as the ligand. Under these optimized conditions, a number of alkyl halides (3 equiv.) were compatible with 1 equiv. of other alkyl bromides, which tolerated functional groups including free hydroxyls (Fig. 2). More interestingly, bromocyclopentane and isopropyl bromide also displayed fairly good selectivity when coupling with 1 equiv. of 4-bromo-1-tosylpiperidine, although the secondary bromides were structurally similar. Both cyclic and open-chain secondary alkyl bromides (limiting reagents) demonstrated good reactivities with 3 equiv. of primary alkyl bromides and iodides. The most notable selectivity was observed in the coupling of 2-(3-bromopropyl)isoindoline-1,3-dione (1 equiv.) with 1-butenylbromide (3 equiv.) which generated the product in 92% yield, suggesting that the chemoselectivity can be tuned by variation of substituents on the alkyl halides.
 | ||
| Fig. 2 Selected examples of the synthesis of alkyl–alkyl products using Ni/Zn and Ni/(Bpin)2 reductive conditions. | ||
Although the reaction mechanism is still under investigation, the radical nature for both alkyl coupling partners was evidenced in the ring-closure and ring-opening of the radical clocks shown in eqn (1).10a
 | (1) |
The requirement for three equivalents of a second alkyl halide in excess in the Ni/Zn reductive coupling indicates that the catalyst moderately differentiates the alkyl coupling partners, wherein competitive homocoupling is severe.10b,c Improvement of the chemoselectivity by Ni/ligand screening is not successful. However, when the reductant was changed to (Bpin)2, the coupling of secondary alkyl bromides (1 equiv.) with primary bromides led to much improved coupling efficiency, in which only 1.5 equiv. of primary bromides were employed (conditions 2, Fig. 2).11 Unfunctionalized alkyl bromides, e.g. cyclohexylbromide, were not effective. However 1.5 equiv. of cyclohexyl iodide could be used to couple with 1 equiv. of primary alkyl bromide. The excellent chemoselectivity is attributed to the formation of the Ni–Bpin complex which effectively differentiates the alkyl coupling partners either by steric or by electronic reasons, or both. Indeed, the coupling of 1-iodo-2,2-dimethylpropane (more hindered 1°-alkyl iodide) and an ester-tethered bromopentane (less hindered) afforded the product in 80% yield (Fig. 2).
A catalytic cycle proposed in Scheme 3 involves borylation of the NiI–X complex (A) to generate a Bpin–NiI species B, which undergoes one electron transfer to (or halide abstraction from) R1–X leading to an R1 radical and a NiII intermediate C. Rapid combination (cage-rebound) of the two species affords Bpin–NiIII(R1)–Br (complex D) (Scheme 3). Subsequent elimination of Bpin–X (X = Br, OMe, etc.) gives a R1–NiI species E, which was oxidatively added by R2–Br, involving a second rapid combination (cage-rebound) of an R2 radical and a NiII complex F to produce a R1–NiIII–R2 intermediate G. Reductive elimination of G gives the product and regenerates the NiI–X complex A.11
 | ||
| Scheme 3 A proposed double oxidative mechanism involving a cage-rebound process for the Ni/B2(pin)2 conditions. | ||
It should be noted that Fu and Liu have disclosed interesting Ni-catalyzed borylation of alkyl halides utilizing very similar reaction conditions to those in Fig. 2 except for solvents and bases. In contrast to Fu's catalytic proposal, elimination of R1–Bpin from complex D may become energetically disfavoured under our reaction conditions.12
The Ni/Zn reductive coupling strategy was applicable to the cyclization of alkyl dihalides (Fig. 3).13 Due to competitive oligomerization, pre-organization of dihalides was required. Under NiI2/ligand catalytic conditions, five and six-membered rings could be facilely constructed. The more challenging seven-membered ring could also be obtained in nearly 40% yield.
 | ||
| Fig. 3 Cyclization of alkyl dihalides. | ||
Although the reductive cross-coupling methods are effective for a variety of alkyl halides, the use of methyl halides, particularly the easily accessible methyl iodide, was not satisfactory, possibly due to the rapid dimerization or formation of MeZnX. With the Ni/(Bpin)2 conditions, this problem could be solved, using MeOTs as the methylation source (Fig. 4).14 The use of NiCl2 as the precatalyst was pivotal, indicating the possible in situ conversion of MeOTs into MeCl, which avoids the side reactions at high concentrations by pre-addition of MeI.
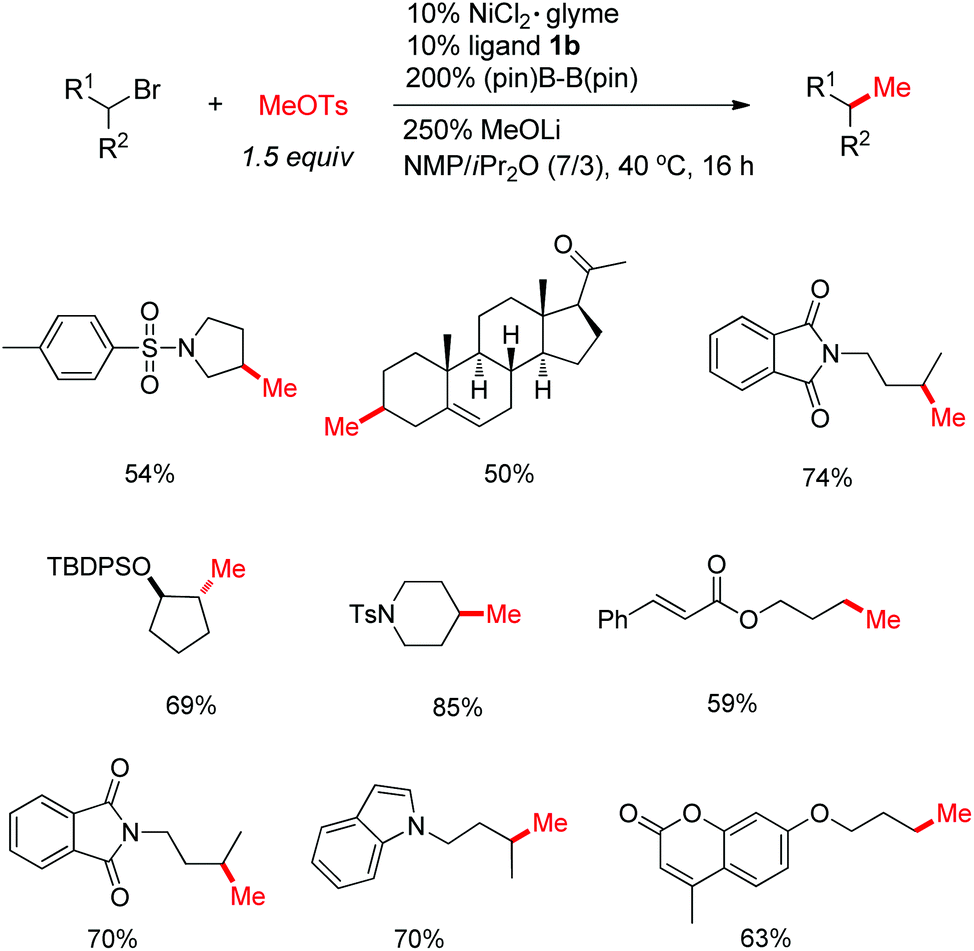 | ||
| Fig. 4 Methylation of alkyl halides with MeOTs. | ||
The challenging selectivity issues in the coupling of two different alkyl halides could be significantly overcome when allylic type electrophiles were used as the coupling partners. The allylation of primary and secondary alkyl halides under Ni(COD)2/Zn conditions generated the allylated alkanes in good to excellent yields, although for alkyl bromides, ligand 1b and MgCl2 were necessary while ligand 3c and CuI were inevitable for alkyl iodides (Table 1).15 The reaction conditions tolerated functional groups including free alcohol and phthalimide. When unsymmetrical allylic carbonates were subjected to allylic alkylation conditions, addition of alkyl to the less hindered allylic carbons took place. The use of cis or trans-alkenes always resulted in trans-products (Table 1, entries 3–6). Alkyl iodides and bromides in many cases displayed distinctive coupling reactivities, e.g. for 3-methyl allylic carbonate, alkyl bromides appeared to be more efficient (Table 1, entries 3–6), whereas opposite results were disclosed for 1,1′-dimethyl allylic carbonate (Table 1, entries 9 and 10).
|
|
|||
|---|---|---|---|
| Entry | Allylic substrates | Product | Yield |
| 1 |

|

|
R1 = H, X = Br: 62% (ligand = iPr-PyBox 3c) |
| 2 | R1 = Me, X = Br: 62% | ||
| 3 |

|
X = I: 65% | |
| 4 | X = Br: 82% | ||
| 5 |

|
As above | X = I: 40% |
| 6 | X = Br: 89% | ||
| 7 |

|
As above | X = I: 88% |
| 8 | X = Br: 90% | ||
| 9 |

|

|
X = I: 80% |
| 10 | X = Br: 70% | ||
| 11 |

|

|
X = Br: 82% |

In the course of our studies, we noticed that for individual allylic alkylation, such as primary alkyl bromide in Table 1, entry 1, further tuning the reaction parameters (ligand 3c, CuI and MgCl2 both 20 mol%) enabled optimal reaction yields. Weix later revealed that under NiCl2(dme)/1b/Mn conditions, the coupling of 1.5 equiv. of 3-aryl allylic acetate with a variety of alkyl halides was also effective, wherein the catalyst loading can be reduced to 5 mol%.16
Although the insight into the reaction mechanism requires more experimental studies, the formation of an allylic Ni complex may be involved as one of the key intermediates, which is faster than oxidative addition of alkyl halides to low valent Ni. One electron reduction with Zn giving an allyl–Ni(I) intermediate, which undergoes a similar oxidative addition process (cage-rebound) with alkyl halides to that disclosed in Negishi coupling (Scheme 2) generating an allyl–NiIII–alkyl intermediate. Subsequent reductive elimination gives the allylated products and Ln–NiI that can be reduced by Zn to regenerate Ni0 and allow the catalytic process to continue (Scheme 4).15
 | ||
| Scheme 4 Proposed double oxidative addition catalytic cycle for allylation of alkyl halides. | ||
3. C(sp3)–C(sp2) bond formation
The success in the reductive cross-coupling of two C(sp3)–X electrophiles suggests that a subtle chemoselectivity difference between the two intrinsically similar coupling partners can be adjusted with the suitable choices of catalytic conditions. In principle, the reactivity gap is enhanced between an alkyl and a C(sp2)–X electrophile, and hence should result in better chemoselectivity. For instance, if a similar mechanism for alkyl/alkyl coupling reactions operates for the coupling of alkyl/aryl halides (Scheme 5), one would anticipate that oxidative addition of an aryl halide to Ni0 to form Ar–NiII should be more favoured than to form Ralkyl–NiII. Subsequent key steps may involve reduction of the Ar–NiII intermediate to Ar–NiI that is more prone to the oxidative addition by alkyl halides than aryl ones to generate Ralkyl–NiIII–Ar through a cage rebound process. | ||
| Scheme 5 Proposed double oxidative addition mechanism for arylation of alkyl halides. | ||
3.1. Arylation of alkyl halides
Weix in 2010 disclosed that aryl iodides could effectively couple with primary halides to offer the alkyl–aryl products in good yields with excellent chemoselectivity wherein equimolar coupling partners were used (Scheme 6). The drawbacks of the reaction conditions comprise the use of expensive phosphine ligands and less effective coupling yields for secondary alkyl halides, although improved conditions using 5b were achieved without enhancing the coupling efficiency.17a,b | ||
| Scheme 6 Weix's catalytic conditions for arylation of alkyl halides. | ||
In Weix's mechanistic studies on the arylation of alkyl halides, a cage-rebound mechanism (Scheme 5) is not operative. Instead, addition of an Ralkyl radical to Ar–NiII–X that forms Ar–NiIII–Ralkyl(X) is key (radical chain). Upon reductive elimination, Ralkyl–Ar is generated associated with NiI–X that can abstract a halide from Ralkyl–Y to form NiII–X(Y) and an alkyl radical. While the Ralkyl radical diffuses to the solution to combine with Ar–NiII–X, NiX2 can be reduced to Ni0 by Mn (Scheme 7). Oxidative addition of ArX to Ni0 forms ArNiII and continues the catalytic process. The alkyl radical is initially formed by halide abstraction of Ar–NiII–X with Ralkyl–Y which also results in an Ar–NiIII–(X)Y intermediate.17a,b
 | ||
| Scheme 7 Weix's radical chain mechanism for arylation of alkyl halides. | ||
We recently conducted the DFT calculations (gas phase) based on the coupling of cyclohexyl and phenyl bromides. The results suggested that the rate limiting step for both Weix's radical chain (Scheme 7) and the double oxidative addition (Scheme 5) mechanisms was the addition of the cyclohexyl radical to PhNiII, with the energy barrier of 10.42 kcal mol−1. As a consequence, the DFT studies cannot exclude the feasibility of the double oxidative addition process.18a A close calculation based on the Ni-catalyzed coupling of the photoredox-generated benzyl radical with aryl halides revealed that addition of the more stable benzyl radical to either Ni0 and ArNiII is likely to give Bn–NiI and ArNiIIIBn, respectively, with energy barriers lower than 5 kcal mol−1 for both. Subsequent oxidative addition of ArBr to the BnNiI would lead to the ArNiIIIBn species. Therein the reductive elimination becomes rate-determining with an energy barrier of ∼10 kcal mol−1.18b Given the small energy difference between the possible mechanisms, subtle changes of the reaction parameters (e.g., concentrations) may favor one of the reaction pathways.
At the same time, we focused our effort on the coupling of secondary alkyl halides with aryl halides. By addition of 1 equiv. of pyridine as an additive, the use of ligand 5b alone offered the coupling products in high yield at room temperature.19 The reaction conditions however are not as effective as those for primary halides reported by Weix and Peng.17,19 A variety of functional groups including free amines and alcohols were tolerated. The electron-rich indole and electron-deficient quinoline moieties were also compatible (Fig. 5).
 | ||
| Fig. 5 Selected examples of arylation of secondary alkyl halides. | ||
Recently, Molander indicated that heteroaromatic halides could be competently incorporated using slightly modified Ni-catalyzed reductive conditions.20 The same group also demonstrated the viability for the coupling of 3-bromo-2,1-borazaronaphthalenes (a vinyl halide) with primary and secondary alkyl iodides, generating boron-containing alkylated heterocycles. Lastly, the coupling of heteroaromatic halides with alkyl tosylates was successfully developed which avoided the use of alkyl halides. Similar to our discovery for the coupling of MeOTs, the exchange of halides with tosylates that forms alkyl halides in situ is believed to be the key.21
3.2. Acylation of alkyl halides
By virtue of the reductive coupling strategy, unsymmetrical ketones could be achieved by exposure of alkyl halides to acid derivatives under NiCl2/Zn conditions.22 Although an early study indicated that coupling of pyridyl carboxylate with essentially primary alkyl iodides enabled the formation of ketones,22e the more accessible acid chlorides was not disclosed. With 4,4′-dimethyl-2,2′-bipyridyl (5d) or phenanthrene 1,10-phenanthroline (6) types of ligands, aroyl chlorides and anhydrides were both suited for acylation of alkyl iodides, whereas alkyl bromides only match the reactivity of acid anhydrides (Fig. 6). MgCl2 was indispensable for increasing the reaction yield. For alkanoyl chlorides, the reaction conditions further required tuning of the solvents and ligands as evident in Weix's studies on their coupling with alkyl iodides.23 Notably, the alkyl–aryl ketone synthesis can be scaled up. For instance 1-(4-(tert-butyl)phenyl)-2-methylpropan-1-one could be prepared in 75% yield (3.6 grams).22d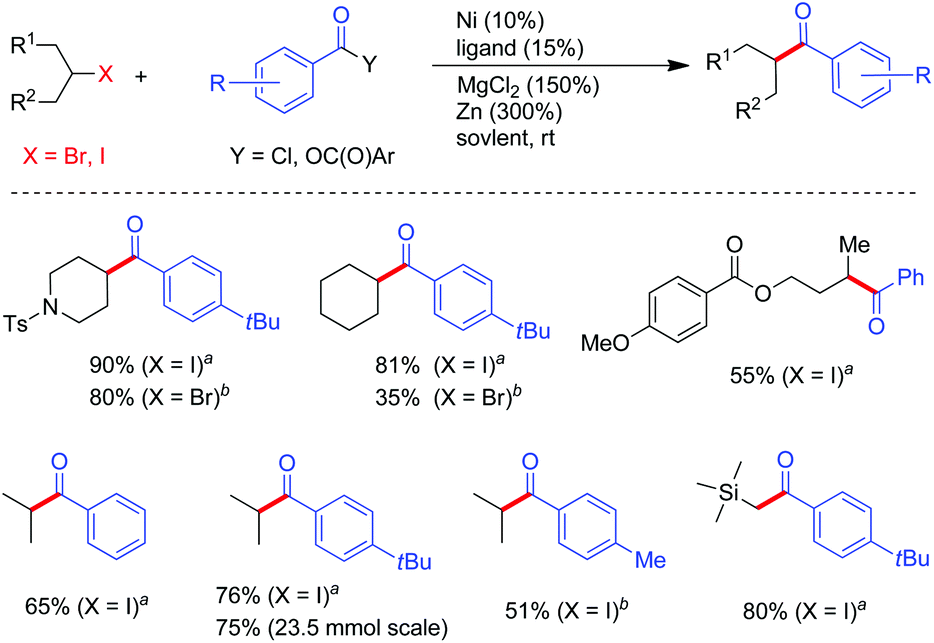 | ||
Fig. 6 Selected examples of acylation of alkyl halides with acid chlorides and anhydrides. (a) Y = Cl, Conditions A: alkyl iodides (1 equiv.), ArCOCl (2.0 equiv.), Ni(acac)2 (10%), ligand 6 (15%), MgCl2 (150%), Zn (300%), CH3CN, 25 °C. (b) Y = OC(O)Ar, Conditions B: alkyl halides (1 equiv.), (ArCO)2O (2 equiv.), Ni(cod)2 (10%), ligand 6 (15%) and CH3CN for X = I; 5d and CH3CN/DMF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) for X = Br, MgCl2 (150%), Zn (300%), 25 °C. :7) for X = Br, MgCl2 (150%), Zn (300%), 25 °C. | ||
Coupling of alkyl halides with in situ activated aryl acids by formation of anhydrides in the presence of MgCl2 and Boc2O has also been achieved, allowing the direct use of acids to generate ketones.22c High coupling yields were observed for the reaction of alkyl bromides with aryl acids wherein more easy-to-handle Ni(acac)2 and ligand 5b served as the pre-catalyst. A wide range of alkyl halides and acids could effectively provide the ketones even for the acids bearing electron-withdrawing groups (conditions C, Fig. 7).
 | ||
| Fig. 7 Selected examples of acylation of alkyl halides with in situ activated acids. (a) Reaction conditions C: R–Br (0.15 mmol, 100%), Ni(acac)2 (5%), Zn (300%), MgCl2 (250%) 5b (7%), CH3CN/DMF 1/4 (1 mL), 25 °C. (b) CN3CN/DMF (1/4, 0.5 mL). (c) 10% Ni(acac)2 and 15% 5b were used. (d) Reaction conditions D: R–I (0.15 mmol, 100%), acid (150%), Ni(acac)2 (10%), 6 (12%), Boc2O (200%), Zn (300%), MgCl2 (150%), CH3CN/THF (v/v = 4:6, 1 mL), 25 °C. (e) 0.5 mL of solvents were used. (f) Reaction conditions E: the same as conditions D except for alkyl bromides, 5b and DMF/THF (v/v = 2:3, 0.5 mL) at 20 °C. | ||
For alkyl acids, fine selection of the ligands and solvents is important, allowing efficient construction of alkyl–alkyl ketones (Fig. 7, conditions D and E).24 For alkyl iodides, a mixture of solvents CH3CN/THF (v/v, 4:6) was used, whereas for alkyl bromides, the solvents DMF/THF were used (v/v, 2:3) with the concentration of the alkyl bromides (limiting reagents) being doubled.24 We reasoned that both solvents and ligands were key factors at least for tuning the rates of anhydride formation.
The applicability of the reductive synthesis of ketones to acyl-C-glycosides was manifested in the coupling of 1-glycosyl bromide with acids and their derivatives.24 It should be noted that preparation of either glycosyl or acyl nucleophiles is a challenge. Hence, the previously reported methods to acyl-C-glycosides generally require multi-step protocols by avoiding direct involvement of classic nucleophiles. Under Ni(acac)2/5b reductive coupling conditions, a wide set of alkyl acids were suitable for acylation of glucosyl, galactosyl and mannosyl bromides in high yield. While moderate and good α-selectivities were observed for glucosides and galactosides, respectively, mannosides were produced in good yields with only α-anomers which suffer from rapid elimination upon column purification. Interestingly, when aryl acids were subjected to the reaction conditions, no products were observed. The addition of BuN4I, however, induced the reaction for aryl acid derivatives.25 The optimized reaction conditions required Ni(ClO4)2·6H2O/5a to be the precatalyst where acid anhydrides were more effective than in situ activated acids. Likewise, the aroyl C-glycosides displayed similar α-selectivities as well as coupling yields to those for alkanoyl analogs (Fig. 8).
 | ||
| Fig. 8 Selected examples of the synthesis of acyl C-glycosides. | ||
The acylation event may again operate through a double oxidative addition or a radical chain mechanism. The former involves oxidative addition of anhydrides to Ni0 generating the R1C(O)–NiII–OC(O)R1 (I) intermediate which may be reduced to R1C(O)–NiI (II).24 Subsequent oxidative addition of alkyl halides to intermediate II offers a RC(O)NiIII–Ralkyl (IV). This process may involve rapid combination of an alkyl radical and a NiII intermediate (III) (cage-rebound, Scheme 8, cycle A). The alternative radical chain process may go through a similar proposal for arylation of alkyl halides (Scheme 7), involving combination of intermediate I with an alkyl radical that is diffused to the bulk solution after halide abstraction from alkyl halides by Ln–NiIX (Scheme 8, cycle B). Reductive elimination of the resultant R1C(O)–NiIII(OC(O)R)–X (II′) gives the acylation products and Ln–NiIX. Reduction of Ln–NiII (IV′) to Ni(0) allowed continuous generation of intermediate I.
 | ||
| Scheme 8 Double oxidative addition (cycle A) vs. radical chain mechanism (cycle B) for acylation of alkyl halides. | ||
If a radical chain mechanism operates, the initial generation of an alkyl radical may arise from halide abstraction from R–X with complex I to give a NiIII species. Indeed, the stoichiometric reaction of 4-bromo-1-tosylpiperidine with a iPrC(O)–NiII–OC(O)iPr complex derived from treatment of (iPrCO)2O with Ni(0) and 5b gave the desired ketone product in 90% yield (eqn (2)).24
 | (2) |
Similar to the studies on the Ni-catalyzed reductive arylation of alkyl halides,17a a linear dependence of the ratio of linear/ring closure products (Fig. 9) on the concentration of the catalyst loading was detected for the coupling of 6-iodohex-1-ene with 3-phenylpropanoic acid.24 This result signifies possible diffusion of the Ralkyl radical from a cage arising from the reaction of complex III′ with Ralkyl–X (Scheme 8, cycle B), which subsequently binds with the NiII complex (I) to give intermediate (II′). A radical-cage-rebound process in cycle A (namely, rapid combination of alkyl radicals with III) would otherwise be accepted if the ratio of the two products remains constant. However, without direct evidence of isolatable NiI and NiIII complexes, the proposed mechanism at this stage is subject to modifications, although a radical chain mechanism appears to be more plausible. In fact, some controversial evidence may reinforce this notion. First, reduction of Ln–NiBr2 by Zn is a very slow process which did not occur even after 3 h, possibly due to the heterogeneous environment.24 The addition of MgCl2 promotes the reduction rate which however is unnecessary in many cases.17,24 Second, the presence of radical inhibitors such as BHT did not decrease the yields for ketone synthesis, which is somewhat difficult to explain if an alkyl radical diffuses to a bulk solution to form a Ni-radical cage.
 | ||
| Fig. 9 Dependence of the ratio of cyclization/linear products on catalyst loading using a radical clock. | ||
Evidence of the radical nature of the alkyl substrates was confirmed by exposure of the deuterated allyl-pendent alkyl bromide to the reaction conditions, which produced 1:1 mixture syn/anti (Ha, Hb) cyclization/coupling ketone products.24 This experiment unambiguously rules out a possible alkyl–Ni/alkene insertion mechanism where a single isomer is predicted (Scheme 9).
 | ||
| Scheme 9 Radical cyclization vs. Ni-concerted insertion mechanism. | ||
A recent Ni-catalyzed reductive trapping of CO2 with C(sp3) electrophiles developed by Martin should be noted.26 The formation of alkyl acids has been achieved from unactivated primary alkyl halides, allylic acetates, benzylic ethers and intramolecular cyclization of alkynyl alkyl bromides. The remarkable regioselectivities that are tuneable using a bidentate and a tetradentate ligand allow effective construction of branch and linear homoallylic acids.
4. More challenging tertiary alkyl halides in the reductive coupling chemistry
The coupling of tertiary alkyl nucleophiles or electrophiles generally displays remarkably distinctive reactivities from their primary and secondary analogues, which generally require a tremendous amount of innovative efforts to obtain high coupling efficiency even though similar coupling protocols are applied.24 This is possible due to the much enhanced steric bulkiness that promotes β-H elimination and induces reluctance for oxidative addition as well as reductive elimination.Although the reductive coupling concept has enabled successful coupling of primary and secondary alkyl halides with other electrophiles, the compatibility of tertiary alkyl halides remained unexplored until we developed quaternary ketone synthesis. With 1.7 equiv. of alkyl acids in excess, we demonstrated that a wide array of tertiary alkyl halides were competent, generating the ketones in good to excellent yields. A suitable choice of solvents proved to be important for the reaction conditions (Fig. 10). Noteworthy is that no isomerization side reactions took place similar to the Ni-catalyzed arylation of tertiary Grignard reagents.27 The mild reaction conditions led to excellent functional group tolerance. More interestingly, the coupling of 1,4-cis-(4-bromo-4-methylcyclohexyl)benzene with 3-phenylpropanoic acid displayed excellent diastereoselectivity (dr > 19:1) by affording the ketone bearing 1-acyl trans to 4-phenyl (Scheme 10). This observation differs from the reported Ni-catalyzed Suzuki arylation where a 1:1 mixture of diastereomers was obtained.28
 | ||
| Fig. 10 Selected examples of acylation of tertiary alkyl bromides with alkyl acids. | ||
 | ||
| Scheme 10 Highly diastereoselective synthesis of 1-methyl-4-phenylcyclohexyl)-3-phenylpropan-1-one. | ||
5. Outlook on the future directions of the reductive coupling of alkyl electrophiles
The pronounced progress in the recent reductive coupling of alkyl halides with other electrophiles has led the synthetic community to reconsider the importance of this powerful approach to the construction of C–C bonds. It also, however, sparks a question where this type of chemistry will head in the next stage.The compatibility of tertiary alkyl halides with many other electrophiles, in particular aryl and alkyl halides, is one of the fundamental challenges to be addressed. Changes of the coupling partners may significantly alter the reactivities of tertiary alkyl electrophiles, which may in turn spur side reactions such as β-elimination and isomerization of the tertiary alkyl groups.
A next important issue lies in the asymmetric reductive coupling of alkyl halides. The current observations on the reductive C(sp3)–C(sp3) and C(sp3)–C(sp2) bond formation consistently suggest that alkyl radicals are involved for alkyl halides. As a consequence, control of enantioselectivity is complicated since radical chemistry is notoriously difficult in asymmetric synthesis. One of the strategies, however, disclosed by Reisman showed that using benzylic halides, acylation and vinylation could successfully afford high enantioselectivities.29 On the basis of the radical chain mechanisms in the acylation and arylation of alkyl halides, the results suggest that chiral C(sp2)–NiII–L*n intermediates enable facial discrimination of the upcoming addition of more stable radical species.18b Unfortunately, no enantiomeric results have been observed for unactivated alkyl halides.
In an early study on our allylic arylation chemistry, we also observed 10% ee in regioselective synthesis of (E)-methyl 4-(4-phenylbut-3-en-2-yl)benzoate using a chiral pybox ligand 3b (Scheme 11).30 This result is consistent with the observation that more stable alkyl intermediates are suitable for achieving asymmetric outcomes under the reductive coupling conditions.
 | ||
| Scheme 11 Asymmetric allylation of aryl bromides with an allylic acetate. | ||
While the development of asymmetric reductive coupling contributes to the understanding of the profound steric effects of the reductive coupling chemistry, the reaction mechanisms are far from being well-established. The studies on arylation and acylation of alkyl halides that favor the radical chain mechanism as opposed to the double oxidative addition process may not be applicable to other conditions involving variation of ligands or reaction types such as in alkylation and allylation of alkyl halides. Understanding the reaction mechanism would on the other hand facilitate the development of new types of reactions pertaining to new electrophiles and incorporating unusual reagents using different metal and reducing reagents, which may significantly expand the future directions of this interesting field. Indeed, trapping alkyl radicals with an ArNiII complex as in the radical chain mechanism (Scheme 7) for the arylation of alkyl halides has evoked dual catalytic systems. In addition to the Ni-catalyzed/photoredox methods,31 alkyl radicals can be generated via Ti-catalyzed epoxide ring opening and cobalt co-catalyzed nucleophilic addition/homolysis from Bn-OMs.32
6. Conclusions
In summary, the recent advances in the Ni-catalyzed reductive coupling of alkyl halides with other electrophiles have resulted in a different view on the synthesis of a variety of important C–C bonds. Under Ni-catalyzed conditions the most challenging chemoselectivity issues between two structurally similar alkyl electrophiles can now be resolved using (Bpin)2 as the terminal reductant, which is much more effective than Ni/Zn conditions.33 The intrinsically high chemoselectivity gaps between alkyl electrophiles and aryl, acyl, and allylic electrophiles and even CO2 on the other hand merit the development of more practical synthesis of relevant alkyl-based products. In particular, alkyl–aryl products can be produced using equimolar alkyl and aryl halides. In some cases, almost quantitative results are generated.Although important mechanistic evidence in favor of the radical chain catalytic process has been established for the arylation and acylation of alkyl halides, the current understanding of this important research field is still in the infant stage. In particular, a cage rebound process cannot be excluded in the formation of C(sp3)–C(sp3) bonds that may involve the reaction of a R–NiI–Ln species with an alkyl halide. Further efforts to gain insights into the reaction details should provide more instructive principles for the future development of this interesting field. Finally, the reductive coupling protocol may find more practical applications in synthetic chemistry since alkyl and other electrophiles are generally more readily accessible with no need for pre-preparation of organometallic nucleophiles.
Acknowledgements
Financial support was provided by the Chinese NSF (no. 20972094, 21172140 and 21372151), the Program for Professor of Special Appointment at Shanghai Institutions of Higher Learning (Dongfang Scholar) Shanghai Education Committee.Notes and references
- For selected reviews on coupling of alkyl halides, see: (a) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS PubMed; (c) X. Hu, Chem. Sci., 2011, 2, 1867 RSC; (d) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC.
- For a review on alkyl-metallics, see: R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- For selected examples of Ni-catalyzed reactions, see: (a) J. Choi, P. Martin-Gago and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 12161 CrossRef CAS PubMed. For Suzuki reactions, see: (b) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed. For Pd-catalyzed reactions, see: (c) J. H. Kirchhoff, C. Dai and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 1945 CrossRef CAS.
- For selected examples of the coupling of tertiary R–MgX catalyzed by Cu: (a) P. Ren, L. A. Stern and X. L. Hu, Angew. Chem., Int. Ed., 2012, 51, 9110 CrossRef CAS PubMed; (b) C.-T. Yang, Z.-Q. Zhang, J. Liang, J.-H. Liu, X.-Y. Lu, H.-H. Chen and L. Liu, J. Am. Chem. Soc., 2012, 134, 11124 CrossRef CAS PubMed. Co: (c) T. Iwasaki, H. Takagawa, S. P. Singh, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2013, 135, 9604 CrossRef CAS PubMed.
- A. K. Steib, T. Thaler, K. Komeyama, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 3303 CrossRef CAS.
- (a) A. Wurtz, Ann. Chim. Phys., 1855, 44, 275–312 Search PubMed; (b) A. Wurtz, Ann. Chem. Pharm., 1855, 96, 364–375 CrossRef; (c) F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS PubMed.
- (a) T. Moragas, A. Correa and R. Martin, Chem. – Eur. J., 2014, 20, 8242 CrossRef CAS PubMed; (b) C. E. I. Knappke, S. Grupe, D. Gartner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem. – Eur. J., 2014, 20, 6828 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (d) J. Y. Nédélec, J. Perichon and M. Troupel, Top. Curr. Chem., 1997, 185, 141 CrossRef.
- J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726 CrossRef CAS PubMed.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed.
- (a) X. L. Yu, T. Yang, S. L. Wang, H. L. Xu and H. G. Gong, Org. Lett., 2011, 13, 2138 CrossRef CAS; (b) M. R. Prinsell, D. A. Everson and D. J. Weix, Chem. Commun., 2010, 46, 5743–5745 RSC; (c) S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383–386 RSC.
- H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022 RSC.
- (a) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693 CrossRef CAS PubMed; (b) J. Yi, J.-H. Liu, J. Liang, J.-J. Dai, C.-T. Yang, Y. Fu and L. Liu, Adv. Synth. Catal., 2012, 354, 1685–1691 CrossRef CAS PubMed.
- W. Xue, H. Xu, Z. Liang, Q. Qian and H. Gong, Org. Lett., 2014, 16, 4984 CrossRef CAS.
- Z. Liang, W. Xue, K. Lin and H. Gong, Org. Lett., 2014, 16, 5620 CrossRef CAS PubMed.
- Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Chem. – Eur. J., 2012, 16, 808 CrossRef PubMed.
- L. L. Anka-Lufford, M. R. Prinsell and D. J. Weix, J. Org. Chem., 2012, 77, 9989–10000 CrossRef CAS PubMed.
- (a) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS PubMed; (b) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed; (c) C. Yan, Y. Peng, X. Xu and Y. Wang, Chem. – Eur. J., 2012, 18, 6039 CrossRef CAS PubMed.
- (a) Q. Ren, F. Jiang and H. Gong, J. Organomet. Chem., 2014, 770, 130 CrossRef CAS PubMed; (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed.
- S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352 CrossRef CAS PubMed.
- (a) G. A. Molander, K. M. Traister and B. T. O'Neill, J. Org. Chem., 2014, 79, 5771 CrossRef CAS PubMed; (b) G. A. Molander, S. R. Wisniewski and K. M. Traister, Org. Lett., 2014, 16, 3692 CrossRef CAS PubMed.
- G. A. Molander, K. M. Traister and B. T. O'Neil, J. Org. Chem., 2015, 80, 2907 CrossRef CAS PubMed.
- (a) F. Wu, W. Lu, Q. Qian, Q. Ren and H. Gong, Org. Lett., 2012, 14, 3044 CrossRef CAS PubMed; (b) W. Lu, Z. Liang, Y. Zhang, F. Wu, Q. Qian and H. Gong, Synthesis, 2013, 2234 CAS; (c) H. Yin, C. Zhao, H. You, K. Lin and H. Gong, Chem. Commun., 2012, 48, 7034 RSC; (d) W. Lu, Z. Liang, Y. Zhang, F. Wu, Q. Qian and H. Gong, Synthesis, 2013, 2234–2240 CAS; (e) M. Onaka, Y. Matsuoka and T. Mukaiyama, Chem. Lett., 1981, 10, 531 CrossRef.
- A. C. Wotal and D. J. Weix, Org. Lett., 2012, 14, 1476 CrossRef CAS PubMed.
- C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed.
- X. Jia, X. Zhang, Q. Qian and H. Gong, Chem. Commun., 2015, 51, 10302 RSC.
- (a) T. Leon, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef CAS PubMed; (b) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212 CrossRef CAS PubMed; (c) T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702 CrossRef CAS PubMed; (d) X. Wang, Y. Liu and R. Martin, J. Am. Chem. Soc., 2015, 137, 6476 CrossRef CAS PubMed.
- Coupling of tertiary R–MgX catalyzed by Ni: (a) C. Lohre, T. Dröge, C. Wang and F. Glorius, Chem. – Eur. J., 2011, 17, 6052 CrossRef CAS PubMed; (b) A. Joshi-Pangu, C.-Y. Wang and M. R. Biscoe, J. Am. Chem. Soc., 2011, 133, 8478 CrossRef CAS PubMed.
- Coupling of tert-alkyl halides with Ar-9BBN: S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed.
- (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442 CrossRef CAS PubMed; (b) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365 CrossRef CAS PubMed.
- X. Cui, S. Wang, Y. Zhang, W. Deng, Q. Qian and H. Gong, Org. Biomol. Chem., 2013, 11, 3094 CAS.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS.
- (a) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48 CrossRef CAS PubMed; (b) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2015, 137, 3237 CrossRef CAS PubMed; (c) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2014, 6, 1115 RSC.
- High chemoselectivity between alkyl tosylates and bromides can be achieved through a Cu-catalyzed in situ Kumada process, see: J.-H. Liu, C.-T. Yang, X.-Y. Lu, Z.-Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem. – Eur. J., 2014, 20, 15334–15338 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |