DOI:
10.1039/C3RA47370H
(Review Article)
RSC Adv., 2014,
4, 17028-17038
Polymeric micelles as drug delivery vehicles
Received
15th January 2014
, Accepted 29th March 2014
First published on 31st March 2014
Abstract
Though much progress has been made in drug delivery systems, the design of a suitable carrier for the delivery of hydrophobic drugs is still a major challenge for researchers. The use of micellar solutions of low molecular weight surfactants has been one of the popular methods for the solubilization of hydrophobic drugs; however, such surfactants suffer from high critical micelle concentration and concomitant low stabilities. In contrast to surfactants of low molecular masses, polymeric micelles are associated with general advantages like higher stability, tailorability, greater cargo capacity, non-toxicity and controlled drug release. Therefore, the current review article is focused on the engineering of the core of polymeric micelles for maximum therapeutic effect. For enhanced drug encapsulation capacity and getting useful insights into the controlled release mechanism we have reviewed the effects of temperature and pH on responsive polymeric micelles. The article also presents important research outcomes about mixed polymeric micelles as better drug carriers in comparison to single polymeric micelles.
1. Introduction
Drug delivery using micellar solutions of amphiphiles is an effective way of delivering drugs to their targets. Due to the hydrophobic environment of the core of micelles, water insoluble drugs can easily be solubilized and thus loaded for delivery at the required targets. Targeted drug delivery systems are developed to produce minimum drug degradation and loss, prevent harmful side effects, increase drug bioavailability and enhance the amount of drugs at the required zone of interest. A variety of drug carriers such as soluble polymers, insoluble natural and synthetic polymers, micro particles, cells, cell ghosts, lipoproteins, liposomes, and amphiphilic polymers based micellar systems are extensively used.1–3 These drug delivery systems are associated with advantages and shortcomings. Low molecular weight surfactants are commonly used as drug delivery devices; however, due to large CMC values, their micelles suffer from thermodynamic and kinetic instability. Lipoproteins are used for the delivery of antitumor drugs because tumors require low density lipoproteins.4 However, the use of lipoprotein is questionable as drug-incorporated lipoproteins could also be recognized by healthy cells and hence they pose competition with natural lipoproteins for receptor sites on tumors.5 Liposomes are used as potential drug delivery agents due to their ability of protecting drugs from loss, targeting the drug to the site of action and thus reducing the toxicity or side effects.6 However, liposomes show no remarkable advancement due to inherent problems such as low encapsulation efficiency, rapid leakage of water soluble drugs in the presence of blood components and poor storage stability. Among the drug carriers, polymeric micelles show remarkable potential due to their large solubilization power, more loading capacity, and higher stability in blood stream, therapeutic potential and longevity. In aqueous system, the polymeric micelles are considered as amphiphilic with hydrophobic part excluded from aqueous environment. The applications of polymeric micelles can be linked with their unique core–shell architecture in which the hydrophobic part provides a space for the encapsulation of hydrophobic drugs, protein or DNA through physical or chemical binding modes. The hydrophilic part of the polymeric micelles is important due to its brush like architecture which allows the hydrophilic part to protect the hydrophobic part from the biological invasion. In addition, the hydrophilic shell minimizes protein adsorption on micelle. As the threshold of renal clearance of nanoparticles is ∼5.5 nm (ref. 7) and the size of polymeric micelles is above the threshold for filtration by kidneys, so, polymeric micelles have maximum drug loading capacity and ability to carry many drugs with prolonged circulation times8–10. These release nano-carriers in blood and passively accumulate in sites with leaky vasculature (e.g., solid tumors and sites of inflammation) because of the enhanced permeability and retention effect.9,10 Besides wide applications of polymeric micelles, there are a number of challenges which must be resolved before they can be used as potential drug carriers. These include further improvement in drug loading efficiency, stability in blood after injection, and making transport facile through the cell membrane.11 The chemical flexibility of triblock copolymers offers the opportunity of engineering of both the core and shell of polymeric micelles to achieve optimum delivery requirement for effective pharmaceutical applications. The simultaneous engineering of the core and shell have led to the development of multifunctional polymeric micelles which integrate several functions in one nano-formulation thus, providing an infinite control over spatial and temporal drug delivery.12 The enhanced accumulation of anticancer drugs in tumor interstitium can be achieved by the optimization of nanoparticles size.13 It is a great challenge for the present researchers to achieve nanocarrier with smallest size. Scientists are using a number of ways for enhancing the accumulation of polymeric micelles loaded drugs in the interstitium of tumor, however, ligand mediated strategy is considered as smart drug delivery system.14 This system is still insufficient because of the reasons of variable EPR in different patients. Therefore, super EPR stratagem has been introduced for enhanced accumulation of drug loaded nanoparticles.15 Based on all these considerations our review article is focused on the detailed characterization, engineering of micellar core, drug loading and release and pharmaceutical applications of micelles.
2. Characterization
Micelles are characterized by the measurement of turbidity, CMC and aggregate size. Nonionic micellar dispersions become turbid at a lower temperature than ionics. The clouding phenomenon is a direct consequence of recognition of larger particles.16 A variety of techniques such as interfacial tension, conductivity, osmotic pressure etc., are employed for the determination of CMC.17 However, in case of polymeric micelles these methods may not be effective due to very low CMC values. Light scattering technique is a powerful tool however; it can only predict the onset of micellization if the CMC occurs in the concentration range where this technique is sensitive. For block copolymers in water, the CMC region lies beyond the sensitivity of this technique.18 Gel permeation chromatography also has its limitations in the determination of CMC of polymeric micelles due to adsorption of polymer on the column.19 Pyrene fluorescence is one of the best options for the determination of CMC of polymeric micelles. Fluorescence spectra of pyrene are highly sensitivity to minor changes in solution and polarity of the probe micro environment.20 On increasing polymer concentration, the apparent pyrene concentration remains unchanged, while its fluorescence intensity increases tremendously after CMC. Upon micellization, the hydrophobic pyrene molecules effectively accumulate at the micellar core by partitioning from the aqueous surrounding phase.21 Based on this partitioning phenomenon of pyrene molecules, the CMC can be easily determined from the plot of fluorescence intensity and concentration of pluronics. The intersection of the lower horizontal and the slope tangent is taken as the CMC of the system.22 The hydrodynamic diameter of polymeric micelles is possible to be determined by dynamic light scattering (DLS) method.23,24 The average diameters of some plain micelles are given in Table 1. As DLS method is not effective for the determination of multimodal size distribution (MSD)25 so, atomic force microscopic technique, capable of differentiating single, aggregate and fused particles is used for the determination of MSD. The size distribution of pluronics F127 and PEG2000–DSPE micelles reported in literature23 reveal that the mean size of plain micelles vary between 1.3 and 21.6 nm with sizes of PEG–DSPE micelles larger than pluronics micelles. In case of PEG–DSPE micelles, the particle size increases as the PEG chain lengthens. The length of PO and EO chains and their ratios are related with the mean diameter of pluronics micelles.23 As is obvious from Tables 1 and 2, plain micelles are of smaller size than drug loaded micelles. After drug incorporation, the mean size increases from 3.3 to 5.6 nm in pluronics F127 micelles (at a polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mTPP ratio of 10:0.5) and from 13.9 to 29.5 nm in PEG2000–DSPE micelles (at a polymer:mTPP ratio of 10:1). The increase in micelle size after mTPP incorporation is attributed to the encapsulation of the drug in the hydrophobic core of polymeric micelle. In some formulations, such as mTPP-loaded pluronics micelle at 10:1 polymer:drug ratio, high particle size such as 168 nm is obtained due to micelles aggregation. Zhang et al.,26 entrapped a sparingly soluble drug, ibuprofen (IBU), into the core of micelles of poly(methyl methacrylate-co-methacrylic acid)-b-poly(poly-(ethylene glycol) methyl ether monomethacrylate) [P(MMA-co-MAA)-b-PPEGMA] via dialysis method and found the morphologies of the micelles to be spherical by SEM and TEM. The dramatic result of their experiments was the very high drug entrapment efficiency of 90%. The SEM and TEM images of micelles in the absence and presence of IBU shown in Fig. 1 clearly reveal the micelles to get swollen by the incorporation of the drug. Kohori et al., also developed a polymeric micelle carrier system with a swollen hydrophobic core suitable for the encapsulation of a large amount of drug.27 In contrast to micellar swelling, some authors have reported a decrease in micellar size by the incorporation of drugs. They have attributed this peculiar characteristic decrease in micellar size to lowering in aggregation number, erosion and hydrolysis.28–30 Sharma and Bhatia documented that in the presence of anti-inflammatory drugs, naproxen and indomethacin, the cores and coronas of the micelles of Pluronic F127 decrease in size by 0.7 nm and 1.1 nm respectively.28 A stunning result of their experiments was the decrease in aggregation number of Pluronic F127 micelles from 89 to about 52 in the presence of naproxen, and 51 in the presence of indomethacin. These results reflect the fact that in the presence of drugs as solute, only a few surfactant molecules result in a single micelle formation. Since the authors used Pluronic F127 above the critical micelle concentration, so the remaining surfactant molecules would have involved in the formation of more micelles.
:mTPP ratio of 10:0.5) and from 13.9 to 29.5 nm in PEG2000–DSPE micelles (at a polymer:mTPP ratio of 10:1). The increase in micelle size after mTPP incorporation is attributed to the encapsulation of the drug in the hydrophobic core of polymeric micelle. In some formulations, such as mTPP-loaded pluronics micelle at 10:1 polymer:drug ratio, high particle size such as 168 nm is obtained due to micelles aggregation. Zhang et al.,26 entrapped a sparingly soluble drug, ibuprofen (IBU), into the core of micelles of poly(methyl methacrylate-co-methacrylic acid)-b-poly(poly-(ethylene glycol) methyl ether monomethacrylate) [P(MMA-co-MAA)-b-PPEGMA] via dialysis method and found the morphologies of the micelles to be spherical by SEM and TEM. The dramatic result of their experiments was the very high drug entrapment efficiency of 90%. The SEM and TEM images of micelles in the absence and presence of IBU shown in Fig. 1 clearly reveal the micelles to get swollen by the incorporation of the drug. Kohori et al., also developed a polymeric micelle carrier system with a swollen hydrophobic core suitable for the encapsulation of a large amount of drug.27 In contrast to micellar swelling, some authors have reported a decrease in micellar size by the incorporation of drugs. They have attributed this peculiar characteristic decrease in micellar size to lowering in aggregation number, erosion and hydrolysis.28–30 Sharma and Bhatia documented that in the presence of anti-inflammatory drugs, naproxen and indomethacin, the cores and coronas of the micelles of Pluronic F127 decrease in size by 0.7 nm and 1.1 nm respectively.28 A stunning result of their experiments was the decrease in aggregation number of Pluronic F127 micelles from 89 to about 52 in the presence of naproxen, and 51 in the presence of indomethacin. These results reflect the fact that in the presence of drugs as solute, only a few surfactant molecules result in a single micelle formation. Since the authors used Pluronic F127 above the critical micelle concentration, so the remaining surfactant molecules would have involved in the formation of more micelles.
Table 1 Properties of different polymers
| Polymers |
Molecular mass |
CMC (M) |
Micelle size (nm) |
| Pluronic F127 |
12600 |
6.9 × 10−5 |
3.3 |
| Pluronic P85 |
4600 |
2.3 × 10−4 |
1.5 |
| Pluronic F68 |
8400 |
1.6 × 10−4 |
1.3 |
| PEG750–DSPE |
1528 |
1.0 × 10−5 |
5.7 |
| PEG2000–DSPE |
2806 |
1.2 × 10−5 |
13.9 |
| PEG5000–DSPE |
5801 |
1.4 × 10−5 |
21.6 |
Table 2 Micelle sizes after drug incorporation, percentage of incorporated drugs and polymer:drug ratios used for loading mTPP in polymeric micelles
| Polymers |
Polymer/mTPP |
Drug incorporated (%) |
Drug weight in micelle (%) |
Micelle size (nm) |
| Pluronic F127 |
10:0.5 |
90.1 ± 1.74 |
4.29 ± 0.083 |
5.60 |
| 10:1 |
7.59 ± 0.162 |
0.690 ± 0.015 |
168 |
| 10:2 |
1.80 ± 0.126 |
0.300 ± 0.021 |
33.8 |
| Pluronic P85 |
10:0.5 |
4.87 ± 0.852 |
0.232 ± 0.041 |
21.9 |
| 10:1 |
2.64 ± 0.484 |
0.240 ± 0.044 |
20.6 |
| 10:2 |
0.810 ± 0.189 |
0.135 ± 0.031 |
20.3 |
| Pluronic F68 |
10:0.5 |
4.12 ± 0.789 |
0.196 ± 0.038 |
20.5 |
| 10:1 |
2.82 ± 0.310 |
0.257 ± 0.029 |
29.9 |
| 10:2 |
2.13 ± 0.113 |
0.355 ± 0.019 |
79.7 |
| PEG750–DSPE |
10:0.5 |
0.340 ± 0.020 |
0.0162 ± 0.001 |
12.9 |
| 10:1 |
1.04 ± 0.288 |
0.0942 ± 0.026 |
10.9 |
| 10:2 |
0.244 ± 0.010 |
0.0406 ± 0.0017 |
13.5 |
| PEG2000–DSPE |
10:0.5 |
93.3 ± 2.26 |
4.44 ± 0.11 |
38.5 |
| 10:1 |
95.4 ± 7.80 |
8.68 ± 0.71 |
29.5 |
| 10:2 |
24.3 ± 0.502 |
4.05 ± 0.084 |
41.0 |
| PEG5000–DSPE |
10:0.5 |
82.1 ± 2.27 |
3.91 ± 0.11 |
11.1 |
| 10:1 |
19.5 ± 0.447 |
1.77 ± 0.041 |
19.3 |
| 10:2 |
8.16 ± 0.226 |
1.36 ± 0.038 |
19.9 |
 |
| | Fig. 1 SEM and TEM images of blank (a and c) and IBU-loaded (b and d) P(MMA3k-co-MAA4.5k)-b-PPEGMA5k assembled micelles.26 | |
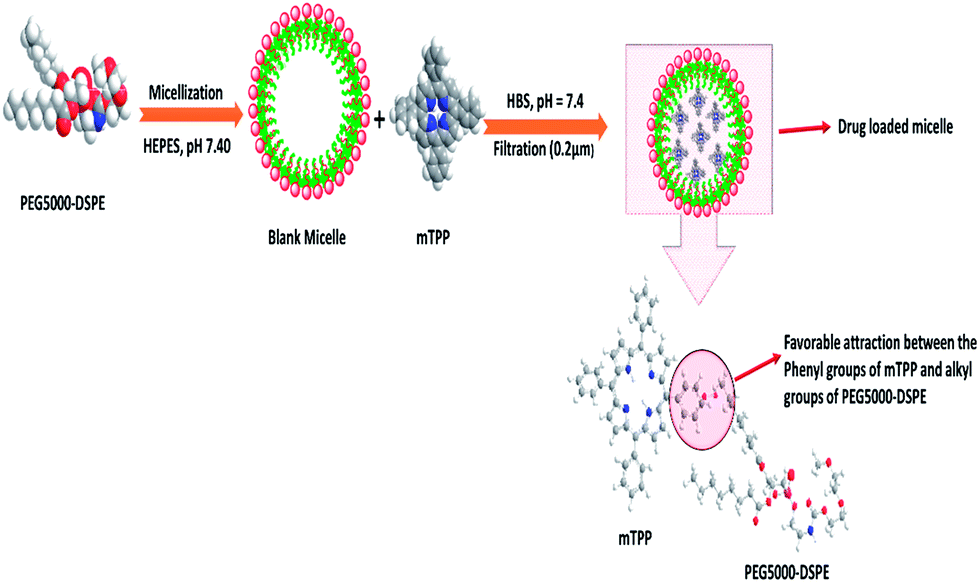
An examination of Tables 1 and 2 reveals that the size of PEG5000–DSPE micelle at low polymer:mTPP ratio (10:0.5) is smaller than the blank micelle but remains constant at high polymer:mTPP ratio (10:1 and 10:2). The decrease in the micellar size of PEG5000–DSPE by the incorporation of mTPP drug has been explained on the basis of favorable interactions between phenyl groups of mTPP and alkyl group of PEG5000–DSPE as shown in Fig. 2. Bronich et al.,31 reported the incorporation of cisplatin and paclitaxel dual drugs into the polypeptide based micelles of polyethylene glycol-block-polyglutamic acid-block-polyphenyl alanine. They determined the sizes of blank and drug loaded micelles as 90 ± 1.2 and 76 ± 4.0 nm respectively. This decrease in size of the drug loaded micelle was attributed to the neutralization and condensation of poly(glutamic acid) units with cisplatin incorporation. Similarly C. J. Hai et al.,32 also reported the decrease in micellar size after the incorporation of norcantharidin in polymeric micelles of poly(ethylene glycol)–poly(caprolactone).
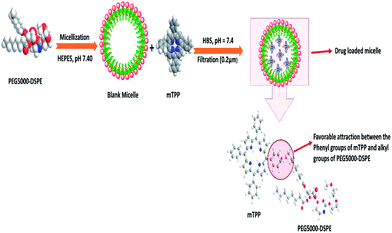 |
| | Fig. 2 Schematic of the decrease in micellar size after drug incorporation. PEG5000–DSPE represents polyethylene glycol–distearoyl phosphatidyl ethanolamine, HEPES (4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid), mTPP (meso-tetraphenyl porphine) and HBS (HEPES Buffered Saline). | |
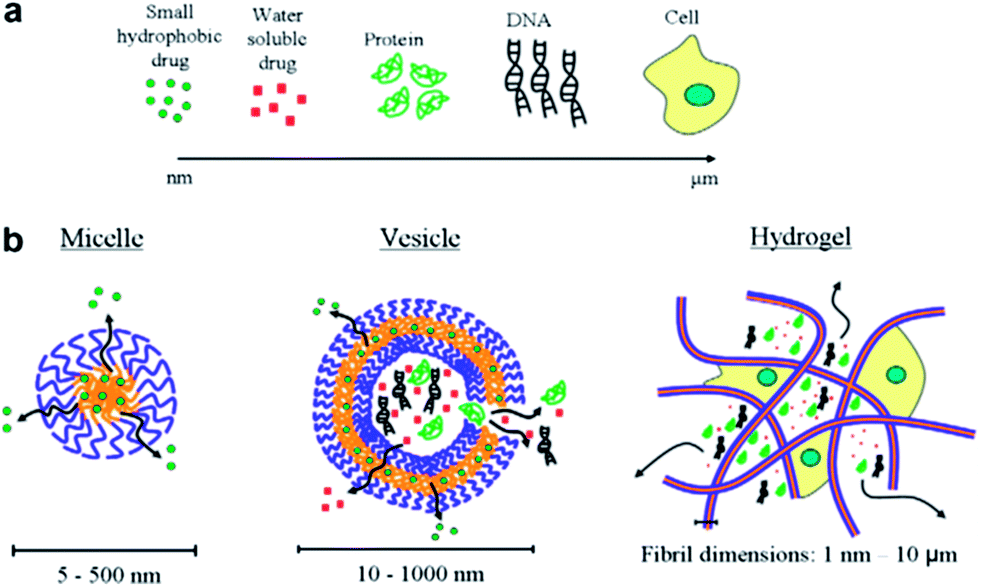
Polymeric micelles excel other drug carriers due to their small size and lower CMC values.33–37 The average hydrodynamic diameter of spherical pluronics micelle is approximately 2 to 30 nm and aggregation number of 10 to 100.38–42 Micelles of single type pluronics copolymer dominated all drug delivery efforts until recent years, but lately the binary systems drew the utmost attention. The negative aspects (comparatively low drug loading, larger particle size and low stability) of mono micellar systems are compensated by the mixing of different polymers to generate mixed micellar systems.43 For example, mixed micelles of poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG5000-b-PCLx) and 1,2-distearoyl-sn-glycero-3-phosphor ethanolamine-N-methoxy poly(ethylene glycol) not only incorporate considerably higher levels of amphotericin B than the PEG5000-b-PCLx micelles but also produce small sized and thermodynamically stable micellar structures.44 With respect to micelles from pluronics, doxorubicin-loaded mixed micellar system from pluronics L61 and F127 is the first micellar formulation to reach clinical trials for cancer chemotherapy.45 Gao et al., developed mixed micelles of pluronics P105 and tocopheryl polyethylene glycol 1000 succinate and found these as more stable and efficient solubilization system for camptothecin.46 Binary system of pluronics P105 and L101 for the incorporation of paclitaxel (PTX) was developed for multidrug resistance tumors.47 Wei et al., reported the loading of PTX onto pluronics P123 and F127 (denoted as P123 and F127, respectively) mixed polymeric micelles that demonstrated the enhancement of antitumor efficacy in MDR human lung tumor cell line A-549.48 The pluronics mixed micelles can significantly increase the blood circulation time of PTX.49 These mixed micelles upon further modification (for selective targeting of cancer cells) via folate-conjugation enhanced their uptake via a receptor-mediated endocytosis.50 Oh et al., reported binary mixing of several hydrophilic (F127, P105, F87, P85, and F68) and hydrophobic (L121, L101, L81, and L61) pluronics.51 Amongst all the tested combinations, mixture of F127 and pluronics L121 form small sized particles and stable dispersions upon sonication or heating, with a 10-fold higher solubilization capacity for sudan (III) dye as compared to F127 micelles. In an attempt to prepare a high solubilization capacity system without extra input of energy, Lee et al., prepared mixed micelles of P123 and L121.52 The particle size for various ratios ranged from 79 to 1014 nm without sonication and 34–140 nm with the aid of sonication. Nevertheless, none of the mixed micellar systems achieved a particle size below 30 nm which is highly desirable for pharmaceutical formulations.53 A comparison of different types of drug delivery systems along with the types of drugs is presented in Fig. 3.
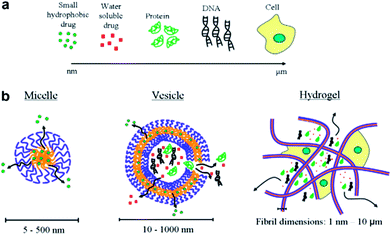 |
| | Fig. 3 (a) Common therapeutics with size range from nanometer to micrometer and (b) therapeutics carriers. | |
3. Engineering of the micellar core
The core of polymeric micelles is engineered for the development of formulations that could achieve therapeutic drug levels upon systemic administration. The miscibility between polymers and drugs plays an important role in drug loading efficiency of polymeric micelles.54 The level of drug encapsulation mainly depends on the extent of hydrophobic interaction between the drug and micellar core. The results of molecular simulation studies, supported by empirical data,55 suggest a significant role of polar interactions and hydrogen bonding between the drug molecules (containing hydrogen-bond forming groups in their structure) and the micellar core in defining the degree of drug solubilization by polymeric micelles. In practice, the length of the hydrophobic block and the type and level of substituents on it have been found to affect the loading efficiency of specific drugs in polymeric micelles.56,57 Drug loading efficiency also depends upon the aggregation number of the block copolymers. Micelles with greater aggregation numbers show more loading capacity.58–62 The comparison of the effect of modification in core structure on the drug loading capacity of polymeric micelles can be seen in Table 3. Polymeric micelles should be stable enough to give maximum retention time to drug in the target zone without having any side effect until its removal from the body. Thermodynamic and kinetic parameters can be used to understand the stability of a micellar system. The thermodynamic stability is directly linked with CMC. The CMC of the polymeric micelles mainly depend upon the hydrophobic character of the molecule. Polymers with long hydrophobic chain show lower stability as compared to polymers with long hydrophilic chain19 as listed in Table 4. The data show that increasing the hydrophobic part of the polymer, the CMC decreases while the stability is enhanced. Those polymers which have low CMC can retain their stability even in very dilute forms within the blood circulatory system. In contrast to thermodynamic stability, kinetic stability is related to the dissociation of polymeric micelles into single chain at concentration below their CMC values. Fundamentally, kinetic stability depends upon the physical state of the core, amount of solvent inside the core, the size of hydrophobic block and the hydrophobic/hydrophilic ratio. It is challenging to control the early elimination of the micelles due to their interactions with blood components.63,64 The micellar stability can be increased by the reduction of CMC, increase in intra-micellar interactions, and covalent cross linking of the micelle core. The CMC of the polymeric micelle can be reduced by increasing the hydrophobic character of the polymer. For example, the attachment of various fatty acids to the core of PEO–P(Asp) micelles was shown to decrease their CMC.65 The kinetic stability of the polymeric micelles can be achieved by the modification of micellar core with structures capable of forming intra-micellar structures, electrostatic interaction and covalent cross linking. The introduction of benzyl groups to PEO–PCL has been reported to increase the rigidity of the micelle core due to intra-micellar interactions.60 Similarly the micelle stability can be enhanced by electrostatic ionic interactions through the formation of polyion complexes.66–68 Covalent cross linking is also one of the important ways of increasing the stability of the polymeric micelles. This type of stability can be attained by thermal and photo-induced polymerization.69–75 Recently click chemistry is introduced for core cross linking.76 Although this strategy provides sufficient stability to polymeric micelles but the clearance of such micelles from the body is a serious issue. To solve this problem researchers have introduced the method of photo reversible cross linking phenomena.77,78
Table 3 Effect of modified core structure on the drug loading capacity
| Polymer |
Substituted core structure |
Drug |
Drug loading capacity |
Reference |
| PEO–P(Asp) |
Fatty acid |
Amphotericin B |
13 times higher as compared to benzyl core structure |
62 |
| PEO–PCL |
Cholesteryl |
Amphotericin B |
1.8 times increase |
63 and 64 |
| Stearyl |
Amphotericin B |
2.1 times increase |
63 and 64 |
| Palmitoyl |
Amphotericin B |
2.3 times increase |
63 and 64 |
| Carboxyl |
Amphotericin B |
2.7 times increase |
63 and 64 |
| Benzyl |
Cucurbitacin I |
1.7 times increase |
65 |
| Benzyl |
Cucurbitacin B |
3 times increase |
65 |
| Cholesteryl |
Cucurbitacin I |
More as compared to benzyl substituents |
66 |
| PEO–PVBODENA |
PVBODENA |
PTX |
37% (w/w) |
42 |
| PEO–PDLLA |
PDLLA |
PTX |
20% (w/w) |
42 |
Table 4 CMC of block copolymers
| Polymer |
CMC (mg L−1) |
Reference |
| PEG-b-PEYM45 |
5.5 |
67 |
| PEG-b-PEYM79 |
2.1 |
67 |
| PEG-b-PEYM98 |
1.3 |
67 |
| PEG113–P[(MTC-OBn)5-(MTC-OU)5] |
63.1 |
68 |
| PEG113–P[(MTC-OBn)8-(MTC-OU)8] |
55.5 |
68 |
| PEG113–P[(MTC-OBn)13-(MTC-OU)13] |
39.8 |
68 |
| PEG113–P[(MTC-OBn)19-(MTC-OU)19] |
10.1 |
68 |
4. Drug loading and release
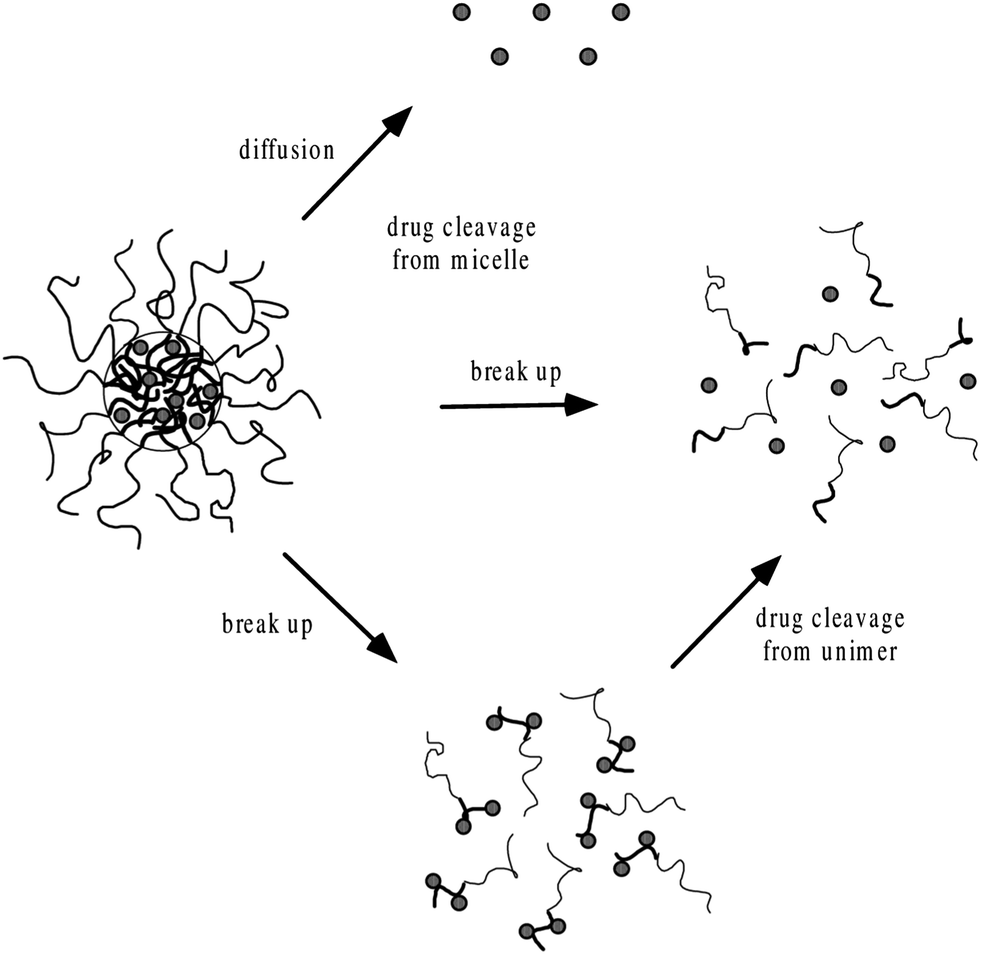
The insoluble drugs can be encapsulated in the micellar core by chemical conjugation or by physical entrapment through dialysis or emulsification. The simple equilibration of the drug and micelles in water may not result in high levels of incorporated drug.79,80 In chemical conjugation technique, the formation of covalent bond between the specific group of the drug and the hydrophobic core of the micelles cause incorporation of the hydrophobic drug inside the polymeric micelle core. Such bonds cause steric hindrance and resistance to enzymatic cleavage.81 In comparison to the chemical method, physical method is more favorable for drug incorporation.82,83 Polyionic compounds can be incorporated through the formation of polyion complex micelles.84,85 Physical entrapment of drugs is generally done by dialysis or oil-in-water emulsion procedure. In dialysis, the drug and polymer are brought from the selective solvent to a solvent that is selective only for the hydrophilic part of the polymer. By the replacement of good solvent with selective one, the hydrophobic portion of the polymer associates to form the micellar core, thus, incorporating insoluble drug. Extending the dialysis over several days can ensure the complete removal of the organic solvent. The oil-in-water emulsion method consists of preparing an aqueous solution of the copolymer to which a solution of the drug in a water-insoluble volatile solvent is added in order to form an oil-in-water emulsion. The micelle–drug conjugate is formed as the solvent evaporates. The main advantage of the dialysis procedure over the latter method is that the use of potentially toxic solvents can be avoided. Working on the incorporation of DOX in PEO–PBLA micelles Kwon et al., found that emulsification method is more efficient than dialysis.80 The drug loading procedure may affect the distribution of a drug within the micelle. The chemical stability of the DOX incorporated into polymeric micelles can be explained on the basis of protection from aqueous environment79 and the increased resistance of plasmid DNA in polyion complex micelles against enzymatic degradation.85 The incorporation efficiency depends on the initial amount of drug added. After maximum loading capacity, drug gets precipitated.86,79 The drug loading efficiency also depends upon the aggregation number of the polymeric micelles. Micelles with high aggregation number cause more solubility of the given drugs in the inner core.87 Drug release from polymeric micelles can be controlled by engineering the polymeric core in such a way to enhance the interaction of drug with core of the micelle. The drugs get released from micellar core by two major pathways i.e., dissociation of the micelle followed by the separation of the drug from monomers and drug–polymer bond breakage within the micelle followed by diffusional escape from the delivery system (Fig. 4).8,88 The nature of release mechanism is explained on the basis of Pappas's equation.89| | |
log(Mt/M∞) = nlogt + logk
| (2) |
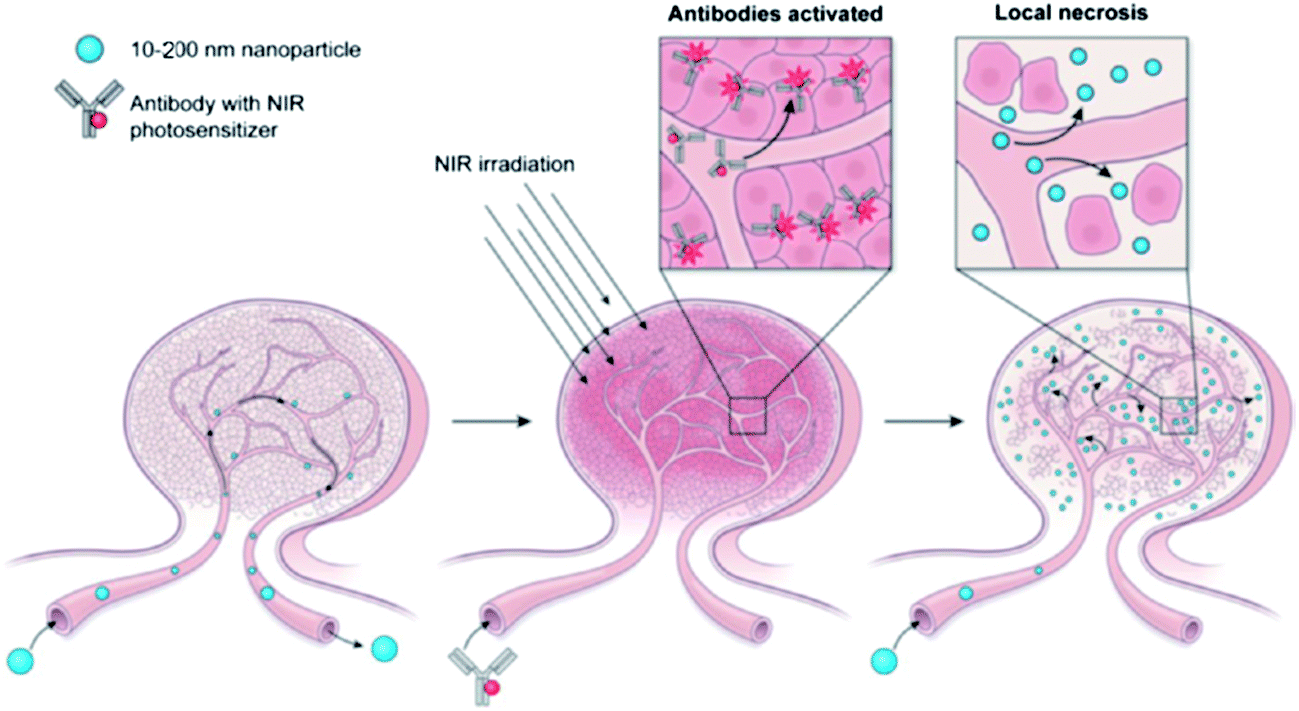
where Mt and M∞ are the absolute cumulative amount of drug released at time t and infinite time, k is the rate constant and n is release exponent which indicates the mechanism of the drug release. If n = 0.45, then the mechanism will be diffusion controlled and if n = 0.89, then it will be swelling controlled release. In case of n between 0.45 and 0.89, the drug release mechanism is of anomalous transport type. The data in Table 5 show that in the case of Asp the values of n are greater than 0.45 for all pH values so the release mechanism for Asp is anomalous transport. On the other hand, in case of DOX, the n values are very close to 0.45 which predict the release mechanism to be diffusion controlled. Drug release can be related with internal stimuli such as pH. These strategies are applicable for controlled drug release in acidic environment of the tumor or endosomes inside the tumor cell. Drug release of pure PTX and PTX loaded micelles in media of pH 5.0 and pH 7.0 reported in literature90 show rapid release of free PTX in comparison to loaded micelles. The release of the free PTX at both pH values was found the same. The release of PTX from polymeric micelles showed strong dependence on the composition of the hydrophobic core of the micelle. For PEO-b-PCL, the release was minimum at both pH 5.0 and pH 7.0. The variation of composition of the polymers at pH 5.0 caused significant effect on the release rate of PTX from polymeric micelle. Before six hours incubation, the release rate for all types of polymers was the same but after six hours incubation, the release rate affected strongly.90 Gillies et al., reported a method of controlled drug release, in which a pH sensitive nanovehicle is developed in such a way that the hydrophobic group remains attached to one of the block of the copolymer via an acid sensitive linkage. The hydrophobic block of the micelle upon hydrolysis gets converted into hydrophilic part and hence, destabilizes the micelle, thus, providing a way of drug release.91 The phenomenon of controlled drug release requires a precise study of maximum therapeutic efficacy that can be obtained by the factors which control the drug concentration levels, dosing intervals and drug retention in tumor. The cancer cells exposed to small amount of drug over long period show more sensitivity to chemotherapy than those targeted with higher drug dose but for short time. In this context, polymeric drug conjugates with pH-dependent tunable drug release have been proposed to allow spatial and temporal control of drug delivery for maximum therapeutic effect in cancer treatment.92 To obtain maximum therapeutic effect in cancer treatment, DOX is conjugated to the P(Asp) part of the PEO–P(Asp) with the help of different spacers such as glycine (Gly) or 4-aminobenzoate (Abz) through a hydrazone linkage. The drug release format of both Gly and Abz micelles is pH dependent and tunable. The role of spacers is important in terms of the stability of polymer micelle in combination with block copolymer chain lengths. Kataoka et al.,13 investigated the platinum based drug release from sub-100 nm micelles of poly(ethylene glycol)-block-poly(glutamic acid) as shown in our designed schematic of Fig. 5. The sub-100 nm micelles were used for platinum based drug release with hypo vasculature tumor (poor permeable tumor) and hyper vasculature tumor (high permeable tumor). On the basis of the obtained results they concluded that with hyper vasculature tumor there is no size dependency of the drug loaded micelles but for hypo vasculature tumor, only drug loaded micelles with size less than 50 nm can penetrate well. Sano et al.,15 were the first to offer explanation to the super-enhanced permeability and retention effect (SUPR) by a scheme shown in Fig. 6. They explained the SUPR with the help of photo immunotherapy (PIT) which is a light mediated treatment based on an antibody–photosensitizer conjugate. It was concluded that under the effect of PIT, particles with size 10–200 nm can easily accumulate at the target side while minimizing nontargeted side effects associated with conventional anticancer drugs.
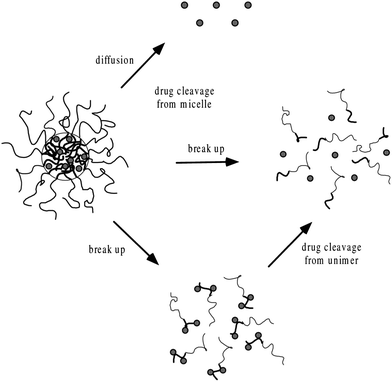 |
| | Fig. 4 Mechanisms of drug release from polymeric micelles.8,88 | |
Table 5 Release exponents and log of rate constants at different pH for PVA/micelle at 37 °C
| Drug |
pH |
log k |
n |
| Asp |
4.0 |
1.55 |
0.58 |
| 5.5 |
1.56 |
0.53 |
| 7.4 |
1.54 |
0.56 |
| 8.4 |
1.58 |
0.53 |
| DOX |
4.0 |
1.07 |
0.43 |
| 5.5 |
0.91 |
0.48 |
| 7.4 |
0.81 |
0.48 |
| 8.4 |
0.72 |
0.46 |
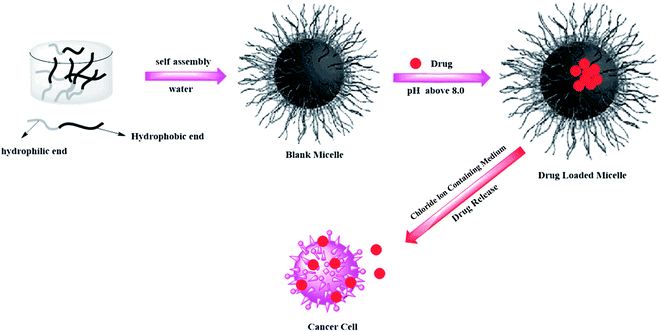 |
| | Fig. 5 Schematic showing loading and release of platinum based drug from polymeric micelles. | |
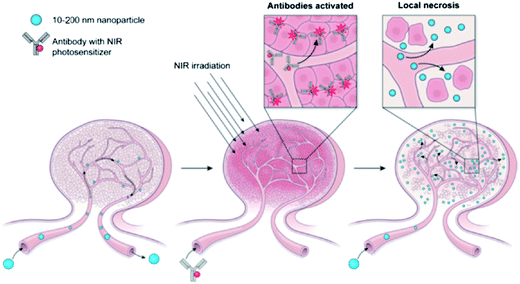 |
| | Fig. 6 Super-enhanced permeability and retention effect (SUPR).15 | |
The most challenging task in polymer preparation is to link the hydrophilic and hydrophobic parts because this linkage is crucial regarding the rate of drug release from micellar core. The rate of drug release mainly depends on the hydrolysable chemical bond between the drug and polymer.8 A stable bond leads to a deep penetration of the drug into micellar interior. Polymers having ester or amide bonds are useful choices as these bonds exhibit excellent stability for hydrolysis in the absence of enzymes under physiological conditions. The report of Bilgicer93 about the half-lives of poly(esters) and poly(amides) as 3.3 and 8.3 years offers evidence about the stability of ester and amide bonds. The cleavage of such bonds results in the production of acid as one of the products which lowers the pH and self-catalyzes the hydrolytic degradation.93
5. Pharmaceutical applications
The investigation of drug targeting mechanism is important for pharmaceutical applications of polymeric micelles as well as other drug carriers. Passive drug targeting mechanism involves the micelles spontaneous penetration into the interstitium through leaky vasculature. The drug efficiency can be enhanced by the polymeric micelles by targeting the specific cell and organs without accumulation in the healthy tissues. Through intravenous administration, the polymeric micelles show prolonged circulation time due to small size and hydrophilic shell that minimizes the uptake by mono phagocytic system (MPS). Moreover, these micelles can be prevented through renal excretion due to their high molecular weight. Indeed, intact polymeric micelles have been recovered from plasma several hours after intravenous injection.94,95 However, liposomes with similar surface characteristics seem to have a longer circulation time than micelles, possibly because extravasations of liposomes from the vasculature are more difficult due to their larger size.83 The capacity of polymeric micelles to reach regions of the body that are poorly accessible to liposomes has been exemplified by Trubetskoy and Torchilin.82 They showed that after subcutaneous injection in the dorsum of rabbit hind-paw, polymeric micelles exhibit higher accumulation in the primary lymph node than liposomes and reach the systemic circulation after massage of the lymph node. As for other drug carriers, plasmatic half-life and uptake of polymeric micelles by the MPS depend on the molecular weight and density of the hydrophilic shell.93,94 Polymeric micelle-incorporated drugs may accumulate into tumors to a greater extent than free drugs and show a reduced distribution in non-targeted areas such as heart.94 Accumulation of polymeric micelles in malignant or inflamed tissues may be due to an increased vascular permeability and impaired lymphatic drainage.96,97 The tumor vessels are more leaky and less perm selective than normal vessels. Large pores exist and may account for the perivascular accumulation of macromolecules and colloidal drug carriers.98,99 However, there is no consistent evidence for the differences in the bio distribution pattern. Zhang et al.,100 were not able to demonstrate any difference between the bio distributions of paclitaxel loaded into MePEO–PDLLA micelles versus paclitaxel solubilized in cremophor. These two formulations also showed similar in vitro distribution between the lipoprotein and lipoprotein deficient fraction of plasma.101 Several in vivo studies have shown that polymeric micelles are able to improve the efficiency of anticancer drugs against leukemia102,103 and solid tumors.100,104 Strict comparisons between the activities of free vs. incorporated drugs are sometimes difficult to be made because efficacy experiments are often carried out at the maximum tolerated dose which may be different for the two formulations.100,102,104,105 The mechanism that governs the pharmaceutical activity of drug loaded polymeric micelles is more complicated than a simple accumulation of the carrier in the targeted area. For instance, an early study by Kabanov et al.106 showed that the neuroleptic activity of intraperitoneally administered haloperidol is increased by more than two orders of magnitude after its incorporation into PEO–PPO–PEO micelles coupled with brain specific antibodies. In this particular case, the enhancement of drug efficacy was attributed to specific targeting107 and/or increased permeability of the drug through biological membranes given by the polymeric amphiphiles.108 Yu et al.,109 were able to increase the in vitro antifungal efficiency of amphotericin B while at the same time decreasing its hemolytic activity by loading the drug into polymeric micelles. It was suggested that polymeric micelles could stabilize amphotericin B against auto-oxidation and/or enhance membrane perturbation of fungal cells. Prolonged exposure due to slow drug release may also be involved in the action mechanism of polymeric micelles. Drugs can be released directly from micelles by diffusion or consequently to the dissociation of the micelle into free polymeric chains.110 Ideally, insoluble drugs must be slowly released from polymeric micelles because uncontrolled release due to weak micellar stability results in the intra-vascular precipitation of the drug. Controlled released patterns have been demonstrated for several micelle preparations. Polymeric micelle formulations are generally associated with a lower toxicity which allows the administration of doses higher than those found to be toxic for the free drug. For instance, the activity of DOX on tumors is limited by its toxicity. In C26 tumor bearing mice, the administration of 20 mg kg−1 of doxorubicin results in toxic deaths, while of 5 mg kg−1 dose is not efficient in inhibiting tumor growth. Thus, the maximum tolerated dose is estimated as 10 mg kg−1. However, incorporation in PEO–P(Asp) micelles permits the administration of doses as high as 50 mg kg−1. Interestingly, the antitumor activity against subcutaneous mouse colon adenocarcinoma 26 is the result of physical entrapment of the drug in micelle, since chemically bound DOX shows no significant anti-tumor effect, probably because chemically-attached DOX is not released due to the absence of hydrolysable link between the drug and hydrophobic chains of the core. The passive drug targeting mechanism can be activated by binding specific ligands (antibodies, sugars) to the water exposed termini of the hydrophilic part of the polymeric micelles or by introducing a polymer sensitive probe to variation in temperature or pH.111–114 An in vitro study on DOX incorporated into PNIPA–polybutyl methacrylate micelles shows that below the LCST (338 °C), the micelle formulation expresses lower cytotoxicity than free DOX towards bovine aorta endothelial cells.115 However, at temperatures above the LCST, the activity of the micelle–drug conjugate is greater than free DOX. Also, the release of DOX from the micelles reach 80% after 15 h at 408 °C, while it remains under 20% at temperatures below 338 °C.115 pH-sensitive micelles serve for the delivery of drugs to tumors, inflamed tissues or endosomal compartments, since they all are associated with a lower pH than normal tissues.116–119
6. Conclusion
Polymeric micelles are the best alternative drug carriers in comparison to other micellar systems. Mixed polymeric micelles are endowed with the incorporation of considerably higher levels of drugs, increased blood circulation time and thermodynamic stability. Engineering of the polymeric micelle core leads to maximum drug loading capacity and longevity. The length of hydrophobic blocks and the nature of substituents present in the core mainly control the drug loading capacity of polymeric micelles. The insoluble drugs can be encapsulated in the micellar core by chemical conjugation or by physical entrapment. In comparison to chemical methods, physical methods are more favorable for drug incorporation. Physical entrapment of drugs is generally done by dialysis or oil-in-water emulsification. The dialysis is preferred over emulsion technique as in the former method the use of potentially toxic solvents is avoided. The passive drug targeting mechanism can be activated by binding specific ligands to the water exposed termini of hydrophilic part of the polymeric micelles or by introducing a polymer probe sensitive to variation in temperature and/or pH. The best method of controlled drug release involves the hydrolytic conversion of the hydrophobic block of micelle to hydrophilic part that could destabilize the micelle for throwing its load at the targeted site. The phenomenon of controlled drug release requires a precise study of maximum therapeutic efficacy that is possible to be achieved by the factors which control drug concentration levels, dose intervals and drug retention in the targeted zone.
Acknowledgements
The authors gratefully acknowledge the financial support of Higher Education Commission Islamabad, University of Toronto Scarborough, Canada, Quaid-i-Azam University and Higher Education, Government of KPK, Pakistan.
References
- P. Couvreur, F. Puisieux and N. Ano, Adv. Drug Delivery Rev., 1995, 10, 141 CrossRef.
- D. D. Lasic, Nature, 1996, 380, 561 CrossRef CAS PubMed.
- M. Yokoyama, Novel passive targetable drug delivery with polymeric micelles, Academic Press, San Diego, 1998, pp. 193 Search PubMed.
- M. S. Baboli, G. Favre, P. Canal and G. Soula, Br. J. Cancer, 1993, 68, 319 CrossRef.
- R. A. Firestone, Bioconjugate Chem., 1994, 5, 105 CrossRef CAS.
- R. E. Pagano, A. J. Schroit and D. K. Struck, in Liposomes: From physical structure to therapeutic applications, ed. C. G. Knight, Elsevier, Amsterdam, 1981, pp. 323 Search PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangion, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS PubMed.
- H. M. Aliabadi and A. Lavasanifar, Expert Opin. Drug Delivery, 2006, 3, 139 CrossRef CAS PubMed.
- G. K. Kwon and M. L. Forrest, Drug Dev. Res., 2006, 67, 15 CrossRef CAS PubMed.
- K. Kataoka, G. S. Kwon, M. Yokoyama, T. Okano and Y. Sakurai, J. Controlled Release, 1993, 24, 119 CrossRef CAS.
- S. Kim, Y. Shi, J. Y. Kim, K. Park and J. X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49 CrossRef CAS PubMed.
- X. B. Xiong, F. Arash, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Biotechnol., 2011, 6, 815 CAS.
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and O. , K. Kataoka, ACS Nano, 2013, 7, 8583 CrossRef CAS PubMed.
- K. Sano, T. Nakajima, P. L. Choyke and H. Kobayashi, ACS Nano, 2013, 7, 717 CrossRef CAS PubMed.
- M. Kozlov, N. Melik-Nubarov, E. Batrakova and A. Kabanov, Macromolecules, 2000, 33, 3305 CrossRef CAS.
- K. Nakamura, E. Ryuichi and M. Takeda, J. Polym. Sci., 1976, 14, 1287 CAS.
- I. Astafieva, X. Zhong and F. A. Eisenberg, Macromolecules, 1993, 26, 7339 CrossRef CAS.
- M. Yokoyama, T. Sugiyama, T. Okano, Y. Sakurai, M. Naito and K. Kataoka, Pharm. Res., 1993, 10, 895 CrossRef CAS.
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Langmuir, 1993, 9, 945 CrossRef CAS.
- A. V. Kabanov and V. Y. Alakov, Crit. Rev. Ther. Drug Carrier Syst., 2002, 19, 1 CrossRef CAS.
- I. Kabanov, I. Nazarova, E. Astafieva, V. Batrakova, A. Alakhov, V. Yaroslavov and V. Kabanov, Macromolecules, 1995, 28, 2303 CrossRef.
- Z. Sezgin, N. Yuksel and T. Baykara, Eur. J. Pharm. Biopharm., 2006, 64, 261 CrossRef CAS PubMed.
- G. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1997, 48, 195 CrossRef CAS.
- J. Xia, H. Zhang, D. R. Rigsbee, P. L. Dubin and T. Shaikh, Macromolecules, 1993, 26, 2759 CrossRef CAS.
- Y. Q. Yang, L. S. Zheng, X. D. Guo, Y. Qian and L. J. Zhang, Biomacromolecules, 2011, 12, 116 CrossRef CAS PubMed.
- F. Kohori, M. Yokoyama, K. Sakai and T. Okano, J. Controlled Release, 2002, 78, 155 CrossRef CAS.
- P. K. Sharma and S. R. Bhatia, Int. J. Pharm., 2004, 278, 361 CrossRef CAS PubMed.
- R. Jalil and J. R. Nixon, J. Microencapsulation, 1990, 7, 297 CrossRef CAS PubMed.
- R. Basak and R. Bandyopadhyay, Langmuir, 2013, 29, 4350 CrossRef CAS PubMed.
- S. S. Desale, S. M. Cohen, Y. Zhao, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2013, 171, 339 CrossRef CAS PubMed.
- C. S. Fang, L. W. Fen, W. Z. Yong, L. Qiang and C. J. Hai, Pharmazie, 2012, 67, 781 Search PubMed.
- P. T. Vladimir, J. Controlled Release, 2001, 73, 137 CrossRef.
- D. A. Chiappetta and A. Sosnik, Eur. J. Pharm. Biopharm., 2007, 66, 303 CrossRef CAS PubMed.
- J. D. Jonkman-de Vries, K. P. Flora, A. Bult and J. H. Beijnen, Drug Dev. Ind. Pharm., 1996, 22, 475 CrossRef CAS.
- A. J. Tim, J. Verweij, W. J. Loos and A. Sparreboom, Clin. Pharmacokinet., 2003, 42, 665 CrossRef PubMed.
- M. C. Jones and J. C. Leroux, Eur. J. Pharm. Biopharm., 1999, 48, 101 CrossRef CAS.
- K. Kataoka, G. S. Kwon, M. Yokoyama, T. Okano and Y. Sakurai, J. Controlled Release, 1993, 24, 119 CrossRef CAS.
- A. Yokoyama, Y. Satoh, T. Sakurai, Y. Okano, T. Matsumura and K. K. Kataoka, J. Controlled Release, 1998, 55, 219 CrossRef.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137 CrossRef CAS.
- B. G. Yu, T. Okano, K. Kataoka and G. Kwon, J. Controlled Release, 1998, 53, 131 CrossRef CAS.
- Y. Kadam, U. Yerramilli and A. Bahadur, Colloids Surf., B, 2009, 72, 141 CrossRef CAS PubMed.
- Z. Attia, J. Ong, P. Hedrick, P. Lee, P. Rachel, P. Hammond and Y. Yang, Curr. Opin. Colloid Interface Sci., 2011, 16, 182 CrossRef PubMed.
- V. G. Kwon, Langmuir, 2006, 23, 9723 Search PubMed.
- V. Alakhov, E. Klinski, S. Li, G. Pietrzynski, A. Venne, E. Batrakova, T. Bronitch and A. Kabanov, Colloids Surf., B, 1999, 16, 113 CrossRef CAS.
- Y. Gao, L. Li and G. Zhai, Colloids Surf., B, 2008, 64, 194 CrossRef CAS PubMed.
- Y. Wang, L. Yu, L. Han, X. Sha and X. Fang, Int. J. Pharm., 2007, 337, 63 CrossRef CAS PubMed.
- Z. Wei, J. Hao, S. Yuan, Y. Li, W. Juan, X. Sha and X. Fang, Int. J. Pharm., 2009, 376, 176 CrossRef CAS PubMed.
- Z. Wei, S. Yuan, Y. Chen, S. Yu, J. Hao, J. Luo, X. Sha and X. Fang, Eur. J. Pharm. Biopharm., 2010, 75, 341 CrossRef PubMed.
- W. Zhang, Y. Shi, Y. Chen, J. Ye, X. Sha and X. Fang, Biomaterials, 2011, 32, 2894 CrossRef CAS PubMed.
- K. Oh, T. Bronich and A. Kabanov, J. Controlled Release, 2004, 94, 411 CrossRef CAS PubMed.
- E. Lee, Y. Oh, Y. Youn, M. Nam, B. Park, J. Yun, J. Kim, H. Song and K. Oh, Colloids Surf., B, 2011, 82, 190 CrossRef CAS PubMed.
- V. Alakhov and A. Kabanov, Expert Opin. Invest. Drugs, 1998, 7, 1453 CrossRef CAS PubMed.
- R. Nagarajan, M. Barry and E. Ruckenstein, Langmuir, 1986, 2, 210 CrossRef CAS.
- S. K. Patel, A. Lavasanifar and P. Choi, Biomacromolecules, 2009, 10, 2584 CrossRef CAS PubMed.
- K. K. Jette, D. Law, E. A. Schmitt and G. S. Kwon, Pharm. Res., 2004, 21, 1184 CrossRef CAS.
- X. T. Shuai, H. Ai, N. Nasongkla, S. Kim and J. M. Gao, J. Controlled Release, 2004, 98, 415 CrossRef CAS PubMed.
- J. E. Chung, M. Yokoyama, K. Suzuki, T. Aoyagi, Y. Sakurai and T. Okano, Colloids Surf., B, 1997, 9, 37 CrossRef CAS.
- J. Lavasanifar, S. Samuel, G. S. Sattari and B. Kwon, Pharm. Res., 2002, 19, 418 CrossRef.
- F. A. Lavasanifar, Macromol. Biosci., 2010, 10, 648 CrossRef PubMed.
- F. A. Lavasanifar, Colloids Surf., B, 2010, 8, 313 Search PubMed.
- O. Molavi, Z. Ma, A. Mahmud, A. Alshamsan, J. Samuel, R. Lai, G. S. Kwon and A. Lavasanifar, Int. J. Pharm., 2008, 347, 118 CrossRef CAS PubMed.
- T. Rupei, J. Weihang and W. Chun, Macromol. Chem. Phys., 2011, 212, 1185 CrossRef PubMed.
- Y. Chuan, B. Amalina, A. Ebrahim, P. K. T. Jeremy, K. Xiyu and G. Shujun, Biomaterials, 2012, 33, 2971 CrossRef PubMed.
- L. J. Samuel and G. S. Kwon, Colloids Surf., B, 2001, 22, 115 CrossRef.
- H. K. Kataoka, Macromolecules, 1995, 28, 5294 CrossRef.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6797 CrossRef CAS.
- J. F. Gohy, S. K. Varshney, S. Antoun and R. Jerome, Macromolecules, 2000, 33, 9298 CrossRef CAS.
- M. Iijima, Y. Nagasaki, T. Okada, M. Kato and K. Kataoka, Macromolecules, 1999, 32, 1140 CrossRef CAS.
- J. Q. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790 CrossRef CAS.
- J. H. Kim, K. Emoto, M. Iijima, Y. Nagasaki, T. Aoyagi, T. Okano, Y. Sakurai and K. Kataoka, Polym. Adv. Technol., 1999, 10, 647 CrossRef CAS.
- X. T. Shuai, T. Merdan, A. K. Schaper, F. Xi and T. Kissel, Bioconjugate Chem., 2004, 15, 441 CrossRef CAS PubMed.
- J. Tao and G. J. Liu, Macromolecules, 1997, 30, 2408 CrossRef CAS.
- T. G. J. Liu, J. F. Ding and M. L. Yang, Macromolecules, 1997, 30, 4084 CrossRef.
- K. Procházka, M. K. Baloch and Z. Tuzar, Makromol. Chem., 1979, 180, 2521 CrossRef PubMed.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718 RSC.
- Q. Jin, X. Liu, G. Liu and J. Ji, Polymer, 2010, 51, 1311 CrossRef CAS PubMed.
- L. J. Samuel and G. S. Kwon, J. Controlled Release, 2002, 79, 165 CrossRef.
- R. S. Stock and W. H. Fay, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1393 CrossRef CAS PubMed.
- G. S. Kwon, M. Naito, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Pharm. Res., 1995, 12, 192 CrossRef CAS.
- K. Ulbrich, C. Konak, Z. Tuzar and J. Kopecek, Makromol. Chem., 1987, 188, 1261 CrossRef CAS PubMed.
- V. S. Trubetskoy and V. P. Torchilin, STP Pharma Sci., 1996, 6, 79 Search PubMed.
- V. Weissig, K. R. Whiteman and V. P. Torchilin, Pharm. Res., 1998, 15, 1552 CrossRef CAS.
- K. Kataoka, H. Togawa, A. Harada, K. Yasugi, T. Matsumoto and S. Katayose, Macromolecules, 1996, 29, 8556 CrossRef CAS.
- S. Katayose and K. Kataoka, J. Pharm. Sci., 1998, 87, 160 CrossRef CAS PubMed.
- M. Yokoyama, A. Satoh, Y. Sakurai, T. Okano, Y. Matsumara, T. Kakizoe and K. Kataoka, J. Controlled Release, 1998, 55, 219 CrossRef CAS.
- S. A. Hagan, G. A. Coombes, M. C. Garnett, S. E. Dunn, M. C. Davies, L. Illum, S. S. Davis, S. E. Harding, S. Purkiss and P. R. Gellert, Langmuir, 1996, 12, 2153 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169 CrossRef CAS.
- Z. M. Miao, S. X. Zhang and R. X. Zhuo, Biomacromolecules, 2006, 7, 2020 CrossRef CAS PubMed.
- M. Shahin and L. Afsaneh, Int. J. Pharm., 2010, 389, 213 CrossRef CAS PubMed.
- L. Bromberg, Macromolecules, 1998, 31, 6148 CrossRef CAS.
- A. W. Alani, Y. Bae, D. A. Rao and G. S. Kwon, Biomaterials, 2010, 31, 1765 CrossRef CAS PubMed.
- B. Bilgicer, Biomedical Engineering, Class notes Biomolecular Topics in Engineering, March 2, 2009 Search PubMed.
- G. Kwon, S. Suwa, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1994, 29, 17 CrossRef CAS.
- A. Rolland, J. E. O'Mullane, P. Goddard, L. Brookman and K. Petrak, J. Appl. Polym. Sci., 1992, 44, 1195 CrossRef CAS PubMed.
- H. Maeda, L.
W. Seymour and Y. Miyamoto, Bioconjugate Chem., 1992, 3, 351 CrossRef CAS.
- R. K. Jain, Adv. Drug Delivery Rev., 1997, 26, 71 CrossRef CAS.
- F. Yuan, M. Leunig, S. K. Huang, D. A. Berk, D. Papahadjopoulos and R. K. Jain, Cancer Res., 1994, 54, 3352 CAS.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752 CAS.
- X. Zhang, H. M. Burt, G. Mangold, D. Dexter, D. Von Hoff, L. Mayer and W. L. Hunter, Anti-Cancer Drugs, 1997, 8, 686 CrossRef PubMed.
- M. Ramaswamy, X. Zhang, H. M. Burt and K. M. Wasan, J. Pharm. Sci., 1997, 86, 460 CrossRef CAS PubMed.
- M. Yokoyama, M. Miyauchi, N. Yamada, T. Okano, Y. Sakurai, K. Kataoka and S. Inoue, Cancer Res., 1990, 50, 1700 Search PubMed.
- X. Zhang, H. M. Burt, D. Von Hoff, D. Dexter, G. Mangold, D. Degen, A. M. Oktaba and W. L. Hunter, Cancer Chemother. Pharmacol., 1997, 40, 81 CrossRef CAS.
- M. Yokoyama, T. Okano, Y. Sakurai, H. Ekimoto, C. Shibazaki and K. Kataoka, Cancer Res., 1991, 51, 3229 CAS.
- M. Yokoyama, S. Fukushima, R. Uehara, K. Okamoto, K. Kataoka, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 50, 79 CrossRef CAS.
- A. V. Kabanov, V. P. Chekhonin, V. Y. Alakhov, E. V. Batrakova, A. S. Lebedev, N. S. Melik-Nubarov, S. A. Arzhakov, A. V. Levashov, G. V. Morozov, E. S. Severin and V. A. Kabanov, FEBS Lett., 1989, 258, 343 CrossRef CAS.
- A. V. Kabanov, E. V. Batrakova, N. S. Melik-Nubarov, N. A. Fedoseev, T. Y. Dorodnich, V. Y. Alakhov, V. P. Chekhonin, I. R. Nazarova and V. A. Kabanov, J. Controlled Release, 1992, 22, 141 CrossRef CAS.
- S. Schreier, S. V. P. Malheiros and E. Paula, Biochim. Biophys. Acta, Biomembr., 2000, 1508, 210 CrossRef CAS.
- B. G. Yu, T. Okano, K. Kataoka, S. Sardari and G. S. Kwon, J. Controlled Release, 1998, 56, 285 CrossRef CAS.
- K. Kataoka, G. S. Kwon, M. Yokoyama, T. Okano and Y. Sakurai, J. Controlled Release, 1993, 24, 119 CrossRef CAS.
- C. S. Cho, M. Y. Chang, H. C. Lee, S. C. Song, M. Goto and T. Akaike, Proc. Int. Symp. Controlled Release Bioact. Mater., 1998, 25, 721 Search PubMed.
- S. Cammas, K. Suzuki, C. Sone, Y. Sakurai, K. Kataoka and T. Okano, J. Controlled Release, 1997, 48, 157 CrossRef CAS.
- J. E. Chung, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 53, 119 CrossRef CAS.
- O. Meyer, D. Papahadjopoulos and J. C. Leroux, FEBS Lett., 1998, 42, 61 CrossRef.
- J. E. Chung, M. Yamato, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, Proc. Int. Symp. Controlled Release Bioact. Mater., 1998, 25, 380 Search PubMed.
- G. Helmlinger, F. Yuan, M. Dellian and R. K. Jain, Nat. Med., 1997, 3, 77 CrossRef PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373 CAS.
- D. C. Litzinger and L. Huang, Biochim. Biophys. Acta, 1992, 1113, 201 CrossRef CAS.
- M. K. Pratten and J. B. Lloyd, Makromol. Chem., 1985, 186, 725 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.