
Selective bromination of 2,5-bis(2-thienyl)pyrroles and solid-state polymerization through the β-carbon of pyrrole†
Abstract
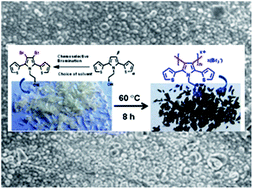
Bromination of 2,5-bis(2-thienyl)pyrrole with NBS in AcOH–THF prefers the β-position of pyrrole over the α-position of thiophene. A DFT study and experiments in different solvent systems suggest the important role of the solvent in tuning the selectivity. Unusual solid-state polymerization (SSP) of β,β′-dibrominated 2,5-bis(2-thienyl)pyrrole was observed through the β-C of pyrrole to afford the bromine doped conjugated polymer. DSC, CHN and SEM analyses were carried out to study the SSP process.
 Please wait while we load your content...
Please wait while we load your content...

