Metal-free aerobic C–H oxidation of cyclic enones†‡
P. A.
Peixoto§
,
M.
Cormier
,
J.
Ekosso Epane
,
A.
Jean
,
J.
Maddaluno
and
M.
De Paolis
*
Laboratoire COBRA, CNRS UMR 6014 & FR 3038, Université et INSA de Rouen, 76821-Mont Saint-Aignan, France. E-mail: michael.depaolis@univ-rouen.fr
First published on 16th June 2014
Abstract
A metal-free procedure is described for the aerobic and complete C–H methylene oxidation of Hajos–Parrish enones to versatile dihydroindenediones. The synthetic utility of these substrates was illustrated by converting them into highly substituted indanes after an intramolecular Friedel–Crafts conjugate addition. Importantly, the aerobic oxidation was compatible with substrates sensitive to radicals.
Introduction
The construction of an all-carbon quaternary stereocenter in a polycyclic framework is a challenging field of investigation. Methyl hydrindenones, such as chiral cyclohexenones, are representative of this type of framework for which the preparation is facilitated by the desymmetrization of prochiral triketones derived from 2-methyl-1,3-cyclopentadione in the presence of an organocatalyst.1 Further developments were reported to access the benzyl or allyl substituted hydrindenones from the corresponding 2-alkyl-1,3-cyclopentadiones, providing a rich platform for innovative and diversified synthetic developments.2 In the context of a project of aerobic and regioselective γ-oxidation of the Hajos–Parrish's enones, we view these structures containing aromatic substituents such as 1a as particularly interesting scaffolds (Scheme 1A). Indeed, selectively oxidizing the C3 position is a challenge: sp3 benzylic carbons may undergo intramolecular H-abstraction while aromatic rings or the allyl group may suffer from intramolecular radical attack.3 It is nevertheless a rewarding task due to the functionalized and polycyclic cyclopentenone obtained after mesylate elimination. Furthermore, a conversion of 1a into dihydroindenediones 3a would give access not only to unique hydrindenones but also to highly substituted indanes after intramolecular Friedel–Crafts conjugate addition. Hence, the preparation of steroid subunits and simplified analogues of natural products such as aplykurodinone-1 (4),4 gomerone C (5)5 and magellanine (6)6 could be projected (Scheme 1B). Herein, we report a metal-free methodology based on the generation of a dienolate of Hajos–Parrish's enones containing various benzylic and allylic substituents to promote the regioselective aerobic oxidation, allowing direct access to versatile dihydroindenediones.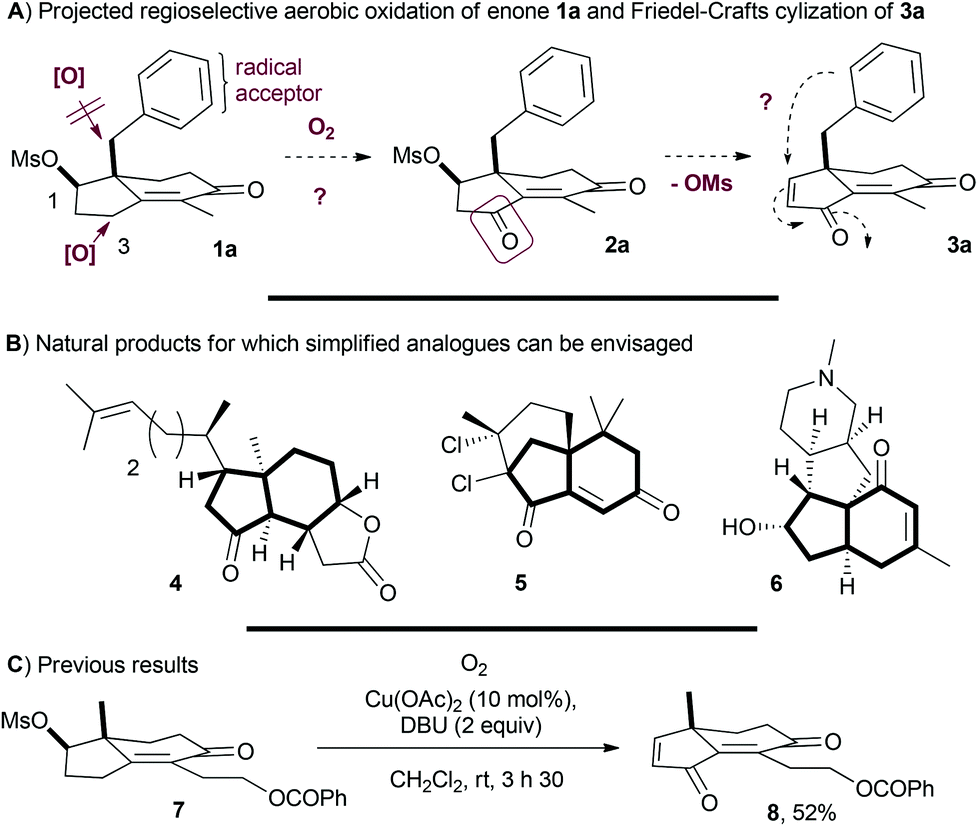 | ||
| Scheme 1 | ||
Metal-free C–H aerobic α-oxidation of electron-withdrawing moieties presents several advantages. In addition to the simplicity of air or oxygen as the oxidant and the absence of metals, the preformation of an alkyl or silyl enol ether is not required as in the typical Rubottom oxidation.7 The added-value of this methodology was demonstrated on ketones, nitriles, imines, esters and enones, leading to the corresponding peroxides, hydroxyls or fragmented products such as ketones depending on the moieties in the vicinity of the peroxide or the presence of reductants.8 Interestingly, when γ-oxidation of enones was performed, the corresponding peroxides or hydroxyls were obtained instead of the ketones.9 For the synthetic study of aplykurodinone-1 (4), we described the γ-aerobic oxidation of the enone 7 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and a catalytic amount of Cu(OAc)2 to produce the dienedione 8 in 52% yield (Scheme 1C).4b The extension of this protocol to enones containing benzylic appendages such as 1a was investigated and the results are presented herein.
Results and discussion
The preparation of mesylate 1a from 1,3-cyclopentadione and benzaldehyde was carried out first (Scheme 2A). Reductive coupling (81%) with the Hantzsch ester followed by quaternization (82%) with ethylvinyl ketone and Robinson annulation (88%) with p-toluenesulfonic acid delivered the enone 9a. Then, chemoselective reduction and mesylation afforded 1a in 93% yield (2 steps). In line with our previous protocol optimized for substrates without benzylic appendages, we began treating 1a with DBU in the presence of Cu(OAc)2 in CH2Cl2 at rt under an atmosphere of oxygen (Scheme 2B). As a result, a complex mixture of products was formed from which 3a could be isolated in no more than 10%. In order to limit side reactions, a copper-free process was tested. Harsher conditions were required to oxidize 1a but, interestingly, with better results. Hence, 3a was isolated in 22% yield by heating 1a at 120 °C for 5 days with DBU (1 equiv.) in distillated toluene under an atmosphere of oxygen.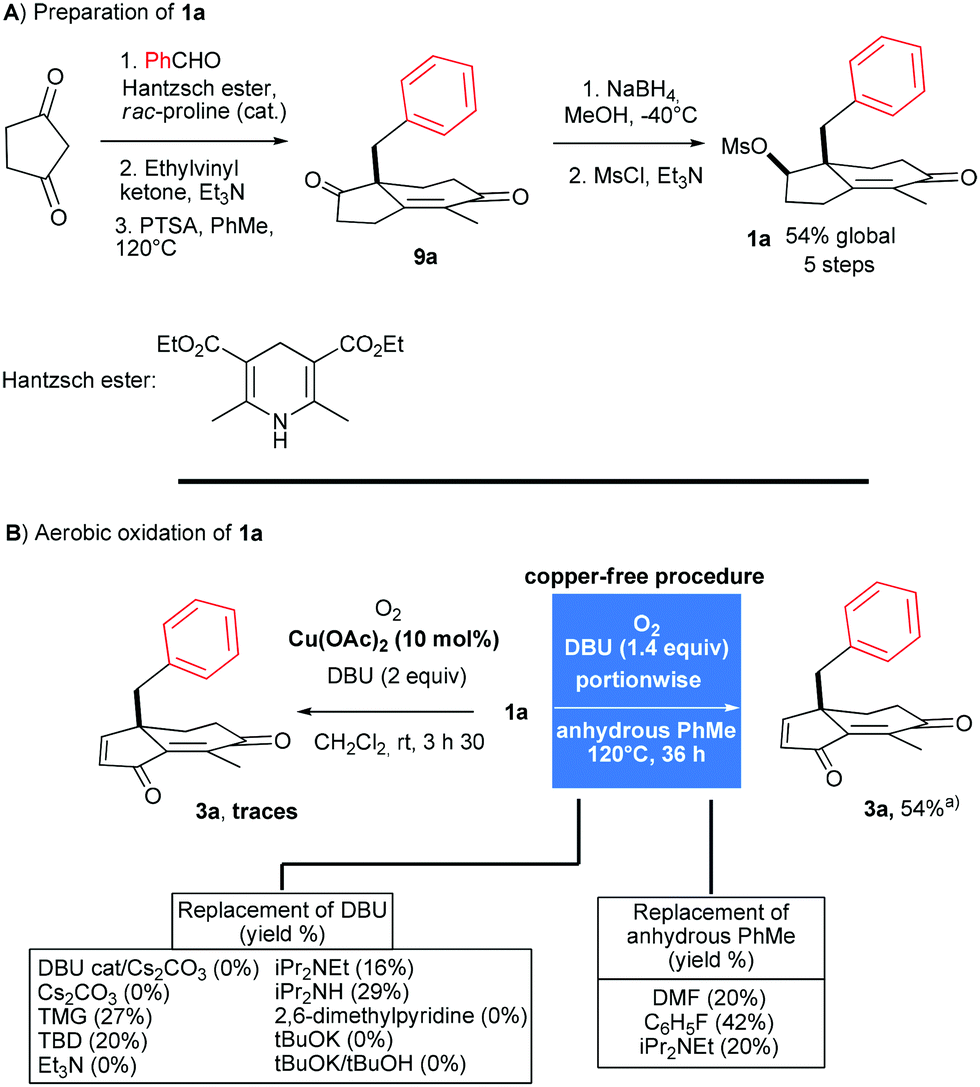 | ||
| Scheme 2 PTSA: p-toluenesulfonic acid; TMG: N,N,N′,N′-tetramethylguanidine; TBD: 1,5,7-triazabicyclo[4.4.0]dec-5-ene. aAverage yield of two experiments. | ||
Increasing the amount of DBU (1.4 equiv.) completed the reaction in 36 h and 3a was isolated in 41% yield.10 Increasing further to 1.5 equiv. the amount of DBU led to lower yield and we assumed that 3a, as a Michael acceptor, was to some extent sensitive to nucleophiles such as DBU.11 While less nucleophilic bases than DBU are available, the strength and tolerance to oxygen are key requirements and a screening of bases such as guanidines, trialkylamines, lutidine, alkoxides or inorganic bases led to lower conversion or no oxidation at all (see Scheme 2B). Somewhat counterintuitively, the solvent in which oxygen is more soluble such as DMF or fluorobenzene was less efficient to promote the reaction, indicating that the polarity and oxygen solubility have to be finely tuned.12 To reduce the concentration of DBU and limit therefore the risk of degradation, a sub-stoichiometric amount of DBU was introduced. After screening of conditions, involving different concentrations, amounts of base, induction time and the use of a syringe pump, we found that adding 0.2 equiv. of DBU to 1a followed by heating (12 h) and portionwise addition of DBU (0.2 equiv. every 1.5 h, for a total of 1.4 equiv.) provided the best result. Proceeding in this way, we were able to isolate 3a in 54% yield after 36 h (Scheme 2B). It is noteworthy that the C3 methylene position is oxidised exclusively to the corresponding ketone during the process. Importantly, this procedure has been repeated several times by different experimentalists and provided consistent results.
Once the optimization was completed, we investigated the compatibility of the chemistry with various substituents at the neopentyl position. Hence, mesylates 1b–e containing naphthyl, p-methoxyphenyl, o-bromophenyl and o-nitrophenyl substituents were prepared and tested (see ESI‡ for details). Remarkably, dihydroindenediones 3b–e were obtained with uniform efficiency in yields ranging from 43 to 54% with the exception of the o-nitrophenyl derivative 3e which was isolated in 31% yield (Scheme 3). Applying the optimized protocol to the allyl-substituted 1f led to the oxidised product 3f in 35% yield. Still, substituted with an allyl group, 3f is a promising substrate for synthetic transformations. As expected for alkane substituted substrates, 1g and 1h were converted more efficiently into 3g (46%) and 3h (50%). From a practical point, the crude mixture obtained was very clean in all cases, facilitating the purification of 3a–h.13
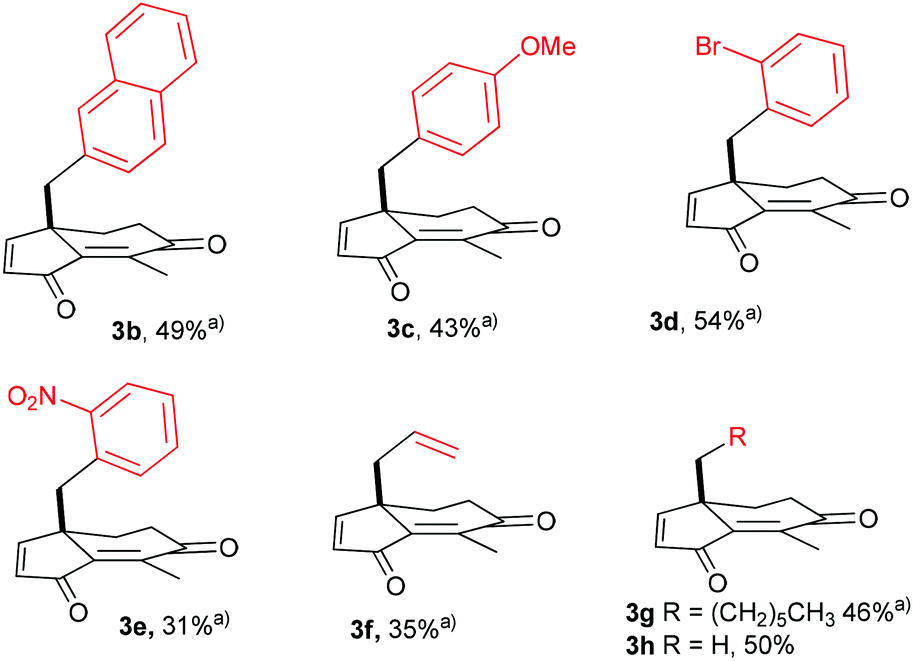 | ||
| Scheme 3 Direct preparation of 3b–h from the corresponding mesylates 1b–h using the optimized protocol. aAverage yield of two experiments. | ||
Even though no detailed mechanistic study was conducted, some observations can be made from experiments. Hence, the oxidation at C3 appears to be the first step since there is no elimination of the mesylate when the reaction is conducted in the absence of oxygen.
To begin with, the formation of the thermodynamic dienolate 10a (R1 = Ph, R2 = Me) can be reasonably postulated (Scheme 4A). A first mechanism could involve a single electron transfer (SET) to oxygen to produce the radical species 11a and 12a which, after combination with the superoxide radical anion, could form the peroxides 13a and 14a. As a Mislow–Evans rearrangement of allylic sulfoxide, allylic peroxide 14a could undergo [2,3]-sigmatropic rearrangement to produce 13a.
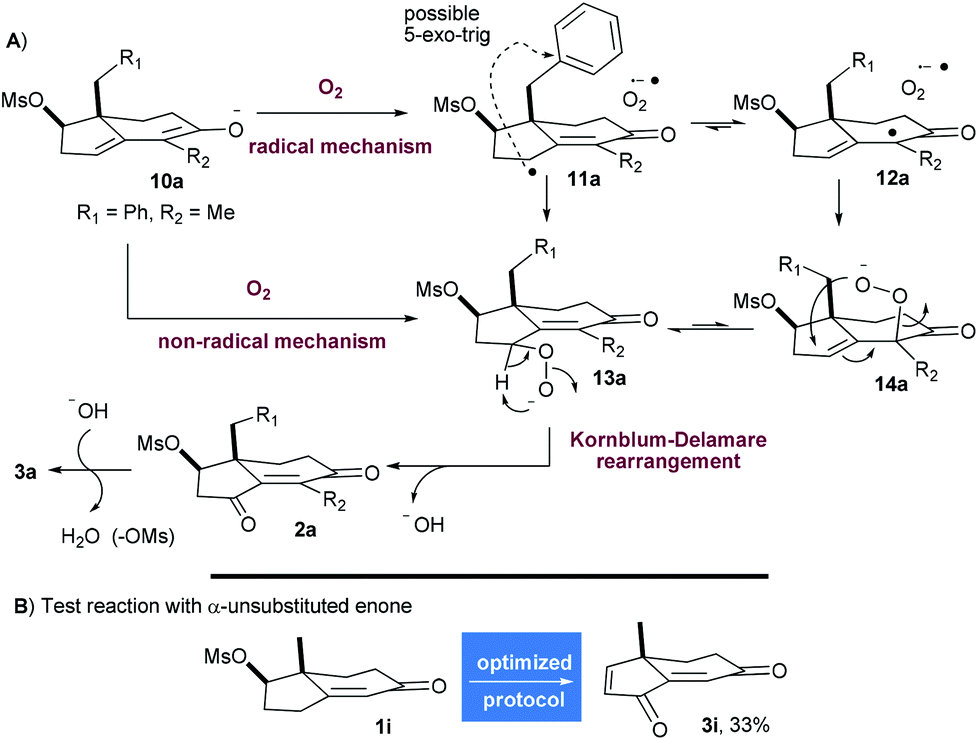 | ||
| Scheme 4 Mechanistic discussions. | ||
To explain the unusual formation of a ketone at C3, a Kornblum–DeLamare rearrangement of peroxide 13a by intra- or intermolecular deprotonation (with DBU) could be invoked to generate 2a.14 The production of hydroxide would trigger the elimination of mesylate and conclude the process. As postulated by Gersmann, an alternative non-radical mechanism could involve the direct combination of dienolate 10a and oxygen to provide the regioisomeric peroxides 13a and 14a.9b,c With the benzylic substrate 1a and the methyl substrate 1h being oxidised with the same efficiency (54% of 3a and 50% of 3h), it seems unlikely that radical species are involved since 1,5-hydrogen atom abstraction from the benzylic position of 11a/12a or radical 5-exo-trig cyclization (see Scheme 4A) could probably occur. These side reactions would be favored on arenes substituted with electron withdrawing or donating groups stabilizing radical species. In this regard, the smooth oxidation of compounds 1c and 1d containing p-methoxy and o-bromo substituents into 3c (43%) and 3d (54%) uphold the hypothesis that an anionic scenario may predominate. Moreover, the process was unaffected by the exclusion of light. Therefore, it seems plausible that an anionic pathway is active during this process even if we cannot completely rule out a competitive and parallel radical cage mechanism being operative since the oxidation of o-nitrobenzylic and allylic substituted enones 1e and 1f was less efficient, but these two appendages are very sensitive to radicals and bases.
The regioselectivity of the reaction on enones that are not α-substituted such as 1i was next investigated (Scheme 4B). Indeed, the Kornblum–DeLamare rearrangement of peroxide 14i (R2 = R1 = H) could lead to unstable 1,2-diketone under these conditions. To investigate this scenario, 1i was oxidised under the optimized conditions delivering 3i in 33% yield. In comparison, the α-substituted enone 1h was converted into 3h in 50% yield, indicating clearly that α-substituted enones are more efficiently oxidised in the γ-position with this protocol.
To illustrate the utility of these scaffolds, an internal Friedel–Crafts conjugate addition was implemented (Scheme 5). After screening of Brønsted and Lewis acids, the best results were obtained with AlCl3.15 Hence, 3a was efficiently converted into indane 15a (84%) featuring an all-carbon quaternary center embedded into a tetracyclic framework. Similarly, the naphthyl derivative 3b provided regioselectively 15b in 61% yield by reaction at the sp2 carbon with the highest electronic density.
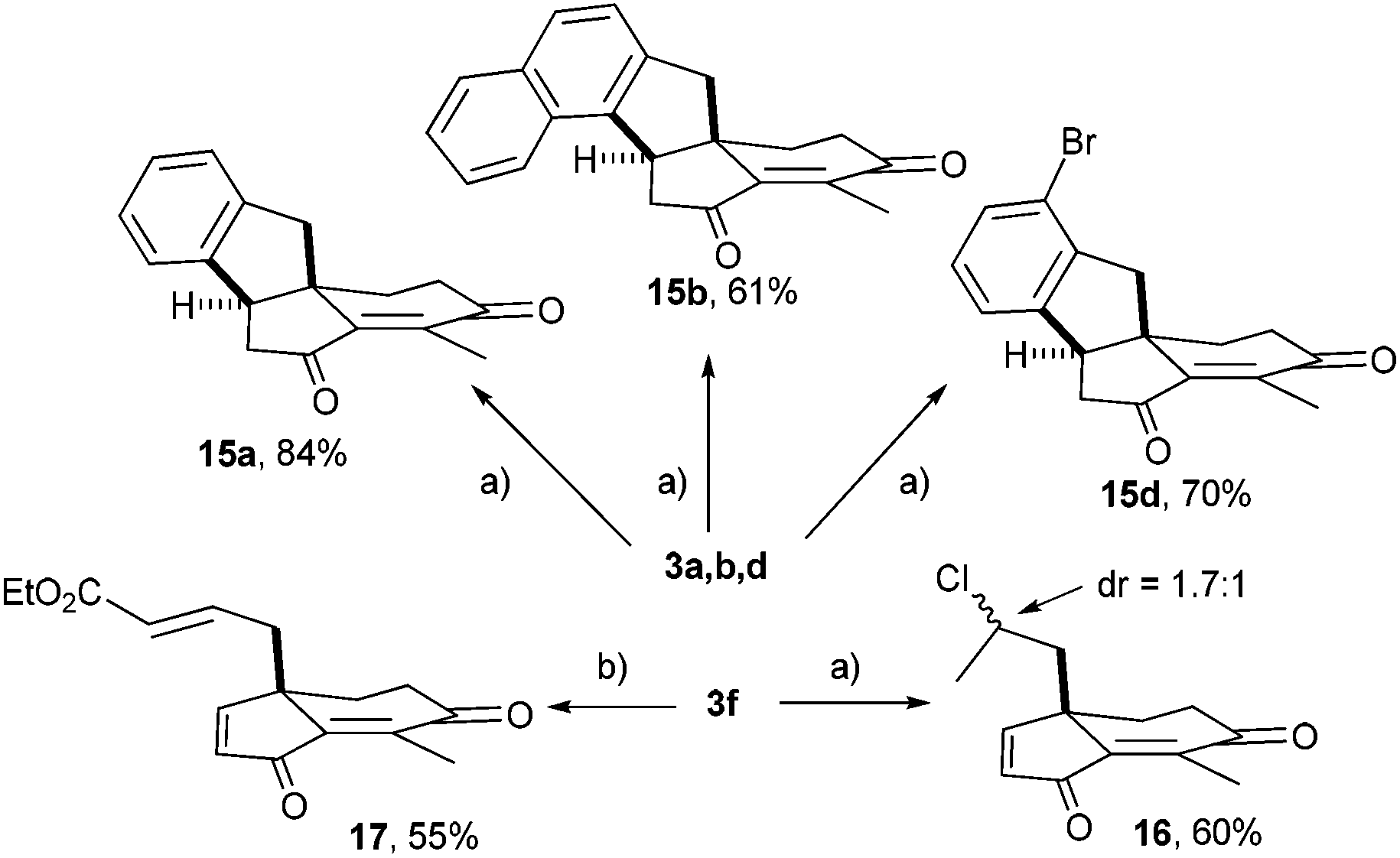 | ||
| Scheme 5 Reagents and conditions: (a) AlCl3 (5 equiv.), CH2Cl2, rt; (b) Grubbs 2nd generation (2 mol%), ethyl acrylate (6 equiv.), CH2Cl2, rt. | ||
Interestingly, 3d was converted into versatile 15d (70%), a compound calling for various C–Br modifications toward molecular diversity. Unexpectedly, similar treatment of allylic substrate 3f did not produce the cyclized product but yielded the Markovnikov hydrochloration adduct 16 (60%) with a modest stereoselectivity (dr = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7). As a further illustration of the potential of 3f, cross-metathesis afforded acrylate 17 (55% yield not optimized) as a promising template for double Michael reactions.
:1.7). As a further illustration of the potential of 3f, cross-metathesis afforded acrylate 17 (55% yield not optimized) as a promising template for double Michael reactions.
Conclusion
In summary, an expedient and simple route to difficult-to-access dihydroindenediones containing various benzylic and allylic groups is described. Based on the aerobic oxidation of the dienolate of Hajos–Parrish's enones, the reaction proceeds with medium efficiency (31–54%) but remains useful in view of the number of transformations involved. Indeed, at least four transformations including deprotonation, oxygenation, rearrangement and elimination are occurring in one flask with an average yield of 75–85% for each one of them. The process led to the complete oxidation of the C–H methylene and is compatible with substrates sensitive to radicals. Further synthetic transformations of the oxidised products include their conversion into indanes displaying a highly substituted carbon network.Experimental
All reactions were carried out under a nitrogen or an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Dry methylene chloride and toluene were respectively obtained by distillation from CaH2 and from sodium. Aerobic oxidations were carried out in a two-neck flask fitted with a vertical condenser with a doubled balloon of oxygen at the top. After prolonged storage, mesylates 1a–i were dried before engaging them in the oxidative process by azeotropic water removal with dry PhMe or by filtration on a pad of SiO2. Reactions were monitored by thin-layer chromatography (TLC) on Merck silica gel plates with QF-254 indicator followed by one of these staining reagents: ammonium molybdate or potassium permanganate. Merck silica gel (60, 40–63 μm) was used for flash column chromatography. NMR experiments were recorded on Bruker Advance DMX-300 or -200 instruments and calibrated using residual undeuterated solvent as an internal reference (7.26 and 77.00 ppm for 1H and 13C NMR in CDCl3). IR spectra were recorded on a PerkinElmer Spectrum 100 FT-IR-spectrometer with major peaks reported. High-resolution mass spectra (HRMS) were recorded by the mass spectrometry service at the IRCOF on an LCT Premier XE benchtop orthogonal acceleration time-of-flight (TOF) mass spectrometer (Waters Micromass).General procedure for the preparation of dienedione
To a stirred solution of mesylates 1a–i (1.0 equiv.) in toluene (0.1 M) at rt was added DBU (0.2 equiv.). The flask was first flushed with O2 and then equipped with a vertical condenser fitted with a balloon of O2. The resulting mixture was then stirred for 10 h at 110 °C (oil bath at 120 °C). After 10 h, 0.2 equiv. of DBU was successively added every 1.5 h (6 × 0.2 equiv., 1.4 equiv. in total). Once 1.4 equiv. of DBU were added, the reaction mixture was stirred at reflux for another 17 h before being concentrated in vacuo. Flash column chromatography of the residue afforded dienediones 3a–i.
3a: According to the general procedure, the reaction of mesylate 1a (201 mg, 0.60 mmol) and DBU (7 × 18 μL, 0.84 mmol) in toluene (6 mL) provided 3a (83 mg, 55%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.5 (cyclohexane–EtOAc, 7:3); IR (film) ν 3027, 2937, 1690, 1668, 1639, 1451, 1181, 820, 738, 701, 469 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.25 (d, J = 6.1 Hz, 1H), 7.07–7.32 (m, 3H), 6.78–6.95 (m, 2H), 6.17 (d, J = 6.1 Hz, 1H), 2.71–3.01 (m, 3H), 2.58 (ddd, J = 19.4, 6.2, 1.5 Hz, 1H), 2.19 (ddd, J = 13.3, 5.8, 1.5 Hz, 1H), 2.10 (s, 3H), 1.86 (ddd ∼ dt, J = 13.3, 6.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 200.0, 196.8, 162.8, 150.4, 135.7, 134.9, 134.3, 130.0 (2C), 128.4 (2C), 127.0, 47.8, 43.0, 33.9, 29.0, 9.8; HRMS (API): calcd for C17H17O2+ [M + H]+: 253.1229, found 253.1227
3b: According to the general procedure, the reaction of mesylate 1b (354 mg, 0.92 mmol) and DBU (7 × 28 μL, 1.29 mmol) in toluene (9.2 mL) provided 3b (136 mg, 49%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.45 (cyclohexane–EtOAc, 65:35); IR (film) ν 2965, 1692, 1670, 1258, 1072, 1012, 816, 734, 475 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.65–7.88 (m, 3H), 7.39–7.51 (m, 3H), 7.35 (d, J = 6.1 Hz, 1H), 7.08 (dd, J = 8.4, 1.7 Hz, 1H), 6.23 (d, J = 6.1 Hz, 1H), 3.15 (d, J = 13.4 Hz, 1H), 2.92–3.10 (m, 2H), 2.69 (ddd, J = 19.3, 6.1, 1.2 Hz, 1H), 2.26 (ddd, J = 13.3, 5.9, 1.2 Hz, 1H), 2.21 (s, 3H), 1.93 (ddd ∼ dt, J = 13.3, 6.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 200.1, 196.7, 162.8, 150.5, 134.9, 134.3, 133.2, 133.2, 132.2, 128.7, 128.0, 127.9, 127.6, 127.5, 126.3, 125.9, 47.9, 43.1, 34.0, 28.9, 9.8; HRMS (API): calcd for C21H19O2+ [M + H]+: 303.1385, found 303.1387.
3c: According to the general procedure, the reaction of mesylate 1c (200 mg, 0.55 mmol) and DBU (7 × 17 μL, 0.77 mmol) in toluene (5.5 mL) provided 3c (66 mg, 43%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.4 (cyclohexane–EtOAc, 65:35); IR (film) ν 2921, 2854, 1692, 1670, 1613, 1510, 1445, 1305, 1245, 1176, 1030, 816, 516 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 6.1 Hz, 1H), 6.87 (d, J = 8.7 Hz, 2H), 6.73–6.82 (m, 2H), 6.23 (d, J = 6.1 Hz, 1H), 3.76 (s, 3H), 2.81–3.01 (m, 3H), 2.63 (ddd, J = 19.3, 6.1, 1.2 Hz, 1H), 2.20 (ddd, J = 13.3, 5.8, 1.2 Hz, 1H), 2.15 (s, 3H), 1.92 (ddd ∼ dt, J = 13.3, 6.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 200.1, 196.8, 162.9, 158.5, 150.5, 134.9, 134.1, 131.0 (2C), 127.6, 113.7 (2C), 55.1, 47.9, 42.1, 33.9, 28.9, 9.8; HRMS (API): calcd for C18H19O3+ [M + H]+: 283.1334, found 283.1351.
3d: According to the general procedure, the reaction of mesylate 1d (200 mg, 0.48 mmol) and DBU (7 × 14 μL, 0.67 mmol) in toluene (4.8 mL) provided 3d (85 mg, 54%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.4 (cyclohexane–EtOAc, 65:35); IR (film) ν 2977, 2937, 1669, 1441, 1179, 1126, 1063, 1023, 950, 830, 764, 445 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.49 (d, J = 6.0 Hz, 1H), 7.45 (dd, J = 8.0, 1.4 Hz, 1H), 7.14 (td, J = 7.5, 1.4 Hz, 1H), 7.02 (td, J = 8.0, 1.6 Hz), 6.87 (dd, J = 7.5, 1.6 Hz, 1H), 6.12 (d, J = 6.0 Hz, 1H), 3.38 (d, J = 13.4 Hz, 1H), 3.09 (d, J = 13.4 Hz, 1H), 2.99 (ddd, J = 19.3, 13.5, 5.8 Hz, 1H), 2.67 (ddd, J = 19.3, 6.1, 1.5 Hz, 1H), 2.31 (ddd, J = 13.5, 5.8, 1.5 Hz, 1H), 2.12 (s, 3H), 2.06 (ddd ∼ dt, J = 13.3, 6.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 199.9, 196.4, 161.3, 149.2, 135.1, 135.0, 134.8, 133.1, 132.2, 128.8, 127.1, 125.8, 48.5, 41.1, 33.9, 30.8, 9.8; HRMS (ESI): calcd for C17H16O2Br+ [M + H]+: 331.0334 and 333.0313, found 331.0339 and 333.0320.
3e: According to the general procedure, the reaction of mesylate 1e (569 mg, 1.5 mmol) and DBU (7 × 45 μL, 2.1 mmol) in toluene (15 mL) provided 3e (136 mg, 31%) as a pale yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.35 (cyclohexane–EtOAc, 6:4); IR (film) ν 2915, 2848, 1691, 1674, 1522, 1462, 1349, 1171, 1080, 841, 827, 784, 747, 710, 472 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.86 (dd, J = 7.8, 1.3 Hz, 1H), 7.44 (td, J = 7.5, 1.5 Hz, 1H), 7.36 (d, J = 6.0 Hz, 1H), 7.36 (td, J = 7.8, 1.5 Hz, 1H), 6.94 (dd, J = 7.5, 1.3 Hz, 1H), 6.06 (d, J = 6.0 Hz, 1H), 3.58 (d, J = 13.3 Hz, 1H), 3.52 (d, J = 13.3 Hz, 1H), 2.94 (ddd, J = 19.4, 13.5, 5.8 Hz, 1H), 2.70 (ddd, J = 19.4, 6.1, 1.4 Hz, 1H), 2.32 (ddd, J = 13.5, 5.8, 1.4 Hz, 1H), 2.12 (s, 3H), 2.11 (ddd ∼ dt, J = 13.5, 6.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 199.5, 196.1, 160.9, 150.0, 148.7, 135.4, 135.1, 133.7, 132.7, 130.7, 128.4, 125.2, 48.6, 38.2, 33.8, 31.2, 9.8; HRMS (API): calcd for C17H16NO4+ [M + H]+: 298.1079, found 298.1079.
3f: According to the general procedure, the reaction of mesylate 1f (150 mg, 0.52 mmol) and DBU (7 × 16 μL, 0.73 mmol) in toluene (10 mL) provided 3f (36 mg, 35%) as a pale yellow amorphous solid (cyclohexane–EtOAc/8:2). Rf = 0.5 (cyclohexane–EtOAc, 6:4); IR (film) ν 2925, 2855, 1694, 1674, 1638, 1451, 1178, 1099, 1072, 1021, 824 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.44 (d, J = 6.1 Hz, 1H), 6.34 (d, J = 6.1 Hz, 1H), 5.50–5.71 (m, 1H), 5.11 (d, J = 10.0 Hz, 1H), 5.03 (dd, J = 16.9, 1.3 Hz, 1H), 2.76 (ddd, J = 19.4, 13.3, 5.7 Hz, 1H), 2.55 (ddd, J = 19.4, 6.2, 1.4 Hz, 1H), 2.46 (dd, J = 13.8, 7.9 Hz, 1H), 2.31 (dd, J = 13.9, 7.1 Hz, 1H), 2.21 (ddd, J = 13.3, 5.7, 1.4 Hz, 1H), 2.12 (s, 3H), 1.91 (ddd ∼ dt, J = 13.3, 6.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 200.2, 197.2, 162.8, 150.2, 135.1, 134.3, 132.3, 119.6, 46.8, 41.1, 33.6, 29.0, 9.8; HRMS (API): calcd for C13H15O2+ [M + H]+: 203.1072, found 203.1072.
3g: According to the general procedure, the reaction of mesylate 1g (357 mg, 1.0 mmol) and DBU (7 × 30 μL, 1.4 mmol) in toluene (10 mL) provided 3g (117 mg, 46%) as a pale yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.4 (cyclohexane–EtOAc, 8:2); IR (film) ν 2925, 2856, 1694, 1674, 1638, 1456, 1178, 1072, 825 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.43 (d, J = 6.1 Hz, 1H), 6.35 (d, J = 6.1 Hz, 1H), 2.75 (ddd, J = 19.2, 13.2, 5.8 Hz, 1H), 2.54 (ddd, J = 19.2, 6.1, 1.5 Hz, 1H), 2.17 (ddd, J = 13.2, 5.8, 1.5 Hz, 1H), 2.12 (s, 3H), 1.90 (ddd ∼ dt, J = 13.2, 6.1 Hz, 1H), 1.31–1.74 (m, 1H), 1.20 (br s, 10H), 0.95–1.10 (m, 1H), 0.84 (t, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 200.4, 197.5, 163.4, 150.9, 135.0, 133.6, 46.8, 36.5, 33.7, 31.6, 29.8, 29.7, 28.9, 25.2, 22.4, 13.9, 9.7; HRMS (API): calcd for C17H25O2+ [M + H]+: 261.1855, found 261.1861.
3h: According to the general procedure, the reaction of mesylate 1h (256 mg, 1.0 mmol) and DBU (7 × 30 μL, 1.4 mmol) in toluene (10 mL) provided 3h (88 mg, 50%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.45 (cyclohexane–EtOAc, 6:4); IR (film) ν 2921, 2870, 1693, 1664, 1641, 1336, 1217, 1149, 1007, 950, 827, 468 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J = 6.0 Hz, 1H), 6.25 (d, J = 6.0 Hz, 1H), 2.78 (ddd, J = 19.1, 12.9, 6.1 Hz, 1H), 2.57 (ddd, J = 19.1, 5.8, 2.1 Hz, 1H), 2.10 (s, 3H), 2.04 (dd, J = 6.1, 2.1 Hz, 1H), 1.98 (ddd ∼ dt, J = 12.9, 5.8 Hz, 1H), 1.32 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 200.2, 197.4, 165.2, 150.9, 133.6 (2C), 43.5, 33.9, 31.4, 23.8, 9.6; HRMS (ESI): calcd for C11H13O2+ [M + H]+: 177.0916, found 177.0910.
3i: According to the general procedure, the reaction of mesylate 1i (100 mg, 1.0 mmol) and DBU (7 × 18 μL, 1.4 mmol) in toluene (4 mL) provided 3i (22 mg, 33%) as a yellow amorphous solid (cyclohexane–EtOAc/7:3). Rf = 0.45 (cyclohexane–EtOAc, 6:4); IR (film) ν 2928, 1680, 1351, 1173, 1074, 958, 845, 818, 530 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.60 (dd, J = 6.0, 1.0 Hz, 1H), 6.34 (d, J = 6.0 Hz, 1H), 6.27 (br s, 1H), 2.80 (ddd, J = 19.0, 13.3, 5.7 Hz, 1H), 2.56–2.66 (m, 1H), 2.14 (ddd, J = 12.9, 5.6, 2.0 Hz), 2.05 (dd, J = 13.1, 5.5 Hz, 1H), 1.41 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 199.1, 195.7, 166.4, 159.1, 133.2, 121.4, 43.1, 34.7, 32.3, 23.6; HRMS (ESI): calcd for C10H9O2− [M − H]−: 161.0603, found 161.0600.
General procedure for the Friedel–Crafts conjugate addition
To a stirred solution of dihydroindenediones 3a–c in CH2Cl2 at rt was added AlCl3 (5 equiv.). The resulting reaction mixture was stirred until completion (monitored by TLC) before being quenched with HCl (1 N, aq. sol.). The layers were separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were brined, dried (Na2SO4) and concentrated in vacuo. Flash chromatography of the crude afforded indanes 15a–c or chloroalkanes 16 and 16′.
15a: According to the general procedure, the reaction of 3a (50 mg, 0.2 mmol) and AlCl3 (133 mg, 1.0 mmol) in CH2Cl2 (4 mL) provided 15a (42 mg, 84%) as a yellow oil (cyclohexane–EtOAc/8:2). Rf = 0.60 (cyclohexane–EtOAc, 7:3); IR (film) ν 2915, 1711, 1671, 1619, 1160, 1138, 1058, 813, 782, 760, 470, 425 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.09–7.27 (m, 5H), 3.29 (t, J = 9.7 Hz, 1H), 3.20 (d, J = 16.5 Hz, 1H), 2.93 (t, J = 4.6 Hz, 1H), 2.86 (d, J = 9.7 Hz, 1H), 2.67 (ddd, J = 19.4, 14.2, 5.4 Hz, 1H), 2.48 (ddd, J = 19.4, 4.8, 2.0 Hz, 1H), 2.23 (dd, J = 19.6, 9.0 Hz, 1H), 2.07 (s, 3H), 2.01–2.14 (m, 1H), 1.92 (ddd ∼ dt, J = 14.2, 4.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 208.1, 200.7, 151.2, 145.1, 140.3, 136.2, 127.4, 127.2, 125.4, 124.7, 52.4, 50.8, 45.3, 39.5, 33.8, 33.3, 10.0; HRMS (API): calcd for C17H17O2+ [M + H]+: 253.1229, found 253.1230.
15b: According to the general procedure, the reaction of 3b (20 mg, 0.07 mmol) and AlCl3 (47 mg, 0.35 mmol) in CH2Cl2 (5 mL) provided indane 15b (17 mg, 80%) as a yellow oil (cyclohexane–EtOAc/8:2). Rf = 0.60 (cyclohexane–EtOAc, 65:35); IR (film) ν 3055, 2921, 2848, 1712, 1672, 1618, 1378, 1186, 1159, 1137, 1058, 811, 782, 730, 467 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.86–7.94 (m, 1H), 7.76 (t, J = 7.6 Hz, 2H), 7.45–7.58 (m, 2H), 7.38 (d, J = 8.4 Hz, 1H), 3.86 (t, J = 9.3 Hz, 1H), 3.47 (d, J = 16.6 Hz, 1H), 3.12–3.27 (m, 2H), 2.83 (ddd, J = 18.3, 14.1, 5.4 Hz, 1H), 2.60 (ddd, J = 18.3, 4.9, 2.1 Hz, 1H), 2.37 (dd, J = 19.7, 8.7 Hz, 1H), 2.24 (ddd, J = 14.1, 5.4, 2.1 Hz, 1H), 2.20 (s, 3H), 2.16 (ddd ∼ dt, J = 14.1, 4.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 208.3, 200.8, 151.2, 140.9, 137.3, 136.2, 133.1, 130.1, 128.7, 128.2, 126.6, 125.4, 123.7, 123.5, 52.4, 49.3, 44.8, 40.8, 34.1, 33.9, 10.1; HRMS (API): calcd for C21H19O2+ [M + H]+: 303.1385, found 303.1385.
15d: According to the general procedure, the reaction of 3d (10 mg, 0.03 mmol) and AlCl3 (28 mg, 0.21 mmol) in CH2Cl2 (5 mL) provided 15d (7 mg, 70%) as a yellow powder (cyclohexane–EtOAc/8:2). Rf = 0.50 (cyclohexane–EtOAc, 7:3); IR (film) ν 2919, 2854, 1713, 1673, 1449, 1180, 1129, 1062, 959, 780 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 7.9 Hz, 1H), 7.21 (d, J = 7.4 Hz, 1H), 7.06–7.14 (m, 1H), 3.48 (t, J = 9.4 Hz, 1H), 3.38 (d, J = 17.1 Hz, 1H), 2.90–3.05 (m, 2H), 2.74 (ddd, J = 19.6, 14.1, 5.3 Hz, 1H), 2.59 (ddd, J = 18.3, 4.8, 1.9 Hz, 1H), 2.32 (dd, J = 19.6, 8.8 Hz, 1H), 2.21 (ddd, J = 13.1, 5.3, 1.9 Hz, 1H), 2.16 (s, 3H), 2.09 (ddd ∼ dt, J = 14.1, 4.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 208.3, 200.8, 151.2, 140.9, 137.3, 136.2, 133.1, 130.1, 128.7, 128.2, 126.6, 125.4, 123.7, 123.5, 52.4, 49.3, 44.8, 40.8, 34.1, 33.9, 10.1; HRMS (API): calcd for C17H16O2Br+ [M + H]+: 331.0334 and 333.0313, found 331.0331 and 333.0307.
16: According to the general procedure, the reaction of dihydroindenedione 3f (24 mg, 0.12 mmol) and AlCl3 (80 mg, 0.6 mmol) in CH2Cl2 (5 mL) provided a stereoisomeric mixture of diastereoisomer 16 (6 mg, 21%) and 16′ (11 mg, 39%) as a colorless oil (cyclohexane–EtOAc/8:2 to 7:3). 16: Rf = 0.5 (cyclohexane–EtOAc, 6:4); IR (film) ν 3072, 2927, 2848, 1695, 1665, 1445, 1316, 1184, 1020, 844, 828, 710, 615, 506, 404 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.63 (d, J = 6.1 Hz, 1H), 6.42 (d, J = 6.1 Hz, 1H), 3.84 (m, 1H), 2.83 (ddd, J = 19.5, 13.2, 5.6 Hz, 1H), 2.60 (ddd, J = 19.5, 6.2, 1.2 Hz, 1H), 2.46 (ddd, J = 13.2, 5.6, 1.2 Hz, 1H), 2.32 (dd, J = 15.2, 8.5 Hz, 1H), 2.15 (s, 3H), 1.97 (dd, J = 15.2, 3.8 Hz, 1H), 1.88 (ddd ∼ dt, J = 13.2, 6.2 Hz, 1H), 1.44 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 200.1, 196.9, 162.6, 149.9, 135.8, 134.8, 54.3, 46.8, 46.4, 33.9, 30.4, 27.1, 10.0; HRMS (API): calcd for C13H16O2Cl [M + H+]: 239.0839, found 239.0834. 16′: Rf = 0.35 (cyclohexane–EtOAc, 6:4); IR (film) ν 2926, 1674, 1186, 825, 617 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58 (d, J = 6.1 Hz, 1H), 6.41 (d, J = 6.1 Hz, 1H), 3.78–4.05 (m, 1H), 2.69 (ddd, J = 19.3, 13.2, 5.3 Hz, 1H), 2.57 (ddd, J = 19.3, 6.6, 1.8 Hz, 1H), 2.21 (ddd, J = 13.2, 5.3, 1.8 Hz, 1H), 2.14 (s, J = 3.6 Hz, 3H), 2.08–2.17 (m, 2H), 1.95 (ddd ∼ dt, J = 13.2, 6.6 Hz, 1H), 1.50 (d, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 199.8, 196.7, 161.5, 150.5, 135.6, 133.8, 53.9, 46.7, 46.3, 33.7, 31.0, 26.5, 9.9; HRMS (API): calcd for C13H16O2Cl [M + H]+: 239.0839, found 239.0845.
17: To a stirred solution of 3f (24 mg, 0.060 mmol) and ethyl acrylate (40 μL, 0.360 mmol) in CH2Cl2 (5 mL) at rt was added Grubbs II catalyst (10 mg, 0.0012 mmol). The resulting mixture was stirred for 12 h before being concentrated in vacuo. Flash column chromatography (cyclohexane–EtOAc, 70:30) of the crude afforded acrylate 17 (18 mg, 55%) as a yellow oil. Rf = 0.55 (cyclohexane–EtOAc, 60:40); IR (film) ν 3029, 2927, 1653, 1352, 1331, 1173, 1017, 940, 843, 743, 702 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.49 (d, J = 6.1 Hz, 1H), 6.76 (m, 1H), 6.39 (d, J = 6.1 Hz, 1H), 5.83 (d, J = 15.5 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 2.53–2.81 (m, 3H), 2.38 (dd, J = 14.0, 7.4 Hz, 1H), 2.23 (dd, J = 13.2, 4.3 Hz, 1H), 2.14 (s, 3H), 1.94 (ddd ∼ dt, J = 13.2, 6.4 Hz, 1H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 199.7, 196.5, 165.5, 161.9, 149.4, 141.8, 135.6, 135.0, 125.6, 60.6, 46.7, 39.5, 33.5, 28.9, 14.2, 9.9; HRMS (API): calcd for C16H19O4+ [M + H]+: 275.1283, found 275.1276.
Acknowledgements
This work has been partially supported by CNRS, Rouen University, INSA Rouen, Labex SynOrg (ANR-11-LABX-0029) and Région Haute-Normandie (CRUNCh network). Région Haute-Normandie and “Ministere de l'Education Nationale et de la Recherche” are acknowledged for a postdoctoral fellowship (PAP) and for a doctoral fellowship (MC). Dr Laurent El Kaïm (ENSTA, CNRS) is acknowledged for helpful discussions.Notes and references
- (a) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1973, 38, 3239 CrossRef CAS. For reviews on the generation of stereogenic quaternary carbon, see: (b) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed; (c) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef; (d) H. Kotsuki and N. Sasakura, in New and Future Developments in Catalysis — Catalysis for Remediation and Environmental Concerns, ed. S. Suib, Elsevier, 2013, p. 563 Search PubMed; (e) For a recent review on organocatalytic construction of chiral cyclohexenone, see: X. Yang, J. Wang and P. Li, Org. Biomol. Chem., 2014, 12, 2499 RSC.
- (a) J. W. J. Kennedy, S. Vietrich, H. Weinmann and D. E. A. Brittain, J. Org. Chem., 2008, 73, 5151 CrossRef CAS PubMed; (b) D. B. Ramachary and M. Kishor, Org. Biomol. Chem., 2008, 6, 4176 RSC; (c) J. Xu, L. Trzoss, W. K. Chang and E. A. Theodorakis, Angew. Chem., Int. Ed., 2011, 50, 3672 CrossRef CAS PubMed.
- For an example of 1,4-radical aryl migration, see: (a) L. El Kaïm, L. Grimaud and E. Vieu, Org. Lett., 2007, 9, 4171 CrossRef PubMed. For an enlightening review on the process, see: (b) A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649 CrossRef CAS.
- (a) Y. Zhang and S. J. Danishefsky, J. Am. Chem. Soc., 2010, 132, 9567 CrossRef CAS PubMed; (b) For an asymmetric synthesis based on the Hajos–Parrish enone, see: P. A. Peixoto, A. Jean, J. Maddaluno and M. De Paolis, Angew. Chem., Int. Ed., 2013, 52, 6971 CrossRef CAS PubMed.
- N. Huwyler and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 13066 CrossRef CAS PubMed.
- (a) G. C. Hirst, T. O. Johnson Jr. and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 2992 CrossRef CAS; (b) For a synthetic approach based on the Hajos–Parrish ketone, see: R. A. Murphy and R. Sarpong, Org. Lett., 2012, 14, 632 CrossRef CAS PubMed.
- G. M. Rubottom, M. A. Vazquez and D. R. Pelegrina, Tetrahedron Lett., 1974, 15, 4319 CrossRef.
- (a) W. von E. Doering and R. M. Haines, J. Am. Chem. Soc., 1954, 76, 482 CrossRef CAS; (b) D. H. R. Barton, S. K. Pradhan, S. Sternhell and J. F. Templeton, J. Chem. Soc., 1961, 255 RSC; (c) H. G. Aurich, Tetrahedron Lett., 1964, 5, 657 CrossRef; (d) R. Hanna and G. Ourisson, Bull. Soc. Chim. Fr., 1967, 3742 CAS; (e) H. H. Wasserman and B. H. Lipshutz, Tetrahedron Lett., 1975, 16, 4611 CrossRef; (f) A. Donetti, O. Boniardi and A. Ezhaya, Synthesis, 1980, 1009 CrossRef CAS; (g) H. H. Wasserman, B. H. Lipshutz, A. W. Tremper and J. S. Wu, J. Org. Chem., 1981, 46, 2991 CrossRef CAS; (h) R. Gigg and R. Conant, J. Chem. Soc., Chem. Commun., 1983, 465 RSC; (i) B. Heckmann, C. Alayrac, C. Mioskowski, S. Chandrasekhar and J. R. Falck, Tetrahedron Lett., 1992, 33, 5205 CrossRef CAS; (j) M. Tona, M. Guardiola, L. Fajarí and A. Messeguer, Tetrahedron, 1995, 51, 10041 CrossRef CAS; (k) T. Watanabe and T. Ishikawa, Tetrahedron Lett., 1999, 30, 7795 CrossRef; (l) M. Bois-Choussy, M. De Paolis and J. Zhu, Tetrahedron Lett., 2001, 42, 3427 CrossRef CAS; (m) J. Capra and T. Le Gall, Synlett, 2010, 441 CAS; (n) A. D. Santos, L. El Kaïm and L. Grimaud, Org. Biomol. Chem., 2013, 11, 3287 Search PubMed; (o) Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 548 CrossRef CAS PubMed; (p) For a recent review on nitroxide-promoted aerobic oxidation see: S. Wertz and A. Studer, Green Chem., 2013, 15, 3116 RSC.
- (a) R. Howe and F. J. McQuillin, J. Chem. Soc., 1958, 1513 RSC; (b) H. R. Gersmann, H. J. W. Nieuwenhuis and A. F. Bickel, Tetrahedron Lett., 1963, 4, 1383 CrossRef; (c) H. R. Gersmann and A. F. J. Bickel, J. Chem. Soc. B, 1971, 2230 RSC; (d) A. L. García-Cabeza, R. Marín-Barrios, R. Azarken, F. J. Moreno-Dorado, M. J. Ortega, H. Vidal, J. M. Gatica, G. M. Massanet and F. M. Guerra, Eur. J. Org. Chem., 2013, 8307 CrossRef.
- To accelerate the reaction, the use of microwave was investigated with oxygen-saturated toluene as a solvent. Under these conditions, we observed a conversion of 12% at 120 °C (2 h) and 33% conversion at 180 °C (3 h) yielding 10% of 3a. We thank a referee for this suggestion.
- Working in wet PhMe decreased the yield of 3a to 28%. For the nucleophilic reactivity of DBU, see: B. Maji, M. Baidya, J. Ammer, S. Kobayashi, P. Mayer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2013, 3369 CrossRef CAS.
- Permanent bubbling of oxygen during the reaction did not improve the yield of the process which was also erratic. For a compilation on the solubility of oxygen, see: R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163 CrossRef CAS PubMed.
- No side product was observed in the crude of these experiments but the progressive formation of an insoluble gum was observed during the reaction. 1H NMR analysis of this gum did not give any conclusive insights into the degradative pathways.
- (a) N. Kornblum and H. E. DeLamare, J. Am. Chem. Soc., 1951, 73, 880 CrossRef CAS; (b) M. Zhang, N. Liu and W. Tang, J. Am. Chem. Soc., 2013, 135, 12434 CrossRef CAS PubMed; (c) I. T. Chen, I. Baitinger, L. Schreyer and D. Trauner, Org. Lett., 2014, 16, 166 CrossRef CAS PubMed.
- For a recent example of cyclization by Friedel–Crafts conjugate addition with AlCl3, see: S. BouzBouz and M. Sanselme, Tetrahedron Lett., 2009, 50, 5884 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria. |
| ‡ Electronic supplementary information (ESI) available: Procedures for the preparations of 1a–i and spectra of all compounds. See DOI: 10.1039/c4qo00125g |
| § Current address: Institut Européen de Chimie et Biologie, 2 rue Robert Escarpit, 33607 Pessac Cedex, France. |
| This journal is © the Partner Organisations 2014 |